

Abstract
Summary. The study investigated the hepatitis B virus (HBV) genotypic resistance profile in 1803 nucleos(t)ide analogue (NA)‐experienced Chinese patients with chronic HBV infection. Serum HBV DNA was extracted, and the reverse transcriptase region was analysed by a high‐sensitive direct PCR sequencing and verified by clonal sequencing if necessary. Drug‐resistant mutations were detected in 560 of the 1803 patients, including 214 of 490 patients who received lamivudine (LAM), 35 of 428 patients who received adefovir (ADV), five of 18 patients who received telbivudine and 306 of 794 patients who received various sequential/combined NA therapies. ADV‐resistant mutations were detected in 36 of 381 patients who received LAM and then switched‐to ADV in contrast to one of 82 patients who received ADV add‐on LAM. Entecavir (ETV)‐resistant mutations were detected not only in LAM‐ and ETV‐treated patients but also in LAM‐treated ETV‐naïve patients. Double mutations rtM204I and rtL180M were detected more frequently in genotype C than in genotype B virus, and patients infected with this mutant had higher alanine transaminase levels than those infected with mutant containing the rtM204I substitution alone. Multidrug‐resistant HBV strains were identified in eight patients, including two novel strains with mutational patterns rtL180M + A181V + S202G + M204V + N236T and rtL180M + S202G + M204V + N236T. The results provide new information on HBV genotypic resistance profiles in a large cohort of Chinese patients with chronic HBV infection and may have important clinical implication for HBV drug resistance management in China.
Keywords: drug resistance, hepatitis B virus, mutation, nucleoside and nucleotide analogues
Abbreviations:
- ADV
adefovir
- ADV‐R
adefovir‐resistant mutations
- ALT
alanine aminotransferase
- CHB
chronic hepatitis B
- CHB‐LC
chronic hepatitis B‐related liver cirrhosis
- Coexist‐R
co‐existence of ADV‐R and LAM‐R or ETV‐R
- ETV
entecavir
- ETV‐R
entecavir‐resistant mutations
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- LAM
lamivudine
- LAM‐R
lamivudine‐resistant mutations
- LdT
telbivudine
- MDR
multidrug‐resistant
- NA
nucleos(t)ide analogue(s)
- PCR
polymerase chain reaction
- RT
reverse transcriptase
Introduction
Hepatitis B virus (HBV) chronic infection afflicts about 350 million people worldwide, of whom 93 million live in China [1, 2]. Morbidity and mortality in chronic hepatitis B (CHB) are linked to persistent viral replication and evolution to CHB‐related liver cirrhosis (CHB‐LC) or hepatocellular carcinoma (HCC) [3]. In China, four nucleos(t)ide analogues (NA), i.e. lamivudine (LAM), adefovir (ADV), entecavir (ETV) and telbivudine (LdT) are currently approved for the treatment of HBV infection, whilst recently developed tenofovir (TDF) is still unavailable. Treatment of CHB and CHB‐LC is aimed at suppressing viral replication to the lowest possible level, and thereby halting the progression of liver disease. However, viral resistance is the main drawback of long‐term antiviral therapy [4, 5, 6]. Suboptimal treatment regimens and drug‐resistant viral infection can increase the incidence of drug resistance and may favour the selection of multidrug‐resistant (MDR) HBV [7, 8].
The resistance mutations are located in the reverse transcriptase (RT) region of the HBV polymerase gene. The rtM204I and rtM204V are classic LAM‐resistant mutations and often coexist with compensatory mutations (rtV173L and rtL180M) [9, 10, 11]. The rtN236T and rtA181V are two well‐recognized ADV‐resistant mutations [12, 13], and some purported mutations such as rtV84M, rtV214A, rtQ215S, rtL217R and rtI233V may reduce susceptibility to ADV, although these are still controversial [13, 14, 15, 16]. Substitutions in rtT184, rtS202 or rtM250 in conjunction with LAM‐resistant mutations result in ETV resistance [17, 18, 19]. Cross‐resistance (usually rtM204I) also exists between LdT and LAM [20]. In addition, rtA181T seems to be an atypical substitution associated with LAM and ADV selection and may reduce the typical extent of virologic breakthrough [21].
Genotypic antiviral resistance is designated by the presence of unique nucleotide and corresponding deduced amino acid mutations in the drug target gene that have been previously demonstrated to be associated with antiviral resistance. The incidence of genotypic resistance is related to viral factors, host factors and treatment characteristics and is also affected by the methods used for detection of resistance mutations and the patient population being studied [22]. Several methods have been used for typing HBV genetic drug‐resistant mutations, each with individual advantages and disadvantages [1, 22]. Direct polymerase chain reaction (PCR) sequencing is the most popular method owing to the abundant information it provides. However, it is also considered less sensitive for typing samples with low viral load (<2000 IU/mL) and minor mutant subpopulations (<20%) [22].
As more antiviral strategies become available for the treatment of CHB and CHB‐LC, the risk and patterns of resistant and cross‐resistant emergence are diverse. Nonoptimal strategies based on the sequential use of NA increase the development of MDR strains [23]. Knowledge on the incidence and patterns of drug‐resistant mutants is valuable for clinicians and would assist clinical monitoring and management of the resistance. To date, data are largely derived from a few clinical trials and cohorts with limited drug resistance profiling. This study is intended to investigate population‐based profiles of HBV genotypic resistance in Chinese patients, with an improved direct PCR sequencing assay.
Patients and methods
Patients
Serum samples were collected from 1803 CHB and CHB‐LC patients who visited Beijing 302 Hospital during July 2007–March 2009. The standard for diagnoses of CHB and CHB‐LC was based on the Chinese Management Scheme of Diagnostic and Therapy Criteria of Viral Hepatitis [24] and have been described elsewhere [25, 26]. The male/female ratio was 1524/279. Average age was 37.2 ± 13.6 years. At the time of sampling for HBV genotyping, all patients were HBsAg positive, and 1203 (66.7%) patients were HBeAg positive. The median (Q1, Q3) of the alanine transaminase (ALT) level was 38 (24, 70) U/L; and the median (Q1, Q3) of the HBV DNA level was 2.8 × 104 (1.7 × 103, 7.5 × 105) IU/mL. All patients had received anti‐HBV NA (LAM, ADV, ETV and LdT) monotherapy, combination or sequential therapy for a minimum of 3 months. Written informed consents for the analysis were obtained from every patient. The study was approved by the ethics committee of Beijing 302 Hospital.
