

Abstract
Human metapneumovirus (hMPV) is a pathogen of the respiratory tract with a worldwide distribution. The purpose of this study was to identify hMPV as the cause of acute respiratory diseases in children admitted at Spedali Civili, a public hospital in Brescia, Italy. Eight hundred forty‐six nasopharyngeal aspirate samples negative for the presence of other common respiratory viruses were tested for the presence of hMPV RNA by reverse transcription‐polymerase chain reaction. Of the 846 samples, 79 (9.3%) were positive for hMPV. Polymerase chain reaction products, obtained by amplification of the partial nucleotide sequence of gene F, were sequenced and compared with sequences deposited in GenBank. All four hMPV subtypes were identified, including the proposed subtype A2 sublineages “A” and “B”. In successive epidemic seasons, large outbreaks of hMPV alternated with small outbreaks in a biannual pattern. This local study provides further evidence that hMPV infection should be considered as a reason for hospital admission for acute respiratory disease in children. J. Med. Virol. 84:511–516, 2012. © 2011 Wiley Periodicals, Inc.
Keywords: molecular epidemiology, respiratory infection
INTRODUCTION
Human metapneumovirus (hMPV) is a member of the Paramyxoviridae family belonging to the Metapneumovirus genus of the Pneumovirinae subfamily [van den Hoogen et al., 2001; Boivin et al., 2002]. Respiratory tract infections due to hMPV are very common in young children worldwide [Falsey et al., 2003; Maggi et al., 2003; Ebihara et al., 2004; Pierangeli et al., 2007; Carneiro et al., 2009]. Retrospective analyses of nasopharyngeal specimens have identified hMPV as the etiological cause of a significant portion of “orphan” acute respiratory‐tract infections. The clinical symptoms of hMPV are similar to those caused by the respiratory syncytial virus (RSV), ranging from upper respiratory disease to bronchiolitis and pneumonia [van den Hoogen et al., 2003; Caracciolo et al., 2008]. Human MPV has a seasonal distribution with a high prevalence mostly in the winter‐spring period [Kashiwa et al., 2004; Caracciolo et al., 2008].
Based on genetic differences, hMPVs are separated into two groups, A and B, with each group divided into genetic subgroups 1 and 2. More recently, phylogenetic analyses indicated further division of subgroup A2 into two new genetic clusters (sublineages), designated A2a and A2b [Huck et al., 2006]. Among all the sublineages of hMPV, the A2 sublineage has the greatest diversity [Biacchesi et al., 2003].
Local epidemiological studies of hMPV are extremely important for a comprehensive understanding of the current status of human infections and for developing control strategies. To date, no Italian studies have evaluated the circulation rate and incidence of hospital admissions due to hMPV over several consecutive epidemics.
The purpose of the present study was to document the incidence of hMPV as a possible causative agent of acute respiratory disease in samples collected from children admitted to a public pediatric hospital in Brescia over five consecutive epidemiologic seasons (2005–2010) and to assess hMPV circulation patterns, isolated hMPV strains were analyzed phylogenetically during the entire observation period (2005–2010).
MATERIALS AND METHODS
Study Participant and Samples
A total of 2,441 nasopharyngeal aspirate specimens were collected from the beginning of October to the end of April over five consecutive epidemic seasons (from 2004–2005 to 2009–2010) from patients who presented at the pediatric division of Spedali Civili in Brescia. Children were eligible for inclusion if younger than 15 years of age and if admitted for acute respiratory infection or an acute respiratory disease with a symptom onset fewer than 5 days before hospital admission. The acute respiratory disease classification was based on World Health Organization guidelines [WHO, 1992]. Samples (n = 846) that tested negative for RSV A and B; influenza viruses (FLU) A and B; parainfluenza viruses (PIV) 1, 2, and 3; and adenoviruses (AdV) based on immunofluorescence studies (Light Diagnostics™, Chemicon, Temecula, CA), as well as for the presence of human rhinoviruses (HRV) by a reverse transcription‐polymerase chain reaction (PCR), were tested for the presence of hMPV RNA. Samples were also tested for the presence of pathogenic bacteria. Positive bacterial cultures were considered an exclusion criterion.
hMPV Detection
Viral RNA was extracted from 200 µl of nasopharyngeal aspirate with the RNAeasy Kit (QIAGEN, Milan, Italy), according to the manufacturer's instructions. A set of primers that amplify a 506‐bp fragment of the hMPV fusion F gene [Huck et al., 2006] was used for the detection of hMPV. The test sensitivity was 100 copies of hMPV genome equivalents, as calculated using an external standard curve. The standard curve was obtained by cloning the 506 bp PCR amplicon of the hMPV F gene into the pGEMeasy vector (Promega, Milan, Italy). Specificity was evaluated by post‐amplification sequencing of each positive sample.
