

Abstract
Respiratory infection is extremely common and a major cause of morbidity and mortality worldwide. The airway epithelium has an important role in host defense against infection and this is illustrated in this review by considering infection by respiratory viruses. In patients with asthma or chronic obstructive pulmonary disease, respiratory viruses are a common trigger of exacerbations. Rhinoviruses (RV) are the most common virus type detected. Knowledge of the immunopathogenesis of such RV‐induced exacerbations remains limited, but information is available from in vitro and from in vivo studies, especially of experimental infection in human volunteers. RV infects and replicates within epithelial cells (EC) of the lower respiratory tract. EC are an important component of the innate‐immune response to RV infection. The interaction between virus and the intracellular signaling pathways of the host cell results in activation of potentially antiviral mechanisms, including type 1 interferons and nitric oxide, and in the producton of cytokines and chemokines [interleukin (IL)‐1β, IL‐6, IL‐8, IL‐11, IL‐16, tumor necrosis factor α, granulocyte macrophage‐colony stimulating factor, growth‐regulated oncogene‐α, epithelial neutrophil‐activating protein‐78, regulated on activation, normal T expressed and secreted, eotaxin 1/2, macrophage‐inflammatory protein‐1α], which influence the subsequent induced innate‐ and specific‐immune response. Although this is beneficial in facilitating clearance of virus from the respiratory tract, the generation of proinflammatory mediators and the recruitment of inflammatory cells result in a degree of immunopathology and may amplify pre‐existing airway inflammation. Further research will be necessary to determine whether modification of EC responses to respiratory virus infection will be of therapeutic benefit.
Keywords: asthma, COPD, respiratory virus infections, immune response
INTRODUCTION
Infection of the respiratory tract is extremely common. It may involve bacteria, viruses, fungi, or combinations of these pathogens. This review will focus on viral infections. Respiratory viral syndromes include common cold, pharyngitis, tracheobronchitis, croup, bronchiolitis, or pneumonia, and viruses implicated include influenza, parainfluenza, enteroviruses, adenovirus, respiratory syncytial virus (RSV), rhinoviruses (RV), and coronaviruses. It is likely that additional viruses exist but await identification. A human metapneumovirus [1] was recently discovered to be the cause of symptoms in young children ranging from upper respiratory tract (URT) disease to severe bronchiolitis and pneumonia. In a subsequent Japanese seroprevalence study for metapneumovirus, all children had been exposed by age 10 [2]. Further attention has been focused on respiratory virus infection as a result of the current epidemic of severe acute respiratory syndrome, for which the causative agent appears to be a novel coronavirus [3].
Pathology resulting from virus infection is influenced by host factors, including age, previous infection or immunization, pre‐existing respiratory or systemic disease, and immunosuppression/compromise [4]. Disease severity is dependent on direct, harmful effects of the virus and tissue damage as a result of the host antiviral immune response. Some immunopathology may be unavoidable if the virus is to be eradicated. An ideal immune response results in early elimination of virus with a minimum of harm to the host.
Virus infections are a major trigger of exacerbations of obstructive airways diseases such as asthma and chronic obstructive pulmonary disease (COPD), and new treatments are urgently needed to reduce the considerable associated morbidity and mortality. Just how the immunopathology of respiratory virus infection interacts with the pre‐existing immunopathology of such diseases is currently not well understood. Data concerning viral immunopathology are available from studies of natural infection, from in vitro studies predominantly concentrating on epithelial cell (EC) culture or preparations of peripheral blood mononuclear cells (PBMC), and from in vivo experimental infection using animal models or human volunteers. RV are the most frequent virus type identified during exacerbations, and RV infections in asthma and COPD are the major focus of our center's research. This article will concentrate on data available for RV infection in normal, healthy individuals and in patients with obstructive airways diseases, but it must be emphasized that the immunopathogenesis of disease as a result of other virus types may be very different. The response of cells of the epithelium of the respiratory tract following virus infection will have a major influence on the subsequent host‐immune response, and this will be considered in detail in this review.
EPIDEMIOLOGY
Acute wheezing illness in infancy
Infection by respiratory viruses is a common cause of wheezing episodes in infancy and as discussed below, of exacerbations in asthmatic children. Exposure to viruses is reflected in the rapid accumulation of antibodies to RV in particular [5] and RSV [6, 7] in the first years of life. Seventy percent of wheezing episodes in the first year are associated with viral respiratory infection [8]; RSV, RV, and influenza B are most frequently cultured [9]. In some children, RSV causes bronchiolitis, a potentially serious lower respiratory tract (LRT) illness with a significant mortality [10], which is associated with an increased risk of subsequent asthma [11]. The potential causative and/or protective role of viruses in the development of subsequent asthma remains controversial and has been recently reviewed [12, 13].
Respiratory viruses as triggers of asthma exacerbations in older children and adults
Asthma is a disease of major importance affecting 20–33% of children in the United Kingdom. The health costs of this condition are enormous in terms of time off school, GP consultations, hospital admissions, and mortality. Viral infections are a major asthma trigger in older children and adults. Their role may have been underestimated in early studies, limited to viral culture and serology and lacking sensitive methods for detection of RV and coronaviruses. The introduction of molecular biology [polymerase chain reaction (PCR), in situ hybridization, and in situ PCR] has implicated viruses in the majority of exacerbations. Direct evidence implicating viruses comes from studies showing increased detection during asthma attacks [9, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34]. The highest rates of detection are from prospective studies with sampling as soon as symptoms commence. In a 1‐year longitudinal study performed in Southampton, UK, upper respiratory viral infections were associated with 80–85% of asthma exacerbations in school‐age children. One hundred eight children, aged 9–11, were selected by questionnaire on the basis of episodic wheeze or cough, and diary cards were used to monitor respiratory symptoms and peak flows. Viruses were detected in 80% of reported episodes of reduced peak expiratory flow (PEF), in 80% of reported episodes of wheeze, and in 85% of reported episodes of upper respiratory symptoms, cough, wheeze, and a fall in PEF. The most commonly identified virus type was RV. In a related study, viral URT infections (URTI) were strongly associated in time with hospital admissions for asthma in children and adults [35]. The half‐monthly rates of URTI were compared with the half‐monthly rates for hospital asthma admissions for the same time period for the hospitals serving the areas from which the cohort of schoolchildren was drawn. Strong correlations were found between the seasonal patterns of URTI and hospital asthma admissions; this relationship was stronger for pediatric than for adult admissions. URTI and asthma admissions were more frequent during periods of school attendance (87% of pediatric and 84% of total admissions) than during the school holidays. Up to 44% of exacerbations in adult asthmatics was associated with the presence of respiratory virus infection [36, 37]. Virus detection between exacerbations when asymptomatic is only 3–12%. In contrast, transtracheal aspirates in adult exacerbations [38] yield sparse bacterial cultures with no correlation to clinical illness and no difference from normal subjects. If an asthmatic child develops infection in which RV, coronavirus, or RSV is cultured, the risk of associated asthma attack is 50–70% [39]. In asthmatics, the predominant viruses are RV, RSV, and parainfluenza. RV is detected in 50% of virus‐induced asthma attacks. Adenoviruses, enteroviruses, and coronaviruses are also detected but less frequently. Influenza is found during annual epidemics. Although metapneumovirus has been associated with URT symptoms, it has not been associated with asthma in a small study of children with asthma [40].
People with atopic asthma are not at greater overall risk of RV infection than healthy individuals but suffer from more frequent LRTI and have more severe and longer‐lasting LRT symptoms [41]. Seventy‐six cohabiting couples were recruited in Southampton, UK; one person in every couple had atopic asthma, and one was healthy. Participants completed daily diary cards of URT and LRT symptoms and measured PEF twice daily. Every 2 weeks, nasal aspirates were taken and examined for RV, which was detected in 10.1% of samples from participants with asthma and 8.5% of samples from healthy participants. Groups did not differ in the frequency of URTI or the severity or duration of symptoms associated with RV infection. However, first, RV infection was associated more frequently with LRTI in participants with asthma than in healthy individuals, and LRT symptoms associated with RV infection were significantly more severe and longer‐lasting in asthmatics than in healthy controls.
COPD
Increasing interest in the clinical features and pathogenesis of COPD reflects the worldwide importance of the disease. More than 14 million patients are affected in the United States alone. It is predicted to become the third leading cause of death worldwide by 2020. National and global initiatives have been launched, and management guidelines have been published [42]. Fewer studies have examined the epidemiology of virus infection in COPD. Many exacerbations of COPD occur without the hallmarks of bacterial infection‐increased volume or purulence of sputum. Between 33% and 70% of exacerbations are associated with symptoms of the common cold. The frequency of exacerbations requiring hospitalization is higher in the winter, and one explanation for this could be the increased frequency of respiratory viruses at this time of the year. A recent study followed COPD patients attending the London Chest Hospital over a 16‐month period [43]. The effects of respiratory viral infection on the time course of COPD exacerbation were examined by monitoring changes in systemic inflammatory markers in stable COPD and at exacerbation. Eighty‐three patients with COPD recorded daily PEF and any increases in respiratory symptoms. Nasal samples and blood were taken for respiratory virus detection by culture, PCR, and serology, and plasma fibrinogen and serum interleukin (IL)‐6 were determined at stable baseline and exacerbation. Sixty‐four percent of exacerbations were associated with a cold, occurring up to 18 days before exacerbation. Seventy‐seven viruses (58.2% RV) were detected in 39.2% of 168 COPD exacerbations in 53 patients. Again, earlier studies relying on serology and virus culture have quoted lower virus detection rates of 15–20% [44, 45, 46, 47]. Viral exacerbations were associated with increased dyspnea, a higher total symptom count at presentation, a longer median symptom recovery period of 13 days, and a tendency toward higher plasma fibrinogen and serum IL‐6 levels. Non‐RSV respiratory viruses were detected in 16% and RSV, in 23.5% of 68 stable COPD patients. Where RSV was identified in patients with stable COPD, these individuals had a higher plasma fibrinogen and serum IL‐6, a higher pCO2, and increased frequency of exacerbations. This suggests that low‐grade, persistent RSV infection contributes to COPD severity, patients with more severe COPD are less able to clear RSV from the airway, or that in some patients, RSV is resulting in detectable, inflammatory changes but no significant symptoms.
Persistent infection with other viruses has been associated with the progression of COPD. In particular, adenovirus appears to persist in a latent form in which viral proteins are produced without replication of complete virus. Such latent infection may amplify lung inflammation as a result of cigarette smoke [48]. Another example is human immunodeficiency virus (HIV), which appears to predispose to smoking‐induced pulmonary emphysema. In a group of 114 HIV‐seropositive individuals, 15% had high‐resolution computed tomography scan evidence of emphysema as compared with 2% of seronegative controls. If smokers (greater than 12 pack years) only were considered, the respective frequencies were 37% and 0%. Amongst smokers, HIV seropositivity was associated with an increased percentage of CD8+ bronchoalveolar lavage (BAL) lymphocytes [49].
COPD is characterized by an accelerated decline in lung function and periods of acute deterioration in symptoms. Frequent exacerbations of COPD may accelerate the decline in lung function [50]. Over 4 years, PEF, forced expiratory volume in 1 s (FEV1), and symptoms were measured at home daily by 109 patients with COPD. Patients with frequent exacerbations had a significantly faster decline in FEV1 and PEF than infrequent exacerbators and were more often admitted to the hospital with longer lengths of stay. Frequent exacerbations were a consistent feature within a patient for different years of the study and were associated with an increased rate of bacterial colonization of the lower airway when stable [51].
The immunology of virus infection in COPD is not well understood. Less data are available than for virus infection in asthma, as this has not yet been the subject of human experimental infection studies. Such studies are clearly needed in view of the increasing evidence for a major role for viruses in causing COPD exacerbations.
