

Abstract
Objectives
There is considerable evidence that implicates dysregulation of type I interferon signaling (or type I IFN signature) in the pathogenesis of systemic sclerosis (SSc). Interferon regulatory factor 7 (IRF7) has been recognised as a master regulator of type I IFN signalling. The objective of this study was to elucidate the role of IRF7 in dermal fibrosis and SSc pathogenesis.
Methods
SSc and healthy control skin biopsies were investigated to determine IRF7 expression and activation. The role of IRF7 in fibrosis was investigated using IRF7 knockout (KO) mice in the bleomycin-induced and TSK/+mouse models. In vitro experiments with dermal fibroblasts from patients with SSc and healthy controls were performed.
Results
IRF7 expression was significantly upregulated and activated in SSc skin tissue and explanted SSc dermal fibroblasts compared with unaffected, matched controls. Moreover, IRF7 expression was stimulated by IFN-α in dermal fibroblasts. Importantly, IRF7 co-immunoprecipitated with Smad3, a key mediator of transforming growth factor (TGF)-β signalling, and IRF7 knockdown reduced profibrotic factors in SSc fibroblasts. IRF7 KO mice demonstrated attenuated dermal fibrosis and inflammation compared with wild-type mice in response to bleomycin. Specifically, hydroxyproline content, dermal thickness as well as Col1a2, ACTA2and interleukin-6 mRNA levels were significantly attenuated in IRF7 KO mice skin tissue. Furthermore, IRF7 KO in TSK/+mice attenuated hydroxyproline content, subcutaneous hypodermal thickness, Col1a2 mRNA as well as α-smooth muscle actin and fibronectin expression.
Conclusions
IRF7 is upregulated in SSc skin, interacts with Smad3 and potentiates TGF-β-mediated fibrosis, and therefore may represent a promising therapeutic target in SSc.
INTRODUCTION
Systemic sclerosis (scleroderma, SSc) is a multi-system autoimmune disease characterised by vasculopathy, fibrosis and immune system dysregulation. The aetiology of SSc is unknown. There are currently no targeted treatment options for the fibrotic complications of SSc, and disease-related mortality remains high.1 Type I interferons (IFNs) are key regulators of the innate immune system. They modulate immune cell differentiation, proliferation and cytokine production. IFN excess is evident in the blood and skin of a large percentage of patients with SSc.2,3 The development of SSc has been reported in patients undergoing IFN treatment.4 Furthermore, a randomised, placebo-controlled trial of subcutaneous IFN-α injection in patients with early SSc showed that treatment with IFN-α resulted in worsening lung function and less improvement in skin fibrosis scores, suggesting a pathologic effect.5 We first described a prominent transcript pattern of upregulated type I IFN-inducible genes, that is, the IFN signature, in peripheral blood cells from patients with SSc, a finding that has been confirmed by several other groups.6–8 A more recent study showed that type I IFN-producing plasmacytoid dendritic cells (pDC) and toll-like receptor 8 (TLR8) are involved in the pathogenesis of SSc.9 However, the exact roles of type I IFN signalling in SSc pathogenesis, including the mechanisms by which dysregulated type I IFN signalling contribute to fibrosis are unclear.
TGF-β has long been recognised as a key mediator of fibrosis in SSc.10 In a previous study, IFN-α2b increased TGF-β1 production and secretion in hepatocytes, suggesting a positive cross-talk between IFN and TGF-β signalling through smad2/3 activation.11 Among IFN signalling pathways, interferon regulatory factors (IRFs) are best characterised as transcriptional regulators of type I IFNs and IFN-inducible genes and play a pivotal role in the regulation of many facets of the innate and adaptive immune response.12 This family comprises nine members: IRF1, IRF2, IRF3, IRF4 (also known as LSIRF, PIP or ICSAT), IRF5, IRF6, IRF7, IRF8 (also known as ICSBP) and IRF9 (also known as ISGF3γ).13,14 As transcription factors, IRFs recognise the IFN-stimulated response element (ISRE) in the promoter region of target inflammatory genes including IFN-α, IL-6 and TNF-α.15–17 Specifically, IRF7 has been recognised as a master regulator of type I IFN signalling. Once phosphorylated, activated IRF7 moves into the nucleus, where it binds to the ISRE site of target genes and leads to secretion of type I IFN and other cytokines.18 IRF7 also has been identified as a Smad3 interacting protein, a key component of TGF-β signalling for collagen production.19 Furthermore, we and others have reported that single nucleotide polymorphisms in the IRF7 gene, including the functional non-synonymous variant rs1131665, are associated with SSc.20,21 Upregulation of IRF7 gene expression in peripheral blood cells from patients with SSc has been reported.6 More importantly, in our large-scale, unbiased, global skin and blood gene expression studies, we found that IRF7 was the top predicted transcription factor responsible for the dysregulated gene expression observed in patients with SSc.22 Altogether, these data implicate an important role for IRF7 in SSc pathogenesis. However, the specific mechanistic role of IRF7 in the fibrotic complications of SSc is unknown.
In the present study, we investigated the role of IRF7 in SSc skin fibroblasts and in two different murine models, the bleomycin-induced skin fibrosis model and TSK/+congenic skin fibrosis model.
METHODS
Patients with SSc and controls
Patients with SSc and unaffected controls were recruited from the Rheumatology Division at the University of Texas Health Science Center at Houston. Three mm punch biopsies were obtained from the extensor forearm using standard methods. Formalin-fixed paraffin-embedded skin tissue was processed for histology and immunohistochemistry (additional details are included in the online supplementary methods section). Fibroblasts from skin tissue were cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum and four to six passage fibroblasts were used for experiments.23 All patients with SSc fulfilled the 2013 classification criteria for systemic sclerosis.24 Online supplementary tables S1 and S2 show the patient and control characteristics. All study subjects provided written informed consent.
Bleomycin-induced skin fibrosis mouse model
IRF7 knockout (KO) mice congenic on the C57BL/6J background were acquired from Riken Japan (Tokyo, Japan). Female C57BL/6J wild-type mice aged 8 weeks were purchased from Jackson Laboratory (Bar Harbor, Maine, USA). Mice were housed in autoclaved cages and fed sterile food and water. Bleomycin (0.02 units/day) (Teva Parenteral Medicines, Irvine, California, USA) dissolved in phosphate-buffered saline (PBS) or PBS alone was administered by subcutaneous injection to the back skin of mice aged 8 weeks 6 days per week daily for 4 weeks. On day 7 or 28, mice were sacrificed and lesional skin was harvested for hydroxyproline, total RNA, histology and immunohistochemistry.
Statistical analysis
Experimental groups were compared by t-test or Mann-Whitney U test depending on the distribution of residuals. Data are presented as mean±SD or SE of the mean (SEM),25 when t-test was used. Dot plot with median, as well as IQR was shown if Mann-Whitney U test was used. Two-tailed p values <0.05 were considered to be statistically significant.
More information on material and methods is provided in the online supplementary materials.