Sequencing and Phylogenetic Analysis
The amplified fragment (nt 3624–4130) was purified with Wizard SV Gel and PCR Clean‐Up System (Promega) and sequenced at the CRIBI BMR Genomics Sequence Facility (Padua, Italy). The phylogenetic analysis was performed on a 435‐bp fragment of the F gene (amino acid position: 222–367) in 62 of the 79 hMPV isolates and compared with 9 reference sequences downloaded from the NCBI data base. All the sequences in the dataset were aligned using CLUSTAL X software, then edited manually with the Bioedit software (version 7.0.9). The phylogenetic tree was estimated using the PAUP* package. The Tamura‐Nei (TrN93) model of nucleotide substitution, incorporating maximum likelihood estimates of base composition and the shape parameter (α) of a gamma distribution (Γ) model of among‐site rate variation was used as it consistently gave a much higher likelihood value using Modeltest v.3–7 implemented in PAUP*. A maximum likelihood tree was estimated under this model using tree bisection‐reconnection branch swapping. The statistical robustness and reliability of the branching order within each phylogenetic tree were confirmed by bootstrap analysis using 1,000 replicates for the Neighbor‐Joining tree and by the Zero Branch Length Test for the maximum likelihood tree [Posada and Crandall, 1998]. The software MEGA 4.1 allowed us to calculate the genetic distances among different clades using the model of nucleotide maximum composite likelihood, including transitions and transversions. The substitution rates were defined as equal among sites, but different among lineages. Gaps were treated by pairwise‐deletion, that is, ignoring only the gaps involved in the comparison of a pair of sequences.
Statistical Analysis
Categorical variables were compared by the chi‐square test. A P‐value of less than 0.05 was considered statistically significant.
RESULTS
Epidemiology of hMPV Infection
During the five consecutive seasons of the study, hMPV was detected in 9.3% (79/846) of specimens that tested negative for other respiratory viruses and in 3.2% (79/2,441) of all specimens analyzed. Of the 79 hMPV RNA‐positive patients, 33 (41.8%) were female and 46 (58.2%) male, with a male:female ratio 1.4:1 (P = 0.44, chi‐square test) (Table I).
Table I.
Characteristics of Clinical Specimens Tested for Human Metapneumovirus
| Parameter | Number of specimens (%) | |
|---|---|---|
| Positive hMPV samples (n = 79) | Samples analyzed (n = 846) | |
| Gender | ||
| Male | 46 (9.5) | 484 (57.2) |
| Female | 33 (9.1) | 362 (42.8) |
| Age group (years) | ||
| <1 | 35 (10.5) | 334 (39.5) |
| 1–5 | 31 (7.7) | 379 (44.8) |
| 5–10 | 10 (11.1) | 90 (10.6) |
| >10 | 3 (7.0) | 43 (5.1) |
| Symptoms | ||
| Pneumonia | 4 (33.3) | 12 (1.4) |
| BPN | 18 (10.6) | 170 (20.1) |
| Bronchiolitis | 11 (10.4) | 106 (12.5) |
| Laryngotracheitis | 2 (7.4) | 27 (31.9) |
| ARD | 44 (8.3) | 531 (62.8) |
ARD, acute respiratory diseases.
The most common clinical symptom observed in patients infected with hMPV was a generic respiratory tract illness (38/79, 48.1%). Cases of bronchiolitis (12/79, 15.2%), bronchopneumonia (18/79, 22.8%), and pneumonia (5/79, 6.3%) were also observed (Table I).