EXPERIMENTAL RV INFECTION IN ASTHMA
Respiratory virus infection in the nasal mucosa and URT has been investigated extensively [52, 53]. Recently, the effects of viruses in the LRT have been studied, but detailed knowledge of the pathogenetic mechanisms involved remains limited. Experimental virus infection in humans is limited by safety concerns [54]. Most studies focus on experimental inoculation of RV in allergic rhinitic, mild asthmatic individuals, or normal controls [55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67]. This provides a useful model of natural virus infection in asthma and offers advantages of patient selection and monitoring under controlled conditions before, during, and after infection of RV‐induced effects, including symptomatology, changes in medication use, lung function, airway pathology, and immunology. Overall, clinical, physiological, and cellular responses to experimental RV infection in asthma appear mild in comparison with natural colds. One reason for this is that reporting symptoms during natural virus infection is biased toward severe episodes; mild or subclinical infections are not reported. One disadvantage of such infection models is the selection of subjects with only mild asthma who are perhaps those less likely to show significant changes in lung function or immunology than those with severe disease.
RV infection of the lower airway
Influenza, parinfluenza, RSV, and adenovirus are well‐recognized causes of lower airway syndromes such as pneumonia and bronchiolitis and are capable of replication in the lower airway. Until recently, a problem with the experimental RV infection model was the uncertainty as to whether RV infection occurred in the lower airway as well as in the URT. Although nasopharyngeal contamination cannot be ruled out, RV has been detected in lower airway specimens such as sputum [23], tracheal brushings [64], and BAL [68] by reverse transcriptase‐PCR and culture. RV has been cultured in cell lines of bronchial EC origin [69, 70], and replication has been demonstrated in primary culture of bronchial EC [71, 72]. Preference of RV for culture at 33°C has been used as an argument against lower airway infection, but there is now evidence that replication does occur at temperatures found in the lower airway [73]. In fact, thermistor studies show that only in the peripheral lung does airway temperature rise above 35ºC [74]. Finally, in situ hybridization shows RV in bronchial biopsies of subjects following experimental infection [71], as previously demonstrated in nasal washing cells [75] and nasal biopsies [52]. This evidence strongly supports a direct lower respiratory epithelial reaction as the initial event in the induction of RV‐mediated asthma exacerbations.
Physiological effects of experimental RV infection
In normal, healthy volunteers, it is difficult to detect changes in lung physiology following experimental RV infection [76]. Subjects with asthma and/or allergic rhinitis exhibit increased pathophysiological effects as a result of RV infection as compared with nonatopic, nonasthmatic controls. With detailed monitoring, it is possible to detect reductions in PEF [77] and home recordings of FEV1 in atopic asthmatic patients in the acute phase of experimental RV16 infection [62]. There is an enhanced sensitivity to histamine and allergen challenge after RV16 inoculation in nonasthmatic, atopic rhinitic subjects [57, 67]. RV16 increases asthma symptoms, coinciding with an increase in the maximal bronchoconstrictive response to methacholine up to 15 days after infection [58]. There, increase in sensitivity to histamine in asthmatic subjects infected with RV16 is most pronounced in those with severe cold symptoms [63].
COMPONENTS OF THE ANTIVIRAL IMMUNE RESPONSE AND THE ROLE OF THE AIRWAY EC
Current concepts of a typical antiviral‐immune response, as reviewed in detail elsewhere [78, 79], result from research in human volunteers and patients but also in experimental animals, especially inbred mice. Results of animal studies may not be directly applicable to the outbred human population, but ethical considerations often limit direct investigation of the human immune system.
A typical response is a combination of nonspecific (innate) and specific immunity. Nonspecific elements include phagocytes such as neutrophils and macrophages, which engulf and destroy viruses; natural killer (NK) cells, which recognize and destroy virus‐infected cells on the basis of reduced human leukocyte antigen class I expression; cells including NK cells, neutrophils, macrophages, mast cells, basophils, and EC, which release cytokines such as interferons (IFNs) with immunoregulatory or antiviral actions; and components of body fluids such as complement, defensins, and surfactant proteins, which are capable of neutralizing viruses independently of or in combination with antibodies.
There is little information on the role of complement in immunity to RV. Recent data suggest that the RV 3C protease cleaves the complement factors C3 and C5, which may interfere with the destuction of virus‐infected cells [80]. For other viruses, for example, influenza, the complement system forms an important link between the innate‐ and specific‐immune systems. Mice deficient for the third component of complement are highly susceptible to primary influenza, showing reduced priming of T helper cells (Th) and cytotoxic T cells in lung‐draining lymph nodes and severely impaired recruitment into the lung of virus‐specific CD4+ and CD8+ effector T cells producing IFN‐γ [81]. Activation of the complement cascade may be necessary for the function of other innate, antiviral proteins such as serum mannose‐binding protein [82].
The α and β defensins are small, cationic, antimicrobial peptides, which have the capacity to kill bacteria, fungi, and enveloped viruses by disruption of the microbial membrane. In vivo, they are probably most important in phagocytic vacuoles and on the surface of skin and mucosal epithelia. In addition to their direct antibiotic role, defensins are increasingly found to have immunomodulatory actions [83] and to play a role in cell recruitment through activation of certain chemokine receptors, for example, hBD3 and CCR6 on dendritic cells (DC).
Specific immunity involves production of antibody by B lymphocytes and the activities of cytotoxic T cells following processing and presentation of viral antigens by additional cells of the immune system, the most important of which are probably DC. Immunological memory modifies the overall response to reinfection by previously encountered virus and alters the timing and magnitude of contributions as a result of different components. In primary infection, virus multiplies in the respiratory tract, reaching peak levels at approximately day 2. At this time, type I IFNs are first detected, peaking at day 3 and falling to become undetectable by day 8. IFNs activate NK cells, first detectable at day 3 and peaking at day 4. In addition to destruction of virally infected cells, NK cells release cytokines including IFN‐γ, which activate additional inflammatory cells in the airway including macrophages. Such nonspecific immune mechanisms are essential in early defense against virus in the first few days. In addition, the innate‐immune system plays a role in stimulating specific immunity and may influence the nature of the specific response, for example, whether this is characterized by type 1 or type 2 cytokines ( Fig. 1 ).
Figure 1.
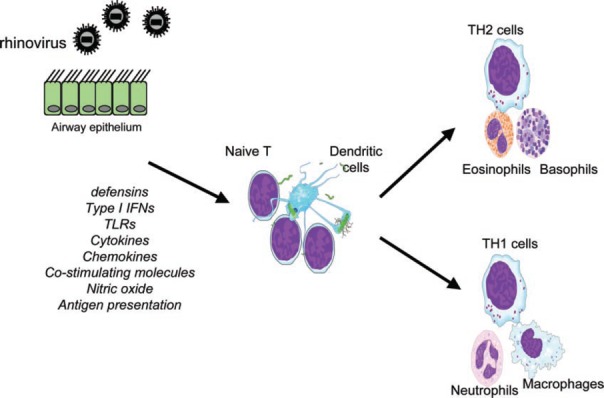
EC can be considered a component of the innate‐immune response to RV infection. Following RV infection, EC respond by the production of mediators with antiviral activity such as type 1 IFNs and nitric oxide (NO). EC production of cytokines and chemokines and the up‐regulation of major histocompatibility complex (MHC) class I and costimulatory molecules (Co stim mols) promote the recruitment of a range of inflammatory cells and through antigen presentation to lymphocytes, stimulate specific immunity. The nature of the EC response to virus infection may influence T cell/DC interaction and type 1/type 2 balance. TLRs, Toll‐like receptors.
Meanwhile, viral antigens are processed locally and in regional lymph nodes by DC and are presented to T cells. CD4+ and CD8+ T cells are detectable at days 4 and 6. CD8+ cytotoxic T cell responses peak at day 7 and then generally decline to become undetectable by day 14. However, memory CD4+ and CD8+ responses may persist for life. T cell recruitment is dependent on the production of chemokines and on alterations in the expression of adhesion molecules on the endothelium of inflamed tissues. Time is also required to generate B cell responses. Mucosal immunoglobulin A (IgA) may be detected at day 3, serum IgM at days 5–6, and IgG at days 7–8, increasing in amount and avidity over the next 2–3 weeks. IgA falls to undetectable levels over 3–6 months. Serum IgG may remain detectable for life. Specific immune mechanisms such as CD8+ T cells and Ig are responsible for the eradication of infectious virus usually by 7 days after infection.
Second infection with the same virus results in rapid mobilization of B and T cell‐specific immunity with an earlier T cell peak coinciding with the NK cell peak at days 3–4. If reinfection is with the same serotype, a rapid increase in levels of pre‐existing, neutralizing antibodies may limit viral replication to such an extent that infection is clinically silent. As this results in fewer infected cells, there is relatively less activation of nonspecific immunity, and it may be difficult to detect a CD8+ T cell response.
Following experimental infection of seronegative subjects with RV2 [84], specific antibodies are detectable at 1–2 weeks, reach a maximum at 5 weeks, persist for at least 1 year, and may remain elevated many years after infection. Local, specific antibody levels may be lost more rapidly. High levels of serum‐neutralizing antibody or specific IgA protect against reinfection with the same RV serotype. However, as it appears relatively late, recovery from illness for seronegative hosts, which usually occurs at 7–10 days, must be a result of other components of the immune response. In seropositive subjects, pre‐existing, serum‐neutralizing antibodies to RV39 and to RV‐Hanks modify experimental infections in human subjects [85, 86]. Local IgA and IgG, passing from the vasculature into the pulmonary interstitium, contribute to viral clearance. However, the large number of serotypes means repeated infection with RV, to which an individual may lack appropriate antibodies, is common.
As a consequence of serotype cross‐reactivity, recurrent infection with RV of different serotypes would be expected to modify T cell responses to subsequent infection, enhancing the antiviral response or in the case of asthmatic subjects, perhaps further amplifying allergic inflammation. T cell responses to RV demonstrate MHC class I‐restricted cross‐reactivity between serotypes as a result of specificity for conserved epitopes within the capsid proteins VP 1–3 [87]. RV16‐ and RV49‐specific T cell clones from human peripheral blood demonstrate recognition of serotype‐specific and shared viral epitopes [88]. Vigorous proliferation of and IFN‐γ production by PBMC in response to RV16 in seronegative subjects are associated with reduced viral shedding after innoculation [89].
The airway epithelium is an important component of antiviral defense. In addition to its function as a physical barrier to the entry of viruses, the responses of EC following viral infection, whether or not this results in destruction of the cell, contribute to innate and adaptive antiviral‐immune responses. Information regarding the effects of RV on EC comes from in vivo studies and from in vitro models using cultured, primary airway EC or transformed cell lines of EC origin such as A549, BEAS‐2B, and H292.
In vivo, RV causes some shedding of infected, ciliated EC, but the extent of shedding does not correlate with symptom scores [90]. In addition, the extent of viral infection of the epithelium may be incomplete even in the nose [91]. In vitro studies exposing monolayer cultures of nasal EC to respiratory viruses at 103–104 50% tissue culture infectious dose/ml demonstrate no detectable cytopathic effect with RV or coronavirus in contrast to the extensive destruction with influenza and adenovirus [92]. Ex vivo infection of cells from the URT and LRT suggests RV infects less than 10% of cells in the epithelium [70]. RV cold symptoms may be produced in part by processes independent of the severity of direct, virus‐induced epithelial damage.
Receptors for entry of RV into host cells
Viruses enter into and replicate within airway EC. Entry is dependent on interaction with host‐cell surface proteins, which function as receptors. In the case of the major group RV, this is intercellular adhesion molecule‐1 (ICAM‐1) [93], and antibodies to ICAM‐1 or with soluble (s)ICAM‐1 can block infection [69]. There is only limited expression of ICAM‐1 in airway epithelium before RV infection [94], and this may explain the patchy nature of infection. RV up‐regulates expression of its own receptor ICAM‐1 in vitro and in vivo. Following experimental infection with RV, ICAM‐1 expression is up‐regulated in nasal epithelium within 24 h, declining by day 5 [95]. RV has similar effects on EC from the lower airway. RV has been shown to up‐regulate ICAM‐1 in primary bronchial EC in vitro [96], and ICAM‐1 is up‐regulated in bronchial biopsies following experimental infection of asthmatic subjects with RV16 [97].
