

Highlights
-
•
We describe our prior efforts in open drug discovery for Ebola and Zika virus.
-
•
We summarize the current literature for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
-
•
We detail computational repurposing efforts and results for SARS-CoV-2.
-
•
To be prepared for future outbreaks we argue we need novel broad-spectrum antivirals.
-
•
Limitations of these efforts include funding for experimental validation, and this lags behind the computational work.
Abstract
In the past decade we have seen two major Ebola virus outbreaks in Africa, the Zika virus in Brazil and the Americas and the current pandemic of coronavirus disease (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). There is a strong sense of déjà vu because there are still no effective treatments. In the COVID-19 pandemic, despite being a new virus, there are already drugs suggested as active in in vitro assays that are being repurposed in clinical trials. Promising SARS-CoV-2 viral targets and computational approaches are described and discussed. Here, we propose, based on open antiviral drug discovery approaches for previous outbreaks, that there could still be gaps in our approach to drug discovery.
Introduction
The two major Ebola virus outbreaks in Africa [1], the Zika virus in Brazil [2] and the current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [3], which was declared by the World Health Organization (WHO) as a pandemic 4, 5, have all occurred in the past decade. In each case there is a strong sense of déjà vu for those scientists involved in drug discovery. A lack of available antivirals to treat the infected patients leads to a clamoring to test anything available, and some pharmaceutical companies charge in to offer their drugs. We also seem to see similar patterns in response across outbreaks. There is a rush to be ‘first’ and this sense of priority might not lead to the best or even any outcome for patients. Always, there is the immediate proposal to create a vaccine and pronouncements that this will be available in a short time or by the end of the year of the actual outbreak in question; and it never happens within these optimistic artificial deadlines. Again, we have experienced this with the current outbreak. For Ebola the vaccine was ready for the second outbreak and has now been approved [6] .
Governments are out to calm their populations while, at the same time, needing to be seen to do something that will vanquish the virus. In the case of SARS-CoV-2 it results in pneumonia [7] and shares aspects of pathology and pathogenesis with severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) [8]. SARS-CoV-2, SARS-CoV and MERS-CoV belong to the same family Coronaviridae and genus Betacoronavirus. Several drug companies immediately made their antivirals available. For example, Abbvie made their HIV approved drug lopinavir/ritonavir combination available to the Chinese government [9] . This drug had previously been suggested as promising based on combination study results from a marmoset study [10]. Publications have already appeared describing the use of this drug in a patient in Korea, significantly decreasing the viral load [11]. Simultaneously, Gilead made available remdesivir, which had previously failed in clinical trials for Ebola [12] but had recently shown activity against MERS in rhesus macaques [13]. Remdesivir, chloroquine and several other drugs were tested in vitro against SARS-CoV-2 (Table 1 ) and shown to be active [14]. Some researchers had also suggested as early as January 2020 what treatment options might be most likely and these included lopinavir/ritonavir, remdesivir, favilavir, arbidol, as well as a broad array of nucleoside analogs, neuraminidase inhibitors, peptides, RNA synthesis inhibitors, anti-inflammatory drugs and traditional Chinese medicines 15, 16, 17.
Table 1.
