

Abstract
To verify the hypothesis that cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in neurons of the human spinal cord, we investigated the presence and distribution of CFTR protein and mRNA in different segments of the human spinal cord obtained from autopsies. The techniques employed included reverse transcriptase‐polymerase chain reaction (RT‐PCR) to detect CFTR gene expression, in situ hybridization to detect mRNA distribution, and immunohistochemistry to detect protein distribution. The specificity of these experiments was established with extensive controls. We found widespread and abundant expression of CFTR in neurons of the human spinal cord. CFTR protein and mRNA are localized to the cytoplasm of neurons in all segments of the spinal cord but not to glial fibrillary acidic protein (GFAP)‐positive cells. CFTR is a very important molecule, acting as a chloride channel and regulating many physiological functions, including salt transport, fluid flow, and intracellular ion concentrations. Its mutation causes cystic fibrosis. Our finding of abundant CFTR in the spinal cord suggests that this molecule may be significant in the normal function and pathology of the spinal cord. © 2009 Wiley‐Liss, Inc.
Keywords: in situ hybridization, immunohistochemistry, spinal cord, cystic fibrosis
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP‐dependent chloride channel, and CFTR gene mutation has been shown to cause cystic fibrosis (CF), the leading lethal genetic disease among the Caucasian population (Ismailov et al.,1996; Stutts et al.,1997; Mohler et al.,1999). The primary structure of the CFTR protein consists of two membrane‐spanning domains, two nucleotide‐binding domains, and a regulatory domain (Kerem et al.,1989; Riordan et al.,1989). CFTR is a multifunctional molecule. In addition to being a cAMP‐dependent Cl– channel, CFTR also functions as a cAMP‐dependent water and bicarbonate channel, a membrane recycling regulator, an outwardly rectified Cl– and Na+ channel modulator, and an ATP transporter (Bradbury et al.,1992,1994; Egan et al.,1992; Hasegawa et al.,1992).
CFTR expression was originally identified in epithelial cells of the respiratory, digestive, and urinary systems (Trezise and Buchwald,1991). Initially, CF was thought to be a lung and digestive system disease that affected mainly children and young adults. Progress in understanding the CFTR gene functions and improved disease treatment have extended CF patients' life spans considerably. This has provided greater opportunity to study CF in adults. Consequently, CFTR has also been found to be expressed in the reproductive tract (Rochwerger and Buchwald,1993; Tizzano et al.,1994; Zheng et al.,2004) and immune cells (McDonald et al.,1992). Mulberg and associates (1995,1998) found CFTR expressed in rat brain and human hypothalamus. CF is now understood to be a complex multisystemic disease that affects multiple organs in adults as well as in children.
CF has many clinical manifestations and complications. Symptoms, which may be related to the central nervous system (CNS) in particular, are not well understood. However, at least three independent lines of evidence suggest that there is CNS involvement in CF. 1) More than 50% of lung transplant recipients with CF have CNS complications, including seizures, severe headaches, strokes, or confusional episodes, which is significantly higher than other groups of lung transplant recipients (Goldstein et al.,2000). 2) Expression of CFTR in the hypothalamus is reduced in Alzheimer's disease (Lahousse et al.,2003). 3) Dystrophic axons with detectable amyloid precursor protein have been observed in the brain of CF patients (Cochran et al.,1991). These findings strongly suggest CNS involvement in CF. In this study, we present evidence to demonstrate that CFTR is expressed in most, or perhaps all, neurons of the spinal cord.
MATERIALS AND METHODS
Tissue Collection and Preparation
The spinal cord tissues from six adult human autopsies with no disease involving the CNS were used. The subjects were aged from 24 to 51 years old, with three males and three females. The bodies were kept at 4°C, and the autopsies were performed within 48 hr after death. The segments investigated in this study included cervical (C), thoracic (T), lumbar (L), and sacral (S) spinal cord. For reverse transcription‐polymerase chain reaction (RT‐PCR), fresh spinal cord tissue samples obtained at autopsy were immediately snap‐frozen in liquid nitrogen and stored at −80°C until use. The small intestine, which was reported to contain abundant CFTR in cell membrane of the mucosa cells (Strong et al.,1994), was taken as a positive control from the same cases and treated with the same protocol as for the spinal cord tissue. For immunohistochemistry and in situ hybridization, specimens were fixed in 4% formalin solution for 24 hr and then embedded in paraffin blocks. The pancreas tissue, in which CFTR expression was confirmed, was taken as a positive control for immunohistochemistry.
RNA Extraction and Quantitative RT‐PCR
To evaluate CFTR mRNA expression in the spinal cord and demonstrate its expression at the mRNA level, we performed real‐time RT‐PCR (He et al.,2008). To increase the number of neurons in the samples, we separated the neuron‐rich gray matter of the spinal cord from the white matter. Trizol reagent (Gibco Invitrogen, Carlsbad, CA) was used for extraction of total RNA from the spinal cord according to the manufacturer's protocol. The RNA concentration was calculated according to OD260 and OD280 measured with a spectrophotometer (Unico 2800H, Shanghai, China).