There are two forms of ICAM‐1: murine (m)ICAM‐1, which favors viral infection, and sICAM‐1, which binds and neutralizes virus outside the cell. RV infection of EC alters the balance in favor of further infection by inducing mRNA for mICAM‐1 and suppresses that of sICAM‐1 [98].
The low‐density lipoprotein receptor (LDL‐R) is the receptor for the minor group RV. An antibody to the LDL‐R blocks RV2 infection of primary human tracheal EC (PHTEC). RV2 infection is associated with cytokine induction and up‐regulation of LDL‐R and increases in the transcription factors specificity protein‐1 and nuclear factor (NF)‐κB [99].
ICAM‐1 expression by human nasal EC is up‐regulated in vitro by exposure to a number of inflammatory cytokines and mediators including IL‐1β, IL‐8, IFN‐γ, tumor necrosis factor α (TNF‐α), and the eosinophil‐derived proteins myelin basic protein (MBP) and eosinophil cationic protein (ECP) [100]. IL‐1β, in particular, may be important in RV‐induced induction of ICAM‐1. Antibodies to IL‐1β but not TNF‐α decreased viral replication and ICAM‐1 expression by PHTEC [101]. Not all respiratory EC lines behave in the same way as primary EC‐for example, A549 cells express ICAM‐1 at lower levels constitutively and show up‐regulation by IFN‐γ and TNF‐α but not by ECP or MBP. The effect of IFN‐γ is complex. Although IFN‐γ up‐regulates ICAM‐1 in uninfected cells, this cytokine inhibits ICAM‐1 up‐regulation by RV14 in H292 cells, and its presence results in reduced viral titers [96, 102].
A pre‐existing elevation of ICAM‐1 expression in the asthmatic airway may contribute to increased symptom severity of RV infection. Type 2 cytokines (IL‐4, IL‐5, IL‐10, IL‐13) up‐regulate ICAM‐1 in H‐292 cells [103]. Allergen challenge results in up‐regulation of ICAM‐1 on conjunctival and nasal EC in atopics [104]. In nasal brushing, EC from atopics, basal ICAM‐1 levels were increased relative to nonatopics and elevated in the relevant allergen season. Nasal EC from atopics showed further up‐regulation after in vitro culture with allergen. The highest basal ICAM‐1 was found on nasal polyp EC, and this was increased further after RV14 infection. Viral titers after RV14 infection were significantly higher for polyp EC than for nonatopic and atopic nonpolyp EC [105].
Modification of EC ICAM‐1 expression is therefore of possible therapeutic benefit. In vitro, RV increases expression of ICAM‐1 and vascular cell‐adhesion molecule‐1 in primary bronchial EC (PBEC) cultures and in A549 cells via a mechanism involving NF‐κB [106, 107]. One of the actions of corticosteroids is inhibition of NF‐κB [108]. In A549 cells and in PBEC pretreatment with three corticosteroids, hydrocortisone, dexamethasone, and mometasone furoate inhibit RV16‐induced increases in ICAM‐1 surface expression, mRNA, and promoter activation without alteration of virus infectivity or replication [109]. Dexamethasone suppresses ICAM‐1 in PHTEC and inhibits RV infection [110]. Dexamethasone does not inhibit infection of PHTEC by minor group RV2 [111]. It is disappointing that a study of inhaled corticosteroids in asthmatics before experimental RV infection failed to show reduced virus‐induced ICAM‐1 expression in bronchial biopsies [97], but it is possible that a longer course and/or a higher dose of inhaled steroid or administration of oral steroids might have demonstrated a significant effect.
Other drugs that affect EC ICAM‐1 include reducing agents [112], the H1 receptor antagonists desloratidine/loratidine, which inhibit RV‐induced ICAM‐1 up‐regulation in human primary bronchial epithelial cells (HPBEC) and in A549 cells [113], and erythromycin, which inhibits infection of PHTEC by major group RV14 and minor group RV2 through effects including ICAM‐1 reduction, blockage of RV RNA entry into endosomes, and small reductions in LDL‐R expression [114].
RV induction of EC production of cytokines and chemokines
EC can activate and recruit a variety of other cell types such as lymphocytes, eosinophils, and neutrophils through the production of chemokines and cytokines ( Fig. 2 ). Such cells are important components of the antiviral response but may also contribute to airway inflammation and dysfunction in asthma or COPD. In vitro studies of RV infection in bronchial EC lines and primary human EC have demonstrated the production of a wide range of proinflammatory cytokines such as IL‐1α, IL‐1β, IL‐6, IL‐11, IL‐16, TNF‐α, and GM‐CSF and the chemokines IL‐8, Gro‐α, ΕΝΑ‐78, RANTES, eotaxin 1/2, and MIP‐1α [69, 115, 116, 117, 118, 119]. In vivo, these cytokines can be found in nasal lavage (NL) in association with RV infection [120, 121].
Figure 2.
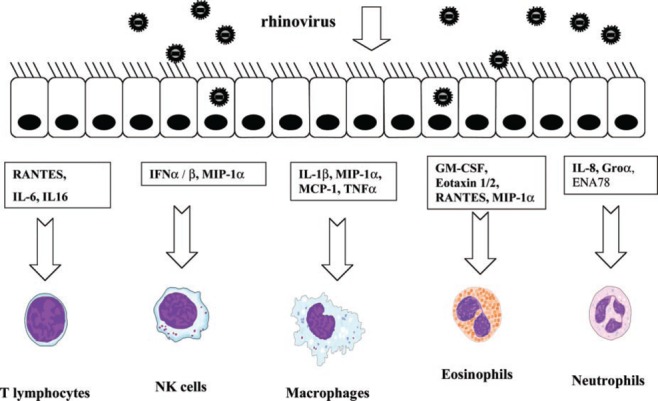
Following RV infection, airway EC produce a range of cytokines and chemokines that promote recruitment and activation of inflammatory cell types including T cells, NK cells, macrophages, eosinophils, and neutrophils. Such responses facilitate clearance of virus but may amplify pre‐existing inflammation and contribute to exacerbation of diseases such as asthma and COPD. RANTES, Regulated on activation, normal T expressed and secreted; MIP‐1α, macrophage‐inflammatory protein‐1α; MCP‐1, monocyte chemoattractant protein‐1; GM‐CSF, granulocyte macrophage‐colony stimulating factor; Groα, growth‐regulated oncogene‐α; ENA78, epithelial neutrophil‐activating protein‐78.
Type 1 IFNs
IFNs are thought to play an important role in innate resistance to viruses [122], acting on virus‐infected cells and surrounding tissues to produce an antiviral state characterized by the expression and antiviral activity of IFN‐stimulated genes (ISGs). There are two main types of IFN: type 1 (IFN‐α, IFN‐β, IFN‐ω, IFN‐τ) and type 2 (IFN‐γ). EC can produce IFN‐α and IFN‐β. There are 14 IFN‐α genes but only 1 IFN‐β gene. IFN‐β synthesis involves NF‐κB, ATF/JUN, and the IFN regulatory factors (IRFs; up to 10 of which are currently identified), activation of which occurs in response to virus‐specific signals including dsRNA, a product of the replication of ssRNA viruses such as RV, RSV, and influenza.
IFN‐β and IFN‐α4 are expressed early through the action of IRF‐3. Activation of the IFN intracellular signaling pathway is required for induction of IRF‐7, which is required for transcription of the full range of IFNs. DNA microarray analysis has shown that following binding to their receptors on target cells, IFNs trigger a complex signaling pathway (mainly Janus tyrosine kinase‐signal transducer and activator of transcription), resulting in the transcription of hundreds of ISGs [123].
Several ISGs have been well studied. These include the dsRNA‐activated serine/threonine protein kinase (PKR), which reduces cellular mRNA translation and transcriptional events, two enzymes involved in mRNA degradation: 2′5′oligoadenylate synthetase and RNase L, the myxovirus resistance (Mx) proteins, and RNA‐specific adenosine deaminase, which is involved in RNA editing. IFNs also up‐regulate cellular expression of MHC classes I and II molecules, therefore increasing antigen presentation to CD8+ and CD4+ T cells and enhancing cellular immune responses.
There is surprisingly little information on RV induction of IFNs in EC. More information is available for influenza. The antiviral Mx proteins inhibit influenza replication at a number of levels. Influenza has evolved mechanisms to resist the actions of IFNs, for example, blocking PKR by the influenza NS1 protein [124].
Proinflammatory cytokines
IL‐1, TNF‐α, and IL‐6 share proinflammatory properties, such as the induction of the acute‐phase response and the activation of T and B lymphocytes. IL‐1 enhances the adhesion of inflammatory cells to endothelium, facilitating chemotaxis [125]. TNF‐α is a potent antiviral cytokine but in vitro, increases the susceptibility of cultured EC to infection by RV14 through up‐regulation of ICAM‐1 [69]. IL‐6 has been shown to stimulate IgA‐mediated immune responses. IL‐11 may also be important in virus‐induced asthma [126]. It appears to cause bronchoconstriction by a direct effect on bronchial smooth muscle [119]. In vivo, IL‐11 is elevated in nasal aspirates from children with colds, levels correlating with the presence of wheezing.
In addition to the induction of IL‐1α and IL‐1β, RV infection results in substantial increases in IL‐1ra in vivo and in vitro. This is a relatively late effect, occurring 48–72 h after infection and may contribute to symptom resolution [127].
EC chemokine production and lymphocyte recruitment
Bronchial biopsies demonstrate increases in cells positive for CD3, CD4, and CD8 within the epithelium and submucosa of normal and asthmatic subjects following experimental RV infection [59]. Such increases coincide with peripheral lymphopenia, suggesting increased recruitment of T cells to the airway.
T cell recruitment into the airway is at least partly under the influence of chemokines, including those whose production by EC is up‐regulated by viruses. The balance of chemokine production by airway EC may influence the nature and the effectiveness of the specific immune response. This balance may in turn be influenced by pre‐existing chronic inflammation, as found in asthma or COPD. It is thought that an effective, antiviral immune response is characterized by the production of type 1 cytokines such as IFN‐γ. There is evidence to suggest that type 1 responses to RV are deficient in individuals with asthma [128]. In a recent study of experimental RV16 infection in subjects with allergic rhinitis or asthma, the balance of airway types 1 and 2 cytokines in induced sputum (IS) induced by viral infection was related to symptoms and viral clearance. An inverse correlation was demonstrated between the ratio of IFN‐γ to IL‐5 mRNA and peak cold symptoms. In addition, subjects with RV16 still detectable 14 days after inoculation had lower IFN‐γ/IL‐5 ratios during the acute cold phase than those who had cleared the virus [129].
Studies of cloned T cells suggest that Th1 and Th2 cells show differential expression of chemokine receptors. There is increased expression of CXCR3 (receptor for IFN‐inducible protein 10, IFN‐inducible T cell‐α chemoattractant, and monokine induced by IFN‐γ) and CCR5 (MIP‐1β) in human Th1 cells and increased expression of CCR4 (thymus and activation‐regulated chemokine and macrophage‐derived chemokine) and to a lesser extent, CCR3 (eotaxin and MCP‐3) in Th2 cells, with selective migration of cells in response to the appropriate chemokines. CCR1 (RANTES, MIP‐1α, MCP‐3) and CCR2 (MCP‐1,2,3,4) were found on Th1 and Th2 cells [130]. Bronchial biopsies from asthmatics show high levels of expression of CCR4 and significant levels of CCR8 by T cells [131].
Increased recruitment of T cells to the airway as a result of virus‐induced chemokine production by EC could amplify pre‐existing allergic inflammation. If the asthmatic airway microenvironment influences the pattern of chemokine expression following virus infection, then this could alter the Th1/Th2 balance of the antiviral immune response.