Compounds screened versus SARS-CoV-2: Vero E6 cells were infected with nCoV- 2019BetaCoV/Wuhan/WIV04/2019 at a multiplicity of infection (MOI) of 0.05 [14]
| Molecule | Class | EC50 (μM) | CC50 (μM) | SI | FDA status |
|---|---|---|---|---|---|
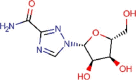 Ribavirin |
Antiviral for hepatitis C | 109.5 | >400 | 3.65 | Approved |
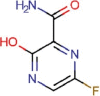
 Penciclovir |
Antiviral for herpesvirus | 95.96 | >400 | 4.17 | Approved |
 Favipiravir |
Broad-spectrum antiviral | 61.88 | >400 | 6.46 | Experimental |
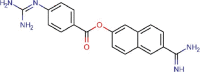 Nafamostat |
Anticoagulant agent | 22.50 | >100 | 4.44 | Approved |
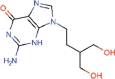
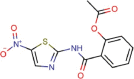 Nitazoxanide |
Anthelmintic agent | 2.21 | 35.53 | 16.76 | Approved |
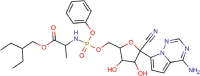 Remdesivir |
Antiviral agent | 0.77 | >100 | 129.87 | Phase II/III |
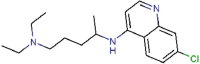 Chloroquine |
Antimalarial agent | 1.13 | >100 | 88.50 | Approved |
The need for other treatment options will be important as we learn more about its transmission [18] and as we see increases in the number of travel-related infections [19]. Already in the past weeks we have seen the rapid spread to Italy and from there to many other countries including Brazil and beyond. Such virus outbreaks are greatly straining the healthcare systems of affected countries and will test every aspect of our infrastructure, supply chains and preparedness [20]. This raises the question of what we can learn from our past drug discovery efforts in an attempt to develop additional drugs for SARS-CoV-2 more efficiently. The acceleration of global SARS-Cov-2 data generation, effectively just in a couple of months, has been remarkable. However, scientific information gaps that impede progress toward small-molecule treatments can still be perceived. Many of these persist from when they were first outlined for Ebola back in 2015 [21]. We can thus make some ‘then-and-now’ comparisons (see Supplementary material Figs. S1–S4 and Table S1 online) as well as share our own experiences of research on these virus outbreaks.
Ebola drug discovery
Our efforts in Ebola drug discovery initially started in 2014 with a computational pharmacophore analysis of a small number of in vitro Ebola active compounds [22]. This was followed by identifying and summarizing all the FDA-approved drugs that could be used against the virus 23, 24 and highlighting various strategies for the next virus outbreak [21]. These steps preceded a pivotal point for us in developing machine learning models for the Ebola virus derived from in vitro data 25, 26.
An early drug [22] identified by these screens was the antimalarial amodiaquine, which was subsequently shown to be associated with decreased mortality [27], as the drug (artesunate/amodiaquine) was used for malaria treatment in some Ebola patients, whereas others took a different malaria medicine (artemether/lumefantrine) [27] . Our Ebola machine learning models were used to select three molecules for in vitro testing [28]. We identified pyronaridine, tilorone and quinacrine as having good in vitro activity (nM) against the Ebola virus [28]. These preliminary data enabled us to obtain funding from the NIH to take one of the compounds through in vivo testing. We also leveraged NIH support to a collaborator to test the other two compounds as well. These molecules were then each tested in the mouse model for Ebola infection and demonstrated significant efficacy 29, 30, 31. Pyronaridine is currently being pursued in larger animal models of Ebola virus infection. Working on computational models through to in vitro testing happened in the space of a few months, whereas it took several years to obtain funding for our first mouse studies. This is by no means a streamlined approach to drug discovery but it was cost effective for the amount of data ultimately generated and led to revitalized interest in these molecules.
The OpenZika drug discovery experience
In 2016 we pulled together a team of researchers in Brazil and the USA to provide some suggestions for an open drug discovery effort for the Zika virus. These included various computational strategies to repurpose molecules and docking into the Zika proteins [32]. We also described resources and molecules that could be prioritized for testing. This was followed by our homology modeling of every Zika virus protein [33] weeks before the first crystal structures for Zika proteins were released. All of this work was performed in the open and it attracted the attention of the IBM World Community Grid team who requested we submit a proposal to them for a Zika project using their distributed computing facilities. This became the OpenZika project led by our team in Brazil and the USA and it involved the docking of millions of molecules from the ZINC database in all Zika virus and related flavivirus protein crystal structures [34] . We have since also performed further modeling of Zika proteins [35] and an analysis of the first 2 years following the outbreak [36]. Our most recent efforts have led to the discovery of an inhibitor for NS3 helicase [37].