Biochemical and serological markers and quantitation of HBV DNA
Serum ALT, HBsAg/anti‐HBs, HBeAg/anti‐HBe, anti‐HBc and other biochemical and serological markers, as well as HBV DNA level were routinely measured or detected in the Central Clinical Laboratory of the Beijing 302 Hospital. HBV DNA level was determined by real‐time quantitative PCR (qPCR) (Fosun Pharmaceutical Co., Ltd., Shanghai, China) with a lower detection limit of 100 IU/mL (≈500 copies/mL).
HBV RT gene amplification and sequencing
Hepatitis B virus gene fragment (nt 54–1278) encompassing the complete RT gene was amplified by nested PCR. The sense and antisense primers for the first‐round PCR were 5′‐AGT CAG GAA GAC AGC CTA CTC C‐3′ (UP3, nt 3146–3167) and 5′‐AGG TGA AGC GAA GTG CAC AC‐3′ (DOWN1, nt 1577–1596), respectively. The sense and antisense primers for the second‐round PCR were 5′‐TTC CTG CTG GTG GCT CCA GTT C‐3′ (UP4, nt 54–75) and 5′‐TTC CGC AGT ATG GAT CGG CAG‐3 (DOWN2, nt 1258–1278), respectively. The first‐round PCR consisted of 10 cycles of 94 °C for 35 s, 59 °C for 35 s (decreasing by 2 °C every other cycle), 72 °C for 70 s; and 30 cycles of 94 °C for 35 s, 56 °C for 35 s, 72 °C for 70 s. The second‐round PCR (conducted in the same tube) consisted of 35 cycles of 94 °C for 25 s, 56 °C for 25 s and 72 °C for 50 s. PCR products were purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Sequencing was performed using the ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequencing data were analysed using the Vector NTI Suite software package (Informax, Frederick, MD, USA).
Mutation analysis
Mutations at 15 locations (including rt80, rt84, rt173, rt180, rt181, rt184, rt194, rt202, rt204, rt214, rt215, rt217, rt233, rt236 and rt250) in the RT gene were analysed. The rtM204I/V was defined as the signature LAM‐resistant mutations (LAM‐R) which also encompassed LdT‐resistant mutations. The rtA181V and rtN236T were defined as the signature ADV‐resistant mutations (ADV‐R). The rtT184A/C/F/G/I/L/M/S, rtS202C/G/I and rtM250I/L/V were defined as the signature ETV‐resistant mutations (ETV‐R) if concomitant with rtM204I/V. Coexistence of ADV‐R and LAM‐R or ETV‐R was defined as Coexist‐R which represents coincidence of detectable HBV mutants resistant to both nucleoside and nucleotide analogues in the viral quasi‐species. MDR mutations were identified from the Coexist‐R samples if the different mutations were co‐located in the same cloned viral sequence. In addition to signature mutations, rtV173L, rtL180M and rtA181T/S were included in drug‐resistant mutational patterns when they were present with signature resistance mutations.
TA cloning
PCR products were purified by QIAquick PCR Purification Kit (Qiagen), incubated with dATP and Taq DNA polymerase and then purified by QIAquick Gel Extraction Kit. The ligation and transformation were performed according to the manufacturer’s instructions of pGEM‐T Vector System II (Promega, Madison, WI, USA). Amplicons were purified by QIAprep Spin Miniprep Kit (Qiagen). The cloned target gene was sequenced with SP6 and T7 primers, and six additional primers were used for the sequencing of full‐length HBV genomes.
HBV genotype analysis
Hepatitis B virus genotype assignment was based on the phylogenetic analysis of the 1225‐bp‐long S/P‐gene sequence (nt 54–1278) as previously described [27, 28].
Statistical analysis
Data are presented as mean ± SD or median (Q1, Q3). Group comparisons were performed using the Wilcoxon or chi‐square tests using SAS 9.0 software (SAS Institute Inc., Cary, NC, USA). A P value of less than 0.05 was considered statistically significant.
Results
Mutation detection in relation to serum HBV DNA levels
For the convenience of evaluation, samples were assigned to four groups based on HBV DNA levels, i.e. <100 IU/mL (group A), ≥100 IU/mL but <2000 IU/mL (group B), ≥2000 IU/mL but <20 000 IU/mL (group C) and ≥20 000 IU/mL (group D). Groups A, B, C and D thus included 272 (15.1%), 393 (21.8%), 313 (17.4%) and 825 (45.8%) samples of the total, respectively. The HBV RT gene was sequenced successfully in 39.3% (107/272), 92.9% (365/393), 98.4% (308/313) and 100% (825/825) of cases in groups A, B, C and D, respectively. Signature resistance mutations were detected in 17.6% (48/272), 32.3% (127/393), 41.9% (131/313) and 30.8% (254/825) in groups A, B, C and D, respectively.
Incidence of drug‐resistant mutations present in various NA‐treated patients
Signature resistance mutations were detected in 560 of 1803 (31.1%) patients, comprising 214 of 490 patients who received LAM, 35 of 428 patients who received ADV, none of 73 patients who received ETV, five of 18 patients who received LdT monotherapies and 306 of 794 patients who received various sequential/combined NA therapies. The incidence of resistance mutations under different NA treatments is summarized in Table 1.
Table 1.