RESULTS
Upregulation and activation of IRF7 in patients with SSc
Because previous studies demonstrated that type I IFN signalling was the most prominent signature in peripheral blood cells from SSc,7 we hypothesised that IRF7 might be upregulated in skin tissue of patient with SSc. Immunohistochemistry confirmed that IRF7 expression was significantly upregulated in SSc skin tissue compared with healthy controls (n=10 per group) (see online supplementary table S1 for demographic and clinical characteristics). Importantly, increased IRF7 phosphorylation was observed in SSc skin compared with healthy controls, indicating increased IRF7 activation (figure 1A). Next, we determined the IRF7-positive fibroblasts and inflammatory cell numbers by morphology (excluding nerve and endothelial cells) in the dermis. IRF7 and phospho-IRF7-positive cell numbers were increased in patients with SSc compared with healthy controls (figure 1A, lower panel). Furthermore, IRF7 and phospho-IRF7 were highly expressed in α-smooth muscle actin (α-SMA)-positive myofibroblasts and CD68-positive macrophages from SSc skin tissues, as evidenced by double immunofluorescent staining (figure 1B). These results demonstrate that IRF7 is upregulated and activated in macrophages and myofibroblasts in the skin of patients with SSc.
Figure 1.

Upregulation and activation of IRF7 in SSc skin tissue. Five-micron thick skin tissue was stained with anti-IRF7 and p-IRF7 antibodies and immunostaining-positive cells (excluding nerve and endothelial cells) number in the dermis was counted in high power field. (A) Representative images. Dot plots with median (red +) and IQR (red −) shows IRF7-positive and p-IRF7-positive cell number inthe per high power field. Red arrows: IRF7 or p-IRF7 staining positive cells. n=10 per group; healthy control vs SSc; *p<0.01 (analysed by Mann-Whitney U test). (B) Immunofluorescence analysis using anti-IRF7 or p-IRF7 and α-SMA antibodies for IRF7-positive or p-IRF7-positive myofibroblast; anti-IRF7 or p-IRF7 and CD68 antibodies for IRF7-positive or p-IRF7-positive macrophages in the SSc and normal control skin. Representative images. Original magnifications: x400 magnifications. Scale bar: 125 μm. n=10. Arrows: IRF7 or p-IRF7 with α-SMA, or IRF7or p-IRF7 with CD68 double staining-positive cells. IRF7, interferon regulatory factor 7; α-SMA, α-smooth muscle actin; SSc, systemic sclerosis.
IRF7 blockade attenuated fibrotic responses to TGF-β and IFN-α in SSc skin fibroblasts in vitro
Considering the prominent IRF7 protein expression and activation in SSc dermal fibroblasts, and the specific role of fibroblasts in the production of extracellular matrix proteins, we next examined IRF7 expression in cultured fibroblasts from patients with SSc (see online supplementary table S2 for demographic and clinical characteristics). Both IRF7 mRNA and protein levels were significantly upregulated in fibroblasts from patients with SSc compared with age-matched and gender-matched healthy controls by quantitative PCR (qPCR) (figure 2A) and western blot analysis, respectively (figure 2B). To characterise the potential upstream regulators of IRF7 in dermal fibroblasts, cultured fibroblasts were stimulated with IFN-α, as well as nine prominent T helper (Th)1/Th2/Th17 cytokines (IFN-γ, TNF-α, TGF-β1, IL-6, CCL-2, IL-10, IL-13, IL-17A, IL-22), followed by measurement of IRF7 mRNA induction by real-time-qPCR. Of the cytokines tested, IFN-α stimulation resulted in the highest IRF7 expression levels (figure 2C).
Figure 2.
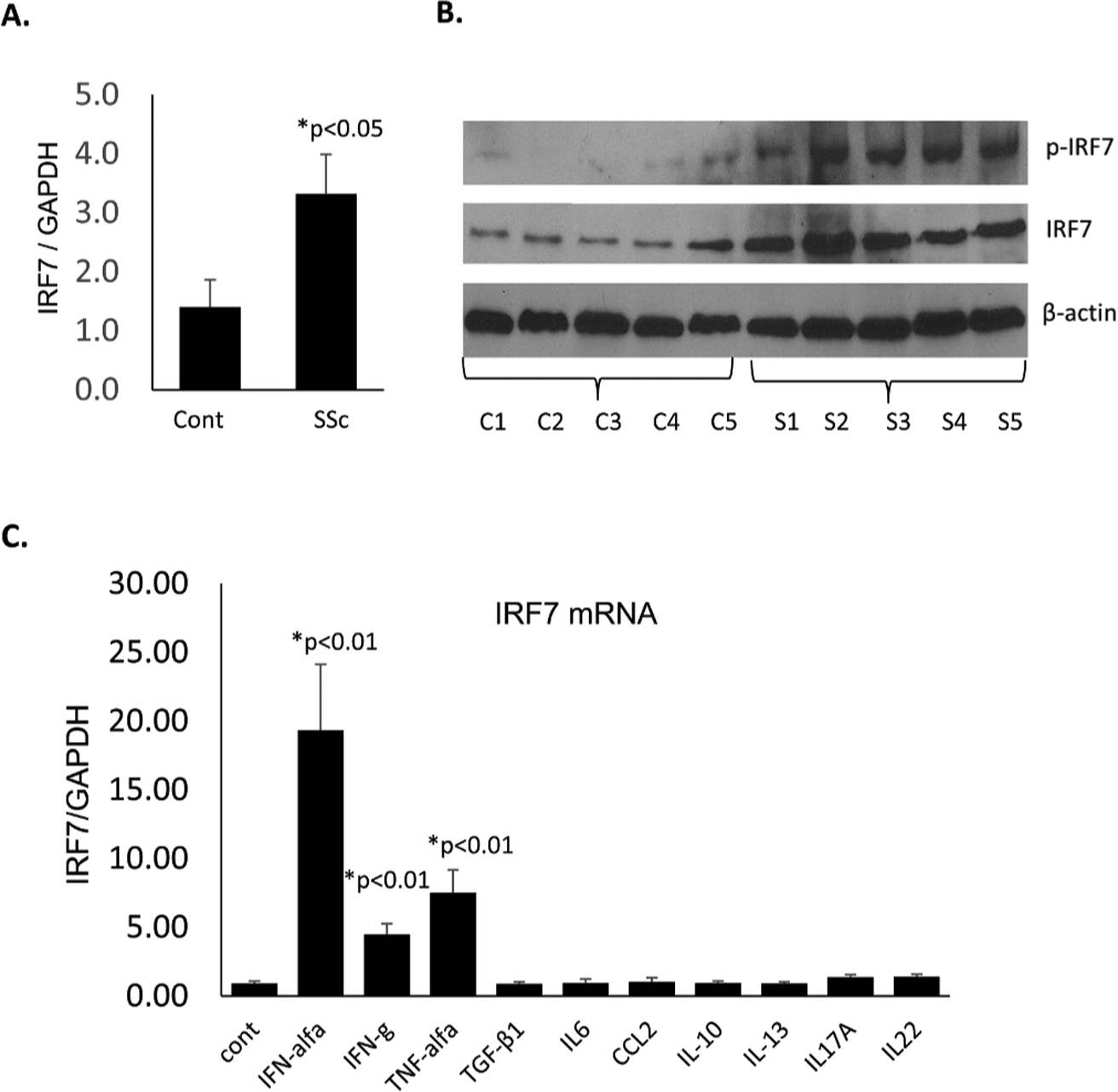
Upregulation and activation of IRF7 in cultured fibroblasts from patients with SSc compared with controls. (A) Early passage (four to six passages) of confluent fibroblasts were harvested and totalRNA was isolated for real-time qPCR. IRF7 expression was normalised against GAPDH expression. The bars represent the mean±SD n=10. *P<0.05 (analysed by t-test). (B) Confluent fibroblasts (four to six passages) were harvested and total protein was isolated for western blot analysis, n=5. (C) Healthy control fibroblasts (four to six passages) were treated with the indicated cytokines for 24 hours. Total RNA was extracted and IRF7 mRNA expression was examined by real-time quantitative PCR. The results were normalised with GAPDH mRNA and IRF7 expression was compared with that in untreated fibroblasts (lane 1); the bars represent the mean±SEM, n=5. *P<0.05 (analysed by t-test). IRF7, interferon regulatory factor 7; SSc, systemic sclerosis.