What we can learn from the OpenZika project to streamline future open science projects would be as follows. We would pick fewer targets to dock molecules into as the OpenZika project created billions of docking results for tens of targets that will take many years to process. We would also suggest that it is best to work with high-resolution crystal structures versus homology models as soon as possible. We would also start with a library of commercially available drugs and antivirals, this would massively limit the number of molecules docked to a few thousand because we found that docking of the ZINC library available at that time led to compounds that were not readily available. Focused libraries are interesting options to perform target-directed virtual screening. Examples are ChemDiv [38] and Asinex [39] databases, which have specific subsets for protease inhibitors, for example facilitating and accelerating the experimental screening. During OpenZika we learnt that having multiple biology labs committed to doing testing in vitro is also very important to have access to experimental results more quickly to validate the computational approach. Working with the IBM World Community Grid did not include any funding. We found it very difficult to obtain NIH funding to support this project from the very beginning (because it was computational) and also attempted small business grants with academic collaborators without success, even though we were leveraging massive computational resources through the World Community Grid. We would recommend that open efforts like this need some funding for in vitro validation. Millions of dollars have been invested by the NIH to fund other Zika research projects since the 2016 outbreak and there is as yet no small molecule of note in the drug discovery pipeline [36]. There is still therefore an unmet medical need for treatments for Zika virus and computational efforts could aid this and be cost-effective.
SARS-CoV-2 drug discovery efforts
The whole genome sequences for SARS-CoV-2 were isolated from patients from several countries, such as Brazil, Canada, China, Germany, Korea and USA, and were made rapidly available at the Global Initiative on Sharing All Influenza Data (GSAID) platform [40]. There were 135 genomes sampled between December 2019 and February 2020. The samples were found to be closely related with few mutations relative to a common ancestor. Preliminary genetic analysis indicates that the Brazilian genome differs by three mutations to the Wuhan reference strain [41]. Two of these mutations are shared with its closest sequence–the Germany strain [41].
In the space of a few weeks after the acknowledgement of the latest coronavirus outbreak, several laboratories started to deposit homology models for the main viral protease 42, 43, and then for all the SARS-CoV-2 proteins using the server I- TASSER [44]. Others performed docking in these models [45] or developed their own models to understand how the virus enters cells by modeling the Spike (S) protein and human angiotensin-converting enzyme 2 (ACE2) protein interaction [46]. At the time of writing there were structures available for the SARS-CoV-2 main protease in complex with an inhibitor N3 (PDB ID 6LU7) [47] and S1 and S2 subunits of Spike glycoprotein receptor-binding domain up (6VSB), the post fusion core subunit (6LXT) and the HR2 domain (6LVN) 48, 49. A useful overview is provided by searching the entire virus sequence against the Protein Data Bank (PDB) (see Supplementary material Fig. S2 online). In comparison, for Zika it took several months from the beginning of the outbreak until there was a single protein available in the PDB [50].
The Spike glycoprotein is responsible for the fusion of the viral membrane with the host-cell membrane. During virus maturation, it is cleaved to its subunits: S1, which attaches the virion to the cell membrane by interacting with the host receptor; and S2, which mediates the fusion of the virion and cellular membranes. A recent study reports that SARS-CoV-2 Spike protein binds ACE2 with higher affinity than SARS-CoV [48].
Because there was only one Cryo-EM structure of SARS-CoV-2 Spike protein with low resolution (3.46 Å) without the human ACE2 protein [48] at the time of writing this paper, we preferred to build a model by homology modeling (see Supplementary material methods online). Using our SARS-CoV-2 Spike protein model and the available SARS-CoV Spike protein crystal structure (PDB: 2AJF), we aligned both structures to compare the residues in the Spike–ACE2 interface residues for both viruses. As can be seen in Fig. 1 , the residues for SARS-CoV are Asn473, Tyr475, Tyr440, Tyr436, Tyr484, Leu472, Tyr442 and Asn479 and for SARS-CoV-2 are Asn167, Tyr169, Tyr133, Tyr129, Gln178, Phe166, Leu135 and Gln173. Moreover, some SARS-CoV-2 residues, such as Gln178, Phe166, Leu135 and Gln173, are not conserved but have the same physical–chemical features, except the residues SARS-CoV-2 Gln178 (polar) and SARS Tyr484 (hydrophobic) (Fig. 1).
Figure 1.

Superposition of SARS-CoV and SARS-CoV-2 spike proteins highlighting the spike–ACE2 interface binding pocket. Zoom in at the residues of SARS-CoV-2 spike S1 Asn167, Tyr169, Tyr133, Tyr129, Gln178, Phe166, Leu135 and Gln173 and the correspondent SARS-CoV spike S1 Asn473, Tyr475, Tyr440, Tyr436, Tyr484, Leu472, Tyr442 and Asn479.
