

Summary
Hepatitis B virus X protein (HBx) acts as a multifunctional protein that regulates intracellular signalling pathways during HBV infection. It has mainly been studied in terms of its interaction with cellular proteins. Here, we show that HBx induces membrane permeabilization independently of the mitochondrial permeability transition pore complex. We generated mitochondrial outer membrane‐mimic liposomes to observe the direct effects of HBx on membranes. We found that HBx induced membrane permeabilization, and the region comprising the transmembrane domain and the mitochondrial‐targeting sequence was sufficient for this process. Membrane permeabilization was inhibited by nonselective channel blockers or by N‐(n‐nonyl)deoxynojirimycin (NN‐DNJ), a viroporin inhibitor. Moreover, NN‐DNJ inhibited HBx‐induced mitochondrial depolarization in Huh‐7 cells. Based on the results of this study, we can postulate that the HBx protein itself is sufficient to induce mitochondrial membrane permeabilization. Our finding provides important information for a strategy of HBx targeting during HBV treatment.
Keywords: hepatitis B virus X protein, membrane permeabilization, viroporin
Abbreviations
- ANT
adenine nucleotide translocator
- GUV
giant unilamellar vesicles
- HAV
hepatitis A virus
- HBV
hepatitis B virus
- HBx
hepatitis B virus X protein
- HCV
hepatitis C virus
- HIV‐1
human immunodeficiency virus type 1
- HTLV‐1
human T‐cell leukaemia virus type 1
- HTLV‐1
human T‐lymphotropic virus type 1
- LUV
large unilamellar vesicles
- MMP
mitochondrial membrane permeability
- NN‐DNJ
N‐(n‐nonyl)deoxynojirimycin
- VDAC
voltage‐dependent anion channel
1. INTRODUCTION
Hepatitis B virus (HBV) is a major cause of hepatitis, and nearly 240 million people are infected worldwide.1 HBV is associated with hepatic fibrosis, cirrhosis and hepatocellular carcinoma.2, 3 HBV is a DNA virus belonging to the Hepadnaviridae family and encodes 7 proteins: 5 structural and 2 nonstructural proteins. The structural proteins include 3 envelope proteins (small, middle and large surface proteins), capsid protein and polymerase. The nonstructural proteins include HBeAg (secreted e antigen) and HBx (hepatitis B virus X protein).4
HBx is a nonstructural protein with a molecular weight of 17.5 kDa. HBx functions as a multifunctional protein that interacts with various transcription factors and cell cycle‐related regulatory proteins, adjusting the intracellular environment to optimal conditions for viral production.5, 6, 7 Interestingly, its intracellular distribution varies depending on its expression level in cells. For example, HBx was detected in the nucleus at a low expression level, while it was distributed throughout the cytoplasm at a high expression level.6 HBx has been reported to target the mitochondrial outer membrane, but not the endoplasmic reticulum, the plasma membrane or lysosomes.6, 8 Mitochondrial outer membrane‐localized HBx is known to alter mitochondrial morphology, fission and aggregation.9 HBx also induces intracellular ROS production 9, 10 and mitochondrial depolarization in hepatoma cells, leading to disruption of mitochondrial respiration and apoptosis. HBx induces mitochondrial membrane permeability, resulting in cytochrome c release and mitochondrial Ca2+ overload that cause apoptosis.11, 12 Although the role of HBx in the mitochondria is not fully understood, it is clear that mitochondrial outer membrane‐localized HBx exerts a different role than nuclear‐targeted HBx, which functions in transcriptional activation.
The viruses that cause liver inflammation have been reported to encode viroporins. Hepatitis A virus (HAV) 2B and hepatitis C virus (HCV) p7 directly induce membrane permeabilization, collectively termed viroporins.13, 14 These are small, nonstructural proteins forming nonselective channels to transport cations and large molecules and have been proposed to modulate the intracellular electrochemical balance of host cells by inducing membrane permeabilization.13, 15 In a related study, HBV was also shown to induce mitochondrial membrane permeabilization.16 However, the viral protein responsible for membrane permeabilization in HBV was not yet fully understood. Here, we explored a novel function for HBx, demonstrating that HBx induces membrane permeabilization independently of mitochondrial proteins.
2. MATERIALS AND METHODS
2.1. Chemicals
Egg L‐α‐phosphatidylcholine (PC), egg L‐α‐phosphatidylethanolamine (PE), liver L‐α‐phosphatidylinositol (PI), brain L‐α‐phosphatidylserine (PS), egg L‐α‐phosphatidic acid (PA) and bovine heart cardiolipin (CL) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 1, 2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine labelled with ATTO 390 (ATTO 390‐DOPE, ATTO fluorescent dye‐labelled phospholipids) was purchased from ATTO (Tokyo, Japan). All other chemicals and anti‐Flag antibody (F3165) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Anti‐HBx antibody (ab2741) was purchased from Abcam (Cambridge, MA, USA). HBx deletion mutants chemically synthesized and purified by high‐performance liquid chromatography were obtained from GL Biochem (Shanghai, China).
2.2. GUV preparation
Ten μL of a mitochondrial outer membrane‐mimic lipid mixture (egg PC: egg PE: liver PI: brain PS: egg PA: heart CL = 54: 29: 13: 2: 1: 1 [weight ratio] dissolved in chloroform: methanol = 4:1 [2 mg/mL]) was dried on indium tin oxide (ITO)‐coated glass at 50°C, and the chamber was filled with 200 mmol L−1 sorbitol. Giant unilamellar vesicles (GUVs) were produced by the application of 2 V peak‐to‐peak and 8 Hz for 2 hours at 36°C using Vesicle Prep Pro (Nanion Technologies GmbH, Munich, Germany). For HBx targeting of GUVs, GUVs were prepared with 0.5% ATTO 390‐DOPE.