Next, we assessed whether IRF7 impacts the TGF-β-induced or IFN-α-induced fibrotic responses in cultured fibroblasts. We pretreated fibroblasts with IRF7 siRNA for 24 hours followed by stimulation with TGF-β for 24 hours or IFN-α for 72 hours. The 72 hours stimulation was selected for IFN-α treatment to replicate the effects of long-term exposure to IFN-α as present in SSc. First, IRF7 gene knockdown was confirmed by IRF7 mRNA expression (figure 3A). IRF7 siRNA treatment attenuated TGF-β1-induced Col1a1, Col1a2 and FN1 mRNA upregulation, and type I collagen and fibronectin expression in SSc fibroblasts (figure 3A–C). On the other hand, IRF7 siRNA pretreatment did not attenuate TGF-β1-induced Col1a1, Col1a2 and FN1 mRNA expression and type I collagen expression in healthy control fibroblasts (figure 3B,C and see online supplementary figure S1). Furthermore, IRF7 siRNA pretreatment abrogated Col1a2 and FN1 mRNA upregulation induced by long-term IFN-α treatment in healthy control fibroblasts (figure 3D). Cumulatively, these data indicate that in a high IFN-α milieu (control fibroblasts stimulated with IFN-α or SSc fibroblasts), IRF7 knockdown decreases the fibrotic response to TGF-β in fibroblasts.
Figure 3.
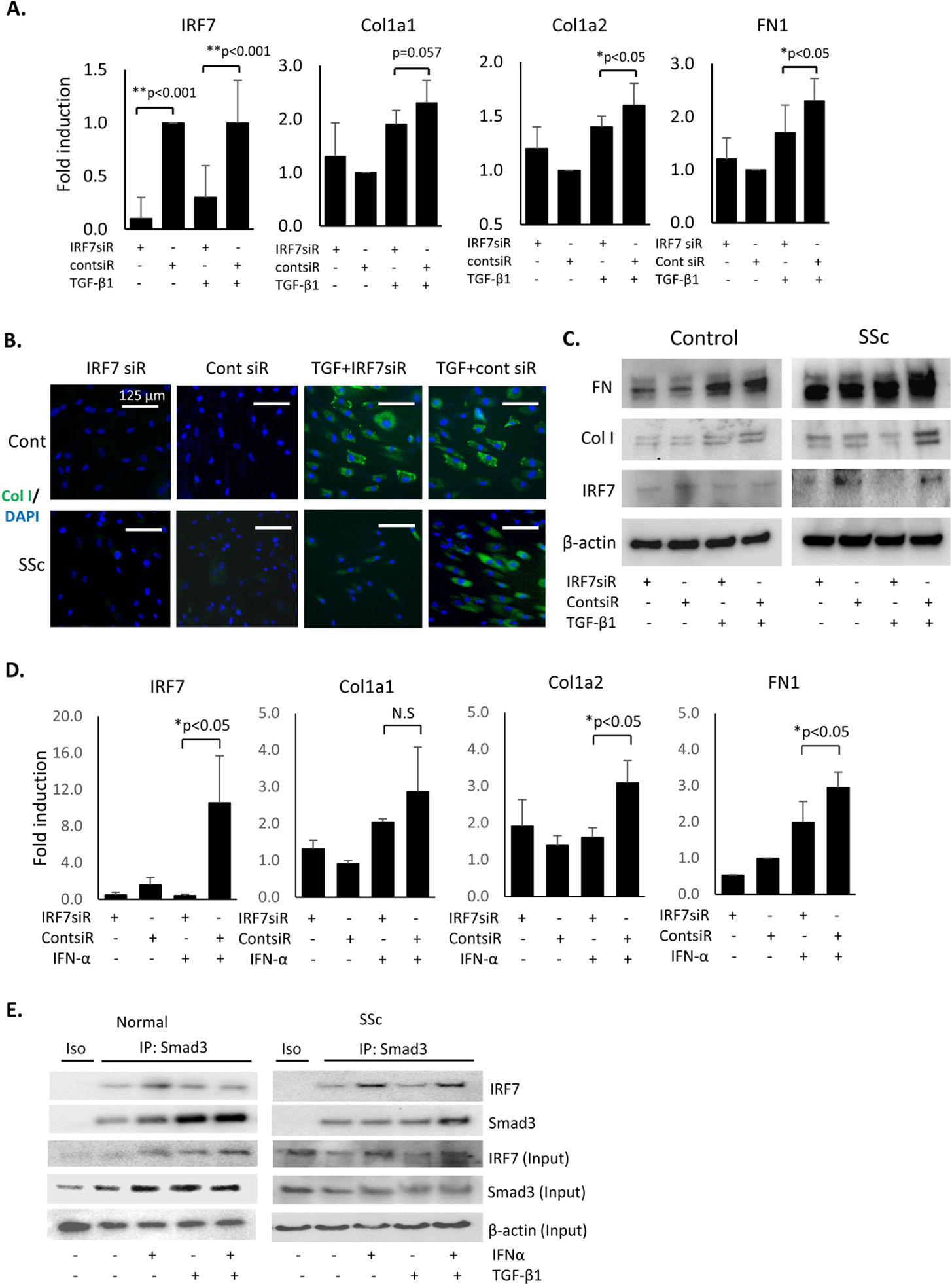
IRF7 siRNA pretreatment blocked Col1a1, Col1a2 and FN1 (encoding fibronectin protein) mRNA expression in SSc dermal fibroblasts. (A) Passages four to six of SSc fibroblasts were pretreated with IRF7 siRNA (20 pmol/mL) for 24 hours followed by treatment with 10 ng/mL of TGF-β1 for 24 hours. Total RNA was extracted and Col1a1, Col1a2, FN1 and IRF7 mRNA expression was examined by real-time quantitative PCR. The results were normalised with GAPDH mRNA; the bars represent the mean±SEM. n=5. **P<0.001; *p<0.05 (analysed by t-test). (B) Immunofluorescence analysis. SSc and healthy control fibroblasts were pretreated with IRF7 or control siRNA (20 pmol/mL) for 24 hours, followed by TGF-β1 treatment (10 ng/mL) for 24 hours, and processed for immunofluorescence staining for type I collagen expression. Representative images. n=5. (C) Passages four tosix of healthy control and SSc fibroblasts were pretreated with IRF7 siRNA for 24 hours followed by treatment with 10 ng/mL of TGF-β1 for 24 hours. Fibroblasts were harvested and total protein was isolated for western blot analysis. (D) Early passage (four to six passages) of confluent healthy control fibroblasts were pretreated with IRF7 or control siRNA for 24 hours, followed by treatment with IFN-α (100 ng/mL) for 72 hours. Total RNA was extracted and Col1a1, Col1a2 FN1 (encoding fibronectin) and IRF7 mRNA expression was examined by real-time quantitative PCR. The results, normalised with GAPDH mRNA, represent the means±SEM. n=5. *P<0.05 (analysed by t-test). (E) Control or SSc fibroblasts were cultured with IFN-α (100 ng/mL), TGF-β1 (10 ng/mL) or IFN-α+TGF-β1 for 24 hours. Total protein was isolated and after immunoprecipitation with Smad3 protein, immunoblotting of IRF7 was performed. Representative images. n=3. IRF7, interferon regulatory factor 7; SSc, systemic sclerosis; TGF, transforming growth factor.
