

Summary
Complementation is a naturally occurring genetic mechanism that has been studied for a number of plus‐strand RNA viruses. Although trans‐complementation is well documented for Flaviviridae family viruses, the first such system for hepatitis C virus (HCV) was only described in 2005. Since then, the development of a number of HCV trans‐complementation models has improved our knowledge of HCV protein functions and interactions, genome replication and viral particle assembly. These models have also been used to produce defective viruses and so improvements are necessary for vaccine assays. This review provides an update on HCV trans‐complementation systems, the viral mechanisms studied therewith and the production and characterization of trans‐encapsidated particles.
Keywords: HCV, protein function, trans‐complementation, trans‐packaging
Abbreviations
- aa
amino acid
- CFU
colony‐forming unit
- ER
endoplasmic reticulum
- FFU
focus‐forming unit
- HCV
hepatitis C virus
- HCVcc
cell‐culture‐grown hepatitis C virus
- I.U.
infectious unit
- IRES
internal ribosomal entry site
- LCS I
low complexity sequence I
- LD
lipid droplet
- NS
non‐structural
- RNA
ribonucleic acid
- wt
wild type
Introduction
Complementation is a naturally occurring genetic mechanism that enables the functional rescue of defective or mutant genomes. This phenomenon can be used for in vitro assays. In conventional complementation experiments, a defective genome or protein is rescued by a wild‐type (wt) copy (Fig. 1). In virology, complementation is a genomic tool used to investigate protein functions and viral particle assembly or establish whether genome replication occurs preferentially through a cis‐acting mechanism. Moreover, this model can also be used to produce defective viruses for various purposes.
Figure 1.
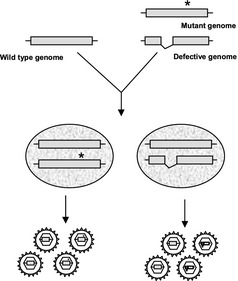
Schematic representation of the trans‐complementation principle in virology. Mutant or defective genomes can be rescued by a wild‐type copy that allows the production of infectious particles capable of a single round of infection.
Flaviviridae are positive‐strand RNA viruses and replicate their genomes in virus‐induced vesicular compartments. These membranous complexes are rather enclosed structures, which tends to limit to exchange of viral RNA and proteins by trans‐complementation 1. Moreover, for a number of plus‐strand RNA viruses from a range of virus families (such as the Picornaviridae (poliovirus), Alphaviridae (Sindbis virus, Semliki Forest virus, and Venezuelan equine encephalitis virus), Coronaviridae (human coronavirus E229) and Flaviviridae (tick‐borne encephalitis virus, Kunjin virus, West Nile virus, and yellow fever virus)), assembly of progeny viruses can be achieved when structural proteins are expressed in trans independently of the RNA molecule that encodes the replicase proteins 2, 3, 4, 5, 6, 7. The particles are infectious but are only capable of a single round of infection. These viral trans‐encapsidation systems may also be useful vaccine delivery systems 8, 9, 10.
Concerning HCV, some researchers have reported the presence of natural HCV subgenomic RNAs in the serum and liver tissue of infected patients. Most of these subgenomic RNAs contain large, in‐frame deletions (from E1 up to NS2) and are always found together with the full‐length wt RNAs 11, 12, 13, 14. The mutant viral genomes persist for at least 2 years and sequence analysis suggests that subgenomic and full‐length HCV RNAs evolve independently 14. The relative abundance and persistence of these subgenomic RNAs in vivo suggests that they are capable of autonomous replication and can be packaged and secreted as infectious viral particles.
The first HCV trans‐complementation studies concerned the replication complex. Next, the development of cell‐culture‐grown hepatitis C virus (HCVcc) systems that can assemble and release of infectious viral particles has made it possible to the study structural regions using trans‐complementation systems. Furthermore, various trans‐packaging systems for HCV subgenomic replicon RNAs have been developed.