2.3. Isolation of the recombinant HBx protein
The HBx protein was obtained as described previously with some modifications.17 Briefly, pET28A‐6x his tag‐HBx was transformed into Escherichia. coli BL21 (DE3) and incubated with 0.5 mmol L−1 isopropyl‐1‐thio‐β‐D‐galactoside (IPTG) for 4 hours at 25°C. Inclusion bodies were harvested and lysed with 8 mol L−1 urea, 10 mmol L−1 dithiothreitol (DTT) and 100 mmol L−1 Tris/NaOH (pH 8) overnight. The HBx protein was purified by nickel‐nitrilotriacetate (Ni‐NTA) agarose (Qiagen, Valencia, CA, USA) and then purified by a PD‐10 desalting column (GE Healthcare, Buckinghamshire, UK). Purified 6x his tag‐HBx was refolded as described previously.17
2.4. LUV permeabilization assay
For large unilamellar vesicles (LUVs) preparation, 50 μL of a MOM‐mimic lipid mixture (10 mg/ mL) was dried and rehydrated with 500 μL of 100 mmol L−1 carboxyfluorescein (CF), 100 mmol L−1 KCl and 10 mmol L−1 HEPES/KOH (pH 7.4). Multilamellar liposomal suspensions were extruded with a 0.1‐μm polycarbonate membrane using an Avanti Mini Extruder (Avanti Polar Lipids) and purified by a PD‐10 column (GE Healthcare). The recombinant HBx protein was added to CF‐enclosed large unilamellar vesicles (CF‐LUVs) in external buffer (100 mmol L−1 KCl and 10 mmol L−1 HEPES/Tris [pH 7.4]), and the CF effluent values were measured using Fluroskan Ascent FL (Thermo Labsystems, UK). CF leakage was calculated using the following formula: , where F = measured fluorescence intensity, F 0 = basal LUVs fluorescence intensity and F max = LUVs treated with 0.5% Triton X‐100.
2.5. Measurement of mitochondrial membrane potential (ΔΨm)
Huh‐7 cells were purchased from the Korea Cell Line Bank (Seoul, Korea). After Huh‐7 cells were transfected with pcDNA3.1 or pcDNA3.1‐Flag HBx, the cells were treated with N‐(n‐nonyl)deoxynojiricin (NN‐DNJ) for 18 hours. For measuring ΔΨm, cells were incubated with 20 nmol L−1 (tetramethylrhodamine ethyl ester) TMRE at 37°C in the dark for 20 minutes. The fluorescence intensity was analysed using a FACSCalibur apparatus (BD Biosciences, San Jose, CA, USA) and quantified by FlowJo software (FlowJo LLC, Ashland, OR, USA). The pcDNA3.1‐Flag HBx was kindly provided by Ding Xue (Addgene plasmid # 42596).
3. RESULTS
3.1. The recombinant HBx protein targets mitochondrial outer membrane‐mimic liposomes
HBx has mainly been studied as a transactivation factor, which interacts with several cellular proteins in host cells. However, the function of HBx on the mitochondrial outer membrane has not been well evaluated. As the exact function of HBx on the mitochondrial membrane is challenging to evaluate in cells, we attempted to investigate HBx function using a recombinant HBx protein and an artificial membrane system. We synthesized recombinant HBx protein in E. coli and recovered its biological activity through a rational refolding method, as previously reported.17 The purity of the refolded recombinant HBx protein was examined via Coomassie blue staining (>95%), and it was further confirmed by anti‐HBx and anti‐His antibodies (Figure 1A,B). As HBx was shown to be inserted into the mitochondrial outer membrane via its transmembrane domain (residues 53‐70), we generated liposomes with a rat liver mitochondrial outer membrane composition, as previously reported.18, 19 To observe the HBx localization in liposomes, we prepared mitochondrial outer membrane‐mimic giant unilamellar vesicles (MOM‐GUVs) including ATTO 390‐DOPE. We treated the recombinant HBx protein for 2 minutes, and HBx‐targeted MOM‐GUVs were then stained with nickel‐nitrilotriacetate conjugated to ATTO 488 fluorescent dye (NTA‐ATTO 488) for the detection of poly‐His‐tagged HBx by fluorescence microscopy. As expected, the recombinant HBx protein was shown to target the MOM‐GUVs (Figure 1C).
Figure 1.