Next, we investigated the interaction of IRF7 with Smad3, a prominent transcription factor in the TGF-β canonical pathway. IRF7 and Smad3 strongly co-localised in SSc fibroblasts, but this strong co-localisation was not observed in unstimulated control fibroblasts, perhaps in part due to lower expression levels of IRF7 and Smad3 (see online supplementary figure S2). Smad3 and IRF7 co-immunoprecipitated in cultured control and SSc fibroblasts, an effect that was enhanced by treatment with IFN-α or the combination of TGF-β plus IFN-α in SSc fibroblasts (figure 3E). Altogether, these results suggest that IRF7 plays a role in potentiating TGF-β-induced fibrosis, possibly through an interaction with Smad3 in fibroblasts.
IRF7 was upregulated in the skin of bleomycin-treated mice and mediated the fibrotic response to bleomycin
To further understand the role of IRF7 in SSc pathogenesis, we used the bleomycin-treated mouse model. C57BL/6 wild-type mice received subcutaneous injections of bleomycin (or PBS as a control) for 6 days a week for 7 or 28 days, then lesional skin tissue was harvested for analysis. IRF7 expression was significantly upregulated in the skin of bleomycin-treated mice compared with control mice (figure 4A). Furthermore, IRF7 activation, as indicated by phosphorylation, was also increased in the skin of bleomycin-injected mice (figure 4A). IRF7 and phospho-IRF7 were observed in α-SMA-positive myofibroblasts and CD68-positive macrophages in the bleomycin-treated fibrotic skin (figure 4B). IRF7 mRNA was also significantly up-regulated in bleomycin-injected lesional skin compare with PBS-injected skin (figure 4C). Thus, bleomycin-induced IRF7 upregulation was similar to that observed in affected skin of patients with SSc.
Figure 4.
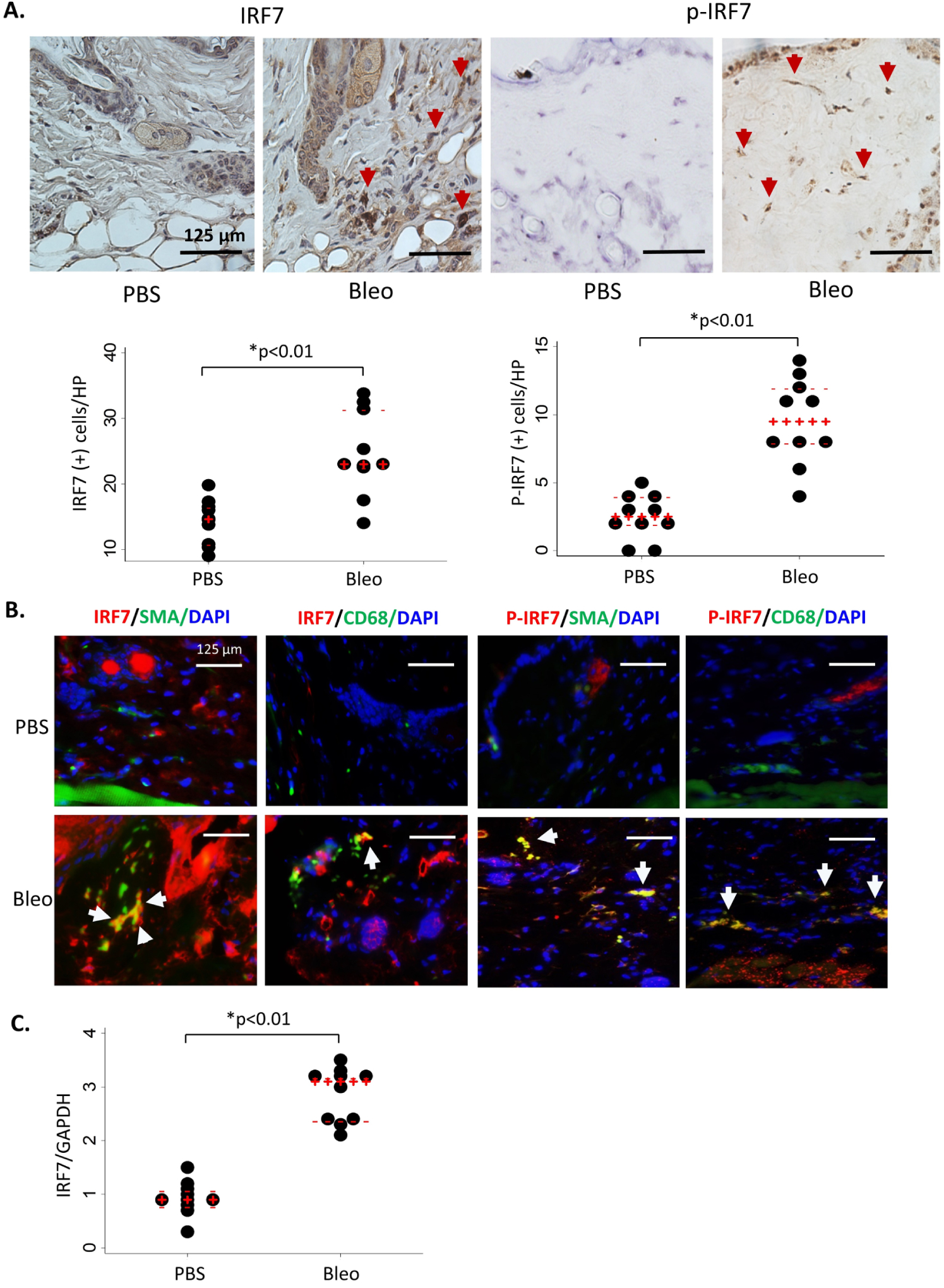
Upregulation and activation of IRF7 in the Bleo-induced skin fibrotic tissues. (A) C57BL/6 wild-type mice received subcutaneous injections of Bleo (6 days per week for 4 weeks) and lesional skin tissue was stained with IRF7 and p-IRF7 antibodies. IRF7-positive or p-IRF7-positive cell (excluding endothelial cells and hair follicle cells) number in the dermis was counted per high power field and analysed. Representative images. Red arrows: IRF7-positive or p-IRF7-positive cells. n=10. Scale bar: 125 μm; PBS vs Bleo; dot plots with median (red +) and IQR (red −) shows IRF7-positive and p-IRF7-positive cell number in the per high power fields.*P<0.01 (analysed by Mann-Whitney U test).(B) Immunofluorescence analysis using anti-IRF7 or p-IRF7 and α-SMA antibodies for IRF7-positive or p-IRF7-positive myofibroblast; anti-IRF7 or p-IRF7 and CD68 antibodies for IRF7-positive or p-IRF7-positive macrophages in the PBS and Bleo-treated lesional skin. Representative images. Original magnifications: x400 magnifications. n=10. Arrows: IRF7 or p-IRF7 with α-SMA, or IRF7 or p-IRF7 with CD68 double staining positive cells. (C) Total RNA from lesional skin was examined for IRF7 mRNA expression by real-time quantitative PCR analysis. The results were normalised with GAPDH mRNA. The results presented by dot plots with median and IQR. n=10. *P<0.01 (analysed by Mann-Whitney U test). Bleo, bleomycin; IRF7, interferon regulatory factor 7; α-SMA, α-smooth muscle actin; PBS, phosphate-buffered saline; SSc, systemic sclerosis.