The reverse transcription reactions were performed with a cDNA Synthesis Kit (K1621; Fermentas, Vilnius, Lithuania) according to manufacturer's protocol. Briefly, 5 μg of total RNA extracted from the tissue was reverse transcribed using random primers. Reverse transcription reactions were performed at 42°C for 60 min.
The CFTR mRNA sequence was obtained from Genbank (NM_000492), and a pair of primers was designed to amplify the specific fragment of CFTR mRNA. The total CFTR mRNA contains 6,132 nucleotides, and the PCR product position is from 473 to 800 nucleotides. The products span two introns of the CFTR gene.
The quantitative PCR (He et al.,2008) was performed with a real‐time PCR Kit (RealMasterMix‐SYBR Green; Tiangen, Beijing, China) following the manufacturer's protocol. Briefly, the SYBR green dye was added into the mix (Mg2+ and Taq DNA polymerase already added), followed by the cDNA template (2 μl RT product for 20 μl reaction volume), and sense and antisense primers (6 μmol for both primers for 20 μl reaction volume). The PCR was carried out by using the DNA Engine Opticon Continuous Fluorescence Detection System (MJ Research, Waltham, MA). The reaction system was optimized and checked by performing standard curves running on serial dilutions of cDNA (R2 > 0.98). The PCRs were optimized also by comparing Cts among duplicate samples (difference of Cts <0.5). The primers for CFTR PCR were as follows: sense primer 5′‐AGGAGGAACGCTCTATCG‐3′, antisense primer 5′‐GCAGACGCCTGTAACAAC‐3′. The PCR product of CFTR should be 328 base pairs. The GAPDH gene was taken as an endogenous control to evaluate the CFTR gene expression levels. The primers for GAPDH were as follows: sense primer 5′‐ACCACAGTCCATGCCATCAC‐3′, antisense primer 5′‐TCCACCACCCTGTTGCTGTA‐3′. The PCR product of GAPDH should be 452 base pairs. Different primer concentrations in the suggested range by the Taq DNA polymerase manufacturer's protocol were tested. The optimal concentrations of primers were used for the quantitative real‐time PCR analysis. The amplification of PCR began with a hot‐start reaction for 10 min at 94°C and then proceeded for 40 cycles at 94°C for 30 sec, 54°C for 30 sec, and 72°C for 30 sec, followed by fluorescent measurements in each cycle. The final cycle was followed by a 72°C extension for 5 min. A melt curve analysis step was done following PCR. The PCRs for CFTR and GAPDH gene were performed with the same conditions. To verify the PCR product and exclude nonspecific amplification, melt curve analysis and agarose gel electrophoresis were performed. A 100‐bp DNA ladder, Marker 1 (MD101; Tiangen), was used as a DNA weigh standard in the agarose gel electrophoresis. To assess whether the difference in mRNA level in the different tissues was statistically significant, one‐way ANOVA was performed.
Plasmid Construction and cRNA Probe Preparation
To analyze the sequence of the PCR product, PCR product was checked for size by agarose gel electrophoresis. Then, the DNA extracted from the gel was subcloned into the pGM‐T (Tiangen) vector, which contained SalI and NcoI restriction sites. The plasmid was then transfected into XLl‐blue Escherichia coli for amplification. Both X‐gal/IPTG and ampicillin selections were performed. The amplified products were sequenced and analyzed with the Basic Local Alignment Search Tool (BLAST) in NCBI to confirm the sequence.
To perform in vitro transcription for cRNA probe synthesis, the plasmid was linearized with SalI restriction enzyme for sense probe generation and NcoI restriction enzyme for antisense probe generation. The antisense probe was generated with SP6 RNA polymerase. The probe was labeled with digoxigenin RNA labeling mixture (Roche Molecular Biochemicals, Mannheim, Germany). A digoxigenin‐labeled cRNA probe specific for SARS virus was used as a negative control, and the specification and construction of this probe have been described in a previous publication (Ye et al.,2007).
Immunohistochemistry
Immunohistochemistry was carried out to localize CFTR protein in human spinal cord. Seventy percent ammonia‐ethanol was used when formalin deposition was found in the tissue. Antigen retrieval was performed by heating the slides at 96°C in sodium citrate buffer (pH 6.0) for 15 min (Kamo et al.,2007). After cooling to room temperature, slides were rinsed in 0.01 M phosphate‐buffered saline (PBS), treated with 0.1% Triton X‐100 PBS for 30 min, and washed with 0.01 M PBS three times for 3 min each. Endogenous peroxidase activity was quenched using 3% hydrogen peroxide incubation for 30 min. Sections were then washed with 0.01 M PBS three times for 3 min each. Specific antibodies to NF (neurofilament, monoclonal; Zymed Laboratories, South San Francisco, CA; dilution 1:100), GFAP (glial fibrillary acidicprotein, monoclonal; Zymed Laboratories; dilution 1:100) and CFTR (M3A7 monoclonal; Neomarker Labvision, Fremont, CA; dilution 1:100; and polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA; dilution 1:200) were used to identify neurons, glial cells, and CFTR reactivities, respectively.