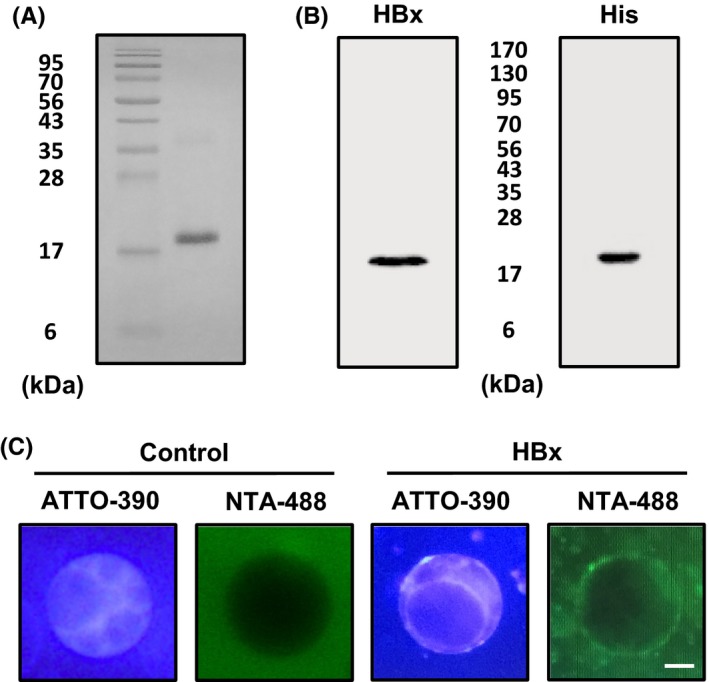
Targeting of the recombinant HBx protein to MOM‐GUVs. A‐B, The purified recombinant 6x‐His‐HBx protein was analysed by Coomassie blue staining and by immunoblot with an anti‐HBx antibody or anti‐His antibody. C, GUVs were prepared at a 54:29:13:2:1:1 molar ratio of PC:PE:PI:PS:PA:CL with 0.5% ATTO 390‐DOPE for the fluorescent labelling of membranes. HBx was treated to GUVs for 5 min, and HBx‐targeted GUVs were incubated with NTA‐ATTO 488 for 5 min to visualize 6x‐His‐HBx on the membranes. The analysis was performed by fluorescence microscopy. The scale bar represents 10 μm
3.2. HBx induces membrane permeabilization
HBx triggers membrane depolarization, probably through interaction with the mitochondrial permeability transition pore.20 In this study, we investigated HBx‐induced membrane permeabilization using MOM‐GUVs without any mitochondrial proteins. First, we created MOM‐GUVs with 200 mmol L−1 sorbitol solution, and they were added to the external buffer containing 100 mmol L−1 KCl and 10 mmol L−1 HEPES/KOH (pH 7.4). Using phase contrast microscopy, we found that MOM‐GUVs became transparent after treatment with HBx in a time‐dependent or concentration‐dependent manner (Figure 2A,B). Loss of contrast around MOM‐GUVs was further analysed by line profiling (Figure 2C). Because MOM‐GUVs were prepared with 200 mmol L−1 sorbitol solution and added to the external buffer containing 100 mmol L−1 KCl and 10 mmol L−1 HEPES/KOH (pH 7.4), this transparency phenomenon implies that the refractive index of the internal solution of MOM‐GUVs became equal to that of the external buffer. This indicates that the internal and external solutions of GUVs were equilibrated through the membrane.
Figure 2.
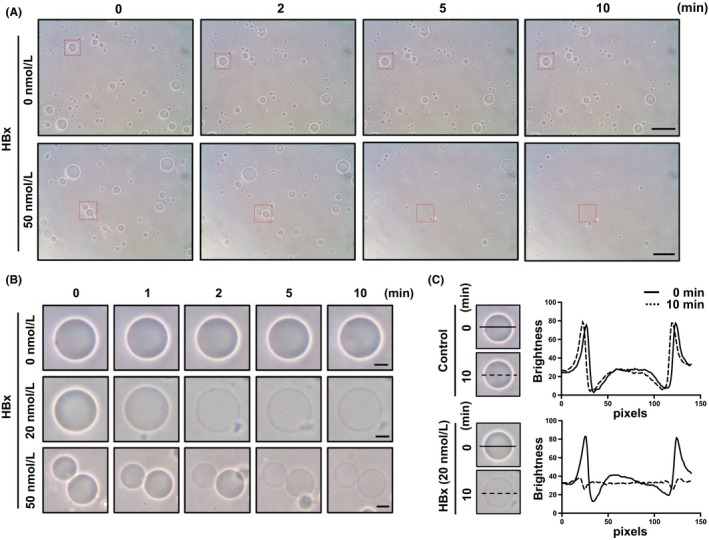
GUV permeabilization induced by HBx. MOM‐GUVs were mixed with external buffer [100 mmol L−1 KCl and 10 mmol L−1 HEPES/KOH (pH 7.4)] and treated with the indicated concentrations of recombinant HBx. A, Images were captured by phase contrast microscopy in a time‐dependent manner. The scale bars represent 50 μm. B, The indicated square regions were magnified. The scale bars represent 10 μm. C, Line profiles of HBx‐treated MOM‐GUVs. The lines represent cross sections of GUVs, and data were analysed by ImageJ software
To quantify the HBx‐induced membrane permeabilization, we performed a liposome permeabilization assay; these are generally used in viroporin studies to evaluate pore‐forming activity.21 As CF is self‐quenched at high concentrations and becomes fluorescent at low concentrations, we made CF‐LUVs with the lipid composition of the mitochondrial outer membrane. Recombinant HBx protein added to CF‐LUVs caused liposome permeabilization in a time‐dependent or concentration‐dependent manner, similar to its activity on GUVs (Figure 3A,B). We also tested the HCV p7 protein (a known cause of membrane permeabilization) as a positive control.19, 22 These results indicated that HBx directly induces membrane permeabilization, independently of other cellular proteins.
Figure 3.