Trans‐Complementation Applied to HCV Replication Models
Trans‐complementation of HCV replication by NS5A
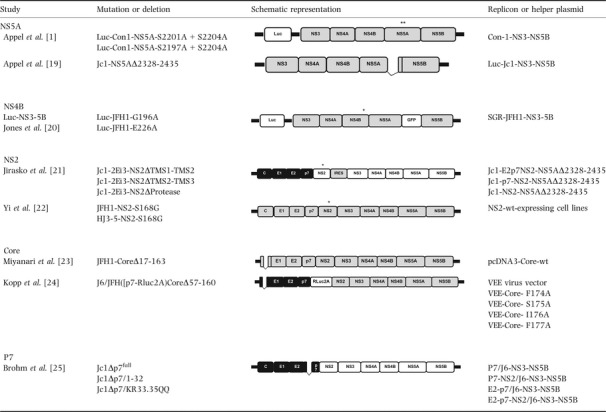
The first trans‐complementation studies were performed in 2005 by Appel et al. by using subgenomic replicons, such as Con‐1 (HCV type 1b, EMBL accession number AJ238799) 15. The researchers looked at whether lethal mutations in non‐structural (NS) HCV genes could be rescued by trans‐complementation 1. A series of replicon RNAs carrying mutations (in the NS3, NS4B, NS5A and NS5B regions) that abolished replication were transfected into Huh‐7 hepatoma cells harbouring autonomous replicating HCV helper RNAs. In this context, only NS5A mutations in the low complexity sequence I (LCS I) domain have been efficiently rescued (Table 1). The required proteins for complementation were NS3‐NS5A, whereas NS5A expressed alone did not restore RNA replication. These findings strongly suggested that other NS HCV proteins specifically act in cis on HCV RNA and, indeed, the hypothesis was confirmed in 2006 by Tong and Malcom 16. Nevertheless, trans‐acting function of NS5A likely would require protein hyperphosphorylation 17.
Table 1.
Schematic representation of replicons used for trans‐complementation, as applied to the replication and assembly processes
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
In 2008, additional studies showed that deletion of NS5A domain III from the Jc1 genome (HCV chimeric type 2a/2a) 18 does not abolish replication but that pseudo‐infectious particles are not produced 19. Therefore, co‐transfection of deleted Jc1 with a JFH1 NS3‐NS5B replicon rescues HCV particle production (Table 1). Taken as a whole, these results have elucidated the key role of NS5A domain III in the HCV assembly process.
Trans‐complementation of HCV replication by NS4B
In 2009, Jones et al. 20 performed the firstly NS4B trans‐complementation using various replicon RNAs carrying various replication‐abolishing mutations in the NS4B C‐terminal domain. The RNAs rescued replication of two G196A and E226A mutants (Table 1). Nevertheless, the complementation efficiency was low – about 1%, vs 24% for the NS5A S232A mutant. Both G196 and E226 are highly conserved amino acids that are predicted to lie within unstructured regions flanking helix 1. The researchers concluded that NS4B complementation was limited.
Trans‐complementation of HCV replication by NS2
Trans‐complementation assays have also been used to study the role of NS2 in viral particle assembly 21. A number of different deletions within trans‐membrane segments (TMS1–3) or the protease domain of the Jc1 (J6/JFH1) chimera genome have been examined (Table 1). Again, replication was seen in the absence of pseudo‐infectious particle production. Three bicistronic helper RNAs (spE2‐p7‐NS2, spp7‐NS2 and spNS2) were built by combining the JFH1 replicase module (UTRs and the NS3–NS5B coding region) and a 5′‐terminal expression module under control of the poliovirus internal ribosomal entry site (IRES). Various cotransfections between Jc1 NS2 mutants and bicistronic helper RNAs were performed in Huh‐7.5 cells. All three NS2 mutants could be rescued by trans‐complementation. Infectivity titres obtained in rescue experiments were about 10‐fold lower than in the Jc1 wt.
At the same time, Yi et al. 22 studied the role of the NS2 serine amino acid (aa) at position 168 (Ser‐168) in the viral assembly and maturation process. They found that substitution of Ser‐168 by an alanine or glycine aa abolished infectious virus production but not polyprotein processing or genome replication (Table 1). However, transfection of Ser‐168 mutant replicon RNA into a cell line expressing wt NS2 rescued the production of infectious virus. In contrast, transfection into a wt H77 (type 1a) NS2‐expressing cell line was unable to rescue virus production. Although H77 and JFH1 NS2‐expressing cells could rescue viral production in the H77‐JFH1 chimera Ser‐168 mutant, this rescue was only partial with wt JFH1 NS2‐expressing cells. The same experiment was performed in a cell line expressing NS2 carrying a 71‐aa deletion in the C‐terminal domain. Infectious particle production was not obtained, indicating that NS2's C‐terminal domain is a major determinant of HCV production. All these results indicate that wt NS2 expression is able to trans‐complement NS2‐defective constructs and is genotype‐ and strain‐dependent.