To further understand the role of IRF7 in the fibrotic response to bleomycin, we compared the response of bleomycin injections in IRF7 KO mice versus control mice. Subcutaneous injection of bleomycin for 7 days induced IL-6, TGF-β1 cytokines along with Col1a2 mRNA upregulation in the wild-type mice skin tissue, and these changes were significantly attenuated in the IRF7 KO mice (see online supplementary figure S3). At 28 days of injection, bleomycin-induced expected fibrotic features including inflammation, increased dermal thickness and collagen accumulation in the wild-type mice skin. These effects were significantly attenuated in IRF7 KO mice (figure 5A). IRF7 KO mice demonstrated attenuated dermal thickness and hydroxyproline content in the lesional skin tissue (figure 5B,C). Bleomycin-induced Col1a2, ACTA2 and IL-6 mRNA expression were attenuated in IRF7 KO mice (figure 5D). TGF-β-stimulated Col1a2 mRNA expression was significantly abrogated in the cultured IRF7 KO mouse fibroblasts but not the Col1a1 mRNA expression (see online supplementary figure S4). These results indicate that lacking of IRF7 led to reduced inflammatory cytokines and fibrotic response in the bleomycin-induced dermal fibrosis animal model.
Figure 5.
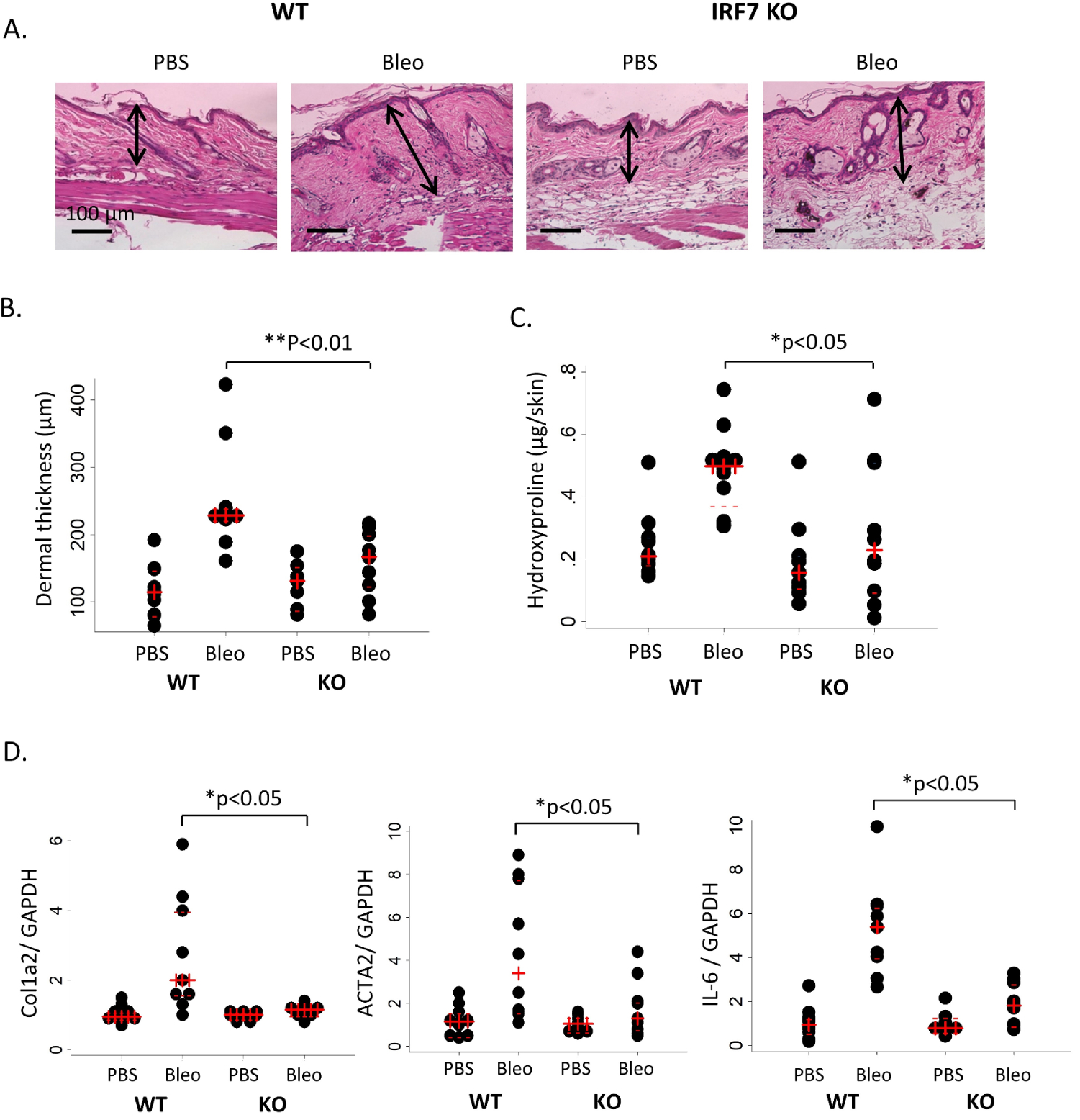
Attenuated skin fibrosis in IRF7 KO mice lesional skin in the Bleo animal model. IRF7 KO and WT mice received daily subcutaneous injections of PBS or Bleo for 6 days per week for 4 weeks, and lesional skin was analysed. (A) Representative images with H&E stain. Original magnification: x100. n=10. Black arrows represent the distance measurement. Scale bar: 100 μm. (B) Quantitation of dermal thickness (n=10) and (C) hydroxyproline content (n=10) demonstrating decreased dermal thickness in IRF7 KO mice injected with Bleo compared with WT mice. The results are shown as dot plots with median (red +) and IQR (red −). *P<0.05, **p<0.01 (analysed by Mann-Whitney U test). (D) Real-time quantitative PCR analysis in the lesional skin from WT and IRF7 KO mice treated with Bleo for 6 days per week for 4 weeks. Results were normalised with GAPDH mRNA and presented by dot plots with median (red +) and IQR (red −) from 10 mice per group. *P<0.05 (analysed by Mann-Whitney U test). IRF7, interferon regulatory factor 7; KO, knockout; PBS, phosphate-buffered saline; WT, wild-type.