EC chemokine production and neutrophil recruitment
Neutrophils are recruited early in response to the production of IL‐8, Gro‐α, and ENA‐78 by EC, and activated neutrophils are a prominent feature of severe asthma. IS in asthmatics and nonasthmatics demonstrates a significant increase in neutrophils at day 4 of a natural cold, correlating with sputum IL‐8 [132]. Similar results were obtained in IS taken 2 and 9 days after experimental RV16 infection in asthmatics [61]. Increased IL‐8 has been found in NL from children with natural colds [121]. Experimental RV16 infection of asthmatics resulted in elevated NL IL‐8, correlating with cold/asthma symptom scores and histamine PC20 [63]. IS from asthmatics with exacerbations has elevated IL‐8 and neutrophilia [133]. A study of experimental infection in asthmatic children also demonstrated elevated IL‐8 and neutrophilia in NL during the acute infection, and levels of neutrophil myeloperoxidase correlated with symptom severity [134]. In asthma exacerbations in asthmatic adults [135], those with virus infection had increased sputum neutrophils, increased neutrophil elastase, and more severe clinical disease. Such studies suggest a prominent role for the neutrophil in tissue damage during virus‐induced asthma.
EC chemokine production and eosinophil recruitment
Eosinophils are increased in bronchial epithelium in biopsies taken from normal and asthmatic volunteers following experimental RV infection; in a small study, eosinophilic inflammation persisted for up to 6 weeks in asthmatic subjects [59]. In allergic rhinitics, experimental RV infection increases BAL eosinophils following segmental allergen challenge, again persisting for 6 weeks [66], and increased levels of ECP are found in the sputum of RV‐infected subjects [61]. Eosinophils accumulate in the airway under the influence of IL‐5, GM‐CSF, IL‐8, RANTES, and eotaxin [136]. Of these, only IL‐5 is not produced by airway EC in vitro after infection by RV. Expression of RANTES is increased in nasal secretions of children with natural virus‐induced asthma [137]. RANTES is up‐regulated in primary nasal EC cultures by RSV [138] and RV [72]. GM‐CSF is important in bone marrow eosinophil production and in eosinophil survival [136], but levels are not increased during viral URTI [137, 139]. Levels of eotaxin in NL rise after experimental RV16 infection [140]. These data suggest a pathogenic role for eosinophils in virus‐induced asthma. However, a protective role is also possible. In allergic rhinitic subjects infected with RV after high‐dose allergen challenge, the severity and duration of cold symptoms were inversely related to the NL eosinophil count before infection [141]. Eosinophils may contribute to viral antigen presentation. Eosinophils pretreated with GM‐CSF bind RV16 via ICAM‐1 and present viral antigen to RV16‐specific T cells, inducing proliferation and secretion of IFN‐γ [142]. Eosinophils have antiviral actions in parainfluenza‐infected guinea pigs [143]. Eosinophil‐derived neurotoxin and ECP have RNase activity and reduce RSV infectivity [144]. The role of the eosinophil in the antiviral immune response thus requires further evaluation.
NO
NO may be important in a range of respiratory diseases [145] including asthma [146] and in virus infection [147]. NO is produced by the constitutive enzymes, NO synthase (NOS) 1 and NOS 3 and by the inducible, calcium‐independent NOS 2 expressed by airway EC [148] and macrophages. NO may have beneficial and harmful actions. It is a gaseous signaling molecule that regulates many aspects of airway biology, including the modulation of airway and vascular smooth‐muscle tone, causing relaxation via the activation of guanylyl cyclase and increased levels of intracellular cyclic guanosine monophosphate and promoting mucociliary clearance through effects on ciliary motility and reduced mucous viscosity [149]. Conversely, NO may combine with superoxide generated by macrophages and neutrophils in inflammatory infiltrate to produce peroxynitrite [150]. Such reactive species are a powerful weapon against bacteria and fungi but are toxic to host cells and may contribute to immunopathology.
In asthma, there is increased NOS 2 expression and an elevated level of exhaled NO [151] that falls with corticosteroid therapy; the level of exhaled NO correlates with sputum eosinophilia and methacholine responsiveness [152]. In contrast, in stable COPD, exhaled NO levels are no different from normal subjects [153], although there may be an increase during exacerbations [154]. In fact, in vitro cigarette smoke reduces cytokine‐induced NOS 2 mRNA expression in the LA‐4 murine cell line, in A549 cells, and in HPBEC [155].
NOS 2 appears at least to be associated with inflammation. Its expression is induced by proinflammatory cytokines including IL‐1β, TNF‐α, IFN‐γ, and lipopolysaccharide and down‐regulated by IL‐4, IL‐10, and transforming growth factor‐β [147]. NO production may be reduced indirectly by these suppressor cytokines through induction of arginase, which competes for l‐arginine, the substrate for NOS 2 [156]. NO suppresses T cell proliferation and Th1 cytokine production, including IFN‐γ, and favors the development of a Th2 response with eosinophilia. NO appears to promote eosinophil chemotaxis [157] and to inhibit eosinophil apoptosis [158]. This interaction between NOS 2 expression and type 1/type 2 balance in the airway may contribute to deficient antiviral immunity in asthma.
The relative importance of the beneficial antimicrobial activity of NO versus the potentially disadvantageous suppression of IFN‐γ is dependent on the specific pathogen, and this will determine the use of NO‐based therapy. NOS 2 knockout mice show an increased susceptibility to infections [159]. Release of NO may be important for NK cell‐mediated target cell killing [160].
NO may also possess antiviral activity. In vitro, RV induces NOS expression in HPBEC [161]. There is increased expression of NOS 2 mRNA in cultured HPBEC after RV16 infection [162]. NO inhibits RV‐induced production of IL‐6, IL‐8, and GM‐CSF and viral replication in a human respiratory EC line [162, 163]. RSV also induces NOS 2 and increases nitrite levels in supernatant from A549 cells, from HPBEC culture, and in BAL fluid from RSV‐infected BALB/c mice, effects opposed by IL‐4 and dexamethasone but unaffected by IL‐13 or IFN‐γ [164]. Replication of RSV in Hep2 cells is inhibited following transfection with a retroviral construct containing NOS, and the NOS inhibitor, NG‐methyl‐l‐arginine, abolishes this inhibition [165]. Replication of influenza A and B in Mabin‐Darby kidney cells is severely impaired by the NO donor, S‐nitroso‐N‐acetylpenicillamine [166]. Influenza A and synthetic dsRNA induce NOS 2 in human airway EC, and this induction appears dependent on activation of PKR [167].
Overall, there is evidence that in vivo, increased lower airway NO production may be of benefit in virus‐induced asthma exacerbations. Work in a guinea pig model suggests that one mechanism for increased airway hyper‐responsiveness during respiratory virus infection is through inhibition of NOS enzymes and a loss of NO‐related relaxation of airway smooth muscle [168]. Studies of human asthmatics would also suggest that NO has a protective role in virus‐induced exacerbations. Following experimental RV16 infection, patients with the greatest increase in exhaled NO had smaller increases in histamine airway responsiveness [161].
It is interesting that studies of viral URTI have failed to demonstrate an increase in nasal NO after experimental RSV, RV, and influenza infections [169]. In normal subjects, experimental influenza infection increased oral NO 8 days postinfection but had no effect on nasal NO [170]. This raises the possibility that during respiratory virus infection, induction of NO is selective for the LRT.
Antigen presentation
EC may also contribute to the specific immune response following virus infection by acting as antigen‐presenting cells, particularly during secondary respiratory viral infections. EC express MHC class I and the costimulatory molecules B7‐1 and B7‐2, and this expression is up‐regulated in vitro by RV16 [171].
Signaling pathways
The responses of airway EC to virus infection are consequences of the interactions between virus and the intracellular signaling pathways of the host cell [172]. Knowledge of the mechanisms involved for RV is currently very limited. Activation of signaling pathways may be dependent on cell‐surface receptor (ICAM‐1, LDL‐R) binding or may occur during viral replication within the cell. The need for replicative virus is demonstrated by the inhibition of RV induction of EC cytokines after UV inactivation. One product of replication, common to ssRNA viruses such as RV, RSV, and influenza, is dsRNA [173], which has been shown to activate components of signaling pathways including dsRNA‐dependent PKR, NF‐κB, and p38 mitogen‐activated protein kinase with resultant induction of IL‐8 and RANTES. Activation of EC by dsRNA may be direct or indirect through the IFN system as discussed above. It has also been suggested that dsRNA may activate EC through binding to TLR3 [174].
SUMMARY
The role of the airway epithelium in host defense has been discussed with reference to studies of the interaction between EC and viruses, in particular, RV. Respiratory virus infection is a common and clinically important problem. In patients with asthma or COPD, viruses are a common trigger of exacerbations and a major cause of morbidity and mortality. RV are the most common virus type detected. Knowledge of the immunopathogenesis of such RV‐induced exacerbations remains limited, but information is available from in vitro and from in vivo studies, especially of experimental infection in human volunteers. RV infects and replicates within EC of the LRT. EC are an important component of the innate‐immune response to RV infection. The interaction between virus and the intracellular signaling pathways of the host cell results in activation of potentially antiviral mechanisms such as IFN and NO and in the production of cytokines and chemokines that influence the subsequent induced innate‐ and specific‐immune response. Although this is beneficial in facilitating clearance of virus from the respiratory tract, the generation of proinflammatory mediators and the recruitment of inflammatory cells result in a degree of immunopathology and in patients with asthma or COPD, may amplify pre‐existing airway inflammation. Further research will be necessary to determine whether modification of EC responses to respiratory virus infection will be of therapeutic benefit.
ACKNOWLEDGMENTS
S. D. Message has been supported by a Medical Research Council Annual Training Fellowship. This work has received further support from a British Lung Foundation Programme Grant.