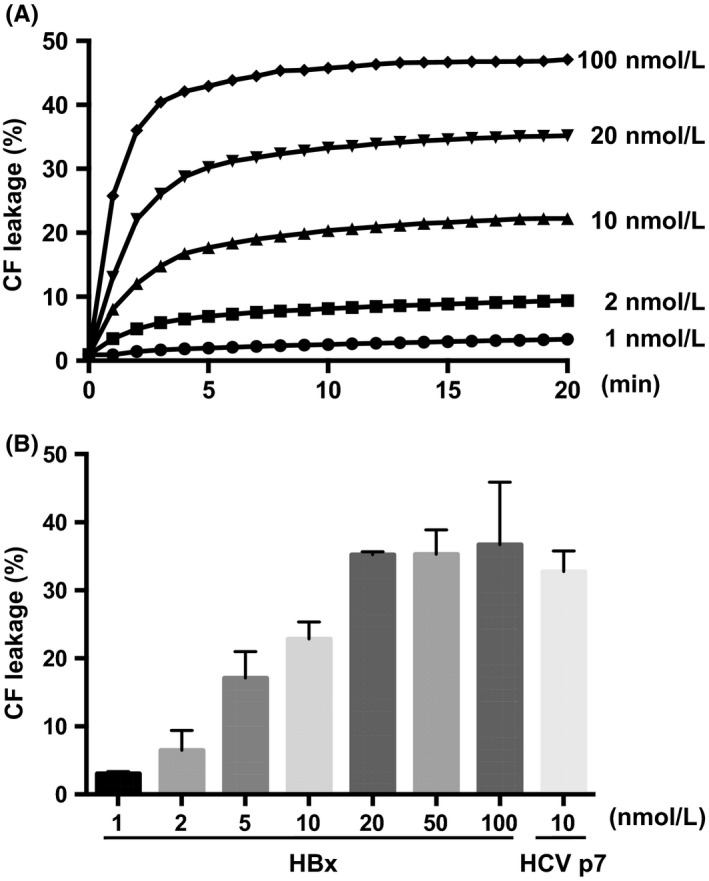
Quantification of HBx‐induced liposome permeabilization. A, Recombinant HBx was added to CF‐enclosed MOM‐LUVs, and the mixture was analysed by spectrophotometry over a time course to measure CF release. B, HBx‐induced liposome permeabilization was quantified at 20 min. HCV p7 was used for a positive control
3.3. HBx‐induced membrane permeabilization is inhibited by nonselective channel blockers and a viroporin inhibitor
We hypothesized that the liposome permeabilization induced by HBx could be nonselective permeabilization by protein‐lipid pore formation, similar to the activity of some viroporins (eg the severe acute respiratory syndrome coronavirus E protein).23 In most cases, viroporin‐induced pore formation is nonselective or weakly selective for cations.23 Therefore, we examined the inhibitory effect of gadolinium ion (Gd3+) and lanthanum ion (La3+), both known as nonselective channel blockers.24, 25 Interestingly, Gd3+ and La3+ effectively inhibited HBx‐induced liposome permeabilization, but Ca2+ and Mg2+ ions did not (Figure 4A,B). The inhibitory effect of Gd3+ (Figure 4C) and La3+ (Figure 4D) was observed in a concentration‐dependent manner.
Figure 4.
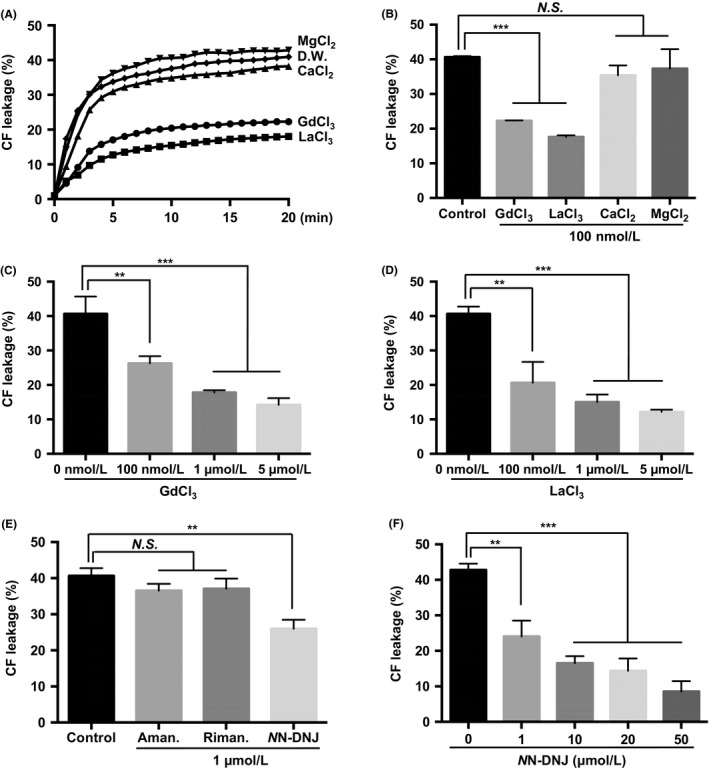
Inhibitory effects of nonselective channel blockers and viroporin inhibitors on HBx‐induced liposome permeabilization. A, CF‐enclosed MOM‐LUVs were incubated with the indicated ions (100 nmol L−1) after exposure to recombinant HBx (20 nmol L−1). B, HBx‐induced liposome permeabilization was quantified at 20 min. Concentration‐dependent inhibitory effects of GdCl3 (C) or LaCl3 (D) on HBx‐induced liposome permeabilization were quantified at 20 min. E, CF‐enclosed MOM‐LUVs were incubated with the indicated viroporin inhibitors (1 μM) after exposure to recombinant HBx (20 nmol L−1) for 20 min. F, Concentration‐dependent inhibitory effects of NN‐DNJ on HBx‐induced liposome permeabilization were quantified at 20 min. **P ≤ 0.01, ***P ≤ 0.001 vs. control by two‑way analysis of variance (ANOVA). N.S.; not significant, Aman., Amantadine; Riman., Rimantadine
We next examined whether the liposome permeabilization caused by HBx was inhibited by well‐known viroporin inhibitors: amantadine, rimantadine and NN‐DNJ. Interestingly, amantadine and rimantadine showed no significant inhibitory effects, while NN‐DNJ efficiently inhibited HBx‐induced liposome permeabilization (Figure 4E). The inhibitory effect of NN‐DNJ was observed in a concentration‐dependent manner (Figure 4F). Taken together, these results indicate that HBx‐induced (nonselective) permeabilization can be inhibited by nonselective channel blockers or by a viroporin inhibitor.