Attenuated fibrosis development in TSK/IRF7 KO congenic mice
To further understand the role of IRF7 in fibrosis, we examined the TSK/+mice as a second animal model, which resembles the fibrotic features of SSc but does not have prominent inflammatory features. We crossbred TSK/+ and IR F7 KO mice to generate a TSK/IRF7 KO double congenic mouse model. All animals used in the experiments were confirmed by genotyping for the fibrillin and IRF7 gene variation (see online supplementary figure S5). Compared with TSK/mice skin tissue, TSK/IRF7 KO double congenic mice showed significantly reduced hypodermal fibrosis (figure 6A). Hypodermal thickness in TSK+/IRF7 KO mice was significantly reduced compared with TSK/+mice (figure 6B). Collagen hydroxyproline content and Col1a2 mRNA levels in the skin were also significantly reduced in TSK/IRF7 KO double congenic mice compared with TSK/+mice (figure 6C,D). Furthermore, α-SMA and fibronectin expression were significantly attenuated in TSK/IRF7 KO mice compared with TSK/+mice (figure 6E). To investigate whether IRF7 and Smad3 interaction was involved in the mechanism of fibrosis in the TSK/+mouse model, we performed immunofluorescence using skin tissue from the TSK/+mouse. The results showed strong co-localisation of Smad3 and IRF7 in TSK/+mice skin tissue. As expected, this was not observed in the TSK+/IRF7 KO double congenic mice (see online supplementary figure S6). These results suggest an interaction between IRF7 and Smad3 in the TSK/+mouse model of skin fibrosis.
Figure 6.
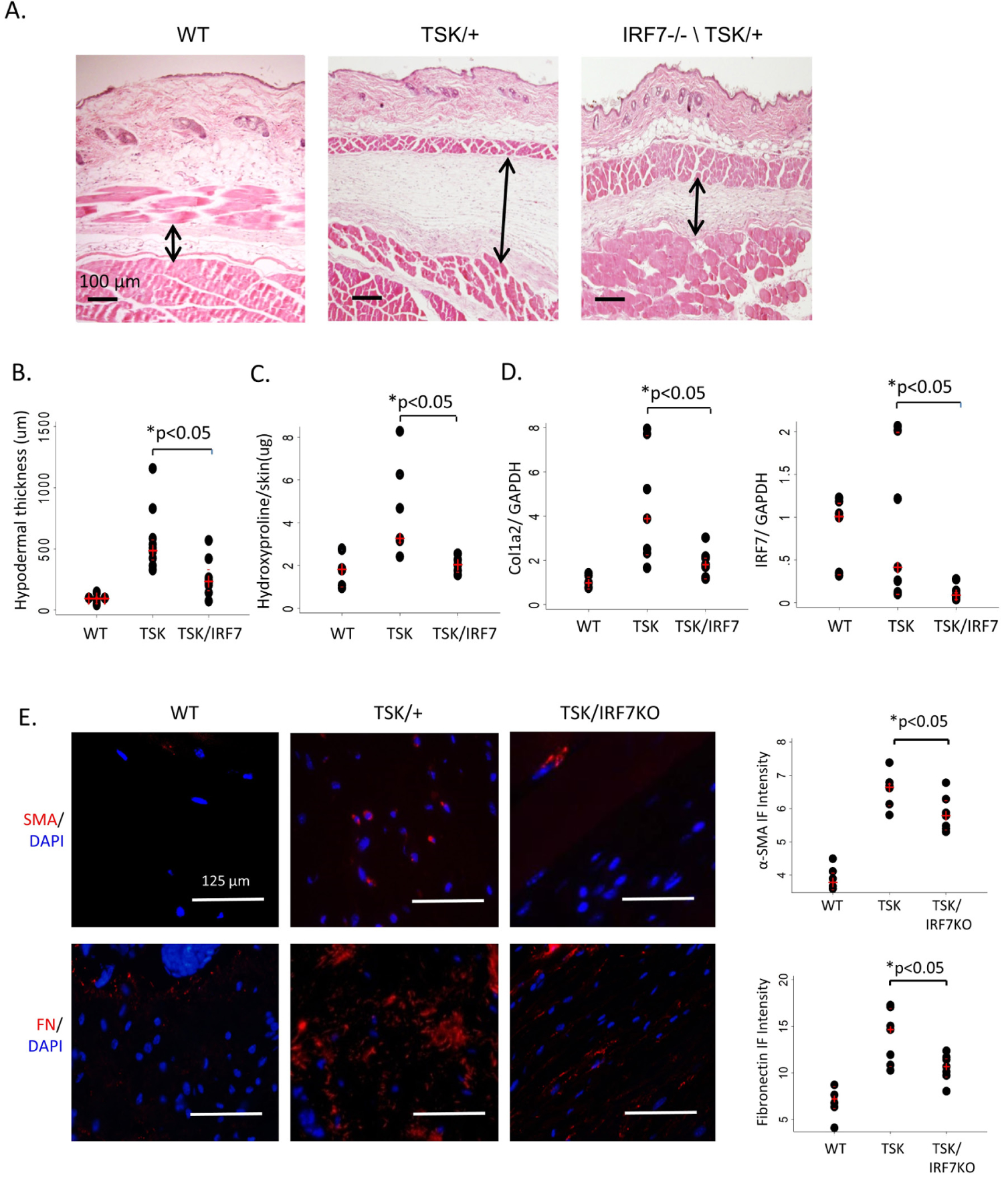
Attenuated skin fibrosis in TSK/IRF7 KO double congenic mice skin. (A) Representative images with H&E stain. Hypodermal thickness was reduced (arrows) in TSK/IRF7 KO mice compared with TSK/+mice. Original magnification: x100. n=7. Black arrows represent the distance measurement. (B) Quantitation of hypodermal thickness demonstrating attenuated hypodermal thickness in TSK/IRF7 KO mice compared with TSK/+mice. The results were given as dot plots with median (red +) and IQR (red −). n=7. *P<0.05 (analysed by Mann-Whitney U test). (C) Hydroxyproline content demonstrating reduced collagen accumulation in TSK/IRF7 KO mice skin compared with TSK/+mice. The results are shown as dot plots with median (red +) and IQR (red −). n=7. *P<0.05 (analysed by Mann-Whitney U test). (D) Total RNA was extracted from back skin of mice and Col1a2 and, IRF7 mRNA expression was determined by real-time quantitative PCR. The results were normalised with GAPDH mRNA. The results presented by dot plots with median (red +) and IQR (red −). n=7. *P<0.05 (analysed by Mann-Whitney U test). (E) Immunofluorescence analysis using anti-FN and α-SMA antibodies in the TSK/IRF7 KO double transgenic mice skin. Representative images (left panels) and intensity analysis of the α-SMA and FN-positive cells in the skin tissue (right panels). The results presented by dot plots with median (red +) and IQR (red −). n=7. *P<0.05 (analysed by Mann-Whitney U test). Original magnifications: x400 magnifications. Scale bar: 125 μm. FN, fibronectin; IRF7, interferon regulatory factor 7; KO, knockout; α-SMA, α-smooth muscle actin; WT, wild-type.