Optimal contrast between the specific labeling and the background for each antigen was achieved by using a PV9000 immunohistochemistry kit containing polyperoxidase anti‐mouse/rabbit IgG (Zymed Laboratories) and a SP9003 immunohistochemistry kit containing polyperoxidase anti‐goat IgG. After incubation with secondary antibody for 30 min at room temperature, 3,3′‐diaminobenzidine (DAB; Zymed Laboratories) or 3‐amino‐9‐ethyl‐carbazole (AEC; Zymed Laboratories) was used to visualize antigen localization. The slides were then counterstained with hematoxylin.
Two negative controls and a positive control were performed. In one negative control, the primary antibody was replaced with buffer, and, in another, the antibody was first neutralized in a test tube with pure CFTR antigen (catalog No. sc‐8909p; Santa Cruz Biotechnology) as a blocking peptide prior to immunostaining. The concentrations of the antibodies and blocking peptide were 1 μg/ml and 5 μg/ml, respectively. Immunostaining for CFTR in human small intestine was used as a positive control, because this tissue has been reported to contain CFTR in mucosa cells (Strong et al.,1994).
In Situ Hybridization
In situ hybridization was performed to localize CFTR mRNA in human spinal cord, on the same set of tissue samples as used for immunohistochemistry. In brief, before hybridization incubation, all solutions were prepared with diethyl pyrocarbonate‐treated water. After deparaffinization and rehydration, tissue sections were heat treated by microwave (Hunter et al.,1999) and were then incubated with a hybridization cocktail containing CFTR probe at 45°C for 18hr (Menard et al.,2007). The slides were washed three times, first with 2× standard saline citrate (SSC) containing 50% formamid, second with 2× SSC, and third with 0.1× SSC at 37°C for 30 min each. After blocking with normal horse serum (1:100), sections were incubated with alkaline phosphatase‐labeled antidigoxigenin antibody (1:500; Roche Diagnostics, Pensburg, Germany) for 1 hr, and the reaction products were colorized with nitro blue tetrazolium/5‐bromo‐4‐choloro‐3‐indolyl phosphate (NBT/BCIP; Promega Corp. Madison, WI), resulting in a purple‐blue signal. Some slides were counterstained with methyl green. Signal specificity was assessed by substituting the probe with an unrelated probe specific for SARS virus with similar nucleotide content and length.
Double Labeling and Staining on Consecutive Tissue Sections
To identify the cell type expressing CFTR by in situ hybridization, labeling combining in situ hybridization with immunohistochemistry was performed. After the colorizing reaction step of in situ hybridization, the slides were heated at 96°C in sodium citrate buffer (pH 6.0) for 10 min to remove the immune complexes and prevent the possible interference of the immune complex with immunostaining of the second antigen (Lan et al.,1995; Kamo et al.,2007). New primary antibodies for cell markers were then added to the same slide and incubated. The individual protocols used were those described above.
To identify the cell type that expresses CFTR, immunostainings of NF and CFTR were performed on consecutive tissue sections. The immune staining results on adjacent sections were compared to establish whether the CFTR‐containing cells were neurons, which should be NF positive.
RESULTS
CFTR mRNA Expression in Human Spinal Cord Detected by Quantitative PCR
CFRT mRNA expression in the spinal cord was detected by quantitative PCR. The melt curve analysis was done, and only single peaks were found in the melt curves in CFTR or GAPDH PCRs. Figure 1A shows the positive results in various segments of human spinal cord. In total RNA extracts, a PCR product of 328 base pairs was identified and was in 100% agreement with CFTR mRNA. No significant difference (P < 0.1) was observed among the different segments of each case or among the different cases. However, the CFTR gene expression in spinal cord was 60–70 times lower than that in the small intestine (Fig. 1A). Figure 1B shows the agarose gel electrophoresis of the PCR products. The negative control yielded no amplification. The PCR results established that C, T, L, and S segments of the spinal cord contained CFTR mRNA.
Figure 1.
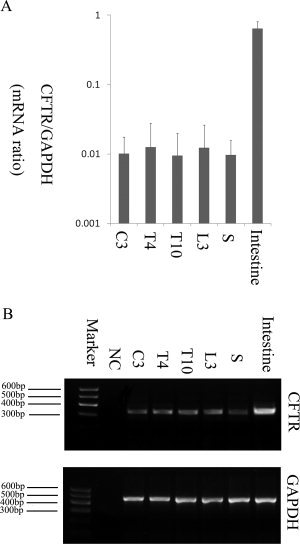
Quantitative PCR analysis of CFTR mRNA expression in the human spinal cord. A: Quantitative PCR results for CFTR mRNA expression level in different spinal cord segments. The dispersion bars are SEM, and six determinations were made. The CFTR mRNA levels in the spinal cord are much lower than that those in the small intestine. B: Predicted PCR product of about 328 base pairs was observed in the agarose gel electrophoresis. Product sequencing showed 100% identity with CFTR mRNA. This product was also observed when RT‐PCR was performed on RNA extracted from human small intestine that was used as a positive control. As a negative control, DEPC‐treated water instead of RNA was used, which gave no band at the expected position. NC, negative control; C3, cervical segment 3; T4, thoracic segment 4; T10 thoracic segment 10; L3, lumbar segment 3; S, sacral segment.