The age range of patients with hMPV infection was 18 days to 173 months (mean age 31.6 months, median age 16.4 months). As expected, most hMPV‐associated hospitalizations occurred in patients under 5 years of age (P < 0.05, chi‐square test), with a slightly higher incidence in the population younger than 1 year of age (35/334, 10.8%) than in the population older than 1 year (44/512, 8.6%; P = 0.39, chi‐square test). The difference in the age distribution of hMPV‐positive patients for each epidemic season is shown in Figure 1. In the 2008–2009 epidemic season, there was a striking prevalence of infection in children older than 1 year (mean age of 25.7 months, median 17.5 months).
Figure 1.
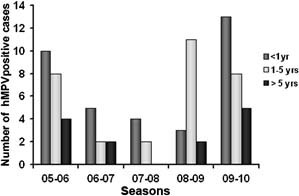
Age distribution of hMPV‐infected children in the five consecutive epidemic seasons evaluated.
An incidence pattern characterized by a biannual alternation between low‐ and high‐intermediate occurrence of hMPV‐associated hospitalization was observed. When only specimens negative for the presence of common respiratory viruses (i.e., RSV A, RSV B, FLU A, FLU B, PIV‐1, PIV‐2, PIV‐3, AdV, and HRV) were analyzed, hMPV was detected most frequently in children hospitalized for acute respiratory diseases in the 2005–2006 epidemic season (22.9%, 22/96). During the following seasons (2006–2007 and 2007–2008), there was a much lower incidence of hMPV‐related hospitalization, with the virus detected in 4.7% (9/192) and 5.7% (6/105) of patients, respectively. Of 163 specimens, 16 (9.8%) were identified as hMPV‐positive during the 2008–2009 observation period, whereas 26 of 290 (9.0%) samples were hMPV‐positive in the 2009–2010 season.
Analysis of the monthly distribution of hMPV incidence revealed a heterogeneous pattern (Fig. 2). Notably, with the exception of the low‐incidence season 2006–2007, hMPV‐associated hospital admission had a 1‐ to 2‐month peak of incidence followed by a month where the incidence decreased dramatically.
Figure 2.
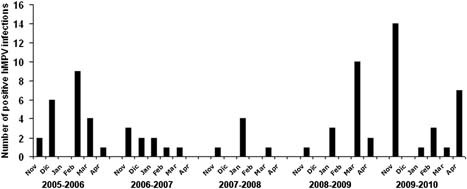
Monthly distribution of hMPV isolation in the five consecutive epidemic seasons evaluated.
Phylogenetic Analysis of hMPV Strains
Genotypes A and B co‐circulated during the different seasons and during the 5 years of study. Subtype A2 was found in 40 of 79 (50.6%) samples, followed by B1 and A1 (14/79 samples, 17.7%), and B2 (11/79 samples, 13.9%). Type A‐positive samples for hMPV were more abundant (54/79) than type B‐positive samples (25/79). Analysis of the results stratified by year (Table II) revealed an alternating 2‐year dominance of the two strains, with serotype A prevailing during the epidemic seasons 2005–2006, 2006–2007, 2009–2010, and serotype B prevailing during the epidemic seasons 2007–2008 and 2008–2009. The hMPV subtype A1 was predominant only in the last season of observation (13 of 14 isolates).
Table II.
Seasonal circulation of hMPV subgroups
| Epidemic season | hMPV subgroup | ||||
|---|---|---|---|---|---|
| A1 (n = 14) | A2a (n = 21) | A2b (n = 19) | B1 (n = 14) | B2 (n = 11) | |
| 2005–2006 | 0 | 11 | 7 | 3 | 1 |
| 2006–2007 | 0 | 5 | 2 | 1 | 1 |
| 2007–2008 | 0 | 0 | 0 | 0 | 6 |
| 2008–2009 | 1 | 2 | 2 | 9 | 2 |
| 2009–2010 | 13 | 3 | 8 | 1 | 1 |
Because of slight differences in the quality of the sequences retrieved for some amplicons, only 62 sequences were adequate for phylogenetic analysis of the hMPV‐positive samples. The sequence alignment (Fig. 3), based on a 435‐bp fragment of gene F sequence, indicated that bootstrap values greater than 75% provided statistical support for clustering the sequences into the five main clades (A1, A2a, A2b, B1, and B2) of hMPV circulating in the Brescia area during the five consecutive epidemic seasons evaluated (Fig. 3). The nucleotide genetic distance between and within clades showed that the B1 clade was more distant from the A2 clade than B2 (0.242 vs. 0.088). The within‐nucleotide genetic distance showed that clade A2 was more heterogeneous than the others (0.029), while the within‐amino acid genetic distance showed that clade B2 was more heterogeneous than the others (0.019).