DISCUSSION
In the current study, we demonstrated that IRF7 mRNA and protein levels were significantly upregulated in the skin and cultured fibroblasts of patients with SSc compared with healthy controls. IRF7 appeared to be expressed in myofibroblasts and macrophages in affected skin tissue. IRF7 knockdown abrogated IFN-α-induced profibrotic gene expression, and IRF7/Smad3 co-localisation was observed in dermal fibroblasts. Moreover, in the bleomycin-induced dermal fibrosis and TSK/+mouse models, IRF7 KO mice had decreased dermal and hypodermal thickness and dermal fibrosis, with associated attenuation in the expression of hydroxyproline and profibrotic genes. These results demonstrate that IRF7 mediates the profibrotic effects of bleomycin, which is thought to induce fibrosis secondary to an inflammatory response, and in TSK/+mice, which is thought to directly induce fibrosis independent of inflammation.
In our previously published skin gene expression study, the skin of patients with SSc had 2754 differentially expressed genes compared with controls. Pathway analysis suggested that IRF7 is the most activated upstream transcription factor inducing the observed aberrant gene expression.22 The top upstream cytokines/growth factors in this analysis were TGF-β1, IFN-α, IFN-γ and IRF5, another transcription factor of the IFN signalling pathway in which SSc-associated polymorphisms have been identified.26 IRF5 was previously shown to mediate the fibrotic response in bleomycin-treated mice through multiple mechanisms, including fibroblast activation, inflammatory cell infiltration, endothelial-to-mesenchymal transition, vascular destabilisation, Th2/Th17 skewed immune polarisation and B-cell activation in this mouse model.27 These data support that type I IFN signalling and its downstream pathway factors like IRF5 and IRF7 are important mediators of dermal fibrosis in animal models of SSc and suggest that IRF7 may play a pathogenic role in SSc.
IRF7, previously known as lymphoid-specific factor, is expressed in fibroblasts, B cells, lymphocytes, pDCs and monocytes. Its expression can be induced by type I IFNs, TNF-α, IL-1β and viral infections.28–32 Inactive IRF7 resides in the cytoplasm. At the early ‘priming’ stage of virus infection, the low level of endogenous IRF7 is phosphorylated and activated by signalling triggered through pathogen recognition receptors and TLRs, and forms a transcriptional complex with nuclear factor-kB or IRF3. This transcriptional complex binds to the virus-response elements in the IFN-α and IFN-β promoters and induces small amounts of type I IFNs.33–35 Through positive feedback, secreted type I IFNs induce synthesis of more IRF7. Later, the newly synthesised IRF7 is activated, accumulates and further upregulates IFN expression in a positive feedback loop. In cultured IRF7 KO mouse embryonic fibroblasts, the viral induction of IFN-α/IFN-β was severely impaired and markedly decreased serum IFN-α levels were observed in the IRF7 KO mice.36 Furthermore, IRF7 induces the pro-inflammatory cytokine IL-6 in pDCs and monocytes.37,38 Importantly, a recent study demonstrated that depletion of pDCs, a major source of IFN-α, in animal models significantly decreased IRF7 mRNA expression and led to attenuated skin fibrosis even when fibrosis was already established.9 These observations demonstrate the importance of IRF7-dependent systemic inflammatory responses.
TGF-β has long been recognised as a key mediator of fibrosis in SSc.10 The C-terminal regions of IRFs, including IRF7, show homology to the C-terminal domains of the Smad family, which may mediate the response to TGF-β.39 Smad3, a key component of TGF-β signalling for stimulation of collagen production, has been identified as an IRF7 interacting protein. Smad3 physically and functionally interacts with IRF7, and TGF-β/Smad3 signalling regulates the transcriptional activity of IRF7.19 On activation, IRF7 forms a transcriptional complex together with other IRFs such as IRF3 or IRF5.33,35 In this study, we further confirmed that IRF7 forms complexes with Smad3. Our in vitro experiments also indicated that IRF7 knockdown abrogated TGF-β-induced fibrosis in SSc fibroblasts regardless of IFN-α stimulation. Taken together, our results suggest a fibroblast-specific function of IRF7, namely in potentiating the TGF-β-mediated fibrotic response (see online supplementary figure S7). However, IRF7 knockdown did not attenuate TGF-β-stimulated collagen accumulation in cultured healthy control skin fibroblasts. This may be explained by considering that TGF-β itself does not stimulate IRF7 accumulation in healthy control fibroblasts (figure 2C, lane 5). On the other hand, IRF7 KO in the non-inflammatory dermal fibrosis model, TSK/+mouse, abrogated the fibrotic response. This finding is contrary to other inflammatory targets such as STAT4 and OX40L whose blockade show an abrogated fibrotic response in the inflammatory bleomycin-induced dermal fibrosis model but not in the TSK/+mouse.40,41 In the present study, we primarily focused on the role of IRF7 in fibroblasts and its potential interplay with the TGF-β pathway. However, a direct link between TGF-β and IFN-α was not tested in the bleomycin-induced skin fibrosis mouse experiments. The possible link is suggested by the ability of IFN-α to upregulate IRF7 expression, the interaction between IRF7 and Smad3 in vitro and reduced fibrosis seen in the IRF7 KO mouse. As an extension of the present study, we plan to investigate the role of IRF7 upregulation in the inflammatory cells and its impact on the exaggerated fibrosis in SSc including experiments with cell-specific conditional KO mouse models in our future studies. Altogether, our data suggest that the upregulation and activation of IRF7 in SSc skin plays an important role in the observed exaggerated inflammation as well as fibrosis, and might provide a link between the prominent IFN signature and activation of TGF-β in SSc. IRF7 may therefore represent a promising novel therapeutic target in SSc.
Supplementary Material
Key messages.
What is already known about this subject?
Patients with systemic sclerosis (SSc) have a prominent interferon (IFN) signature.
Interferon regulatory factor 7 (IRF7) is predicted to be a top regulator of SSc skin gene transcript signature based on previous global gene expression studies.
What does this study add?
IRF7 is overexpressed in SSc fibrotic skin and interact with Smad3, a prominent member of transforming growth factor-β (TGF-β) signalling pathway in dermal fibroblasts.
IRF7 knockdown in SSc dermal fibroblasts abrogated TGF-β-induced profibrotic gene expression.
Knockout of IRF7 ameliorates experimental fibrosis in bleomycin-induced skin fibrosis and TSK/+congenic murine models of SSc.
How might this impact on clinical practice or future developments?
Our studies indicate that IRF7 can potentiate the TGF-β-mediated fibrosis in dermal fibroblasts and might provide a link between the prominent IFN signature and fibrosis in SSc.
Acknowledgements
Parts of this work have been presented as an oral presentation in the concurrent abstract sessions of the American College of Rheumatology Annual Scientific Meetings in 2015 and 2017.