In conclusion, trans‐complementation based on NS regions has shown that (i) NS5A region carrying mutations in the LCS1 domain or deletions in domain III can be complemented by the minimal sequence NS3‐NS5A, (ii) NS2 can be efficiency trans‐complemented by wt NS2 and (iii) NS4B trans‐complementation exists for mutations in unstructured regions but is weak.
Trans‐Complementation Applied to the HCV Assembly Process
Trans‐complementation of HCV multiplication by core
The use of the HCVcc system to produce infectious viral particles has enabled investigation of the core protein's role in the assembly process. It has been shown that Core and 60–90% of the NS proteins were localized within lipid droplets (LDs) 23. Conversely, the lack of core expression in replicating JFH1 cells (JFH1dC) was associated with a diffuse distribution of NS proteins on the endoplasmic reticulum (ER) and the absence of concentration around the LDs. Moreover, LD levels were lower in JFH1dC replicating cells than in wt JFH1 replicating cells. Interestingly, NS proteins again accumulated around LDs when JFH1dC replicating cells were transfected with a plasmid expressing wt Core. These results showed that LD‐associated Core recruits NS proteins from the ER to the LDs. Moreover, Core activity could be restored by trans‐complementation.
In parallel, Kopp et al. 24 used trans‐complementation assays to investigate the role of the core C‐terminal region and to identify the essential residues involved in infectious particle production. Mutations were introduced between residues 170–191 (Table 1). It was found that mutations at positions 170 and 174–177 abrogated pseudo‐infectious particle production, despite the presence of wild‐type replication levels. In contrast, cotransfection of the J6/JFH1 replicon (containing a large deletion in the sequence coding for Core protein, that is residues 57–160) with a full‐length HCV core protein plasmid led to significant levels of pseudo‐infectious particle release. No pseudo‐infectious particles were obtained when using Core 170 and 174–177 mutants plasmids. In this context, trans‐complementation systems have highlighted the importance of these residues in pseudo‐infectious particle production.
Trans‐complementation of HCV multiplication by p7
Brohm et al. 25 developed a complementation system for investigating key determinants that are essential for the optimal function of p7 in HCV infectious particle production. Three Jc1 replicons were constructed: one with a deletion of the entire p7 coding region (Jc1Δp7full), a second lacking residues 1–32 of p7 (Jc1Δp7half) and a third carrying a dual mutation of the highly conserved basic residues KR33/35 to QQ (Jc1‐KR33.35QQ) (Table 1). The replicons Jc1Δp7full and Jc1Δp7half were unable to generate infectious particles. With the Jc1‐KR33.35QQ replicon, 100‐fold fewer infectious particles were released than with the parent Jc1 genome. These defective HCV p7 replicons were then cotransfected with bicistronic JFH1‐derived helper replicons expressing either p7 alone, p7‐NS2, E2‐p7, E2 alone or E2‐p7‐NS2 in the first cistron and JFH1 NS proteins in the second. Jc1Δp7half infectivity was rescued by all helper RNAs expressing p7, whereas the E2 signal sequence had no influence. However, cotransfection of E2‐p7 and E2‐p7‐NS2 helper RNAs yielded higher virus titres. In the case of the Jc1Δp7full replicon, complementation was achieved only by the co‐expression of E2‐p7‐NS2. Jc1‐KR33.35QQ infectivity was rescued by all helper RNAs, although pseudo‐infectious particle production was only increased in the presence of E2‐p7 and E2‐p7‐NS2 helper RNAs. Lastly, no production of infectious particles was seen with the Con1‐p7 (HCV type 1b) helper replicon, which indicated genotype specificity. These results indicate that a p7 protein‐deficient replicon could only be rescued by E2‐p7‐NS2 expression and not by p7 protein alone.
In conclusion, trans‐complementation of the structural region has shown that Core can be trans‐complemented by wt Core and p7 can be trans‐complemented by the E2‐p7‐NS2 sequence and not by p7 expressed alone.
HCV Trans‐Encapsidation Systems
The first trans‐encapsidation systems were described for packaging replicons for flavivirus, including Kunjin virus 2, 3, yellow fever virus 4, tick‐borne encephalitis virus 5 and West Nile virus 6, 7. These virus‐like‐particle‐generating systems enabled the development of vaccine models and the analysis of some aspects of viral assembly and entry 6. Based on these studies, various systems for packaging HCV replicons have been created.
Packaging replicons
Two types of constructs have been used in trans‐encapsidation systems: a minimal replicase complex and the full‐length genome carrying deletions in envelope‐encoding genes (Table 2).