Figure 3.
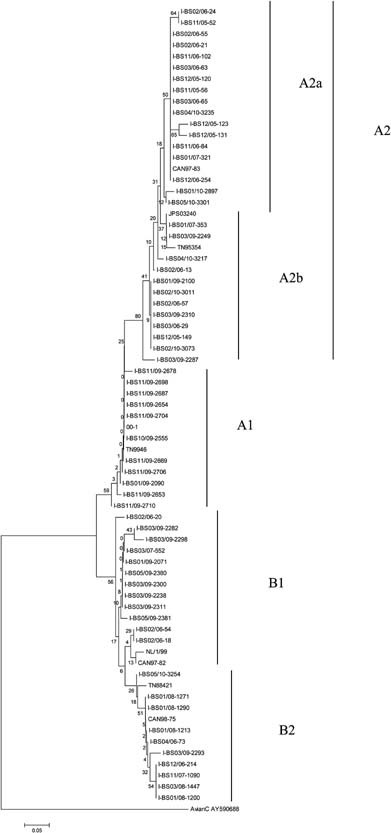
Phylogenetic tree of the hMPV B clade isolates.
Taken together, these data suggest that the selective pressure transient, probably immunologic, influences the prevalence of hMPV strains in different epidemic seasons.
DISCUSSION
The present study examined epidemiological and phylogenic data of hMPV outbreaks over five consecutive epidemic seasons in Brescia, Italy. Human MPV was detected by reverse transcription‐PCR assay as the sole pathogen present in approximately 3.2% of all acute respiratory disease‐suffering patients. In some cases, coinfections might not have been detected because the respiratory samples were not tested for other potential pathogens such as Coronaviruses and Bocaviruses. In addition, in contrast to the PCR method used for hMPV detection, the other respiratory viruses in the present study were examined by immunofluorescence assays. In general, PCR assays are more sensitive than antigen detection methods [Herrmann et al., 2001]. Therefore, it is likely that the true prevalence of hMPV coinfection with the respiratory viruses that were examined by immunofluorescence assays may have actually a higher incidence than the one we have documented.
In the present study, only patients with infections severe enough to be hospitalized were considered. In this experimental setting, the mean age of children hospitalized for hMPV infection was higher than that in similar studies, despite the fact that 33 of 79 hMPV‐positive patients were younger than 1 year. Williams et al. [2006] suggested a possible correlation between older age and upper respiratory disease, which may at least partially explain the finding that the majority of hMPV‐positive samples were obtained from children with generic respiratory symptoms (Table I).
In the geographical area in which this study was performed, the incidence of hospital admission for acute respiratory diseases decreased dramatically from May to October and hMPV infection virtually disappears (data not shown). Consistent with other studies [Caracciolo et al., 2008; Pitoiset et al., 2010], hMPV‐associated hospitalizations were mostly concentrated in a narrow period of time (1–2 months) during the epidemic seasons evaluated.
The hMPV prevalence varies from season to season [Rafiefard et al., 2008; Arnott et al., 2011]. In contrast to most other studies where differences in the annual incidence of hMPV are documented, there was a biannual trend in the incidence of hospitalization due to hMPV infection. Only one 5‐year Swedish study [Rafiefard et al., 2008] reported a 2‐year trend in hMPV incidence. A recent study performed in China described a stable incidence rate during a 3‐year observation period [Xiao et al., 2010]. The discrepancies between reports might be due to geographic variables or to circulation patterns of different viral strains, which may help to maintain better immune surveillance.