Drug‐resistant mutations in patients who received various nucleos(t)ide analogue therapies
| Drug usage* (mutant/total) | LAM‐R | ADV‐R | ETV‐R | Coexist‐R | Drug usage (mutant/total) | LAM‐R | ADV‐R | ETV‐R | Coexist‐R |
|---|---|---|---|---|---|---|---|---|---|
| LAM (214/490) | 208 | – | 5 | 1 | LAM→LAM + ADV + ETV (0/1) | – | – | – | – |
| ADV (35/428) | – | 35 | – | – | LAM→LAM + ETV (0/1) | – | – | – | – |
| ETV (0/73) | – | – | – | – | LAM→LAM + ETV→ETV (1/1) | – | – | 1 | – |
| LdT (5/18) | 5 | – | – | – | LAM→LdT (6/10) | 6 | – | – | – |
| LAM→ADV (136/381) | 87 | 36 | 4 | 9 | LAM + ADV (1/15) | 1 | – | – | – |
| LAM→ETV (35/77) | 25 | – | 10 | – | LAM + ADV→ADV (0/4) | – | – | – | – |
| LAM→ADV→ETV (38/57) | 29 | – | 8 | 1 | LAM + ADV→ADV→ADV + LdT (1/1) | 1 | – | – | – |
| LAM→ETV→ADV (4/7) | 4 | – | – | – | LAM + ADV→ADV→ETV (1/1) | 1 | – | – | – |
| LAM→ADV→ADV + ETV (3/5) | 3 | – | – | – | LAM + ADV→ETV (0/1) | – | – | – | – |
| LAM→ADV→ADV + LdT (2/4) | – | – | 2 | – | LAM→ADV→ETV→ADV→ADV + LAM (1/1) | – | – | – | 1 |
| LAM→ADV→ETV→ADV + ETV (1/1) | 1 | – | – | – | ADV→ETV (1/27) | – | 1 | – | – |
| LAM→ADV→LAM→ADV + ETV (1/1) | 1 | – | – | – | ADV→LAM (3/12) | 3 | – | – | – |
| LAM→ADV→LAM + ADV (5/10) | 3 | – | – | 2 | ADV→ADV + LAM (2/19) | 1 | – | – | 1 |
| LAM→ADV→LdT (8/12) | 7 | 1 | – | – | ADV + LdT (1/3) | – | 1 | – | – |
| LAM→ADV + ETV (2/3) | 2 | – | – | – | ADV→LAM→LAM + ADV (2/3) | 1 | – | 1 | – |
| LAM→ADV + ETV→ADV (1/1) | 1 | – | – | – | ADV→ETV→LdT (0/1) | – | – | – | – |
| LAM→ADV + LdT (1/1) | 1 | – | – | – | ADV→ADV + LdT (0/4) | – | – | – | – |
| LAM→ETV→ETV + ADV (1/3) | 1 | – | – | – | ADV→LAM→ETV (1/1) | 1 | – | – | – |
| LAM→LAM + ADV (32/82) | 30 | 1 | 1 | – | ADV→LdT (8/23) | 3 | 3 | – | 2 |
| LAM→LAM + ADV→ADV (3/7) | 3 | – | – | – | ADV→ADV + ETV (0/1) | – | – | – | – |
| LAM→LAM + ADV→ADV→ETV (1/1) | – | – | 1 | – | ETV→ADV (0/3) | – | – | – | – |
| LAM→LAM + ADV→ETV (1/3) | 1 | – | – | – | ETV→ETV + ADV (0/3) | – | – | – | – |
| LAM→LAM + ADV→ETV→LdT (1/1) | 1 | – | – | – | |||||
| LAM→LAM + ADV→ETV + ADV (1/1) | – | – | 1 | – | Total (560/1803) | 431 | 78 | 34 | 17 |
LAM‐R, ADV‐R and ETV‐R represent lamivudine‐, adefovir‐ and entecavir‐resistant mutations, respectively. Coexist‐R represents coexistence of detectable LAM‐R/ ETV‐R (nucleoside) and ADV‐R (nucleotide) in viral populations.
LdT, telbivudine.
*Each mono‐ or sequential/combined therapy lasted for ≥3 months.
Mutational patterns present in various NA usages
Of LAM‐resistant mutations, rtM204V was usually concomitant with rtL180M ± V173L. By contrast, rtM204I was more often accompanied by rtL80I (36.5%) than rtM204V (3.9%). Of the resistance mutations detected in patients who received monotherapies, rtM204I (32.2%), rtM204V + L180M ± V173L (32.2%) and rtM204I + L180M ± V173L (21.0%) were dominant patterns for LAM; rtN236T + A181T and/or rtA181V (34.3%), rtN236T (31.4%) and rtA181V (28.6%) were dominant patterns for ADV, and mutations containing rtM204I were dominant patterns for LdT. No resistance mutations were detected in patients treated with ETV monotherapy (Table 2). The HBV mutational patterns in patients treated with sequential/combined therapies were more diverse. Amongst them, 29 harboured ETV‐R mutants and 16 harboured Coexist‐R mutants (Table 3).
Table 2.
Drug‐resistant mutations in patients who received nucleos(t)ide analogue monotherapies
| Drug usage* | Major mutational patterns | n | Drug usage | Major mutational patterns | n |
|---|---|---|---|---|---|
| LAM (n = 214/490) | ADV (n = 35/428) | ||||
| LAM‐R | M204I | 69 | ADV‐R | N236T | 11 |
| M204V + L180M | 54 | A181V | 10 | ||
| M204I + L180M | 44 | N236T + A181T | 5 | ||
| M204I/V + L180M | 15 | N236T + A181V | 4 | ||
| M204V + L180M + V173L | 15 | N236T + A181T/V | 2 | ||
| M204I/V + L180M + A181S | 3 | N236T + A181T/S | 1 | ||
| M204I/V + L180M + V173L | 3 | N236T + M250L | 1 | ||
| M204I + L180M + A181T | 2 | A181T/V | 1 | ||
| M204V | 1 | ETV (n = 0/73) | – | – | |
| M204I/V | 1 | ||||
| M204I + L180M + V173L | 1 | LdT (n = 5/18) | |||
| Coexist‐R | M204I + V173M + A181V | 1 | LAM‐R | M204I | 3 |
| ETV‐R | M204I + L180M + V173L + M250L | 1 | M204I + L180M | 1 | |
| M204I + M250L | 1 | M204I/V + L180M | 1 | ||
| M204V + T184S | 1 | ||||
| M204I/V + L180M + T184S | 1 | ||||
| M204V + L180M + S202G | 1 | ||||
LAM‐R, ADV‐R and ETV‐R represent lamivudine‐, adefovir‐ and entecavir‐resistant mutations, respectively. Coexist‐R represents coexistence of detectable LAM‐R/ETV‐R and ADV‐R in viral populations.