Detection and Localization of CFTR by Immunohistochemistry
CFTR immunoreactivity was detected in human spinal cord with both monoclonal and polyclonal antibodies with immunohistochemistry. The cell type that contained CFTR positivity was confirmed to be neurons by positive NF immunostaining in the same cells on consecutive tissue sections (see Fig. 3D,E). Most, or almost all, neurons showed CFTR‐positive signals in the human spinal cord (Fig. 2). These signals were cytoplasmic and resided in soma, axon, and dendrites (Fig. 3A,B). Signal strength varied among neurons, and no positive CFTR signal was found in smaller cells that were GFAP positive, a marker for glial cells. No obvious differences in distribution and staining abundance were observed among the six different human cases analyzed. No specific signal was observed when antigen neutralization was performed or when the primary antibodies were replaced with buffer, thereby establishing the specificity of the technique (Fig. 3F,G).
Figure 3.
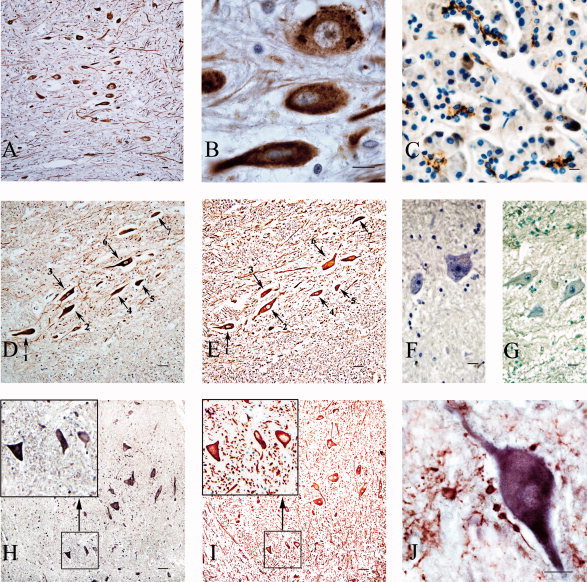
CFTR expressions in neurons of the human spinal cord demonstrated by immunohistochemistry and in situ hybridization. A: Immunohistochemistry results for CFTR in human cervical spinal cord. The brown positive signals indicating CFTR protein presence were developed with DAB. Most neurons (based on visual impression), including motor neurons, show positive signals. B: High‐power picture of a part of the field in A. A and B show the signals in the cytoplasm of neuron bodies, dendrites, and axons. C: Immunohistochemistry results for CFTR in human pancreas as a positive control. The brown positive signals indicating CFTR exist at the glandular lumen of the pancreas exocrine gland. D,E: Immunohistochemistry for CFTR and NF on serial sections taken from a cervical cord segment. The area shown is from the anterior horn. The brown CFTR‐positive signals are localized to the cytoplasm of the neurons. The red positive signals were developed with AEC. The same neurons shown in two consecutive sections is indicated with numbered arrows. CFTR‐positive cells also show NF‐positive signals, indicating that these CFTR‐positive cells are neurons. H,I: Results of in situ hybridization of CFTR and immunohistochemistry of NF on serial sections. H shows the results of in situ hybridization of CFTR mRNA. The purple signal was colorized with alkaline phosphatase and demonstrates that CFTR mRNA is present in the cytoplasm of positive cells. I shows that these positive cells are NF‐positive neurons. The left top corners of each figure show high‐power pictures of the boxed areas. The same three cells show both positive in situ hybridization signals of CFTR and positive immunohistochemistry signals of NF. F: Negative control for immunohistochemistry of CFTR. G: Negative control for CFTR mRNA in situ hybridization. J: Double‐labeling results combining in situ hybridization of CFTR mRNA (purple blue) and immunohistochemistry of GFAP (red), a marker for glial cells. These results demonstrate that CFTR mRNA is present in neurons but not in the GFAP‐positive cells. Scale bars = 20 μm.
Figure 2.
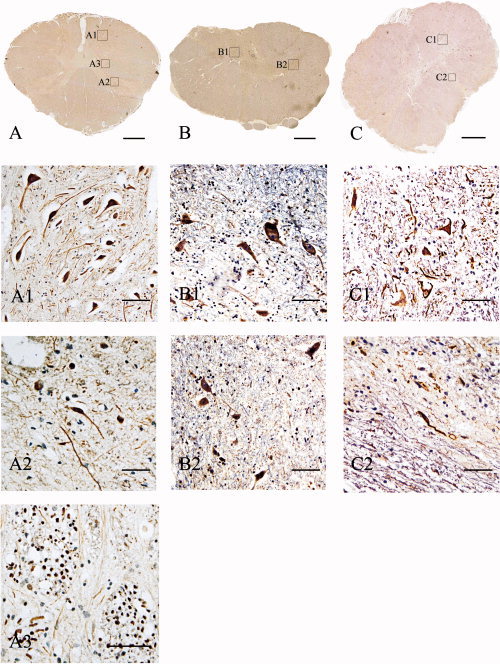
CFTR immunohistochemistry detection in different segments of the human spinal cord. A–C: Immunohistochemistry of the transverse section of the C3, T6, and L1 segments, respectively. The positive cells were not visible at this magnification. Enlarged images of regions in A–C defined by labeled squares are shown in A1–3, B1,2, and C1,2, respectively. A1: Enlarged image of the region labeled in A, showing CFTR immunoreactivity in neurons of the anterior horn of the C3 segment of the spinal cord. A2: Neurons of the posterior horn also contained CFTR positivity. A3: Transverse section of nerve bundles in the spinal cord. The nerve fibers contain CFTR‐positive signals. B: Immunohistochemistry results of the transverse section of T6 segment of the human spinal cord. B1 is a high‐power view of a region labeled in the panel demonstrating CFTR immunoreactivity in neurons in the anterior horn of the T6 segment of human spinal cord. B2: Neurons in the posterior horn of T6 segment are also CFTR positive. C: Transverse section of L1 segment of the human spinal cord. Higher power images of defined regions are shown in C1 (the anterior horn) and C2 (the posterior horn). Scale bars = 0.5 cm in A–C; 20 μm in A1–C2.