Phylogenetic analyses performed on a partial sequence of the F gene indicated that all hMPV subtypes circulated in Brescia during the study period. The data from the present study confirmed five distinct genetic lineages of hMPV, designated A1, A2a, A2b, B1, and B2. Most of the strains in Brescia belonged to the A2 lineage. In particular, of the 62 strains analyzed philogenetically, 11 were classified as A1 lineage, 29 as A2 lineage, 12 as B1 lineage, and 10 as B2 lineage. In addition, hMPV sublineages A2a and A2b, originally proposed based on hMPV sequences from specimens obtained in Germany [Huck et al., 2006], were also detected in the present analysis (17 and 12 isolates, respectively). Other studies performed in Europe as well as in other countries [Ljubin‐Sternak et al., 2008; Arnott et al., 2011] demonstrated the presence of these sublineages, suggesting their worldwide circulation. Many factors potentially influence the circulation of hMPV strains, including virus infectivity, preexisting immunity in the population and antigenic variability; in particular, the extent of antigenic variability between hMPV subgroups is not clear in animals or humans, but the presence of multiple hMPV strains in a single season could affect the design of vaccines or prophylactic antibodies. If antigenic variability leads to partial immune escape, antigenic variation might have an important role in the ability of different virus strains to escape the preexisting immune response. The present analysis of the mean genetic distances revealed that clade A2 was more heterogeneous than the others. Further investigation of clade A2 is needed, especially because a study by Vicente et al. [2006] suggested that genotype A is more pathogenic than genotype B, thereby causing greater clinical severity in children. A correlation between the infecting subtype and disease severity has also been hypothesized for RSV infection. A postulated mechanism for the alternating circulation of different hMPV subgroups is population immune pressure [Oliveira et al., 2009]. Long‐term, prospective, population‐based surveillance is needed to establish whether different subgroups of hMPV vary in virulence and whether circulation patterns are the result of immune pressure.
Acknowledgements
We thank the personnel of the Paediatric Microbiology and Paediatric Units of the Brescia Civic Hospital for their precious help in samples' collection and storage.
REFERENCES
- Arnott A, Vong S, Sek M, Naughtin M, Beauté J, Rith S, Guillard B, Deubel V, Buchy P. 2011. Genetic variability of human metapneumovirus amongst an all ages population in Cambodia between 2007 and 2009. Infect Genet Evol (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacchesi S, Skiadopoulos MH, Boivin G, Hanson CT, Murphy BR, Collins PL, Buchholz UJ. 2003. Genetic diversity between human metapneumovirus subgroups. Virology 315: 1–9. [DOI] [PubMed] [Google Scholar]
- Boivin G, Abed Y, Pelletier G, Ruel L, Moisan D, Côté S, Peret TC, Erdman DD, Anderson LJ. 2002. Virological features and clinical manifestations associated with human metapneumovirus: A new paramyxovirus responsible for acute respiratory‐tract infections in all age groups. J Infect Dis 186: 1330–1334. [DOI] [PubMed] [Google Scholar]
- Caracciolo S, Minini C, Colombrita D, Rossi D, Miglietti N, Vettore E, Caruso A, Fiorentini S. 2008. Human metapneumovirus infection in young children hospitalized with acute respiratory tract disease: Virologic and clinical features. Pediatr Infect Dis J 27: 406–412. [DOI] [PubMed] [Google Scholar]
- Carneiro BM, Yokosawa J, Arbiza J, Costa LF, Mirazo S, Nepomuceno LL, Oliveira TF, Goulart LR, Vieira CU, Freitas GR, Paula NT, Queiróz DA. 2009. Detection of all four human metapneumovirus subtypes in nasopharyngeal specimens from children with respiratory disease in Uberlândia, Brazil. J Med Virol 81: 1814–1818. [DOI] [PubMed] [Google Scholar]
- Ebihara T, Endo R, Kikuta H, Ishiguro N, Ishiko H, Hara M, Takahashi Y, Kobayashi K. 2004. Human metapneumovirus infection in Japanese children. J Clin Microbiol 42: 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsey AR, Erdman D, Anderson LJ, Walsh EE. 2003. Human metapneumovirus infections in young and elderly adults. J Infect Dis 187: 785–990. [DOI] [PubMed] [Google Scholar]