REFERENCES
- 1. van den Hoogen, B. G. , de Jong, J. C. , Groen, J. , Kuiken, T. , de Groot, R. , Fouchier, R. A. , Osterhaus, A. D. (2001) A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ebihara, T. , Endo, R. , Kikuta, H. , Ishiguro, N. , Yoshioka, M. , Ma, X. , Kobayashi, K. (2003) Seroprevalence of human metapneumovirus in Japan. J. Med. Virol. 70, 281–283. [DOI] [PubMed] [Google Scholar]
- 3. Enserink, M. , Vogel, G. (2003) Infectious diseases: deferring competition, global net closes in on SARS. Science 300, 224–225. [DOI] [PubMed] [Google Scholar]
- 4. Malcolm, E. , Arruda, E. , Hayden, F. G. , Kaiser, L. (2001) Clinical features of patients with acute respiratory illness and rhinovirus in their bronchoalveolar lavages. J. Clin. Virol. 21, 9–16. [DOI] [PubMed] [Google Scholar]
- 5. Blomqvist, S. , Roivainen, M. , Puhakka, T. , Kleemola, M. , Hovi, T. (2002) Virological and serological analysis of rhinovirus infections during the first two years of life in a cohort of children. J. Med. Virol. 66, 263–268. [DOI] [PubMed] [Google Scholar]
- 6. Ukkonen, P. , Hovi, T. , von Bonsdorff, C. H. , Saikku, P. , Penttinen, K. (1984) Age‐specific prevalence of complement‐fixing antibodies to sixteen viral antigens: a computer analysis of 58,500 patients covering a period of eight years. J. Med. Virol. 13, 131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glezen, W. P. , Taber, L. H. , Frank, A. L. , Kasel, J. A. (1986) Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 140, 543–546. [DOI] [PubMed] [Google Scholar]
- 8. Wright, A. L. , Holberg, C. J. , Martinez, F. D. , Morgan, W. J. , Taussig, L. M. (1989) Breast feeding and lower respiratory tract illness in the first year of life. Group Health Medical Associates. BMJ 299, 946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duff, A. L. , Pomeranz, E. S. , Gelber, L. E. , Price, G. W. , Farris, H. , Hayden, F. G. , Platts‐Mills, T. A. , Heymann, P. W. (1993) Risk factors for acute wheezing in infants and children: viruses, passive smoke, and IgE antibodies to inhalant allergens. Pediatrics 92, 535–540. [PubMed] [Google Scholar]
- 10. Simoes, E. A. (1999) Respiratory syncytial virus infection. Lancet 354, 847–852. [DOI] [PubMed] [Google Scholar]
- 11. Martinez, F. D. (2003) Respiratory syncytial virus bronchiolitis and the pathogenesis of childhood asthma. Pediatr. Infect. Dis. J. 22 (Suppl. 2), S76–S82. [DOI] [PubMed] [Google Scholar]
- 12. Taussig, L. M. , Wright, A. L. , Holberg, C. J. , Halonen, M. , Morgan, W. J. , Martinez, F. D. (2003) Tucson children's respiratory study: 1980 to present. J. Allergy Clin. Immunol. 111, 661–675. [DOI] [PubMed] [Google Scholar]
- 13. Mallia, P. , Johnston, S. L. (2002) Respiratory viruses: do they protect from or induce asthma? Allergy 57, 1118–1129. [DOI] [PubMed] [Google Scholar]
- 14. Rylander, E. , Eriksson, M. , Pershagen, G. , Nordvall, L. , Ehrnst, A. , Ziegler, T. (1996) Wheezing bronchitis in children. Incidence, viral infections, and other risk factors in a defined population. Pediatr. Allergy Immunol. 7, 6–11. [DOI] [PubMed] [Google Scholar]
- 15. Rakes, G. P. , Arruda, E. , Ingram, J. M. , Hoover, G. E. , Zambrano, J. C. , Hayden, F. G. , Platts‐Mills, T. A. , Heymann, P. W. (1999) Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am. J. Respir. Crit. Care Med. 159, 785–790. [DOI] [PubMed] [Google Scholar]
- 16. Tyrrell, D. A. J. (1965) A collaborative study of the aetiology of acute respiratory infections in Britain 1961–4. BMJ 2, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Disney, M. E. , Matthews, R. , Williams, J. D. (1971) The role of infection in the morbidity of asthmatic children admitted to hospital. Clin. Allergy 1, 399–406. [DOI] [PubMed] [Google Scholar]
- 18. Glezen, W. P. , Loda, F. A. , Clyde, W. A. J. , Senior, R. J. , Sheaffer, C. I. , Conley, W. G. , Denny, F. W. (1971) Epidemiologic patterns of acute lower respiratory disease of children in a pediatric group practice. J. Pediatr. 78, 397–406. [DOI] [PubMed] [Google Scholar]
- 19. Horn, M. E. , Brain, E. , Gregg, I. , Yealland, S. J. , Inglis, J. M. (1975) Respiratory viral infection in childhood. A survey in general practice, Roehampton 1967–1972. J. Hyg. (Lond.) 74, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mitchell, I. , Inglis, J. M. , Simpson, H. (1978) Viral infection as a precipitant of wheeze in children. Combined home and hospital study. Arch. Dis. Child. 53, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Henderson, F. W. , Clyde, W. A. J. , Collier, A. M. , Denny, F. W. , Senior, R. J. , Sheaffer, C. I. , Conley 3rd, W. G. , Christian, R. M. (1979) The etiologic and epidemiologic spectrum of bronchiolitis in pediatric practice. J. Pediatr. 95, 183–190. [DOI] [PubMed] [Google Scholar]
- 22. Horn, M. E. , Brain, E. A. , Gregg, I. , Inglis, J. M. , Yealland, S. J. , Taylor, P. (1979) Respiratory viral infection and wheezy bronchitis in childhood. Thorax 34, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horn, M. E. , Reed, S. E. , Taylor, P. (1979) Role of viruses and bacteria in acute wheezy bronchitis in childhood: a study of sputum. Arch. Dis. Child. 54, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carlsen, K. H. , Orstavik, I. , Leegaard, J. , Hoeg, H. (1984) Respiratory virus infections and aeroallergens in acute bronchial asthma. Arch. Dis. Child. 59, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jennings, L. C. , Barns, G. , Dawson, K. P. (1987) The association of viruses with acute asthma. N. Z. Med. J. 100, 488–490. [PubMed] [Google Scholar]
- 26. Freymuth, F. , Vabret, A. , Brouard, J. , Toutain, F. , Verdon, R. , Petitjean, J. , Gouarin, S. , Duhamel, J. F. , Guillois, B. (1999) Detection of viral, Chlamydia pneumoniae and Mycoplasma pneumoniae infections in exacerbations of asthma in children. J. Clin. Virol. 13, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mertsola, J. , Ziegler, T. , Ruuskanen, O. , Vanto, T. , Koivikko, A. , Halonen, P. (1991) Recurrent wheezy bronchitis and viral respiratory infections. Arch. Dis. Child. 66, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roldaan, A. C. , Masural, N. (1982) Viral respiratory infections in asthmatic children staying in a mountain resort. Eur. J. Respir. Dis. 63, 140–150. [PubMed] [Google Scholar]
- 29. Johnston, S. L. , Pattemore, P. K. , Sanderson, G. , Smith, S. , Lampe, F. , Josephs, L. , Symington, P. , O'Toole, S. , Myint, S. H. , Tyrrell, D. A. , et al. (1995) Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 310, 1225–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berkovich, S. , Millian, S. J. , Snyder, R. D. (1970) The association of viral and mycoplasma infections with recurrence of wheezing in the asthmatic child. Ann. Allergy 28, 43–49. [PubMed] [Google Scholar]
- 31. Lambert, H. P. , Stern, H. (1972) Infective factors in exacerbations of bronchitis and asthma. BMJ 3, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McIntosh, K. , Ellis, E. F. , Hoffman, L. S. , Lybass, T. G. , Eller, J. J. , Fulginiti, V. A. (1973) The association of viral and bacterial respiratory infections with exacerbations of wheezing in young asthmatic children. J. Pediatr. 82, 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minor, T. E. , Dick, E. C. , DeMeo, A. N. , Ouellette, J. J. , Cohen, M. , Reed, C. E. (1974) Viruses as precipitants of asthmatic attacks in children. J. Am. Med. Assoc. 227, 292–298. [PubMed] [Google Scholar]
- 34. Minor, T. E. , Dick, E. C. , Baker, J. W. , Ouellette, J. J. , Cohen, M. , Reed, C. E. (1976) Rhinovirus and influenza type A infections as precipitants of asthma. Am. Rev. Respir. Dis. 113, 149–153. [DOI] [PubMed] [Google Scholar]
- 35. Johnston, S. L. , Pattemore, P. K. , Sanderson, G. , Smith, S. , Campbell, M. J. , Josephs, L. K. , Cunningham, A. , Robinson, B. S. , Myint, S. H. , Ward, M. E. , Tyrrell, D. A. , Holgate, S. T. (1996) The relationship between upper respiratory infections and hospital admissions for asthma: a time‐trend analysis. Am. J. Respir. Crit. Care Med. 154, 654–660. [DOI] [PubMed] [Google Scholar]
- 36. Nicholson, K. G. , Kent, J. , Ireland, D. C. (1993) Respiratory viruses and exacerbations of asthma in adults. BMJ 307, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beasley, R. , Coleman, E. D. , Hermon, Y. , Holst, P. E. , O'Donnell, T. V. , Tobias, M. (1988) Viral respiratory tract infection and exacerbations of asthma in adult patients. Thorax 43, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berman, S. Z. , Mathison, D. A. , Stevenson, D. D. , Tan, E. M. , Vaughan, J. H. (1975) Transtracheal aspiration studies in asthmatic patients in relapse with “infective” asthma and in subjects without respiratory disease. J. Allergy Clin. Immunol. 56, 206–214. [DOI] [PubMed] [Google Scholar]
- 39. Pattemore, P. K. , Johnston, S. L. , Bardin, P. G. (1992) Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin. Exp. Allergy 22, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rawlinson, W. D. , Waliuzzaman, Z. , Carter, I. W. , Belessis, Y. C. , Gilbert, K. M. , Morton, J. R. (2003) Asthma exacerbations in children associated with rhinovirus but not human metapneumovirus infection. J. Infect. Dis. 187, 1314–1318. [DOI] [PubMed] [Google Scholar]
- 41. Corne, J. M. , Marshall, C. , Smith, S. , Schreiber, J. , Sanderson, G. , Holgate, S. T. , Johnston, S. L. (2002) Frequency, severity, and duration of rhinovirus infections in asthmatic and non‐asthmatic individuals: a longitudinal cohort study. Lancet 359, 831–834. [DOI] [PubMed] [Google Scholar]
- 42. Anonymous (1995) Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. American Thoracic Society. Am. J. Respir. Crit. Care Med. 152, S77–S121. [PubMed] [Google Scholar]
- 43. Seemungal, T. , Harper‐Owen, R. , Bhowmik, A. , Moric, I. , Sanderson, G. , Message, S. , Maccallum, P. , Meade, T. W. , Jeffries, D. J. , Johnston, S. L. , Wedzicha, J. A. (2001) Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 164, 1618–1623. [DOI] [PubMed] [Google Scholar]
- 44. Smith, C. B. , Golden, C. A. , Kanner, R. E. , Renzetti Jr., A. D. (1980) Association of viral and Mycoplasma pneumoniae infections with acute respiratory illness in patients with chronic obstructive pulmonary diseases. Am. Rev. Respir. Dis. 121, 225–232. [DOI] [PubMed] [Google Scholar]
- 45. Gump, D. W. , Phillips, C. A. , Forsyth, B. R. , McIntosh, K. , Lamborn, K. R. , Stouch, W. H. (1976) Role of infection in chronic bronchitis. Am. Rev. Respir. Dis. 113, 465–474. [DOI] [PubMed] [Google Scholar]
- 46. Tager, I. , Speizer, F. E. (1975) Role of infection in chronic bronchitis. N. Engl. J. Med. 292, 563–571. [DOI] [PubMed] [Google Scholar]
- 47. Greenberg, S. B. , Allen, M. , Wilson, J. , Atmar, R. L. (2000) Respiratory viral infections in adults with and without chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 162, 167–173. [DOI] [PubMed] [Google Scholar]