Detection and Localization of CFTR mRNA by In Situ Hybridization
To detect and localize CFTR gene expression at the mRNA level, in situ hybridization was carried out. Positive CFTR mRNA in situ hybridization signals were found in the neuronal cytoplasm of most (or all based on visual impression) neuronal cells (Fig. 3H). The distribution pattern of mRNA signal was identical to that found in CFTR protein immunohistochemistry (Fig. 3H,I). Hybridization with a size‐ and GC‐content‐matched unrelated probe as a negative control gave no positive signal (Fig. 3G). The in situ hybridization results demonstrated CFTR mRNA transcription in neurons of the spinal cord.
Labeling in Consecutive Tissue Sections and Double Labeling Combining In Situ Hybridization and Immunohistochemistry
Both consecutive tissue sections and double‐labeling techniques demonstrated that the CFTR‐positive cells are neurons but not glial cells. Immunostaining on consecutive tissue sections for CFTR and NF showed that both antigens were localized to the same cells. Both immunostaining of CFTR protein and in situ hybridization of CFTR mRNA, combined with immunostaining of GFAP, clearly demonstrated that CFTR protein and mRNA were present in a population of cells distinctly different from the GFAP‐positive cells (Fig. 3J).
DISCUSSION
In this study, we have shown that CFTR is present in human spinal cord. RT‐PCR, immunohistochemistry, and in situ hybridization cogently gave the same result. Sequencing of PCR product further confirmed that the amplified product was indeed CFTR, providing evidence that CFTR is expressed in human spinal cord tissues. The CFTR protein and mRNA signals were localized to the cytoplasm of positive cells in the C, T, L, and S spinal cord. These positive cells were confirmed to be neurons but not glial cells by double labeling with neuronal or glial cell markers and on consecutive sections.
Originally, CFTR was thought to be present only in epithelial cells (Kerem et al.,1989; Riordan et al.,1989), and the function of CFTR in these cells has been extensively studied (Hasegawa et al.,1992; Bradbury,1999; Mehta,2005). Recently, expression of CFTR in nonepithelial cells was reported. These nonepithelial cells include the macrophages, lymphocytes, neutrophils, smooth muscle cells, and neurons (Fonknechten et al.,1992; Johannesson et al.,1997; Di et al.,2006; Painter et al.,2006; Vandebrouck et al.,2006). Mulberg and associates (1994) reported expression of CFTR in neurons of the rat brain. They also reported that CFTR was associated with clathrin‐coated vesicles (CCV) in neurons of the hypothalmus of the bovine brain. Bradbury and associates reported CFTR protein within subcellular compartments of the T84 cell line, possibly within synaptic or clathrin‐coated vesicles, which might be involved in neuropeptide transport or other molecular trafficking (Bradbury et al.,1994; Mulberg et al.,1994). However, in the human nervous system, CFTR has previously been reported only in the anterior hypothalamus (Mulberg et al.,1995,1998; Weyler et al.,1999) but not in the spinal cord. Here we found the expression and localization of CFTR in human spinal cord at both the mRNA and the protein levels. This is the first report that CFTR is extensively present in neurons of the human spinal cord.
The significance of CFTR expression in neurons throughout the human spinal cord is twofold. First, it suggests that this molecule may play a role in normal function of the human spinal cord, particularly in view of its previously identified cellular functions and physiology. For example, as a phosphorylation‐dependent epithelial chloride channel, CFTR is expressed in the plasma membrane and membrane of organelles (Denning et al.,1992; Mohamed et al.,1997; Benharouga et al.,2003), and plays a key role in regulating the steady state of cellular electrolytes via regulation of salt transport, fluid flow, and ion concentrations (Egan et al.,1992; Sheppard and Welsh,1999). It has been proposed that CFTR could regulate the activity of membrane proteins through direct protein–protein interactions (Stutts et al.,1995) or by modulating membrane trafficking (Prince et al.,1994). CFTR can also mediate ATP hydrolysis, thereby regulating the flux of glutathione (GSH), which in turn serves in the regulation of protein/enzyme function (Kogan et al.,2003). It also functions as a neuromodulator and neurotransmitter (Janaky et al.,1999). It has been reported that CFTR is critical for cAMP‐dependent regulation of membrane recycling (Bradbury et al.,1992). The interactions among three important molecules, CFTR, adenosine triphosphate, and glutathione, may profoundly affect neuronal function (Aoyama et al.,2008; Yue et al.,2008). After detection of CFTR in the hypothalamus, it was proposed that CFTR might be involved in regulating energy utilization, sexual maturation, and reproductive behavior (Mulberg et al.,1995,1998; Weyler et al.,1999). The results of these studies provide a link between CFTR and neuronal functions of the central nervious system.