The main protease (Mpro) of coronaviruses is a cysteine protease that participates in the viral replication, cleaving the polyprotein. Mpro is organized into three domains and is active in a homodimer form [51]. This protease recognizes peptide substrate P4, P2, P1 and P1′ positions through the S4, S2, S1 and S1′ protein subsites, which are critical for substrate binding [53]. The SARS-CoV-2 Mpro S4 pocket is formed by amino acid residue Pro168, the S2 pocket by Met49, the S1 pocket by Ser1, Phe140, His163 and Glu166 and S1’ pocket by His41, Cys145 and Gly143 residues [80] . We aligned the crystal structures of Mpro of SARS-CoV, MERS-CoV and SARS-CoV-2 to compare the structures and the residues of the catalytic site. The catalytic site for SARS-CoV and SARS-CoV-2 are the same: His41–Cys145 [52], whereas for MERS-Cov it is His41–Cys148 [52] (Fig. 2 ). This catalytic site is localized between the interface of domain I (residues 8–101) and II (102–184). The third domain (residues 201–303) is C-terminal, and consists of a loop region, required for the dimerization 53, 54, 55, 56. Recently, it has been reported that the structures of Mpro of SARS-CoV and SARS-CoV-2 have high identity of 96% [43]. The function and the structural equivalence of Mpro of SARS-CoV, MERS-CoV and SARS-CoV-2 demonstrate that the coronavirus proteases are promising targets for development of broad-spectrum antivirals in these viruses.
Figure 2.

Superposition of SARS-CoV, MERS-CoV and SARS-CoV-2 main proteases, and respective catalytic binding site. SARS-CoV (PDB ID 1Q2W) and SARS-CoV-2 (PDB ID 6LU7) share the same residues at the catalytic site: His41–Cys145, and MERS-CoV (PDB ID 5WKL) His41 and Cys148, situated between domains I and II.
MERS and SARS repurposing
Drug repurposing offers a potentially faster approach to identify drugs that are already approved for other uses and might work for the disease of interest 57, 58, 59. This type of strategy was attempted for MERS-CoV and SARS-CoV 60, 61 and the repurposing of host-based therapies has also been proposed for coronaviruses [62]. Several studies have screened a few hundred molecules and tested the activity against MERS-CoV and SARS-CoV (Table 2 ) 63, 64. The first screen of 348 FDA-approved drugs identified four FDA-approved drugs including chloroquine, chlorpromazine, loperamide and lopinavir [63]. At the same time, a second study screened 290 compounds and identified 27 FDA- and non-FDA-approved drugs including chloroquine among the many hits [64], and the Abl2 kinase inhibitors such as imatinib which impacts the early stages of infection after internalization and endosomal trafficking. This latter molecule appears to work by inhibiting fusion at the viral membrane [65]. Chloroquine is widely used for malaria and loperamide is used for diarrhea. These represent drugs that are readily available in many locations around the world and for which physicians are also familiar with the side-effect profile. The ligand datasets from these and other screens can be used to apply machine learning methods to produce predictive models that can then be used to screen compound libraries and suggest new compounds to test. As an example, we have developed several models for ACE2, SARS-CoV and MERS-CoV using our Assay Central software 66, 67, 68, 69 which uses ECFP6 descriptors and the Bayesian algorithm (Fig. 3 , the experimental details are described in the Supplementary material online; the Bayesian models are also available at www.assaycentral.org). The advantage of this approach is that the method is fast, does not require crystal structures and it enables small-molecule structures to be scored against many models simultaneously.
Table 2.