LdT, telbivudine.
*Each mono‐ or sequential/combined therapy lasted for ≥3 months.
Table 3.
Mutational patterns of ETV and coexist resistance in patients who received sequential/combined nucleos(t)ide analogue therapies
| Drug usage* | Major mutational patterns | n | Drug usage | Major mutational patterns | n |
|---|---|---|---|---|---|
| LAM→ADV (n = 136) | LAM→ADV→ETV (n = 38) | ||||
| ETV‐R | V173L + M204I + M250L | 1 | Coexist‐R | L180M + A181V + M204V | 1 |
| L180M + T184I/L + M204V | 2 | ETV‐R | L180M + S202G + M204V | 5 | |
| V173L + L180M + M204I + M250L | 1 | L180M + T184L/A + M204V | 1 | ||
| Coexist‐R | L180M + A181T + M204I + N236T | 1 | L180M + T184L + M204I/V | 1 | |
| L180M + A181V + M204V + N236T | 1 | L180M + T184S + M204V | 1 | ||
| L180M + A181V + M204I/V | 1 | LAM→ADV→ADV + LdT (n = 2) | |||
| L180M + A181V + M204V | 4 | ETV‐R | L180M + M204I + M250L | 2 | |
| V173L + A181V + M204I | 1 | LAM→LAM + ADV (n = 32) | |||
| A181V + M204I + M250I | 1 | ETV‐R | V173L + M204I + M250L | 1 | |
| LAM→ETV (n = 35) | LAM→LAM + ADV→ADV→ETV (n = 1) | ||||
| ETV‐R | L180M + T184S + M204V | 2 | ETV‐R | L180M + A181G + T184L + M204V | 1 |
| L180M + S202G + M204V | 1 | LAM→LAM + ADV→ETV + ADV (n = 1) | |||
| L180M + T184I + M204I/V | 1 | ETV‐R | L180M + T184I + M204I | 1 | |
| L180M + T184S + M204I | 1 | LAM→LAM + ETV→ETV (n = 1) | |||
| L180M + T184A + M204V | 1 | ETV‐R | L180M + T184L + M204V | 1 | |
| L180M + T184L + S202G + M204V | 1 | ADV→LdT (n = 8) | |||
| M204V + A181S + T184I | 1 | Coexist‐R | L180M + A181V + M204V | 2 | |
| V173L + L180M + T184S + M204I | 1 | ADV→ADV + LAM (n = 2) | |||
| L180M + T184L + M204V | 1 | Coexist‐R | V173L + L180M + A181T/V + M204V | 1 | |
| LAM→ADV→LAM + ADV (n = 5) | ADV→LAM→LAM + ADV (n = 2) | ||||
| Coexist‐R | L180M + M204V + N236T | 1 | ETV‐R | V173L + L180M + M204I + M250L | 1 |
| L180M/I + A181T/V + M204I + M250L | 1 | LAM→ADV→ETV →ADV→ADV + LAM (n = 1) | |||
| Coexist‐R | L180M + A181V + S202G + M204V + N236T | 1 | |||
ETV‐R represents entecavir‐resistant mutations, and Coexist‐R represents coexistence of detectable LAM‐R/ETV‐R and ADV‐R in viral populations.
LAM, lamivudine; ADV, adefovir; ETV, entecavir; LdT, telbivudine.
*Each mono‐ or sequential/combined therapy lasted for ≥3 months.
Genotype and its association with mutational patterns
Hepatitis B virus genotypes C, B and D were assigned in 1351 (84.2%), 240 (15.0%) and 14 (0.8%) of the 1605 successfully sequenced samples, respectively. Identical results were obtained from 58 random samples typed by the INNO‐LiPA Genotyping kit (Innogenetics, Ghent, Belgium). The frequencies of rtM204I with/without rtL180M were significantly different between genotypes C and B (44.6%/55.4%vs 19.4%/80.6%, P < 0.01). Patients harbouring the rtM204I + L180M mutant had higher ALT levels than those harbouring the rtM204I mutant alone [median (Q1, Q3) 41 (28, 69) U/L vs 32 (22, 53) U/L, P < 0.01]. HBV DNA levels were not significantly different between the two groups [median (Q1, Q3) 1.5 × 105 (6.7 × 103, 5.9 × 106) vs 6.8 × 104 (2.5 × 103, 1.5 × 106), P > 0.05].
Analysis of purported mutations
Table 4 summarizes the incidence of the purported mutations under different NA treatment schedules, including rtV84M, rtA181T/S (alone), rtV214A, rtQ215S, rtL217R and rtI233V. The patterns of rtA181T/S together with signature resistance mutations are presented in 2, 3. The incidence of rtV84M, rtA181T and rtV214A was relatively high, whereas the incidence of rtQ215S, rtL217R and rtI233V was quite low. The mutation rtA194T with potential resistance to TDF [29] was not detected.
Table 4.