3.4. NN‐DNJ inhibits HBx‐induced mitochondrial depolarization in Huh‐7 cells
In this study, we showed that NN‐DNJ inhibited HBx‐induced liposome permeabilization. Next, we attempted to examine the NN‐DNJ effect on HBx‐induced mitochondrial depolarization in Huh‐7 cells. We transfected HBx into Huh‐7 cells for 6 hours and treated cells with NN‐DNJ for an additional 18 hours (Figure 5A). Mitochondrial membrane potential was quantified using TMRE, a mitochondrial membrane potential indicator. We found that HBx‐induced mitochondrial depolarization was inhibited by NN‐DNJ (Figure 5B,C), and NN‐DNJ did not change the expression level of HBx (Figure 5D). As demonstrated in Figure 4E, amantadine and rimantadine showed no significant inhibitory effects (Figure 5E). These results indicate that HBx‐induced mitochondrial depolarization could be inhibited by NN‐DNJ, and this may be related to the membrane permeabilization induced by HBx.
Figure 5.
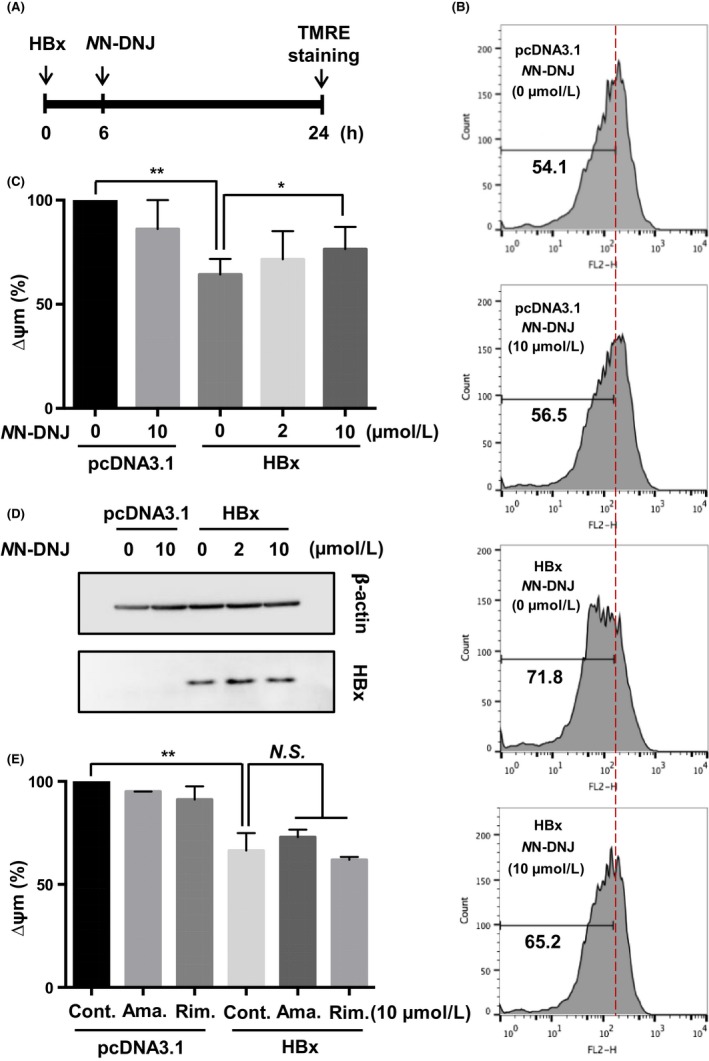
Inhibitory effect of NN‐DNJ on HBx‐induced mitochondrial depolarization. A, Flag HBx was transfected for 6 hrs, and NN‐DNJ was added for an additional 18 hrs. B, C, E, After transfection with vector or HBx for 6 hrs, the Huh‐7 cells were treated with viroporin inhibitors for 18 hrs and then stained with TMRE. Decreased levels of ∆Ψm were analysed by flow cytometry. D, Cell lysates were analysed by immunoblot with anti‐Flag antibody. *P ≤ 0.05, **P ≤ 0.01 vs. control by two‑way analysis of variance (ANOVA). N.S.; not significant, Cont., Control; Ama., Amantadine; Rim., Rimantadine
3.5. The region of HBx with residues 53‐117 is responsible for membrane permeabilization
We investigated the specific region of HBx that accounted for liposome permeabilization. We hypothesized that HBx‐induced membrane permeabilization was not dependent on the transactivation region. Considering that the hydrophobic region of cytochrome c has been shown to insert into membranes to form lipidic pores, we synthesized an HBx deletion mutant (53‐117) without the transactivation domain but with the transmembrane domain (Figure 6A). As expected, the 53‐117 mutant of HBx induced liposome permeabilization‐like wild‐type HBx (Figure 6B). Nonselective channel inhibitors (Gd3+ and La3+) (Figure 6C) and NN‐DNJ (Figure 6D) showed inhibitory effects on the membrane permeabilization induced by the 53‐117 mutant of HBx, but they only partially blocked liposome permeabilization (in contrast to the more robust inhibition of permeabilization seen with wild‐type HBx). These results indicate that residues 53‐117 of HBx are required for membrane permeabilization and that HBx‐induced membrane permeabilization is independent of its transactivation function.