- 48. Hogg, J. C. (1999) Childhood viral infection and the pathogenesis of asthma and chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 160, S26–S28. [DOI] [PubMed] [Google Scholar]
- 49. Diaz, P. T. , King, M. A. , Pacht, E. R. , Wewers, M. D. , Gadek, J. E. , Nagaraja, H. N. , Drake, J. , Clanton, T. L. (2000) Increased susceptibility to pulmonary emphysema among HIV‐seropositive smokers. Ann. Intern. Med. 132, 369–372. [DOI] [PubMed] [Google Scholar]
- 50. Donaldson, G. C. , Seemungal, T. A. , Bhowmik, A. , Wedzicha, J. A. (2002) Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 57, 847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patel, I. S. , Seemungal, T. A. , Wilks, M. , Lloyd‐Owen, S. J. , Donaldson, G. C. , Wedzicha, J. A. (2002) Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 57, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bardin, P. G. , Johnston, S. L. , Sanderson, G. , Robinson, B. S. , Pickett, M. A. , Fraenkel, D. J. , Holgate, S. T. (1994) Detection of rhinovirus infection of the nasal mucosa by oligonucleotide in situ hybridization. Am. J. Respir. Cell Mol. Biol. 10, 207–213. [DOI] [PubMed] [Google Scholar]
- 53. van Benten, I. J. , KleinJan, A. , Neijens, H. J. , Osterhaus, A. D. , Fokkens, W. J. (2001) Prolonged nasal eosinophilia in allergic patients after common cold. Allergy 56, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gwaltney, J. M. J. , Hendley, O. , Hayden, F. G. , McIntosh, K. , Hollinger, F. B. , Melnick, J. L. , Turner, R. B. (1992) Updated recommendations for safety‐testing of viral inocula used in volunteer experiments on rhinovirus colds. Prog. Med. Virol. 39, 256–263. [PubMed] [Google Scholar]
- 55. Halperin, S. A. , Eggleston, P. A. , Beasley, P. , Suratt, P. , Hendley, J. O. , Groschel, D. H. , Gwaltney Jr., J. M. (1985) Exacerbations of asthma in adults during experimental rhinovirus infection. Am. Rev. Respir. Dis. 132, 976–980. [DOI] [PubMed] [Google Scholar]
- 56. Bardin, P. G. , Fraenkel, D. J. , Sanderson, G. , Dorward, M. , Lau, L. C. , Johnston, S. L. , Holgate, S. T. (1994) Amplified rhinovirus colds in atopic subjects. Clin. Exp. Allergy 24, 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lemanske, R. F. J. , Dick, E. C. , Swenson, C. A. , Vrtis, R. F. , Busse, W. W. (1989) Rhinovirus upper respiratory infection increases airway hyperreactivity and late asthmatic reactions. J. Clin. Invest. 83, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cheung, D. , Dick, E. C. , Timmers, M. C. , de Klerk, E. P. , Spaan, W. J. , Sterk, P. J. (1995) Rhinovirus inhalation causes long‐lasting excessive airway narrowing in response to methacholine in asthmatic subjects in vivo. Am. J. Respir. Crit. Care Med. 152, 1490–1496. [DOI] [PubMed] [Google Scholar]
- 59. Fraenkel, D. J. , Bardin, P. G. , Sanderson, G. , Lampe, F. , Johnston, S. L. , Holgate, S. T. (1995) Lower airways inflammation during rhinovirus colds in normal and in asthmatic subjects. Am. J. Respir. Crit. Care Med. 151, 879–886. [DOI] [PubMed] [Google Scholar]
- 60. Grunberg, K. , Kuijpers, E. A. , de Klerk, E. P. , de Gouw, H. W. , Kroes, A. C. , Dick, E. C. , Sterk, P. J. (1997) Effects of experimental rhinovirus 16 infection on airway hyperresponsiveness to bradykinin in asthmatic subjects in vivo. Am. J. Respir. Crit. Care Med. 155, 833–838. [DOI] [PubMed] [Google Scholar]
- 61. Grunberg, K. , Smits, H. H. , Timmers, M. C. , de Klerk, E. P. , Dolhain, R. J. , Dick, E. C. , Hiemstra, P. S. , Sterk, P. J. (1997) Experimental rhinovirus 16 infection. Effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am. J. Respir. Crit. Care Med. 156, 609–616. [DOI] [PubMed] [Google Scholar]
- 62. Grunberg, K. , Timmers, M. C. , de Klerk, E. P. , Dick, E. C. , Sterk, P. J. (1999) Experimental rhinovirus 16 infection causes variable airway obstruction in subjects with atopic asthma. Am. J. Respir. Crit. Care Med. 160, 1375–1380. [DOI] [PubMed] [Google Scholar]
- 63. Grunberg, K. , Timmers, M. C. , Smits, H. H. , de Klerk, E. P. , Dick, E. C. , Spaan, W. J. , Hiemstra, P. S. , Sterk, P. J. (1997) Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin‐8 in nasal lavage in asthmatic subjects in vivo. Clin. Exp. Allergy 27, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Halperin, S. A. , Eggleston, P. A. , Hendley, J. O. , Suratt, P. M. , Groschel, D. H. , Gwaltney Jr., J. M. (1983) Pathogenesis of lower respiratory tract symptoms in experimental rhinovirus infection. Am. Rev. Respir. Dis. 128, 806–810. [DOI] [PubMed] [Google Scholar]
- 65. Calhoun, W. J. , Swenson, C. A. , Dick, E. C. , Schwartz, L. B. , Lemanske, R. F. J. , Busse, W. W. (1991) Experimental rhinovirus 16 infection potentiates histamine release after antigen bronchoprovocation in allergic subjects. Am. Rev. Respir. Dis. 144, 1267–1273. [DOI] [PubMed] [Google Scholar]
- 66. Calhoun, W. J. , Dick, E. C. , Schwartz, L. B. , Busse, W. W. (1994) A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J. Clin. Invest. 94, 2200–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gern, J. E. , Calhoun, W. , Swenson, C. , Shen, G. , Busse, W. W. (1997) Rhinovirus infection preferentially increases lower airway responsiveness in allergic subjects. Am. J. Respir. Crit. Care Med. 155, 1872–1876. [DOI] [PubMed] [Google Scholar]
- 68. Gern, J. E. , Galagan, D. M. , Jarjour, N. N. , Dick, E. C. , Busse, W. W. (1997) Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am. J. Respir. Crit. Care Med. 155, 1159–1161. [DOI] [PubMed] [Google Scholar]
- 69. Subauste, M. C. , Jacoby, D. B. , Richards, S. M. , Proud, D. (1995) Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J. Clin. Invest. 96, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mosser, A. G. , Brockman‐Schneider, R. , Amineva, S. , Burchell, L. , Sedgwick, J. B. , Busse, W. W. , Gern, J. E. (2002) Similar frequency of rhinovirus‐infectible cells in upper and lower airway epithelium. J. Infect. Dis. 185, 734–743. [DOI] [PubMed] [Google Scholar]
- 71. Papadopoulos, N. G. , Bates, P. J. , Bardin, P. G. , Papi, A. , Leir, S. H. , Fraenkel, D. J. , Meyer, J. , Lackie, P. M. , Sanderson, G. , Holgate, S. T. , Johnston, S. L. (2000) Rhinoviruses infect the lower airways. J. Infect. Dis. 181, 1875–1884. [DOI] [PubMed] [Google Scholar]
- 72. Schroth, M. K. , Grimm, E. , Frindt, P. , Galagan, D. M. , Konno, S. I. , Love, R. , Gern, J. E. (1999) Rhinovirus replication causes RANTES production in primary bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 20, 1220–1228. [DOI] [PubMed] [Google Scholar]
- 73. Papadopoulos, N. G. , Sanderson, G. , Hunter, J. , Johnston, S. L. (1999) Rhinoviruses replicate effectively at lower airway temperatures. J. Med. Virol. 58, 100–104. [DOI] [PubMed] [Google Scholar]
- 74. McFadden Jr., E. R. , Pichurko, B. M. , Bowman, H. F. , Ingenito, E. , Burns, S. , Dowling, N. , Solway, J. (1985) Thermal mapping of the airways in humans. J. Appl. Physiol. 58, 564–570. [DOI] [PubMed] [Google Scholar]
- 75. Bruce, C. , Chadwick, P. , al‐Nakib, W. (1990) Detection of rhinovirus RNA in nasal epithelial cells by in situ hybridization. J. Virol. Methods 30, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. de Kluijver, J. , Grunberg, K. , Sont, J. K. , Hoogeveen, M. , Van Schadewijk, W. A. , de Klerk, E. P. , Dick, C. R. , van Krieken, J. H. , Sterk, P. J. (2002) Rhinovirus infection in nonasthmatic subjects: effects on intrapulmonary airways. Eur. Respir. J. 20, 274–279. [DOI] [PubMed] [Google Scholar]
- 77. Bardin, P. G. , Fraenkel, D. , Sanderson, G. , van Schalkwyk, E. M. , Holgate, S. T. , Johnston, S. L. (2000) Peak expiratory flow changes during experimental rhinovirus infection. Eur. Respir. J., 16, 980–985. [DOI] [PubMed] [Google Scholar]
- 78. Whitton, J. L. , Oldstone, M. B. A. (1996) Immune response to viruses In Field's Virology (Fields B. N., Knipe D. N., Howley P. M., eds.), Philadelphia, PA, Lippincott‐Raven, 345–374. [Google Scholar]
- 79. Yewdell, J. W. , Bennink, J. R. (1997) Immune responses to viruses In Clinical Virology (Richman D. R., Whiteley R. J., Hayden F. G., eds.), Oxford, UK, Churchill Livingstone, 271–306. [Google Scholar]
- 80. Amineva, S. P. , Gern, J. E. (2003) Rhinovirus 3C protease cleaves the C3 and C5 complement factors. Am. J. Respir. Crit. Care Med. 167, A212 (abstract). [Google Scholar]
- 81. Kopf, M. , Abel, B. , Gallimore, A. , Carroll, M. , Bachmann, M. F. (2002) Complement component C3 promotes T‐cell priming and lung migration to control acute influenza virus infection. Nat. Med. 8, 373–378. [DOI] [PubMed] [Google Scholar]
- 82. Anders, E. M. , Hartley, C. A. , Reading, P. C. , Ezekowitz, R. A. (1994) Complement‐dependent neutralization of influenza virus by a serum mannose‐binding lectin. J. Gen. Virol. 75, 615–622. [DOI] [PubMed] [Google Scholar]
- 83. Yang, D. , Biragyn, A. , Kwak, L. W. , Oppenheim, J. J. (2002) Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 23, 291–296. [DOI] [PubMed] [Google Scholar]
- 84. Barclay, W. S. , al‐Nakib, W. , Higgins, P. G. , Tyrrell, D. A. (1989) The time course of the humoral immune response to rhinovirus infection. Epidemiol. Infect. 103, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alper, C. M. , Doyle, W. J. , Skoner, D. P. , Buchman, C. A. , Seroky, J. T. , Gwaltney, J. M. , Cohen, S. A. (1996) Prechallenge antibodies: moderators of infection rate, signs, and symptoms in adults experimentally challenged with rhinovirus type 39. Laryngoscope 106, 1298–1305. [DOI] [PubMed] [Google Scholar]
- 86. Alper, C. M. , Doyle, W. J. , Skoner, D. P. , Buchman, C. A. , Cohen, S. , Gwaltney, J. M. (1998) Prechallenge antibodies moderate disease expression in adults experimentally exposed to rhinovirus strain hanks. Clin. Infect. Dis. 27, 119–128. [DOI] [PubMed] [Google Scholar]
- 87. Hastings, G. Z. , Francis, M. J. , Rowlands, D. J. , Chain, B. M. (1993) Epitope analysis of the T cell response to a complex antigen: proliferative responses to human rhinovirus capsids. Eur. J. Immunol. 23, 2300–2305. [DOI] [PubMed] [Google Scholar]
- 88. Gern, J. E. , Dick, E. C. , Kelly, E. A. , Vrtis, R. , Klein, B. (1997) Rhinovirus‐specific T cells recognize both shared and serotype‐restricted viral epitopes. J. Infect. Dis. 175, 1108–1114. [DOI] [PubMed] [Google Scholar]
- 89. Parry, D. E. , Busse, W. W. , Sukow, K. A. , Dick, C. R. , Swenson, C. , Gern, J. E. (2000) Rhinovirus‐induced PBMC responses and outcome of experimental infection in allergic subjects. J. Allergy Clin. Immunol. 105, 692–698. [DOI] [PubMed] [Google Scholar]
- 90. Turner, R. B. , Hendley, J. O. , Gwaltney Jr., J. M. (1982) Shedding of infected ciliated epithelial cells in rhinovirus colds. J. Infect. Dis. 145, 849–853. [DOI] [PubMed] [Google Scholar]