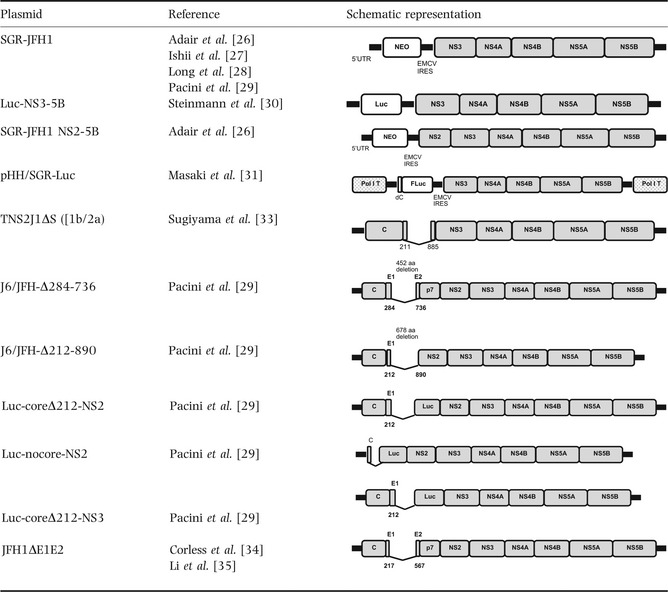
Table 2.
Schematic representation of replicons used in trans‐packaging systems
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The first studies used JFH1‐NS3‐NS5B replicons associated either with a resistance gene (such as neomycin in SGR‐JFH1) 26, 27, 28, 29 or a reporter gene (such as Renilla luciferase in Luc‐NS3‐NS5B) 30. The SGR‐JFH1 replicon enabled the establishment of stable cell lines 26, the titration of colony‐forming units (CFU/mL) 26, 27 and the selection of efficiently replicating cell clones 28. Rapid infectious particle assays 30 could been performed with the Luc‐NS3‐NS5B replicon. The SGR‐JFH1 NS2‐NS5B construct was used by Adair et al. 26 to study NS2's role in the particle assembly process and has yielded the highest infectious titres. Masaki et al. and Suzuki et al. chose to use a JFH‐1 cDNA clone plasmid containing the Pol I promoter, terminator sequences and luciferase gene reporter sequences for directly transfecting cells without the need for a transcription step 31, 32 (Table 1).
Another approach was adopted by Pacini and Sugiyama, using replicons based on the defective genomes found in both serum and liver tissue from chronically HCV‐infected patients. Sugiyama et al. generated a 1b/JFH1‐2a chimera based on defective 1b patient isolate and that carried deletions in the genes coding for the envelope, p7 and NS2 proteins 33. Likewise, Pacini et al. used a J6/JFH1 chimera to build constructs with various deletions in the E1‐E2 glycoprotein region (448 aa) and the E1 to NS2 (674 aa), E2 to p7, core to p7 and E2 to NS2 regions, respectively (Table 2).
Lastly, several research groups have studied replicons carrying deletions (351 aa) in the envelope gene (JFH1ΔE1E2 or J6/JFH1‐ΔE1E2) 29, 30, 34, 35 (Table 2).
Structural protein expression systems
Encapsidation of subgenomic replicons with partially or totally deleted structural regions has been performed in various systems expressing structural proteins in trans (Table 3) in the presence or absence of p7 and NS2 proteins. Some researchers have focused on replicons, helper viruses 29, 30 and direct expression vectors 29, 31, whereas others have established stable cell lines 27, 30, 33 and baculovirus or lentivirus delivery systems 26, 28.
Table 3.
Summary of infectious particles generated by cotransfection of deleted subgenomic replicons with structural proteins expressed in trans. Packaging feasibility is expressed as positive (+) or negative (−). nt: not tested
| Deleted subgenomic replicons | |||||
|---|---|---|---|---|---|
| JFH1‐NS3‐NS5B | JFH1‐NS2‐NS5B | CΔ212‐NS3‐NS5B | JFH1ΔE1E2 | ||
| Structural proteins expressed in trans | Core‐NS3 | + | nt | + | nt |
| Core‐NS2 | + | + | + | + | |
| Core‐p7 | − | + | − | + | |
| Core‐E2 | nt | nt | − | + | |
| E1‐NS2 | nt | nt | + | + | |
| E1‐p7 | nt | − | − | + | |
| E1‐E2 | nt | nt | − | + | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The first trans‐encapsidation assays were performed in Huh7.5 cells via transient transfection with replicons or helper viruses, allowed reproducing in vitro natural trans‐encapsidation mechanism in vivo 29. Similar experiments with heterologous helper viruses (such as Con1/C3 and Jc1 30) have been attempted. However, given the competition between defective replicons and helper viruses, it has been difficult to obtain equal replication and high trans‐encapsidation efficiencies. Moreover, two different markers must been introduced to identify the two encapsidated replicons.