Figure 6.
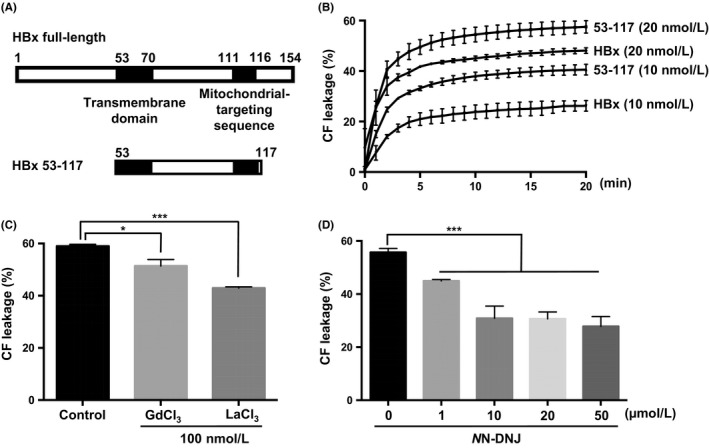
Effect of HBx residues 53‐117 on liposome permeabilization. A‐B, A deletion mutant of HBx (53‐117) containing the transmembrane domain and mitochondrial‐targeting sequence was added to CF‐enclosed MOM‐LUVs and analysed by spectrophotometry over a time course to measure CF release. CF‐enclosed MOM‐LUVs were incubated with (C) the indicated nonselective channel blockers (100 nmol L−1) or (D) NN‐DNJ after exposure to the 53‐117 mutant (20 nmol L−1) for 20 min, and CF release was measured by spectrophotometry *P ≤ 0.05, ***P ≤ 0.001 vs. control by two‑way analysis of variance (ANOVA).
4. DISCUSSION
Viruses alter the function of cellular organelles to enhance viral replication and escape from host cells. One of these organelles is the mitochondrion, and the modulation of this organelle is indispensable for the viral life cycle.26 Mitochondria have a diverse set of functions including energy production, apoptosis and metabolite supply.27 One of the main events during apoptosis is mitochondrial depolarization, which is caused by the opening of a permeability transition (PT) pore.20 The PT pore includes adenine nucleotide translocator (ANT) protein, voltage‐dependent anion channel (VDAC) protein and matrix cyclophilin D protein.28 A variety of viruses control the PT pore to trigger apoptosis. The influenza A virus PB1‐F2 protein interacts with VDAC1 and ANT3 to decrease mitochondrial membrane permeability (MMP), inducing apoptosis by the release of pro‐apoptotic proteins.29 The human immunodeficiency virus type 1 (HIV‐1) viral protein R (Vpr), the Walleye dermal sarcoma virus Orf C protein and the human T‐lymphotropic virus type 1 (HTLV‐1) p13II protein have all been reported to localize in the mitochondria and modulate mitochondrial proteins to induce apoptosis.30, 31, 32 In contrast, some viruses directly induce mitochondrial depolarization through viral‐encoded viroporins. HCV p7, human T‐cell leukaemia virus type 1 (HTLV‐1) p13 and picornavirus 2B proteins detected in mitochondria all induce mitochondrial depolarization.33, 34 In the case of HCV p7, the protein forms hexameric complexes that act as ion channels permeable to cations to enhance the entry and escape of viral particles.19, 35, 36 HBx also reduces the transmembrane potential and induces apoptosis by interacting with VDAC3.20 HBx mainly has been studied in terms of its regulation of signalling cascades, including its interaction with several cellular proteins.
In this study, we evaluated the function of HBx on biological membranes and demonstrated that HBx induces membrane permeabilization. This effect was inhibited by a viroporin inhibitor and by nonselective cation channel blockers (Figure 4). As an artificial membrane system without any cellular proteins was used, we hypothesize that this membrane permeabilization is mediated by HBx only. We excluded the possibility of HBx as an ion channel protein (viroporin) because its predicted secondary structure is far different from that of the viroporins, which have 1 or 2 alpha‐helical transmembrane domains. We questioned the means by which HBx could trigger membrane permeabilization in this artificial liposome system, but to elucidate the exact mechanism, more extensive studies are required. However, we suggest 1 possibility based on the previous studies in which cytochrome c, the protein of the mitochondrial electron transport chain, was reported to induce membrane permeabilization and it shows (or was suggested to have) the “channel‐pore dualism,” which is regarded as a viroporin phenomenon.37, 38 We found that HBx permeated CF, which has a 0.6‐nm Stokes radius (Figure 3). However, HBx is not classified as an ion channel protein.38 Cytochrome c interacts with specific lipids of the mitochondrial inner membrane to induce lipid pore formation.38 It was thus shown to induce membrane permeabilization, releasing CF and 10‐kDa dextran from liposomes. It has been proposed that the hydrophobic region of cytochrome c affects the formation of lipid pores by its insertion into the acyl chains of the lipid bilayer. It is possible that HBx forms a protein‐lipid pore with viroporin‐like activity, and that it induces membrane permeabilization similar to cytochrome c. Consistent with the structural features of cytochrome c, HBx also has a hydrophobic region (residues 54‐70) that may serve as a transmembrane domain for insertion into the mitochondrial outer membrane. It is possible that membrane permeabilization by HBx is similar to that of cytochrome c. These results indicate that HBx may form a nonselective pore to pass ions and small molecules through the mitochondrial outer membrane. However, the mitochondrial outer membrane has a permeability threshold that allows molecules below 1.5 kDa to pass via pore‐forming channels (such as those created by VDAC).39 While these pore‐forming channels are regulated by various cellular factors, HBx‐induced mitochondrial outer membrane permeabilization may not be modulated, thereby inducing uncontrolled membrane permeabilization followed by cell death. Subsequent studies are needed to determine the way in which HBx directly or indirectly affects mitochondrial outer membrane permeability.