- 91. Turner, R. B. , Winther, B. , Hendley, J. O. , Mygind, N. , Gwaltney Jr., J. M. (1984) Sites of virus recovery and antigen detection in epithelial cells during experimental rhinovirus infection. Acta Otolaryngol. Suppl. 413, 9–14. [DOI] [PubMed] [Google Scholar]
- 92. Winther, B. , Gwaltney, J. M. , Hendley, J. O. (1990) Respiratory virus infection of monolayer cultures of human nasal epithelial cells. Am. Rev. Respir. Dis. 141, 839–845. [DOI] [PubMed] [Google Scholar]
- 93. Greve, J. M. , Davis, G. , Meyer, A. M. , Forte, C. P. , Yost, S. C. , Marlor, C. W. , Kamarck, M. E. , McClelland, A. (1989) The major human rhinovirus receptor is ICAM‐1. Cell 56, 839–847. [DOI] [PubMed] [Google Scholar]
- 94. Winther, B. , Greve, J. M. , Gwaltney, J. M. J. , Innes, D. J. , Eastham, J. R. , McClelland, A. , Hendley, J. O. (1997) Surface expression of intercellular adhesion molecule 1 on epithelial cells in the human adenoid. J. Infect. Dis. 176, 523–525. [DOI] [PubMed] [Google Scholar]
- 95. Winther, B. , Arruda, E. , Witek, T. J. , Marlin, S. D. , Tsianco, M. M. , Innes, D. J. , Hayden, F. G. (2002) Expression of ICAM‐1 in nasal epithelium and levels of soluble ICAM‐1 in nasal lavage fluid during human experimental rhinovirus infection. Arch. Otolaryngol. Head Neck Surg. 128, 131–136. [DOI] [PubMed] [Google Scholar]
- 96. Bianco, A. , Spiteri, M. A. (1998) A biological model to explain the association between human rhinovirus respiratory infections and bronchial asthma. Monaldi Arch. Chest Dis. 53, 83–87. [PubMed] [Google Scholar]
- 97. Grunberg, K. , Sharon, R. F. , Hiltermann, T. J. , Brahim, J. J. , Dick, E. C. , Sterk, P. J. , Van Krieken, J. H. (2000) Experimental rhinovirus 16 infection increases intercellular adhesion molecule‐1 expression in bronchial epithelium of asthmatics regardless of inhaled steroid treatment. Clin. Exp. Allergy 30, 1015–1023. [DOI] [PubMed] [Google Scholar]
- 98. Whiteman, S. C. , Bianco, A. , Knight, R. A. , Spiteri, M. A. (2003) Human rhinovirus selectively modulates membranous and soluble forms of its intercellular adhesion molecule‐1 (ICAM‐1) receptor to promote epithelial cell infectivity. J. Biol. Chem. 278, 11954–11961. [DOI] [PubMed] [Google Scholar]
- 99. Suzuki, T. , Yamaya, M. , Kamanaka, M. , Jia, Y. X. , Nakayama, K. , Hosoda, M. , Yamada, N. , Nishimura, H. , Sekizawa, K. , Sasaki, H. (2001) Type 2 rhinovirus infection of cultured human tracheal epithelial cells: role of LDL receptor. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L409–L420. [DOI] [PubMed] [Google Scholar]
- 100. Altman, L. C. , Ayars, G. H. , Baker, C. , Luchtel, D. L. (1993) Cytokines and eosinophil‐derived cationic proteins upregulate intercellular adhesion molecule‐1 on human nasal epithelial cells. J. Allergy Clin. Immunol. 92, 527–536. [DOI] [PubMed] [Google Scholar]
- 101. Terajima, M. , Yamaya, M. , Sekizawa, K. , Okinaga, S. , Suzuki, T. , Yamada, N. , Nakayama, K. , Ohrui, T. , Oshima, T. , Numazaki, Y. , Sasaki, H. (1997) Rhinovirus infection of primary cultures of human tracheal epithelium: role of ICAM‐1 and IL‐1beta. Am. J. Physiol. 273, L749–L759. [DOI] [PubMed] [Google Scholar]
- 102. Sethi, S. K. , Bianco, A. , Allen, J. T. , Knight, R. A. , Spiteri, M. A. (1997) Interferon‐gamma (IFN‐gamma) down‐regulates the rhinovirus‐induced expression of intercellular adhesion molecule‐1 (ICAM‐1) on human airway epithelial cells. Clin. Exp. Immunol. 110, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bianco, A. , Sethi, S. K. , Allen, J. T. , Knight, R. A. , Spiteri, M. A. (1998) Th2 cytokines exert a dominant influence on epithelial cell expression of the major group human rhinovirus receptor, ICAM‐1. Eur. Respir. J. 12, 619–626. [DOI] [PubMed] [Google Scholar]
- 104. Canonica, G. W. , Ciprandi, G. , Pesce, G. P. , Buscaglia, S. , Paolieri, F. , Bagnasco, M. (1995) ICAM‐1 on epithelial cells in allergic subjects: a hallmark of allergic inflammation. Int. Arch. Allergy Immunol. 107, 99–102. [DOI] [PubMed] [Google Scholar]
- 105. Bianco, A. , Whiteman, S. C. , Sethi, S. K. , Allen, J. T. , Knight, R. A. , Spiteri, M. A. (2000) Expression of intercellular adhesion molecule‐1 (ICAM‐1) in nasal epithelial cells of atopic subjects: a mechanism for increased rhinovirus infection? Clin. Exp. Immunol. 121, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Papi, A. , Johnston, S. L. (1999) Respiratory epithelial cell expression of vascular cell adhesion molecule‐1 and its up‐regulation by rhinovirus infection via NF‐kappaB and GATA transcription factors. J. Biol. Chem. 274, 30041–30051. [DOI] [PubMed] [Google Scholar]
- 107. Papi, A. , Johnston, S. L. (1999) Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM‐1) via increased NF‐kappaB‐mediated transcription. J. Biol. Chem. 274, 9707–9720. [DOI] [PubMed] [Google Scholar]
- 108. Barnes, P. J. , Adcock, I. M. (1998) Transcription factors and asthma. Eur. Respir. J. 12, 221–234. [DOI] [PubMed] [Google Scholar]
- 109. Papi, A. , Papadopoulos, N. G. , Degitz, K. , Holgate, S. T. , Johnston, S. L. (2000) Corticosteroids inhibit rhinovirus‐induced intercellular adhesion molecule‐1 up‐regulation and promoter activation on respiratory epithelial cells. J. Allergy Clin. Immunol. 105, 318–326. [DOI] [PubMed] [Google Scholar]
- 110. Ohrui, T. , Yamaya, M. , Sekizawa, K. , Terajima, M. , Yamada, N. , Suzuki, T. , Okinaga, S. , Hoshi, H. , Suzuki, H. , Sasaki, H. , (1996) [Rhinovirus infection and expression of adhesion molecules in human tracheal epithelium.] Nihon Kyobu Shikkan Gakkai Zasshi; Jpn. J. Thorac. Dis. 34 (Suppl.), 121–125. [PubMed] [Google Scholar]
- 111. Suzuki, T. , Yamaya, M. , Sekizawa, K. , Yamada, N. , Nakayama, K. , Ishizuka, S. , Kamanaka, M. , Morimoto, T. , Numazaki, Y. , Sasaki, H. (2000) Effects of dexamethasone on rhinovirus infection in cultured human tracheal epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L560–L571. [DOI] [PubMed] [Google Scholar]
- 112. Papi, A. , Papadopoulos, N. G. , Stanciu, L. A. , Bellettato, C. M. , Pinamonti, S. , Degitz, K. , Holgate, S. T. , Johnston, S. L. (2002) Reducing agents inhibit rhinovirus‐induced up‐regulation of the rhinovirus receptor intercellular adhesion molecule‐1 (ICAM‐1) in respiratory epithelial cells. FASEB J. 16, 1934–1936. [DOI] [PubMed] [Google Scholar]
- 113. Papi, A. , Papadopoulos, N. G. , Stanciu, L. A. , Degitz, K. , Holgate, S. T. , Johnston, S. L. (2001) Effect of desloratadine and loratadine on rhino‐virus‐induced intercellular adhesion molecule 1 upregulation and promoter activation in respiratory epithelial cells. J. Allergy Clin. Immunol. 108, 221–228. [DOI] [PubMed] [Google Scholar]
- 114. Suzuki, T. , Yamaya, M. , Sekizawa, K. , Hosoda, M. , Yamada, N. , Ishizuka, S. , Yoshino, A. , Yasuda, H. , Takahashi, H. , Nishimura, H. , Sasaki, H. (2002) Erythromycin inhibits rhinovirus infection in cultured human tracheal epithelial cells. Am. J. Respir. Crit. Care Med. 165, 1113–1118. [DOI] [PubMed] [Google Scholar]
- 115. Papadopoulos, N. G. , Bates, P. J. , Bardin, P. G. , Papi, A. , Leir, S. H. , Fraenkel, D. J. , Meyer, J. , Lackie, P. M. , Sanderson, G. , Holgate, S. T. , Johnston, S. L. (2000) Rhinoviruses infect the lower airways. J. Infect. Dis. 181, 1875–1884. [DOI] [PubMed] [Google Scholar]
- 116. Papadopoulos, N. G. , Papi, A. , Meyer, J. , Stanciu, L. A. , Salvi, S. , Holgate, S. T. , Johnston, S. L. (2001) Rhinovirus infection up‐regulates eotaxin and eotaxin‐2 expression in bronchial epithelial cells. Clin. Exp. Allergy 31, 1060–1066. [DOI] [PubMed] [Google Scholar]
- 117. Donninger, H. , Glashoff, R. , Haitchi, H. M. , Syce, J. A. , Ghildyal, R. , van Rensburg, E. , Bardin, P. G. (2003) Rhinovirus induction of the CXC chemokine epithelial‐neutrophil activating peptide‐78 in bronchial epithelium. J. Infect. Dis. 187, 1809–1817. [DOI] [PubMed] [Google Scholar]
- 118. Becker, S. , Quay, J. , Soukup, J. (1991) Cytokine (tumor necrosis factor, IL‐6, and IL‐8) production by respiratory syncytial virus‐infected human alveolar macrophages. J. Immunol. 147, 4307–4312. [PubMed] [Google Scholar]
- 119. Einarsson, O. , Geba, G. P. , Zhu, Z. , Landry, M. , Elias, J. A. (1996) Interleukin‐11: stimulation in vivo and in vitro by respiratory viruses and induction of airways hyperresponsiveness. J. Clin. Invest. 97, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Abisheganaden, J. A. , Avila, P. C. , Kishiyama, J. L. , Liu, J. , Yagi, S. , Schnurr, D. , Boushey, H. A. (2000) Effect of clarithromycin on experimental rhinovirus‐16 colds: a randomized, double‐blind, controlled trial. Am. J. Med. 108, 453–459. [DOI] [PubMed] [Google Scholar]
- 121. Lau, L. , Corne, J. , Scott, S. (1996) Nasal cytokines in the common cold. Am. J. Respir. Crit. Care Med. 153, A866 (abstract). [Google Scholar]
- 122. Samuel, C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Katze, M. G. , He, Y. , Gale Jr., M. (2002) Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2, 675–687. [DOI] [PubMed] [Google Scholar]
- 124. Bergmann, M. , Garcia‐Sastre, A. , Carnero, E. , Pehamberger, H. , Wolff, K. , Palese, P. , Muster, T. (2000) Influenza virus NS1 protein counteracts PKR‐mediated inhibition of replication. J. Virol. 74, 6203–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Proud, D. , Gwaltney, J. M. J. , Hendley, J. O. , Dinarello, C. A. , Gillis, S. , Schleimer, R. P. (1994) Increased levels of interleukin‐1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J. Infect. Dis. 169, 1007–1013. [DOI] [PubMed] [Google Scholar]
- 126. Einarsson, O. , Geba, G. P. , Zhou, Z. , Landry, M. L. , Panettieri, R. A. J. , Tristram, D. , Welliver, R. , Metinko, A. , Elias, J. A. (1995) Interleukin‐11 in respiratory inflammation. Ann. N. Y. Acad. Sci. 762, 89–100. [DOI] [PubMed] [Google Scholar]
- 127. Yoon, H. J. , Zhu, Z. , Gwaltney Jr., J. M. , Elias, J. A. (1999) Rhinovirus regulation of IL‐1 receptor antagonist in vivo and in vitro: a potential mechanism of symptom resolution. J. Immunol. 162, 7461–7469. [PubMed] [Google Scholar]
- 128. Papadopoulos, N. G. , Stanciu, L. A. , Papi, A. , Holgate, S. T. , Johnston, S. L. (2002) A defective type 1 response to rhinovirus in atopic asthma. Thorax 57, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gern, J. E. , Vrtis, R. , Grindle, K. A. , Swenson, C. , Busse, W. W. (2000) Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am. J. Respir. Crit. Care Med. 162, 2226–2231. [DOI] [PubMed] [Google Scholar]