Other researchers have chosen structural proteins expressing plasmids (in the presence or absence of p7 and NS2) to perform trans‐encapsidation and determine the minimal sequence required for efficient encapsidation. Various plasmid constructs have been used, such as the pEF6 plasmid (encoding C‐NS2, C‐p7, C‐E2, E1‐NS2, E1‐p7 or E1‐E2 from J6/JFH1 chimeras 29), the pCAGC plasmid (expressing C‐p7 and C‐NS2 regions 31, 32), pCDNA‐E1‐p7 34 and pcDNA3‐JFH1‐E1/E2 35 (Table 2).
In parallel, the use of stable cell lines has improved the yield of pseudo‐infectious particles. With a pEF4 plasmid, Ishii et al. 27 established stable cell lines expressing JFH1 Core‐p7 and Core‐NS2 proteins, respectively. Steinmann et al. 30 cotransfected lentiviral vectors expressing Jc1 Core‐E1 and E2‐NS2 proteins into Huh7.5 and Huh7‐lunet cells, respectively, and obtained high levels of structural protein expression. Conversely, stable cell lines based on defective replicons with resistant genes (such as SGR‐JFH1) can be used to select more efficient cell clones and achieve high levels of replication.
The latter models to have been developed for the expression of complementing structural proteins are baculovirus systems expressing JFH1 Core to p7 or Core to NS2 proteins 26 and lentivirus systems expressing Core to NS2 28.
Trans‐packaging particle production
Although trans‐encapsidation assays have preferentially been performed in human liver‐derived cell lines (i.e. Huh7 and subclones), murine hepatic cell line have also been studied 28. In a two‐step development process, deleted replicons and helper replicons are first validated in transient experiments and then adapted to stable cell lines for standardizing and improving the system. Various researchers have thus shown that trans‐packaging systems are functional (Table 3).
It has been proven that the NS3‐NS5B replicon can be packaged by Core‐NS2 or Core‐NS3 proteins expressed in trans but not by Core‐p7 26, 27, 28, 30, 31. Therefore, NS2 protein is essential for infectious particle production. The most efficient trans‐packaging has been obtained with Core‐NS2 28. In parallel, it was found that the NS2‐NS5B replicon could be packaged by the Core‐p7 region. However, the trans‐packaging efficiency was higher with the NS3‐NS5B replicon and the Core‐NS2 sequence 26.
In the case of full‐length defective genomes, trans‐packaging has been observed with Core‐NS2, Core‐NS3 and E1‐NS2 constructs 29, 33. The efficiency was greatest with Core‐NS2 33.
In turn, the pseudo‐infectious particles’ physical and antigenic properties have been compared with those observed for wt virus 26, 27, 28, 30, 35. The particle density was similar to that seen in HCVcc particles. Neutralization assays with anti‐CD81 27, 28, 29, 30, 31, 35, anti‐E2 26, anti‐SCARB‐1 and anti‐claudin‐1 28 antibodies have shown that pseudo‐infectious particles use both E2 glycoprotein and HCV entry receptors for cell entry. Ishii et al. 27 have observed particles under the electron microscope and have confirmed similarities with HCVcc particles. These pseudo‐infectious particles were capable of a only single round of infection and the defective genome had been clearly encapsidated 27, 30, 35.
Several research groups have developed assays for trans‐packaged particles 26, 27, 28, 30, 35, whereas others have only observed the particles’ features and estimated the efficiency of trans‐encapsidation 29, 31, 33. Various titres of pseudo‐infectious particles have been obtained: 3.4 ± 0.6 102 focus‐forming units (FFU)/mL according to Ishii et al., 104 tissue culture infectivity dose (TCID)50/mL according to Steinmann et al., 50 CFU/mL according to Adair et al., 1.1 103 FFU/mL according to Corless et al. and, lastly, 80 IU/mL (infectious units/mL) in a transient system and 300 IU/mL in stable cell lines, according to Li et al. 26, 27, 30, 34, 35 (Table 4). The comparison of the respective systems’ yields is difficult because of heterogeneity of the infectious units used and differences in detection experiments.
Table 4.