Incidence of purported mutations associated with resistance
| NA usage* | V84M (containing) | A181T or S (alone) | V214A (containing) | Q215S (containing) | L217R (containing) | I233V (containing) |
|---|---|---|---|---|---|---|
| LAM | V84M + A181T (1), V84M + M204I (2) V84M + L80I + L180M + M204I (1) V84M + L180M + M204V ± V173L (2) | A181T (2) | V214A + L180M + M204I (3) V214A + L180M + M204V (1) V214A + M204I (2) | Q215S + L180M + M204I (1) | L217R + L180M + M204I (1) | I233V (1) |
| LAM→ADV | V84M + L80I (2), V84M + M204I (1) V84M + L180M + M204I/V (1) V84M + L180M + A181T + M204I + N236T (1) | A181T (14) A181S (1) | V214A + A181T (1) V214A + A181T + N236T (1) V214A + L180M + M204V (1) V214A + V173L + L180M + M204V (1) V214A (3) | – | L217R + L180M + N236T (1) | – |
| LAM→ETV | V84M + T184I (1), V84M + L180M + M204I (1) V84M + L80I + L180M + T184I + M204I/V (1) | – | V214A + M204I (1) | – | – | – |
| LAM→LAM + ADV | V84M + A181T (1), V84M + A181T + V214A (1) V84M + M204I (1), V84M + A181T + M204I (1) | A181T (3) | V214A + A181T (2) V214A + V84M + A181T (1) | – | – | – |
| LAM + ADV→ADV | – | A181T (1) | V214A (1) | – | – | – |
| ADV | V84M (1), V84M + A181T (1) V84M + L80I + A181T + N236T (1) | A181T (11) | V214A + A181T/V + N236T (1) V214A + A181T (1), V214A (4) V214A + A181T + N236T (1) | Q215S + A181V (1) | L217R (1) | I233V + A181T (1) I233V (3) |
| ADV→LdT | V84M + M204I (1) | A181T (5) | V214A + A181T/V + N236T (1) V214A + A181T (1) | – | – | – |
| LdT | – | A181T (1) | – | – | – | – |
| LAM→ADV→ETV | V84M + L180M + M204V (1) V84M + L80I + M204I (1) V84M + L80I + L180M + M204I (1) | – | – | – | – | – |
| LAM→ADV + ETV | V84M + L180M + M204V (1) | – | – | – | – | – |
| LAM→ADV + ETV→ADV | V84M + M204I (1) | – | – | – | – | – |
| Total | 27 | 38 | 26 | 2 | 3 | 5 |
The incidence number of mutations is indicated in bracket. LAM, lamivudine; ADV, adefovir; ETV, entecavir; LdT, telbivudine; NA, nucleos(t)ide analogues.
*Each mono‐ or sequential/combined therapy lasted for ≥3 months.
MDR mutations
To identify MDR mutations, clonal sequencing (≥20 clones/sample) was performed for the 16 samples identified as Coexist‐R by direct PCR sequencing. The results showed that eight samples contained different monodrug‐resistant mutants, whereas the other eight harboured MDR HBV strains, with or without non‐MDR strain coexistence (Fig. 1). Two novel MDR HBV strains with triple genotypic resistance to LAM, ADV and ETV were identified from one of these samples. The mutational patterns of the two strains were rtL180M + A181V + S202G + M204V + N236T and rtL180M + S202G + M204V + N236T (GenBank accession numbers: GQ402161 and GQ402162), respectively. The emergence of drug‐resistant and MDR HBV strains was closely associated with drug administration schedule and clinical features (Fig. 2).
Figure 1.

Identification of multidrug‐resistant (MDR) mutations. Clonal sequencing identified that eight patients who received sequential NA harboured MDR mutants with signature mutations resistant to both nucleoside (LAM/LdT/ETV) and nucleotide analogues (ADV). The NA treatment schedules are shown at the bottom of the figure. Treatment duration (month) is indicated in brackets. LAM, lamivudine; ADV, adefovir; ETV, entecavir; LdT, telbivudine; NA, nucleos(t)ide analogues.
Figure 2.
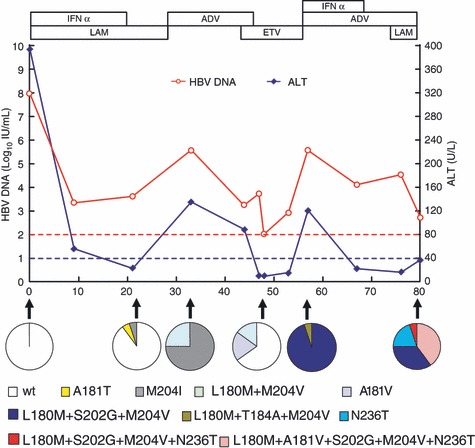
Evolution of resistant HBV strains with clinical features in one case during antiviral treatment. Changes in serum HBV DNA and ALT levels are presented along with successive antiviral therapies. The dynamic changes of wild‐type/resistant mutants were analysed by clonal sequencing (≥20 clones for each sample), and these are depicted as pie charts serially. Red dashes represent lower detection limit for HBV DNA in the clinic (100 IU/mL). Blue dashes represent upper normal limit of ALT (40 U/L). LAM, lamivudine; ADV, adefovir; ETV, entecavir; IFN, interferon; ALT, alanine transaminase; HBV, hepatitis B virus.
Discussion
Hepatitis B virus drug resistance develops in succession as genotypic resistance, phenotypic resistance of virologic breakthrough and biochemical/clinical breakthrough [30, 31]. Early discovery of genotypic resistance allows timely adjustment of therapy strategies to avoid virologic rebound and hepatitis flare and to distinguish suboptimal responses caused by nonviral factors. Satisfying this clinical requirement needs a high‐sensitive resistance‐typing assay. Though direct sequencing is widely used to detect ‘new’ substitutions, and taken as a ‘gold standard’, it is considered as a relatively less‐sensitive assay [22]. We developed a high‐sensitive nested PCR assay allowing us to analyse samples with quite low viral load as we did for the SARS coronavirus [32, 33, 34]. In this study, HBV DNA levels were quantitated in the Central Clinical Laboratory of the hospital, and we performed the direct PCR sequencing assay without knowing the viral load beforehand. Thus, the sequencing success rates in relation to serum HBV DNA levels are substantially objective. A small proportion of samples were positive (i.e., ≥100 IU/mL) in HBV DNA quantitation by routine qPCR but negative in HBV gene amplification for direct sequencing. In fact, this is for the 28/393 samples in group B (≥100 IU/mL but <2000 IU/mL) and the 5/313 samples in group C (≥2000 IU/mL but <20 000 IU/mL), although a lower detection limit of the latter assay was set at 20 IU/mL with a calibration standard. This may be attributed to differences between methodologies, e.g. the direct PCR sequencing assay needs to amplify much longer viral gene fragments than routine qPCR does and thus will be influenced more by virus disintegration. A similar phenomenon was also observed by other investigators [35]. Although line probe assays can detect minor sequences as low as 5% in the virus pool, an HBV DNA concentration no less than 104 IU/mL is required [36]. In our one‐tube PCR assay, all amplicons from the first‐round PCR were subjected to the second‐round PCR. The inhibitory effect of the first‐round PCR products was eliminated by a special technique (Chinese patent application number: 200910092331.1), which contributed the most to the higher sensitivity of this assay. These advantages plus low cost permitted the use of the direct PCR sequencing assay for monitoring of drug resistance in the clinic.