Second, mutation or alteration of this molecule may induce CNS dysfunction in CF or other neural diseases. More than 1,500 mutations of CFTR gene have been identified in CF (Radpour et al.,2008). Different mutations can result in different disorders, including the failure of CFTR trafficking to the cell membrane, reduced channel activity, and abnormal splicing of the CFTR mRNA (McAuley and Elborn,2000). When these mutations of the CFTR gene occur as in CF, the functions of CFTR in neurons may be altered, thus affecting the normal CNS function. Dystrophic axons in the nucleus gracilis and demyelination of the fasciculus gracilis have been reported in CF patients, with 66% developing dystrophic axons (Cavalier and Gambetti,1981). Subclinical extrapyramidal hemosiderosis has also been reported in CF (Wongmongkolrit et al.,1985). Such findings cannot be explained by the pathological changes in the lungs or other organs typically associated with CF. Mutant CFTR may accumulate in the cytoplasm that could block the molecular transmission in the cell (Gentzsch et al.,2007). Accumulation and failure in the transmission of molecules could cause cell dystrophy (Gentzsch et al.,2007). Furthermore, Cl– homeostasis determines the efficacy of synaptic inhibition in the motoneurons (Ueno et al.,2002). CFTR, as a Cl– channel, has been suggested to participate in the control of Cl– homeostasis and neuronal excitability in rat spinal cord (Ostroumov et al.,2007). Our results of expression of CFTR in human spinal cord provide a basis for the relationship between CFTR mutation and the neuronal symptoms seen in CF.
In summary, our study provides evidence for expression CFTR in neurons of the human spinal cord. The potential physiological and clinical significance of this discovery demands further attention.
Acknowledgements
We are grateful to Ruishu Deng, Zhuo Li, Yan Dong, Yan Zhou, Peng Ding, Yingying Zhao, Baokai Yang, Qi Cao, and Lu Yao for their helpful discussions and technical assistance.
REFERENCES
- Aoyama K,Watabe M,Nakaki T. 2008. Regulation of neuronal glutathione synthesis. J Pharmacol Sci 108: 227–238. [DOI] [PubMed] [Google Scholar]
- Benharouga M,Sharma M,So J,Haardt M,Drzymala L,Popov M,Schwapach B,Grinstein S,Du K,Lukacs GL. 2003. The role of the C terminus and Na+/H+ exchanger regulatory factor in the functional expression of cystic fibrosis transmembrane conductance regulator in nonpolarized cells and epithelia. J Biol Chem 278: 22079–22089. [DOI] [PubMed] [Google Scholar]
- Bradbury NA. 1999. Intracellular CFTR: localization and function. Physiol Rev 79( Suppl 1): S175–S191. [DOI] [PubMed] [Google Scholar]
- Bradbury NA,Jilling T,Berta G,Sorscher EJ,Bridges RJ,Kirk KL. 1992. Regulation of plasma membrane recycling by CFTR. Science 256: 530–532. [DOI] [PubMed] [Google Scholar]
- Bradbury NA,Cohn JA,Venglarik CJ,Bridges RJ. 1994. Biochemical and biophysical identification of cystic fibrosis transmembrane conductance regulator chloride channels as components of endocytic clathrin‐coated vesicles. J Biol Chem 269: 8296–8302. [PubMed] [Google Scholar]
- Cavalier SJ,Gambetti P. 1981. Dystrophic axons and spinal cord demyelination in cystic fibrosis. Neurology 31: 714–718. [DOI] [PubMed] [Google Scholar]
- Cochran E,Bacci B,Chen Y,Patton A,Gambetti P,Autilio‐Gambetti L. 1991. Amyloid precursor protein and ubiquitin immunoreactivity in dystrophic axons is not unique to Alzheimer's disease. Am J Pathol 139: 485–489. [PMC free article] [PubMed] [Google Scholar]
- Denning GM,Ostedgaard LS,Cheng SH,Smith AE,Welsh MJ. 1992. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J Clin Invest 89: 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di A,Brown ME,Deriy LV,Li C,Szeto FL,Chen Y,Huang P,Tong J,Naren AP,Bindokas V,Palfrey HC,Nelson DJ. 2006. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8: 933–944. [DOI] [PubMed] [Google Scholar]
- Egan M,Flotte T,Afione S,Solow R,Zeitlin PL,Carter BJ,Guggino WB. 1992. Defective regulation of outwardly rectifying Cl– channels by protein kinase A corrected by insertion of CFTR. Nature 358: 581–584. [DOI] [PubMed] [Google Scholar]
- Fonknechten N,Chelly J,Lepercq J,Kahn A,Kaplan JC,Kitzis A,Chomel JC. 1992. CFTR illegitimate transcription in lymphoid cells: quantification and applications to the investigation of pathological transcripts. Hum Genet 88: 508–512. [DOI] [PubMed] [Google Scholar]