- Herrmann B, Larsson C, Zweygberg BW. 2001. Simultaneous detection and typing of influenza viruses A and B by a nested reverse transcription‐PCR: Comparison to virus isolation and antigen detection by immunofluorescence and optical immunoassay (FLU OIA). J Clin Microbiol 39: 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huck B, Scharf G, Neumann‐Heifelin D, Puppe W, Weigl J, Falcone V. 2006. Novel human metapneumovirus sublineage. Emerg Infect Dis 12: 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwa H, Shimozono H, Takao S. 2004. Clinical pictures of children with human metapneumovirus infection: Comparison with respiratory syncytial virus infection. Jpn J Infect Dis 57: 80–82. [PubMed] [Google Scholar]
- Ljubin‐Sternak S, Santak M, Cepin‐Bogović J, Baće A, Vojnović G, Mlinarić‐Galinović G, Forcić D, Drazenović V, Falsey AR. 2008. Detection of genetic lineages of human metapneumovirus in Croatia during the winter season 2005/2006. J Med Virol 80: 1282–1287. [DOI] [PubMed] [Google Scholar]
- Maggi F, Pifferi M, Vatteroni M, Fornai C, Tempestini E, Anzilotti S, Lanini L, Andreoli E, Ragazzo V, Pistello M, Specter S, Bendinelli M. 2003. Human metapneumovirus associated with respiratory tract infections in a 3‐year study of nasal swabs from infants in Italy. J Clin Microbiol 41: 2987–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira DB, Durigon EL, Carvalho AC, Leal AL, Souza TS, Thomazelli LM, Moraes CT, Vieira SE, Gilio AE, Stewien KE. 2009. Epidemiology and genetic variability of human metapneumovirus during a 4‐year‐long study in Southeastern Brazil. J Med Virol 81: 915–921. [DOI] [PubMed] [Google Scholar]
- Pierangeli A, Gentile M, Di Marco P, Pagnotti P, Scagnolari C, Trombetti S, Lo Russo L, Tromba V, Moretti C, Midulla F, Antonelli G. 2007. Detection and typing by molecular techniques of respiratory viruses in children hospitalized for acute respiratory infection in Rome, Italy. J Med Virol 79: 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitoiset C, Darniot M, Huet F, Aho SL, Pothier P, Manoha C. 2010. Human metapneumovirus genotypes and severity of disease in young children (n = 100) during a 7‐year study in Dijon hospital, France. J Med Virol 82: 1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA. 1998. MODEL TEST: Testing the model of DNA substitution. Bioinformatics 14: 817–818. [DOI] [PubMed] [Google Scholar]
- Rafiefard F, Yun Z, Orvell C. 2008. Epidemiologic characteristics and seasonal distribution of human metapneumovirus infections in five epidemic seasons in Stockholm, Sweden, 2002–2006. J Med Virol 80: 1631–1638. [DOI] [PubMed] [Google Scholar]
- van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med 7: 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen BG, van Doornum GJ, Fockens JC, Cornelissen JJ, Beyer WE, de Groot R, Osterhaus AD, Fouchier RA. 2003. Prevalence and clinical symptoms of human metapneumovirus infection in hospitalized patients. J Infect Dis 188: 1571–1577. [DOI] [PubMed] [Google Scholar]
- Vicente D, Montes M, Cilla G, Perez‐Yarza EG, Perez‐Trallero E. 2006. Differences in clinical severity between genotype A and genotype B human metapneumovirus infection in children. Clin Infect Dis 42: e111–e113. [DOI] [PubMed] [Google Scholar]
- World Health Organization . 1992. International Statistical Classification of Diseases and Related Health Problems, 1989 Revision. Geneva: World Health Organization. [Google Scholar]
- Williams JV, Wang CK, Yang CF, Tollefson SJ, House FS, Heck JM, Chu M, Brown JB, Lintao LD, Quinto JD, Chu D, Spaete RR, Edwards KM, Wright PF, Crowe JE Jr. 2006. The role of human metapneumovirus in upper respiratory tract infections in children: A 20‐year experience. J Infect Dis 193: 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao NG, Xie ZP, Zhang B, Yuan XH, Song JR, Gao HC, Zhang RF, Hou YD, Duan ZJ. 2010. Prevalence and clinical and molecular characterization of human metapneumovirus in children with acute respiratory infection in China. Pediatr Infect Dis J 29: 131–134. [DOI] [PubMed] [Google Scholar]