Hepatitis B virus RT sequences were available in nearly 40% of patients with undetectable HBV DNA by routine qPCR, implying that a very low viraemia may still exist in NA‐treated patients with good virologic response. This offers a circumstantial explanation for frequent and rapid relapse of many NA responders when treatment is discontinued [37, 38]. The resistance mutations were detected in 48 patients with viral load less than 100 IU/mL, suggesting that low viral replication may reduce but not prevent drug resistance development. In clinical practice, HBV DNA quantitation is still more practicable than genotypic resistance testing to monitor drug resistance for patients with good virologic response considering cost‐effectiveness. It was observed that patients with moderate levels of HBV DNA (2000–20 000 IU/mL) had higher resistance mutation incidence (41.9%) than those with higher viral load. One explanation could be that in the presence of the antiviral agent, suboptimal suppression of virus replication may provide greater replication advantage of the mutants against the wild type than poor suppression of virus replication does [39]. Other influence factors may include differences in treatment strategy and duration.
More varied ADV‐R patterns were detected in patients receiving LAM switching‐to ADV than in those received add‐on ADV. This is consistent with the findings by other groups [40, 41, 42]. Interestingly, LAM‐R patterns were also detected frequently in the ADV add‐on group. One possible reason is that ADV has relatively weaker potency to suppress LAM‐resistant mutants whilst LAM has stronger potency to suppress ADV‐resistant ones. The rtM204V was usually concomitant with rtL180M. Therefore, LAM switching‐to LdT is unsuitable once YMDD mutations occurred, as LdT is ineffective against both rtM204I and rtL180M + M204V mutants, though it remains effective against the single rtM204V mutation [6]. Pre‐existing ETV‐R patterns were detected in some LAM‐experienced and ETV‐naïve patients. This may account for primary resistance or rapid failure to ETV treatment in some LAM‐refractory patients.
The incidence of mutational patterns of rtM204I and rtM204V has been reported to be significantly different between genotype B and C HBV [43], but we did not observe any significant differences in this study. We found that the incidence of rtM204I concomitant with rtL180M was significantly higher in genotype C than in genotype B, and patients with rtM204I + L180M had a higher ALT level compared to those with rtM204I alone. As rtL180M can compensate for the replication capacity of the YMDD mutant and higher viral replication may induce stronger immune responses, it is plausible that elevated ALT was associated with the fluctuating cycle of viral replication and hepatic inflammation. Consistent with this is the observation that rtL180M with rtM204I have been reported to decrease serum ALT normalization significantly after ADV therapy [44].
There are several nonsignature mutations that have been associated with NA selection. Amongst these, rtL80I was reported to compensate for the loss of replication efficiency associated with the acquisition of LAM resistance, particularly in the case of rtM204I [45]. Our results were consistent with this view as 85.2% of rtL80I concurred with rtM204I and rtM204I/V. The rtL80I occurred proportionally equally between the presence and absence of compensatory mutations rtL180M ± V173L, but it occurred more frequently in genotype C than in genotype B (7.6%vs 3.7%, P = 0.019). The rtV84M, rtA181T and rtV214A were more common in LAM‐ and/or ADV‐treated patients. By contrast, rtV84M and rtV214A were more likely to be concomitant mutations. The rtQ215S, rtL217R and rtI233V had a lower incidence. The association of these purported mutations with drug resistance needs further clarification.
Multidrug‐resistant strains often arise in suboptimal sequential or combined therapeutic strategy if it does not result in rapid and complete viral suppression, especially when there is a large replication space available for the mutants to spread [7]. Nevertheless, because nucleoside (LAM, ETV and LdT) and nucleotide analogues (ADV) usually have a complementary cross‐resistance profile when they are used in combination and MDR HBV strains usually replicate their genome less efficiently than either wild‐type HBV or monodrug‐resistant mutants do, the MDR HBV strains resistant to both types of NA occur infrequently in the clinic. To date, reports on clinical MDR HBV strains are restricted to double resistance against LAM and ADV [23, 46, 47, 48, 49, 50], if rtA181T/V cross‐resistance alone and anti‐HBs immunoglobulin resistance are excluded. In this study, RT sequences with MDR mutational patterns were identified from eight patients receiving sequential NA. Interestingly, triple resistance against LAM, ADV and ETV was found which to our knowledge has not been reported previously. Dynamic analysis showed that MDR HBV strains developed in close relation to sequential drug administration and in accordance with clinical features. Coexistence of monodrug‐resistant mutants may favour the emergence of MDR strains. Although in vitro phenotyping was not performed in this study, it has been shown that MDR strains with signature mutations to both LAM and ADV (rtM204V + A181V/N236T) have competent replicative capacity in the presence of LAM and ADV and obviously reduced susceptibility to each of the drugs in comparison with wild‐type stains [51, 52].
Unlike in a rigorously designed clinical trial, the patients enrolled in this study were following a variety of NA schedules and treatment duration varied extensively. The duration of treatment might influence the incidence of resistant HBV strains, although the influence was relatively minor in the large population samples of our study. Many patients had received different NA therapies before they came to seek medical care in our hospital, and their samples at baseline and early stage of treatment were unavailable, which makes our investigation hard to be systematic. However, the present population‐based cross‐sectional investigation has the advantage of acquiring an overall HBV resistance profile from clinical practice which makes it greatly informative. Taken together, our results provide new insight into HBV genotypic resistance profiles in a large cohort of Chinese patients with chronic HBV infection, which may have important clinical implications for management of HBV drug resistance in China.
Acknowledgements
This work was supported by grants from the National Key Basic Research Developing Project (2007CB512803) and the Beijing Natural Science Foundation (7091006) and partly by National 11th Five‐Year Special Grand Project for Infectious Diseases (2008ZX10002‐011, 2009ZX10004‐314).
References
- 1. Pawlotsky JM, Dusheiko G, Hatzakis A et al. Virologic monitoring of hepatitis B virus therapy in clinical trials and practice: recommendations for a standardized approach. Gastroenterology 2008; 134: 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ministry of Health of the People’s Republic of China . Issued on 2008‐04‐21. Available at: http://www.moh.gov.cn/sofpro/cms/previewjspfile/mohbgt/cms_0000000000000000144_tpl.jsp?requestCode=33255&CategoryID=4811. European Association for the Study of the Liver (accessed 1 April 2009).