- Gentzsch M,Choudhury A,Chang XB,Pagano RE,Riordan JR. 2007. Misassembled mutant DeltaF508 CFTR in the distal secretory pathway alters cellular lipid trafficking. J Cell Sci 120: 447–455. [DOI] [PubMed] [Google Scholar]
- Goldstein AB,Goldstein LS,Perl MK,Haug MT,Arroliga AC,Stillwell PC. 2000. Cystic fibrosis patients with and without central nervous system complications following lung transplantation. Pediatr Pulmonol 30: 203–206. [DOI] [PubMed] [Google Scholar]
- Hasegawa H,Skach W,Baker O,Calayag MC,Lingappa V,Verkman AS. 1992. A multifunctional aqueous channel formed by CFTR. Science 258: 1477–1479. [DOI] [PubMed] [Google Scholar]
- He HC,Bi XC,Zheng ZW,Dai QS,Han ZD,Liang YX,Ye YK,Zeng GH,Zhu G,Zhong WD. 2008. Real‐time quantitative RT‐PCR assessment of PIM‐1 and hK2 mRNA expression in benign prostate hyperplasia and prostate cancer. Med Oncol (in press). [DOI] [PubMed] [Google Scholar]
- Hunter JM,Sperry EE,Ravilly S,Colin AA. 1999. Thoracoabdominal asynchrony and ratio of time to peak tidal expiratory flow over total expiratory time in adolescents with cystic fibrosis. Pediatr Pulmonol 28: 199–204. [DOI] [PubMed] [Google Scholar]
- Ismailov II,Awayda MS,Jovov B,Berdiev BK,Fuller CM,Dedman JR,Kaetzel M,Benos DJ. 1996. Regulation of epithelial sodium channels by the cystic fibrosis transmembrane conductance regulator. J Biol Chem 271: 4725–4732. [DOI] [PubMed] [Google Scholar]
- Janaky R,Ogita K,Pasqualotto BA,Bains JS,Oja SS,Yoneda Y,Shaw CA. 1999. Glutathione and signal transduction in the mammalian CNS. J Neurochem 73: 889–902. [DOI] [PubMed] [Google Scholar]
- Johannesson M,Bogdanovic N,Nordqvist AC,Hjelte L,Schalling M. 1997. Cystic fibrosis mRNA expression in rat brain: cerebral cortex and medial preoptic area. Neuroreport 8: 535–539. [DOI] [PubMed] [Google Scholar]
- Kamo N,Yasuchika K,Fujii H,Hoppo T,Machimoto T,Ishii T,Fujita N,Tsuruo T,Yamashita JK,Kubo H,Ikai I. 2007. Two populations of Thy1‐positive mesenchymal cells regulate in vitro maturation of hepatic progenitor cells. Am J Physiol Gastrointest Liver Physiol 292: G526–534. [DOI] [PubMed] [Google Scholar]
- Kerem B,Rommens JM,Buchanan JA,Markiewicz D,Cox TK,Chakravarti A,Buchwald M,Tsui LC. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245: 1073–1080. [DOI] [PubMed] [Google Scholar]
- Kogan I,Ramjeesingh M,Li C,Kidd JF,Wang Y,Leslie EM,Cole SP,Bear CE. 2003. CFTR directly mediates nucleotide‐regulated glutathione flux. EMBO J 22: 1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahousse SA,Stopa EG,Mulberg AE,de la Monte SM. 2003. Reduced expression of the cystic fibrosis transmembrane conductance regulator gene in the hypothalamus of patients with Alzheimer's disease. J Alzheimers Dis 5: 455–462. [DOI] [PubMed] [Google Scholar]
- Lan HY,Mu W,Nikolic‐Paterson DJ,Atkins RC. 1995. A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J Histochem Cytochem 43: 97–102. [DOI] [PubMed] [Google Scholar]
- McAuley DF,Elborn JS. 2000. Cystic fibrosis: basic science. Paediatr Respir Rev 1: 93–100. [DOI] [PubMed] [Google Scholar]
- McDonald TV,Nghiem PT,Gardner P,Martens CL. 1992. Human lymphocytes transcribe the cystic fibrosis transmembrane conductance regulator gene and exhibit CF‐defective cAMP‐regulated chloride current. J Biol Chem 267: 3242–3248. [PubMed] [Google Scholar]
- Mehta A. 2005. CFTR: more than just a chloride channel. Pediatr Pulmonol 39: 292–298. [DOI] [PubMed] [Google Scholar]
- Menard A,de Los Santos PE,Graindorge A,Cournoyer B. 2007. Architecture of Burkholderia cepacia complex sigma70 gene family: evidence of alternative primary and clade‐specific factors, and genomic instability. BMC Genomics 8: 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A,Ferguson D,Seibert FS,Cai HM,Kartner N,Grinstein S,Riordan JR,Lukacs GL. 1997. Functional expression and apical localization of the cystic fibrosis transmembrane conductance regulator in MDCK I cells. Biochem J 322: 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ,Kreda SM,Boucher RC,Sudol M,Stutts MJ,Milgram SL. 1999. Yes‐associated protein 65 localizes p62(c‐Yes) to the apical compartment of airway epithelia by association with EBP50. J Cell Biol 147: 879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulberg AE,Wiedner EB,Bao X,Marshall J,Jefferson DM,Altschuler SM. 1994. Cystic fibrosis transmembrane conductance regulator protein expression in brain. Neuroreport 5: 1684–1688. [DOI] [PubMed] [Google Scholar]