The titres of pseudo‐infectious particles obtained in various studies
| Study | Infectious titre | Units | Vector for structural proteins | Systems |
|---|---|---|---|---|
| Ishii et al. 27 | 340 | FFU/mL | pEF4JFHc‐NS2 | Stable cell line |
| Steinmann et al. 30 | 10 000 | TCID50/mL | pWPI‐CE1‐BSD and pWPI‐SpE2p7NS2‐BSD | Stable cell line |
| Adair et al. 26 | 50 | CFU/mL | pFBM[JFH1)C‐p7 | Baculovirus |
| Li et al. 35 | 300 | I.U./mL | pcDNA3‐JFH1‐E1/E2 | Stable cell line |
| Corless et al. 34 | 1100 | FFU/mL | pcDNA‐E1‐p7 | Transient expression system |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Several groups have tried to improve their trans‐packaging systems by introducing the above‐mentioned adaptive mutations into subgenomic replicons 30, 32, 35 or complemented structural protein regions 26. For example, the V2440L mutation has been introduced into C‐terminal region of NS5A and was found to enhance production 36. Using JFH1ΔE1E2 replicons, Li et al.'s group inserted an M1051L mutation into NS3 region and a C2219R mutation into NS5A. They obtained a peak titre of 5 × 103 IU/mL 35. Similar observations have been obtained with baculovirus systems and the introduction of F172C and P173S mutations in the C‐terminal part of Core 26, 37. Lastly, mutations in E2 (N417S), p7 (N765D) and NS2 (Q1012R) were introduced into the core‐NS2 expression plasmid pCAGC and resulted in a more than fourfold increase in pseudoparticle production 32.
In parallel, heterologous trans‐packaging assays have been developed. The first group to do so was Steinmann et al., who tested the JFH1‐Luc‐NS3‐NS5B replicon with Con1/C3 (1b/JFH1) and Jc1 (J6‐2a/JFH1) chimeras. Infectious, trans‐packaging particles were obtained with each of the two chimeras but Jc1 gave the best yield 30. Li et al. 35 chose heterologous trans‐encapsidation of JFH1ΔE1E2 replicons with HCV J6 2, H77 (1a) and Con1 (1b) envelope glycoproteins. Only J6 (2a) envelope proteins were able to rescue pseudo‐infectious particle production. Intergenotype compatibility between envelope proteins and the other proteins may be essential in the assembly process 35.
Conclusions
The new JFH1‐based culture system has enabled the development of trans‐complementation systems that have facilitated studies of (i) genomic and protein functions and (ii) interactions between HCV proteins. Trans‐complementation based on NS regions has shown that (i) NS5A region carrying mutations in the LCS1 domain or deletions in domain III can be complemented by the minimal sequence NS3‐NS5A, (ii) NS2 can be efficiency trans‐complemented by wt NS2 and (iii) NS4B trans‐complementation exists for mutations in unstructured regions but is weak. As expected, genomic trans‐complementation in HCV replication systems is limited and NS HCV proteins act preferentially in cis. These trans‐complementation studies have increased our knowledge of non‐structural proteins’ functions and interactions in HCV. They have demonstrated that NS5A domain III is a major determinant of HCV assembly. NS4B is involved in RNA replication but also contributes to virus assembly and release. NS2 is involved in the viral assembly process; in a late‐post assembly maturation step (perhaps in concert with NS5A) and, it confers infectivity on the HCV particle. Moreover, NS2 generates a strict cis requirement for the core region, to allow efficient trans‐packaging of the subgenomic RNA – revealing the complex interplay between NS2 and core genes 29. Overall, these results have confirmed that NS proteins are involved in viral assembly and release.
Trans‐complementation of the structural region has shown that Core can be trans‐complemented by wt Core. Core recruits NS proteins and replication complexes to LD‐associated membranes. This recruitment is critical for producing infectious viruses and residues 170 and 174–177 are important for the production of infectious viruses. Likewise, p7 can be trans‐complemented by the E2‐p7‐NS2 sequence. p7 is absolutely essential for the production of pseudo‐infectious HCV particles; it can operate independently of an upstream signal sequence and a tyrosine residue close to its conserved, dibasic motif is important for optimal virus production in genotype 2a viruses. These results have clarified the role of Core, p7 and LDs in pseudo‐infectious particle production and have indicated that some steps of virus assembly take place around LDs.
Various trans‐packaging systems have been developed with subgenomic replicons. The structural proteins were expressed in trans using helper replicons, helper plasmids, stable cell lines or viral vectors. Although the respective efficiencies of these systems cannot be compared directly, the titres of trans‐packaged infectious particles (around 102 FFU/mL) were generally lower than wt particle titres (about 103–104 FFU/mL for the JFH1 strain). Nonetheless, these titres have been improved by the introduction of adaptive mutations that favour particle production with defective replicons or structural regions expressed in trans. However, some limits exist with heterologous glycoproteins and intergenotype compatibility seems to be required for some proteins.