- 3. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of chronic hepatitis B. J Hepatol 2009; 50: 227–242. [DOI] [PubMed] [Google Scholar]
- 4. Papatheodoridis GV, Manolakopoulos S, Dusheiko G, Archimandritis AJ. Therapeutic strategies in the management of patients with chronic hepatitis B virus infection. Lancet Infect Dis 2008; 8: 167–178. [DOI] [PubMed] [Google Scholar]
- 5. Dienstag JL. Hepatitis B virus infection. N Engl J Med 2008; 359: 1486–1500. [DOI] [PubMed] [Google Scholar]
- 6. Zoulim F, Perrillo R. Hepatitis B: reflections on the current approach to antiviral therapy. J Hepatol 2008; 48: S2–S19. [DOI] [PubMed] [Google Scholar]
- 7. Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009; 137: 1593–1608. [DOI] [PubMed] [Google Scholar]
- 8. Zoulim F, Buti M, Lok AS. Antiviral‐resistant hepatitis B virus: can we prevent this monster from growing? J Viral Hepat 2007; 14: 29–36. [DOI] [PubMed] [Google Scholar]
- 9. Allen MI, Deslauriers M, Andrews CW et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27: 1670–1677. [DOI] [PubMed] [Google Scholar]
- 10. Ono SK, Kato N, Shiratori Y et al. The polymerase L528M mutation cooperates with nucleotide binding‐site mutations, increasing hepatitis B virus replication and drug resistance. J Clin Invest 2001; 107: 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delaney WEt, Yang H, Westland CE et al. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol 2003; 77: 11833–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Angus P, Vaughan R, Xiong S et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology 2003; 125: 292–297. [DOI] [PubMed] [Google Scholar]
- 13. Borroto‐Esoda K, Miller MD, Arterburn S. Pooled analysis of amino acid changes in the HBV polymerase in patients from four major adefovir dipivoxil clinical trials. J Hepatol 2007; 47: 492–498. [DOI] [PubMed] [Google Scholar]
- 14. Schildgen O, Sirma H, Funk A et al. Variant of hepatitis B virus with primary resistance to adefovir. N Engl J Med 2006; 354: 1807–1812. [DOI] [PubMed] [Google Scholar]
- 15. Pellicelli AM, Barbaro G, Francavilla R et al. Adefovir and lamivudine in combination compared with adefovir monotherapy in HBeAg‐negative adults with chronic hepatitis B virus infection and clinical or virologic resistance to lamivudine: a retrospective, multicenter, nonrandomized, open‐label study. Clin Ther 2008; 30: 317–323. [DOI] [PubMed] [Google Scholar]
- 16. Qi X, Xiong S, Yang H, Miller M, Delaney WE IV. In vitro susceptibility of adefovir‐associated hepatitis B virus polymerase mutations to other antiviral agents. Antivir Ther 2007; 12: 355–362. [PubMed] [Google Scholar]
- 17. Tenney DJ, Levine SM, Rose RE et al. Clinical emergence of entecavir‐resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother 2004; 48: 3498–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tenney DJ, Rose RE, Baldick CJ et al. Two‐year assessment of entecavir resistance in Lamivudine‐refractory hepatitis B virus patients reveals different clinical outcomes depending on the resistance substitutions present. Antimicrob Agents Chemother 2007; 51: 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baldick CJ, Eggers BJ, Fang J et al. Hepatitis B virus quasispecies susceptibility to entecavir confirms the relationship between genotypic resistance and patient virologic response. J Hepatol 2008; 48: 895–902. [DOI] [PubMed] [Google Scholar]
- 20. Lai CL, Leung N, Teo EK et al. A 1‐year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen‐positive chronic hepatitis B. Gastroenterology 2005; 129: 528–536. [DOI] [PubMed] [Google Scholar]
- 21. Warner N, Locarnini S. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatololgy 2008; 48: 88–98. [DOI] [PubMed] [Google Scholar]
- 22. Lok AS, Zoulim F, Locarnini S et al. Antiviral drug‐resistant HBV: standardization of nomenclature and assays and recommendations for management. Hepatology 2007; 46: 254–265. [DOI] [PubMed] [Google Scholar]
- 23. Brunelle MN, Jacquard AC, Pichoud C et al. Susceptibility to antivirals of a human HBV strain with mutations conferring resistance to both lamivudine and adefovir. Hepatology 2005; 41: 1391–1398. [DOI] [PubMed] [Google Scholar]
- 24. Chinese Society of Infectious Diseases and Parasitology , Chinese Society of Hepatology . Management scheme of diagnostic and therapy criteria of viral hepatitis. Zhonghua Gan Zang Bing Za Zhi (Chinese J Hepatol) 2000; 6: 324–329. [Google Scholar]
- 25. Xu D, Fu J, Jin L et al. CD4+CD25+ regulatory T cells in circulation and liver actively influence the antiviral immune response and disease progression in patients with hepatitis B. J Immunol 2006; 177: 739–747. [DOI] [PubMed] [Google Scholar]
- 26. Zou Z, Xu D, Li B et al. Compartmentalization and its implication for peripheral immunologically‐competent cells to the liver in patients with HBV‐related acute‐on‐chronic liver failure. Hepatol Res 2009; 39: 1198–1207. [DOI] [PubMed] [Google Scholar]
- 27. Liu Y, Zhong Z, Zou Z et al. Features and clinical implications of hepatitis B virus genotypes and mutations in basal core promoter/precore region in 507 Chinese patients with acute and chronic hepaittis B. J Clin Virol 2010; 47: 243–247. [DOI] [PubMed] [Google Scholar]