- 130. Bonecchi, R. , Bianchi, G. , Bordignon, P. P. , D'Ambrosio, D. , Lang, R. , Borsatti, A. , Sozzani, S. , Allavena, P. , Gray, P. A. , Mantovani, A. , Sinigaglia, F. (1998) Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Panina‐Bordignon, P. , Papi, A. , Mariani, A. , Di Lucia, P. , Casoni, G. , Bellettato, C. , Buonsanti, C. , Miotto, D. , Mapp, C. , Villa, A. , Arrigoni, G. , Fabbri, L. M. , Sinigaglia, F. (2001) The C‐C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen‐challenged atopic asthmatics. J. Clin. Invest. 107, 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pizzichini, M. M. , Pizzichini, E. , Efthimiadis, A. , Chauhan, A. J. , Johnston, S. L. , Hussack, P. , Mahony, J. , Dolovich, J. , Hargreave, F. E. (1998) Asthma and natural colds. Inflammatory indices in induced sputum: a feasibility study. Am. J. Respir. Crit. Care Med. 158, 1178–1184. [DOI] [PubMed] [Google Scholar]
- 133. Fahy, J. V. , Kim, K. W. , Liu, J. , Boushey, H. A. (1995) Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J. Allergy Clin. Immunol. 95, 843–852. [DOI] [PubMed] [Google Scholar]
- 134. Teran, L. M. , Johnston, S. L. , Schroder, J. M. , Church, M. K. , Holgate, S. T. (1997) Role of nasal interleukin‐8 in neutrophil recruitment and activation in children with virus‐induced asthma. Am. J. Respir. Crit. Care Med. 155, 1362–1366. [DOI] [PubMed] [Google Scholar]
- 135. Wark, P. A. , Johnston, S. L. , Moric, I. , Simpson, J. L. , Hensley, M. J. , Gibson, P. G. (2002) Neutrophil degranulation and cell lysis is associated with clinical severity in virus‐induced asthma. Eur. Respir. J. 19, 68–75. [DOI] [PubMed] [Google Scholar]
- 136. Gleich, G. J. (2000) Mechanisms of eosinophil‐associated inflammation. J. Allergy Clin. Immunol. 105, 651–663. [DOI] [PubMed] [Google Scholar]
- 137. Teran, L. M. , Seminario, M. C. , Shute, J. K. , Papi, A. , Compton, S. J. , Low, J. L. , Gleich, G. J. , Johnston, S. L. (1999) RANTES, macrophage‐inhibitory protein 1alpha, and the eosinophil product major basic protein are released into upper respiratory secretions during virus‐induced asthma exacerbations in children. J. Infect. Dis. 179, 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Saito, T. , Deskin, R. W. , Casola, A. , Haeberle, H. , Olszewska, B. , Ernst, P. B. , Alam, R. , Ogra, P. L. , Garofalo, R. (1997) Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J. Infect. Dis. 175, 497–504. [DOI] [PubMed] [Google Scholar]
- 139. Noah, T. L. , Henderson, F. W. , Henry, M. M. , Peden, D. B. , Devlin, R. B. (1995) Nasal lavage cytokines in normal, allergic, and asthmatic schoolage children. Am. J. Respir. Crit. Care Med. 152, 1290–1296. [DOI] [PubMed] [Google Scholar]
- 140. Greiff, L. , Andersson, M. , Andersson, E. , Linden, M. , Myint, S. , Svensson, C. , Persson, C. G. (1999) Experimental common cold increases mucosal output of eotaxin in atopic individuals. Allergy 54, 1204–1208. [DOI] [PubMed] [Google Scholar]
- 141. Avila, P. C. , Abisheganaden, J. A. , Wong, H. , Liu, J. , Yagi, S. , Schnurr, D. , Kishiyama, J. L. , Boushey, H. A. (2000) Effects of allergic inflammation of the nasal mucosa on the severity of rhinovirus 16 cold. J. Allergy Clin. Immunol. 105, 923–932. [DOI] [PubMed] [Google Scholar]
- 142. Handzel, Z. T. , Busse, W. W. , Sedgwick, J. B. , Vrtis, R. , Lee, W. M. , Kelly, E. A. , Gern, J. E. (1998) Eosinophils bind rhinovirus and activate virus‐specific T cells. J. Immunol. 160, 1279–1284. [PubMed] [Google Scholar]
- 143. Adamko, D. J. , Yost, B. L. , Gleich, G. J. , Fryer, A. D. , Jacoby, D. B. (1999) Ovalbumin sensitization changes the inflammatory response to subsequent parainfluenza infection. Eosinophils mediate airway hyper‐responsiveness, m(2) muscarinic receptor dysfunction, and antiviral effects. J. Exp. Med. 190, 1465–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Domachowske, J. B. , Dyer, K. D. , Adams, A. G. , Leto, T. L. , Rosenberg, H. F. (1998) Eosinophil cationic protein/RNase 3 is another RNase A‐family ribonuclease with direct antiviral activity. Nucleic Acids Res. 26, 3358–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Nevin, B. J. , Broadley, K. J. (2002) Nitric oxide in respiratory diseases. Pharmacol. Ther. 95, 259–293. [DOI] [PubMed] [Google Scholar]
- 146. Fischer, A. , Folkerts, G. , Geppetti, P. , Groneberg, D. A. (2002) Mediators of asthma: nitric oxide. Pulm. Pharmacol. Ther. 15, 73–81. [DOI] [PubMed] [Google Scholar]
- 147. Akaike, T. , Maeda, H. (2000) Nitric oxide and virus infection. Immunology 101, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Donnelly, L. E. , Barnes, P. J. (2002) Expression and regulation of inducible nitric oxide synthase from human primary airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 26, 144–151. [DOI] [PubMed] [Google Scholar]
- 149. Folkerts, G. , Nijkamp, F. P. (1998) Airway epithelium: more than just a barrier. Trends Pharmacol. Sci. 19, 334–341. [DOI] [PubMed] [Google Scholar]
- 150. Bogdan, C. , Rollinghoff, M. , Diefenbach, A. (2000) Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr. Opin. Immunol. 12, 64–76. [DOI] [PubMed] [Google Scholar]
- 151. Kharitonov, S. A. , Yates, D. , Robbins, R. A. , Logan‐Sinclair, R. , Shinebourne, E. A. , Barnes, P. J. (1994) Increased nitric oxide in exhaled air of asthmatic patients. Lancet 343, 133–135. [DOI] [PubMed] [Google Scholar]
- 152. Jatakanon, A. , Lim, S. , Kharitonov, S. A. , Chung, K. F. , Barnes, P. J. (1998) Correlation between exhaled nitric oxide, sputum eosinophils, and methacholine responsiveness in patients with mild asthma. Thorax 53, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rutgers, S. R. , van der Mark, T. W. , Coers, W. , Moshage, H. , Timens, W. , Kauffman, H. F. , Koeter, G. H. , Postma, D. S. (1999) Markers of nitric oxide metabolism in sputum and exhaled air are not increased in chronic obstructive pulmonary disease. Thorax 54, 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Maziak, W. , Loukides, S. , Culpitt, S. , Sullivan, P. , Kharitonov, S. A. , Barnes, P. J. (1998) Exhaled nitric oxide in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 157, 998–1002. [DOI] [PubMed] [Google Scholar]
- 155. Hoyt, J. C. , Robbins, R. A. , Habib, M. , Springall, D. R. , Buttery, L. D. , Polak, J. M. , Barnes, P. J. (2003) Cigarette smoke decreases inducible nitric oxide synthase in lung epithelial cells. Exp. Lung Res. 29, 17–28. [DOI] [PubMed] [Google Scholar]
- 156. Zimmermann, N. , King, N. E. , Laporte, J. , Yang, M. , Mishra, A. , Pope, S. M. , Muntel, E. E. , Witte, D. P. , Pegg, A. A. , Foster, P. S. , Hamid, Q. , Rothenberg, M. E. (2003) Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J. Clin. Invest. 111, 1863–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ferreira, H. H. , Medeiros, M. V. , Lima, C. S. , Flores, C. A. , Sannomiya, P. , Autunes, E. , De Nucci, G. (1996) Inhibition of eosinophil chemotaxis by chronic blockade of nitric oxide biosynthesis. Eur. J. Pharmacol. 310, 201–207. [DOI] [PubMed] [Google Scholar]
- 158. Beauvais, F. , Michel, L. , Dubertret, L. (1995) The nitric oxide donors, azide and hydroxylamine, inhibit the programmed cell death of cytokine‐deprived human eosinophils. FEBS Lett. 361, 229–232. [DOI] [PubMed] [Google Scholar]
- 159. Wei, X. Q. , Charles, I. G. , Smith, A. , Ure, J. , Feng, G. J. , Huang, F. P. , Xu, D. , Muller, W. , Moncada, S. , Liew, F. Y. (1995) Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 375, 408–411. [DOI] [PubMed] [Google Scholar]
- 160. Cifone, M. G. , Ulisse, S. , Santoni, A. (2001) Natural killer cells and nitric oxide. Int. Immunopharmacol. 1, 1513–1524. [DOI] [PubMed] [Google Scholar]
- 161. de Gouw, H. W. , Grunberg, K. , Schot, R. , Kroes, A. C. , Dick, E. C. , Sterk, P. J. (1998) Relationship between exhaled nitric oxide and airway hyperresponsiveness following experimental rhinovirus infection in asthmatic subjects. Eur. Respir. J. 11, 126–132. [DOI] [PubMed] [Google Scholar]
- 162. Sanders, S. P. , Kim, J. , Connolly, K. R. , Porter, J. D. , Siekierski, E. S. , Proud, D. (2001) Nitric oxide inhibits rhinovirus‐induced granulocyte macrophage colony‐stimulating factor production in bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 24, 317–325. [DOI] [PubMed] [Google Scholar]
- 163. Sanders, S. P. , Siekierski, E. S. , Porter, J. D. , Richards, S. M. , Proud, D. (1998) Nitric oxide inhibits rhinovirus‐induced cytokine production and viral replication in a human respiratory epithelial cell line. J. Virol. 72, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kao, Y. J. , Piedra, P. A. , Larsen, G. L. , Colasurdo, G. N. (2001) Induction and regulation of nitric oxide synthase in airway epithelial cells by respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 163, 532–539. [DOI] [PubMed] [Google Scholar]
- 165. Ali‐Ahmad, D. , Bonville, C. A. , Rosenberg, H. F. , Domachowske, J. B. (2003) Replication of respiratory syncytial virus is inhibited in target cells generating nitric oxide in situ. Front. Biosci. 8, a48–a53. [DOI] [PubMed] [Google Scholar]
- 166. Rimmelzwaan, G. F. , Baars, M. M. , de Lijster, P. , Fouchier, R. A. , Osterhaus, A. D. (1999) Inhibition of influenza virus replication by nitric oxide. J. Virol. 73, 8880–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Uetani, K. , Der, S. D. , Zamanian‐Daryoush, M. , de La Motte, C. , Lieberman, B. Y. , Williams, B. R. , Erzurum, S. C. (2001) Central role of double‐stranded RNA‐activated protein kinase in microbial induction of nitric oxide synthase. J. Immunol. 165, 988–996. [DOI] [PubMed] [Google Scholar]
- 168. Folkerts, G. , van der Linde, H. J. , Nijkamp, F. P. (1995) Virus‐induced airway hyperresponsiveness in guinea pigs is related to a deficiency in nitric oxide. J. Clin. Invest. 95, 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Gentile, D. A. , Doyle, W. J. , Belenky, S. , Ranck, H. , Angelini, B. , Skoner, D. P. (2002) Nasal and oral nitric oxide levels during experimental respiratory syncytial virus infection of adults. Acta Otolaryngol. 122, 61–66. [DOI] [PubMed] [Google Scholar]
- 170. Murphy, A. W. , Platts‐Mills, T. A. , Lobo, M. , Hayden, F. (1998) Respiratory nitric oxide levels in experimental human influenza. Chest 114, 452–456. [DOI] [PubMed] [Google Scholar]
- 171. Papi, A. , Stanciu, L. A. , Papadopoulos, N. G. , Teran, L. M. , Holgate, S. T. , Johnston, S. L. (2000) Rhinovirus infection induces major histocompatibility complex class I and costimulatory molecule upregulation on respiratory epithelial cells. J. Infect. Dis. 181, 1780–1784. [DOI] [PubMed] [Google Scholar]
- 172. Mogensen, T. H. , Paludan, S. R. (2001) Molecular pathways in virus‐induced cytokine production. Microbiol. Mol. Biol. Rev. 65, 131–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Gern, J. E. , French, D. A. , Grindle, K. A. , Brockman‐Schneider, R. A. , Konno, S. , Busse, W. W. (2003) Double‐stranded RNA induces the synthesis of specific chemokines by bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 28, 731–737. [DOI] [PubMed] [Google Scholar]
- 174. Alexopoulou, L. , Holt, A. C. , Medzhitov, R. , Flavell, R. A. (2001) Recognition of double‐stranded RNA and activation of NF‐kappaB by Toll‐like receptor 3. Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]