Flaviviridae trans‐complementation studies have enabled the production of vaccine‐like preparations based on trans‐packaging particles. In particular, it has been reported that the envelope glycoprotein Erns in classical swine fever virus (a pestivirus) can be complemented in trans by an SK6 cell line that constitutively expresses this protein 38. Pigs vaccinated with these defective viral particles were protected against a lethal challenge with the virulent Brescia strain. This observation opened up new perspectives in the development of modified, live‐attenuated vaccines. However, there are no reports of in vivo vaccine activity or neutralizing antibody production with HCV trans‐complementation particles. One possible explanation is that immunization studies require high infectious titres (in the order to 107 or 108 FFU/mL) but trans‐packaging particle production is low (about 102 FFU/mL). Additional studies are needed to increase pseudo‐infectious particle yields for vaccine assays.
Lastly, HCV trans‐complementation is a natural phenomenon that can be observed with native defective subgenomic RNAs identified in infected patients and encapsidated by helper viruses.
In conclusion, HCV trans‐complementation systems have been a valuable tool for improving our knowledge of the HCV life cycle. Nevertheless, these systems need to be perfected for use in vaccine assays.
Acknowledgements
This study was funded by the Conseil Régional de Picardie (Hepanovir, grant no. AAP08‐12).
References
- 1. Appel N, Herian U, Bartenschlager R. Efficient rescue of hepatitis C virus RNA replication by trans‐complementation with nonstructural protein 5A. J Virol 2005; 79(2): 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khromykh AA, Varnavski AN, Westaway EG. Encapsidation of the flavivirus kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J Virol 1998; 72(7): 5967–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harvey TJ, Anraku I, Linedale R et al Kunjin virus replicon vectors for human immunodeficiency virus vaccine development. J Virol 2003; 77(14): 7796–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones CT, Patkar CG, Kuhn RJ. Construction and applications of yellow fever virus replicons. Virology 2005; 331(2): 247–259. [DOI] [PubMed] [Google Scholar]
- 5. Gehrke R, Ecker M, Aberle SW, Allison SL, Heinz FX, Mandl CW. Incorporation of tick‐borne encephalitis virus replicons into virus‐like particles by a packaging cell line. J Virol 2003; 77(16): 8924–8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanna SL, Pierson TC, Sanchez MD, Ahmed AA, Murtadha MM, Doms RW. N‐linked glycosylation of west nile virus envelope proteins influences particle assembly and infectivity. J Virol 2005; 79(21): 13262–13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puig‐Basagoiti F, Deas TS, Ren P, Tilgner M, Ferguson DM, Shi PY. High‐throughput assays using a luciferase‐expressing replicon, virus‐like particles, and full‐length virus for West Nile virus drug discovery. Antimicrob Agents Chemother 2005; 49(12): 4980–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anraku I, Harvey TJ, Linedale R et al Kunjin virus replicon vaccine vectors induce protective CD8 + T‐cell immunity. J Virol 2002; 76(8): 3791–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pushko P, Parker M, Ludwig GV, Davis NL, Johnston RE, Smith JF. Replicon‐helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo . Virology 1997; 239(2): 389–401. [DOI] [PubMed] [Google Scholar]
- 10. Zhou X, Berglund P, Rhodes G, Parker SE, Jondal M, Liljestrom P. Self‐replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994; 12(16): 1510–1514. [DOI] [PubMed] [Google Scholar]
- 11. Bernardin F, Stramer SL, Rehermann B et al High levels of subgenomic HCV plasma RNA in immunosilent infections. Virology 2007; 365(2): 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwai A, Marusawa H, Takada Y et al Identification of novel defective HCV clones in liver transplant recipients with recurrent HCV infection. J Viral Hepat 2006; 13(8): 523–531. [DOI] [PubMed] [Google Scholar]
- 13. Noppornpanth S, Smits SL, Lien TX, Poovorawan Y, Osterhaus AD, Haagmans BL. Characterization of hepatitis C virus deletion mutants circulating in chronically infected patients. J Virol 2007; 81(22): 12496–12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yagi S, Mori K, Tanaka E et al Identification of novel HCV subgenome replicating persistently in chronic active hepatitis C patients. J Med Virol 2005; 77(3): 399–413. [DOI] [PubMed] [Google Scholar]
- 15. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999; 285(5424): 110–113. [DOI] [PubMed] [Google Scholar]