In this study, we tested 3 viroporin inhibitors (amantadine, rimantadine and NN‐DNJ). Interestingly, HBx‐induced membrane permeabilization was not inhibited by adamantane derivatives (amantadine and rimantadine), whereas NN‐DNJ inhibited HBx‐induced membrane permeabilization independently of the mitochondrial permeability transition pore complex (Figure 4). Membrane permeabilization induced by the HBx protein was observed in the artificial membrane system, and it now needs to be confirmed in cells. However, it is difficult to confirm this finding in cells because the mechanism behind HBx‐induced mitochondrial dysfunction is very complex and has not been well elucidated yet. Instead, we showed that NN‐DNJ inhibited HBx‐caused mitochondrial depolarization in Huh‐7 cells (Figure 5). Extensive studies remain to fully explain the direct role of HBx in triggering mitochondrial depolarization.
NN‐DNJ has low cytotoxicity and has potential to be a therapeutic agent for HBV.40, 41 NN‐DNJ has been used to inhibit HBV, and its inhibitory concentration 50 (IC50) is in the range of 1‐10 μM. Similarly, our results showed that 10 μM of NN‐DNJ was sufficient to inhibit HBx‐induced mitochondrial depolarization. From the combined results, we can suggest that the NN‐DNJ effect on HBV may be related to membrane permeabilization by HBx, and additional experiments to support this hypothesis are needed. In conclusion, we have demonstrated that HBx induces membrane permeabilization, and that this effect is inhibited by 1 viroporin inhibitor and by 2 triply‐charged lanthanides. It is possible that other viroporin inhibitors or nonselective channel blockers also inhibit HBx activity. In the present study, we identified a novel function for HBx, demonstrating its direct role in inducing mitochondrial membrane permeabilization. Based on the results obtained in this study, we propose that HBx‐induced membrane permeabilization may be an attractive therapeutic target for HBV treatment.
Lee H‐R, Cho YY, Lee GY, You D‐G, Yoo YD, Kim YJ. A direct role for hepatitis B virus X protein in inducing mitochondrial membrane permeabilization. J Viral Hepat. 2018;25:412–420. 10.1111/jvh.12831
Funding information
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C1074) and by grants from the Samjin Pharmaceutical Company (no. 0620171080).
Hye‐Ra Lee and Young Youn Cho contributed equally to this work.
Contributor Information
Y. D. Yoo, Email: ydy1130@korea.ac.kr.
Y. J. Kim, Email: yoonjun@snu.ac.kr.
REFERENCES
- 1. Liang TJ, Block TM, McMahon BJ, et al. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62:1893‐1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liver KAftSot . KASL clinical practice guidelines: management of chronic hepatitis B. Clin Mol Hepatol. 2016;22:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lau JY, Wright TL. Molecular virology and pathogenesis of hepatitis B. Lancet. 1993;342:1311‐1340. [DOI] [PubMed] [Google Scholar]
- 4. Lucifora J, Protzer U. Hepatitis B virus X protein: a key regulator of the virus life cycle. Rijeka, Croatia: INTECH Open Access Publisher; 2012. [Google Scholar]
- 5. Doria M, Klein N, Lucito R, Schneider R. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995;14:4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henkler F, Hoare J, Waseem N, et al. Intracellular localization of the hepatitis B virus HBx protein. J Gen Virol. 2001;82:871‐882. [DOI] [PubMed] [Google Scholar]
- 7. Kim S‐J, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013;9:e1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takada S, Shirakata Y, Kaneniwa N, Koike K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene. 1999;18:6965‐6973. [DOI] [PubMed] [Google Scholar]
- 9. Kim S‐J, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9:e1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zou L‐Y, Zheng B‐Y, Fang X‐F, et al. HBx co‐localizes with COXIII in HL‐7702 cells to upregulate mitochondrial function and ROS generation. Oncol Rep. 2015;33:2461‐2467. [DOI] [PubMed] [Google Scholar]
- 11. Kremsdorf D, Soussan P, Paterlini‐Brechot P, Brechot C. Hepatitis B virus‐related hepatocellular carcinoma: paradigms for viral‐related human carcinogenesis. Oncogene. 2006;25:3823‐3833. [DOI] [PubMed] [Google Scholar]
- 12. Clippinger AJ, Gearhart TL, Bouchard MJ. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF‐κB and the mitochondrial permeability transition pore. J Virol. 2009;83:4718‐4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10:563‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shukla A, Dey D, Banerjee K, Nain A, Banerjee M. The C‐terminal region of the non‐structural protein 2B from Hepatitis A Virus demonstrates lipid‐specific viroporin‐like activity. Sci Rep. 2015;5:15884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez ME, Carrasco L. Viroporins. FEBS Lett. 2003;552:28‐34. [DOI] [PubMed] [Google Scholar]