Funding This study was supported by Scleroderma Foundation new Investigator Grant (Wu), an Arthritis National Research Foundation grant (Skaug), R01AR073284 (Assassi) and DoD W81XWH-16-1-0296 (Assassi).
Footnotes
Competing interests None declared.
Ethics approval The study was approved by the institutional review board of the University of Texas Health Science Center at Houston. The animal protocols were institutionally approved by the University of Texas Health Science Center at Houston Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
REFERENCES
- 1.Elhai M, Meune C, Avouac J, et al. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology 2012;51:1017–26. [DOI] [PubMed] [Google Scholar]
- 2.Skaug B, Assassi S. Type I interferon dysregulation in systemic sclerosis. Cytokine 2019. [DOI] [PubMed] [Google Scholar]
- 3.Wu M, Assassi S. The role of type 1 interferon in systemic sclerosis. Front Immunol 2013;4:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solans R, Bosch JA, Esteban I, et al. Systemic sclerosis developing in association with the use of interferon alpha therapy for chronic viral hepatitis. Clin Exp Rheumatol 2004;22:625–8. [PubMed] [Google Scholar]
- 5.Black CM, Silman AJ, Herrick AI, et al. Interferon-? does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 1999;42:299–305. [DOI] [PubMed] [Google Scholar]
- 6.Tan FK, Zhou X, Mayes MD, et al. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology 2006;45:694–702. [DOI] [PubMed] [Google Scholar]
- 7.York MR, Nagai T, Mangini AJ, et al. A macrophage marker, siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and Toll-like receptor agonists. Arthritis Rheum 2007;56:1010–20. [DOI] [PubMed] [Google Scholar]
- 8.Farina GA, York MR, Di Marzio M, et al. Poly(I:C) drives type I IFN-and TGFβ-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol 2010;130:2583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ah Kioon MD, Tripodo C, Fernandez D, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 2018;10:eaam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lafyatis R Transforming growth factor β—at the centre of systemic sclerosis. Nat Rev Rheumatol 2014;10:706–19. [DOI] [PubMed] [Google Scholar]
- 11.De Luján Alvarez* M, Quiroga* AD, Parody JP, et al. Cross-talk between IFN-α and TGF-β 1 signaling pathways in preneoplastic rat liver. Growth Factors 2009;27:1–11. [DOI] [PubMed] [Google Scholar]
- 12.Tamura T, Yanai H, Savitsky D, et al. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 2008;26:535–84. [DOI] [PubMed] [Google Scholar]
- 13.Mamane Y, Heylbroeck C, Génin P, et al. Interferon regulatory factors: the next generation. Gene 1999;237:1–14. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi T, Ogasawara K, Takaoka A, et al. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 2001;19:623–55. [DOI] [PubMed] [Google Scholar]
- 15.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2001;2:378–86. [DOI] [PubMed] [Google Scholar]
- 16.Platanias LC. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat Rev Immunol 2005;5:375–86. [DOI] [PubMed] [Google Scholar]
- 17.Decker T, Müller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol 2005;5:675–87. [DOI] [PubMed] [Google Scholar]
- 18.Dietrich N, Lienenklaus S, Weiss S, et al. Murine toll-like receptor 2 activationinduces type I interferon responses from endolysosomal compartments. PLoS One 2010;5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qing J, Liu C, Choy L, et al. Transforming growth factor beta/Smad3 signaling regulates IRF-7 function and transcriptional activation of the beta interferon promoter. Mol Cell Biol 2004;24:1411–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmona FD, Gutala R, Simeón CP, et al. Novel identification of the IRF7 region as an anticentromere autoantibody propensity locus in systemic sclerosis. Ann Rheum Dis 2012;71:114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu Q, Zhao J, Qian X, et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum 2011;63:749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Assassi S, Swindell WR, Wu M, et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 2015;67:3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vangipuram M, Ting D, Kim S, et al. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp 2013;77:e3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of rheumatology/European League against rheumatism collaborative initiative. Ann Rheum Dis 2013;72:1747–55. [DOI] [PubMed] [Google Scholar]
- 25.Lydersen S Statistical review: frequently given comments. Ann Rheum Dis 2015;74:323–5. [DOI] [PubMed] [Google Scholar]
- 26.Radstake TRDJ Gorlova O, Rueda B, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet 2010;42:426–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saigusa R, Asano Y, Taniguchi T, et al. Multifaceted contribution of the TLR4-activated IRF5 transcription factor in systemic sclerosis. Proc Natl Acad Sci U S A 2015;112:15136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Pagano JS. IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol Cell Biol 1997;17:5748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.WC A, Moore PA, LaFleur DW, et al. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J Biol Chem 1998;273:29210–7. [DOI] [PubMed] [Google Scholar]
- 30.Rivieccio MA, John GR, Song X, et al. The cytokine IL-1beta activates IFN response factor 3 in human fetal astrocytes in culture. J Immunol 2005;174:3719–26. [DOI] [PubMed] [Google Scholar]
- 31.Sato M, Suemori H, Hata N, et al. Distinct and essential roles of transcriptionfactors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 2000;13:539–48. [DOI] [PubMed] [Google Scholar]
- 32.Farina A, Cirone M, York M, et al. Epstein–Barr virus infection induces aberrant TLR activation pathway and fibroblast-myofibroblast conversion in scleroderma. J Invest Dermatol 2014;134:954–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wathelet MG, Lin CH, Parekh BS, et al. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol Cell 1998;1:507–18. [DOI] [PubMed] [Google Scholar]
- 34.Yang H, Lin CH, Ma G, et al. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J. Biol. Chem 2003;278:15495–504. [DOI] [PubMed] [Google Scholar]
- 35.Barnes BJ, Field AE, Pitha-Rowe PM. Virus-induced Heterodimer Formation between IRF-5 and IRF-7 Modulates Assembly of the IFNA Enhanceosome in Vivo and Transcriptional Activity of IFNA Genes. J. Biol. Chem 2003;278:16630–41. [DOI] [PubMed] [Google Scholar]
- 36.Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005;434:772–7. [DOI] [PubMed] [Google Scholar]
- 37.Sweeney SE. Targeting interferon regulatory factors to inhibit activation of the type I IFN response: implications for treatment of autoimmune disorders. Cell Immunol 2011;271:342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yasuda K, Richez C, Maciaszek JW, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol 2007;178:6876–85. [DOI] [PubMed] [Google Scholar]
- 39.Eroshkin A, Mushegian A. Conserved transactivation domain shared by interferon regulatory factors and Smad morphogens. J Mol Med 1999;77:403–5. [DOI] [PubMed] [Google Scholar]
- 40.Avouac J, Fürnrohr BG, Tomcik M, et al. Inactivation of the transcription factor STAT-4 prevents inflammation-driven fibrosis in animal models of systemic sclerosis. Arthritis Rheum 2011;63:800–9. [DOI] [PubMed] [Google Scholar]
- 41.Elhai M, Avouac J, Hoffmann-Vold AM, et al. OX40L blockade protects against inflammation-driven fibrosis. Proc Natl Acad Sci U S A 2016;113:E3901–E3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.