- 16. Tong X, Malcolm BA. Trans‐complementation of HCV replication by non‐structural protein 5A. Virus Res 2006; 115(2): 122–130. [DOI] [PubMed] [Google Scholar]
- 17. Fridell RA, Qiu D, Valera L, Wang C, Rose RE, Gao M. Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS‐790052. J Virol 2011; 85(14): 7312–7320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lindenbach BD, Evans MJ, Syder AJ et al Complete replication of hepatitis C virus in cell culture. Science 2005; 309(5734): 623–626. [DOI] [PubMed] [Google Scholar]
- 19. Appel N, Zayas M, Miller S et al Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 2008; 4(3): e1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jones DM, Patel AH, Targett‐Adams P, McLauchlan J. The hepatitis C virus NS4B protein can trans‐complement viral RNA replication and modulates production of infectious virus. J Virol 2009; 83(5): 2163–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jirasko V, Montserret R, Appel N et al Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J Biol Chem 2008; 283(42): 28546–28562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yi M, Ma Y, Yates J, Lemon SM. Trans‐complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog 2009; 5(5): e1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miyanari Y, Atsuzawa K, Usuda N et al The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 2007; 9(9): 1089–1097. [DOI] [PubMed] [Google Scholar]
- 24. Kopp M, Murray CL, Jones CT, Rice CM. Genetic analysis of the carboxy‐terminal region of the hepatitis C virus core protein. J Virol 2010; 84(4): 1666–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brohm C, Steinmann E, Friesland M et al Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans‐complementation. J Virol 2009; 83(22): 11682–11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adair R, Patel AH, Corless L, Griffin S, Rowlands DJ, McCormick CJ. Expression of hepatitis C virus (HCV) structural proteins in trans facilitates encapsidation and transmission of HCV subgenomic RNA. J Gen Virol 2009; 90(Pt 4): 833–842. [DOI] [PubMed] [Google Scholar]
- 27. Ishii K, Murakami K, Hmwe SS et al Trans‐encapsidation of hepatitis C virus subgenomic replicon RNA with viral structure proteins. Biochem Biophys Res Commun 2008; 371(3): 446–450. [DOI] [PubMed] [Google Scholar]
- 28. Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 2011; 141(3): 1057–1066. [DOI] [PubMed] [Google Scholar]
- 29. Pacini L, Graziani R, Bartholomew L, De Francesco R, Paonessa G. Naturally occurring hepatitis C virus subgenomic deletion mutants replicate efficiently in Huh‐7 cells and are trans‐packaged in vitro to generate infectious defective particles. J Virol 2009; 83(18): 9079–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. Efficient trans‐encapsidation of hepatitis C virus RNAs into infectious virus‐like particles. J Virol 2008; 82(14): 7034–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Masaki T, Suzuki R, Saeed M et al Production of infectious hepatitis C virus by using RNA polymerase I‐mediated transcription. J Virol 2010; 84(11): 5824–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki R, Saito K, Kato T et al Trans‐complemented hepatitis C virus particles as a versatile tool for study of virus assembly and infection. Virology 2012; 432(1): 29–38. [DOI] [PubMed] [Google Scholar]
- 33. Sugiyama K, Suzuki K, Nakazawa T et al Genetic analysis of hepatitis C virus with defective genome and its infectivity in vitro . J Virol 2009; 83(13): 6922–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corless L, Crump CM, Griffin SD, Harris M. Vps4 and the ESCRT‐III complex are required for the release of infectious hepatitis C virus particles. J Gen Virol 2010; 91(Pt 2): 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li R, Qin Y, He Y et al Production of hepatitis C virus lacking the envelope‐encoding genes for single‐cycle infection by providing homologous envelope proteins or vesicular stomatitis virus glycoproteins in trans. J Virol 2011; 85(5): 2138–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaul A, Woerz I, Meuleman P, Leroux‐Roels G, Bartenschlager R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J Virol 2007; 81(23): 13168–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delgrange D, Pillez A, Castelain S et al Robust production of infectious viral particles in Huh‐7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 2007; 88(Pt 9): 2495–2503. [DOI] [PubMed] [Google Scholar]
- 38. Widjojoatmodjo MN, Van Gennip HG, Bouma A, Van Rijn PA, Moormann RJ. Classical swine fever virus E(rns) deletion mutants: trans‐complementation and potential use as nontransmissible, modified, live‐attenuated marker vaccines. J Virol 2000; 74(7): 2973–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]