- 16. Tan C, Guo H, Zheng M, Chen Y, Huang W. Involvement of mitochondrial permeability transition in hepatitis B virus replication. Virus Res. 2009;145:307‐311. [DOI] [PubMed] [Google Scholar]
- 17. Basu A, Chen WN, Leong SSJ. A rational design for hepatitis B virus X protein refolding and bioprocess development guided by second virial coefficient studies. Appl Microbiol Biotechnol. 2011;90:181‐191. [DOI] [PubMed] [Google Scholar]
- 18. Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic. 2015;16:1‐18. [DOI] [PubMed] [Google Scholar]
- 19. Lee GY, Lee S, Lee H‐R, Do YY. Hepatitis C virus p7 mediates membrane‐to‐membrane adhesion. Biochim Biophys Acta. 2016;1861:1096‐1101. [DOI] [PubMed] [Google Scholar]
- 20. Rahmani Z, Huh K‐W, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage‐dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol. 2000;74:2840‐2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gervais C, Dô F, Cantin A, et al. Development and validation of a high‐throughput screening assay for the hepatitis C virus p7 viroporin. J Biomol Screen. 2011;16:363‐369. [DOI] [PubMed] [Google Scholar]
- 22. StGelais C, Tuthill TJ, Clarke DS, Rowlands DJ, Harris M, Griffin S. Inhibition of hepatitis C virus p7 membrane channels in a liposome‐based assay system. Antiviral Res. 2007;76:48‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nieto‐Torres JL, Verdiá‐Báguena C, Castaño‐Rodriguez C, Aguilella VM, Enjuanes L. Relevance of viroporin ion channel activity on viral replication and pathogenesis. Viruses. 2015;7:3552‐3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kovacs T, Csakany B, Török A, Csillik B. Effect of lanthanum‐induced blockade of calcium channels on nerve regeneration. J Hirnforsch. 1990;32:195‐201. [PubMed] [Google Scholar]
- 25. Ermakov YA, Kamaraju K, Sengupta K, Sukharev S. Gadolinium ions block mechanosensitive channels by altering the packing and lateral pressure of anionic lipids. Biophys J. 2010;98:1018‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anand SK, Tikoo SK. Viruses as modulators of mitochondrial functions. Adv Virol. 2013;2013:738794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815‐833. [DOI] [PubMed] [Google Scholar]
- 29. Zamarin D, García‐Sastre A, Xiao X, Wang R, Palese P. Influenza virus PB1‐F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005;1:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ciminale V, Zotti L, D'Agostino DM, et al. Mitochondrial targeting of the p 13 II protein coded by the x‐II ORF of human T‐cell leukemia/lymphotropic virus type I(HTLV‐I). Oncogene. 1999;18:4505‐4514. [DOI] [PubMed] [Google Scholar]
- 31. Jacotot E, Ravagnan L, Loeffler M, et al. The HIV‐1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191:33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nudson WA, Rovnak J, Buechner M, Quackenbush SL. Walleye dermal sarcoma virus Orf C is targeted to the mitochondria. J Gen Virol. 2003;84:375‐381. [DOI] [PubMed] [Google Scholar]
- 33. Silic‐Benussi M, Marin O, Biasiotto R, D'Agostino DM, Ciminale V. Effects of human T‐cell leukemia virus type 1 (HTLV‐1) p13 on mitochondrial K+ permeability: a new member of the viroporin family? FEBS Lett. 2010;584:2070‐2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su Y‐C, Hong J‐R. Betanodavirus B2 causes ATP depletion‐induced cell death via mitochondrial targeting and complex II inhibition in vitro and in vivo. J Biol Chem. 2010;285:39801‐39810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pavlović D, Neville DC, Argaud O, et al. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long‐alkyl‐chain iminosugar derivatives. Proc Natl Acad Sci. 2003;100:6104‐6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griffin SD, Harvey R, Clarke DS, Barclay WS, Harris M, Rowlands DJ. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine‐sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J Gen Virol. 2004;85:451‐461. [DOI] [PubMed] [Google Scholar]
- 37. Mehnert T, Routh A, Judge P, et al. Biophysical characterization of Vpu from HIV‐1 suggests a channel‐pore dualism. Proteins: Struct, Funct, Bioinf. 2008;70:1488‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bergstrom CL, Beales PA, Lv Y, Vanderlick TK, Groves JT. Cytochrome c causes pore formation in cardiolipin‐containing membranes. Proc Natl Acad Sci. 2013;110:6269‐6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rostovtseva TK, Bezrukov SM. VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr. 2008;40:163‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Block TM, Lu X, Platt FM, et al. Secretion of human hepatitis B virus is inhibited by the imino sugar N‐butyldeoxynojirimycin. Proc Natl Acad Sci. 1994;91:2235‐2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mehta A, Ouzounov S, Jordan R, et al. Imino sugars that are less toxic but more potent as antivirals, in vitro, compared with Nn‐nonyl DNJ. Antiviral Chem Chemother. 2002;13:299‐304. [DOI] [PubMed] [Google Scholar]