- Mulberg AE,Resta LP,Wiedner EB,Altschuler SM,Jefferson DM,Broussard DL. 1995. Expression and localization of the cystic fibrosis transmembrane conductance regulator mRNA and its protein in rat brain. J Clin Invest 96: 646–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulberg AE,Weyler RT,Altschuler SM,Hyde TM. 1998. Cystic fibrosis transmembrane conductance regulator expression in human hypothalamus. Neuroreport 9: 141–144. [DOI] [PubMed] [Google Scholar]
- Ostroumov K,Grandolfo M,Nistri A. 2007. The effects induced by the sulphonylurea glibenclamide on the neonatal rat spinal cord indicate a novel mechanism to control neuronal excitability and inhibitory neurotransmission. Br J Pharmacol 150: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RG,Valentine VG,Lanson NA Jr,Leidal K,Zhang Q,Lombard G,Thompson C,Viswanathan A,Nauseef WM,Wang G,Wang G. 2006. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 45: 10260–10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince LS,Workman RB Jr,Marchase RB. 1994. Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci U S A 91: 5192–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radpour R,Gourabi H,Dizaj AV,Holzgreve W,Zhong XY. 2008. Genetic investigations of CFTR mutations in congenital absence of vas deferens, uterus, and vagina as a cause of infertility. J Androl 29: 506–513. [DOI] [PubMed] [Google Scholar]
- Riordan JR,Rommens JM,Kerem B,Alon N,Rozmahel R,Grzelczak Z,Zielenski J,Lok S,Plavsic N,Chou JL, et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073. [DOI] [PubMed] [Google Scholar]
- Rochwerger L,Buchwald M. 1993. Stimulation of the cystic fibrosis transmembrane regulator expression by estrogen in vivo. Endocrinology 133: 921–930. [DOI] [PubMed] [Google Scholar]
- Sheppard DN,Welsh MJ. 1999. Structure and function of the CFTR chloride channel. Physiol Rev 79( Suppl 1): S23–S45. [DOI] [PubMed] [Google Scholar]
- Strong TV,Boehm K,Collins FS. 1994. Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J Clin Invest 93: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ,Canessa CM,Olsen JC,Hamrick M,Cohn JA,Rossier BC,Boucher RC. 1995. CFTR as a cAMP‐dependent regulator of sodium channels. Science 269: 847–850. [DOI] [PubMed] [Google Scholar]
- Stutts MJ,Rossier BC,Boucher RC. 1997. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A‐mediated regulation of epithelial sodium channel single channel kinetics. J Biol Chem 272: 14037–14040. [DOI] [PubMed] [Google Scholar]
- Tizzano EF,Silver MM,Chitayat D,Benichou JC,Buchwald M. 1994. Differential cellular expression of cystic fibrosis transmembrane regulator in human reproductive tissues. Clues for the infertility in patients with cystic fibrosis. Am J Pathol 144: 906–914. [PMC free article] [PubMed] [Google Scholar]
- Trezise AE,Buchwald M. 1991. In vivo cell‐specific expression of the cystic fibrosis transmembrane conductance regulator. Nature 353: 434–437. [DOI] [PubMed] [Google Scholar]
- Ueno T,Okabe A,Akaike N,Fukuda A,Nabekura J. 2002. Diversity of neuron‐specific K+‐Cl– cotransporter expression and inhibitory postsynaptic potential depression in rat motoneurons. J Biol Chem 277: 4945–4950. [DOI] [PubMed] [Google Scholar]
- Vandebrouck C,Melin P,Norez C,Robert R,Guibert C,Mettey Y,Becq F. 2006. Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir Res 7: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyler RT,Yurko‐Mauro KA,Rubenstein R,Kollen WJ,Reenstra W,Altschuler SM,Egan M,Mulberg AE. 1999. CFTR is functionally active in GnRH‐expressing GT1‐7 hypothalamic neurons. Am J Physiol 277: C563–C571. [DOI] [PubMed] [Google Scholar]
- Wongmongkolrit T,Wyszynski R,Hershey CO,Varnes AW. 1985. Evidence of subclinical extrapyramidal hemosiderosis in cystic fibrosis. Acta Neuropathol 65: 265–269. [DOI] [PubMed] [Google Scholar]
- Ye J,Zhang B,Xu J,Chang Q,McNutt MA,Korteweg C,Gong E,Gu J. 2007. Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am J Pathol 170: 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue HJ,Conrad D,Dimsdale JE. 2008. Sleep disruption in cystic fibrosis. Med Hypoth 71: 886–888. [DOI] [PubMed] [Google Scholar]
- Zheng XY,Chen GA,Wang HY. 2004. Expression of cystic fibrosis transmembrane conductance regulator in human endometrium. Hum Reprod 19: 2933–2941. [DOI] [PubMed] [Google Scholar]