- 28. Ren X, Xu Z, Liu Y et al. Hepatitis B virus genotype and basal core promoter/precore mutations are associated with hepatitis B‐related acute‐on‐chronic liver failure without pre‐existing liver cirrhosis. J Viral Hepat 2010; doi: 10.1111/j.1365-2893.2009.01254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amini‐Bavil‐Olyaee S, Herbers U, Sheldon J, Luedde T, Trautwein C, Tacke F. The rtA194T polymerase mutation impacts viral replication and susceptibility to tenofovir in hepatitis B e antigen‐positve and hepatitis B e antigen‐negative hepatitis B virus strains. Hepatology 2009; 49: 1158–1165. [DOI] [PubMed] [Google Scholar]
- 30. Keeffe EB, Dieterich DT, Pawlotsky JM, Benhamou Y. Chronic hepatitis B: preventing, detecting, and managing viral resistance. Clin Gastroenterol Hepatol 2008; 6: 268–274. [DOI] [PubMed] [Google Scholar]
- 31. Keeffe EB, Dieterich DT, Han SH et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2008 update. Clin Gastroenterol Hepatol 2008; 6: 1315–1341. [DOI] [PubMed] [Google Scholar]
- 32. Xu D, Zhang Z, Wang FS. SARS‐associated coronavirus quasispecies in individual patients. N Engl J Med 2004; 350: 1366–1367. [DOI] [PubMed] [Google Scholar]
- 33. Xu D, Zhang Z, Chu FL et al. Genetic variation of SARS coronavirus in Beijing hospital. Emerg Infect Dis 2004; 10: 789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu D, Zhang Z, Jin L et al. Persistent shedding of viable SARS‐CoV in urine and stool during the convalescent phase of SARS patients. Eur J Clin Microbiol Infect Dis 2005; 24: 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tran N, Berne R, Chann R et al. European multicenter evaluation of high‐density DNA probe arrays for detection of hepatitis B virus resistance mutations and identification of genotypes. J Clin Microbiol 2006; 44: 2792–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Degertekin B, Hussain M, Tan J, Oberhelman K, Lod AS. Sensitivity and accuracy of an updated line probe assay (HBV DR v.3) in detecting mutations associated with hepatitis B antiviral resistance. J Hepatol 2009; 50: 42–48. [DOI] [PubMed] [Google Scholar]
- 37. Hadziyannis SJ, Papatheodoridis GV. Hepatitis B e antigen‐negative chronic hepatitis B: natural history and treatment. Semin Liver Dis 2006; 26: 130–141. [DOI] [PubMed] [Google Scholar]
- 38. Shouval D, Lai CL, Chang TT et al. Relapse of hepatitis B in HBeAg‐negative chronic hepatitis B patients who discontinued successful entecavir treatment: the case for continuous antiviral therapy. J Hepatol 2009; 50: 289–295. [DOI] [PubMed] [Google Scholar]
- 39. Ghany MG, Doo EC. Antiviral resistance and hepatitis B therapy. Hepatology 2009; 49: S174–S184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peters MG, Hann Hw H, Martin P et al. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine‐resistant chronic hepatitis B. Gastroenterology 2004; 126: 91–101. [DOI] [PubMed] [Google Scholar]
- 41. Rapti I, Dimou E, Mitsoula P, Hadziyannis SJ. Adding‐on versus switching‐to adefovir therapy in lamivudine‐resistant HBeAg‐negative chronic hepatitis B. Hepatology 2007; 45: 307–313. [DOI] [PubMed] [Google Scholar]
- 42. Yatsuji H, Suzuki F, Sezaki H et al. Low risk of adefovir resistance in lamivudine‐resistant chronic hepatitis B patients treated with adefovir plus lamivudine combination therapy: two‐year follow‐up. J Hepatol 2008; 48: 923–931. [DOI] [PubMed] [Google Scholar]
- 43. Pan XP, Li LJ, Du WB, Li MW, Cao HC, Sheng JF. Differences of YMDD mutational patterns, precore/core promoter mutations, serum HBV DNA levels in lamivudine‐resistant hepatitis B genotypes B and C. J Viral Hepat 2007; 14: 767–774. [DOI] [PubMed] [Google Scholar]
- 44. Cha CK, Kwon HC, Cheong JY et al. Association of lamivudine‐resistant mutational patterns with the antiviral effect of adefovir in patients with chronic hepatitis B. J Med Virol 2009; 81: 417–424. [DOI] [PubMed] [Google Scholar]
- 45. Warner N, Locarnini S, Kuiper M et al. The L80I substitution in the reverse transcriptase domain of the hepatitis B virus polymerase is associated with lamivudine resistance and enhanced viral replication in vitro. Antimicrob Agents Chemother 2007; 51: 2285–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yim HJ, Hussain M, Liu Y, Wong SN, Fung SK, Lok AS. Evolution of multi‐drug resistant hepatitis B virus during sequential therapy. Hepatology 2006; 44: 703–712. [DOI] [PubMed] [Google Scholar]
- 47. Villet S, Pichoud C, Villeneuve JP, Trepo C, Zoulim F. Selection of a multiple drug‐resistant hepatitis B virus strain in a liver transplanted patient. Gastroenterology 2006; 131: 1253–1261. [DOI] [PubMed] [Google Scholar]
- 48. Shim JH, Suh DJ, Kim KM et al. Efficacy of entecavir in patients with chronic hepatitis B resistant to both lamivudine and adefovir or to lamivudine alone. Hepatology 2009; 50: 1064–1071. [DOI] [PubMed] [Google Scholar]
- 49. Santantonio T, Fasano M, Durantel S et al. Adefovir dipivoxil resistance patterns in patients with lamivudine‐resistant chronic hepatitis B. Antivir Ther 2009; 14: 557–565. [PubMed] [Google Scholar]
- 50. Inous J, Ueno Y, Wakui Y et al. Emergence of multiple drug‐resistant mutants of hepatitis B virus with long‐term lamivudine and adefovir bitherapy. Hepatology 2009; 50: 518A. 19575365 [Google Scholar]
- 51. Brunelle MN, Lucifora J, Neyts J et al. In vitro activity of 2,4‐Diamino‐6‐(phosphonomethoxy)ethoxy]‐pyrimidine against multidrug‐resistant hepatitis B virus mutants. Antimicrob Agents Chemother 2007; 51: 2240–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Villet S, Billioud G, Pichoud C et al. In vitro characterization of viral fitness of therapy‐resistant hepatitis B variants. Gastroenterology 2009; 136: 168–176. [DOI] [PubMed] [Google Scholar]