Compounds identified with in vitro activity against MERS or SARS 63, 64; additional molecules can be found in [61]
| Molecule | Class | MERS EC50 (μM) |
MERS CC50 (μM) |
MERS SI |
SARS EC50 (μM) |
SARS CC50 (μM) |
SARS SI |
Ref. | FDA status |
|---|---|---|---|---|---|---|---|---|---|
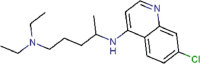 Chloroquine |
Antimalarial | 3.0 | 58.1 | 19.4 | 4.1 | >128 | 31 | [66] | Approved |
 Chlorpromazine |
Antipsychotic | 4.9 | 21.3 | 4.3 | 8.8 | 24.3 | 2.8 | [66] | Approved |
 Loperamide |
Antidiarrheal | 4.8 | 15.5 | 3.2 | 5.9 | 53.8 | 9.1 | [66] | Approved |
 Lopinavir |
Antiviral for HIV | 8.0 | 24.4 | 3.1 | 17.1 | >32 | >2 | [66] | Approved |
 Emetine |
Anti-protozoal and vomiting inducer | 0.014 | 0.051 | [67] | Approved | ||||
 Chloroquine diphosphate |
Antimalarial | 6.275 | 6.538 | [67] | Yes | ||||
 Hydroxychloroquine sulfate |
Antimalarial | 8.279 | 7.966 | [67] | Yes | ||||
 Mefloquine |
Antimalarial | 7.416 | 15.553 | [67] | Yes | ||||
 Amodiaquine |
Antimalarial | 6.212 | 1.274 | [67] | Approved | ||||
 Aloxistatin |
Cysteine protease inhibitor | 1.275 | 0.761 | [67] | Experimental | ||||
 Gemcitabine |
Antineoplastic agent | 1.216 | 4.957 | [67] | Approved | ||||
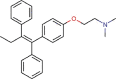 Tamoxifen |
Antineoplastic agent | 10.117 | 92.886 | [67] | Approved | ||||
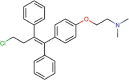 Toremifene |
Antineoplastic agent | 12.915 | 11.969 | [67] | Approved | ||||
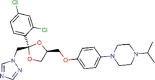 Terconazole |
Antifungal agent | 12.203 | 15.327 | [67] | Approved | ||||
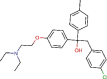 Triparanol |
Cholesterol-lowering agent | 5.283 | [67] | Withdrawn | |||||
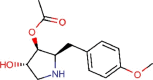 Anisomycin |
Antibacterial agent | 0.003 | 0.191 | [67] | Experimental | ||||
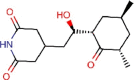 Cycloheximide |
Antifungal agent | 0.189 | 0.043 | [67] | Experimental | ||||
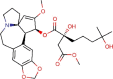 Homoharringtonine |
Antineoplastic agent | 0.0718 | [67] | Approved | |||||
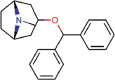 Benztropine |
Anticholinergic agent | 16.627 | 21.611 | [67] | Approved | ||||
 Fluspirilene |
Antipsychotic agent | 7.477 | 5.963 | [67] | Approved | ||||
 Thiothixene |
Antipsychotic agent | 9.297 | 5.316 | [67] | Approved | ||||
 Fluphenazine |
Antipsychotic agent | 5.868 | 21.431 | [67] | Approved | ||||
 Astemizole |
Antihistamine agent | 4.884 | 5.591 | [67] | Withdrawn | ||||
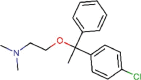 Chlorphenoxamine |
Anticholinergic and antihistamine agent | 12.646 | 20.031 | [67] | Approved | ||||
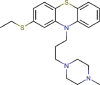 Thiethylperazine |
Antipsychotic agent | 7.865 | [67] | Approved | |||||
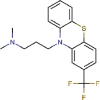 Triflupromazine |
Antipsychotic agent | 5.758 | 6.398 | [67] | Approved | ||||
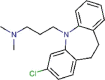 Clomipramine |
Antidepressant agent | 9.332 | 13.238 | [67] | Approved | ||||
 Imatinib |
Antineoplastic agent | 17.689 | 9.823 | [67] | Approved | ||||
 Dasatinib |
Antineoplastic agent | 5.468 | 2.100 | [67] | Approved |
Figure 3.

Fivefold cross validation ROC plots for Bayesian machine learning models using ECFP6 fingerprints for SARS and MERS datasets which can be used for scoring and selecting compounds to test: (a) SARS phenotypic screen; (b) SARS 3C-like proteinase; (c) SARS 3C-like proteinase primary screen from PubChem; (d) SARS replicase polyprotein 1ab; (e) MERS phenotypic screen; (f) angiotensin-converting enzyme 2.
Repurposing collections of compounds
As a well-established principle any SARS-CoV-2 relevant enzyme assay, protein–protein interaction antagonism or viral infectivity determination can be run against a collection of approved drugs. However, it is neither trivial to get a definitive list of these structures in silico prepared for docking or to acquire these as a physical collection ready for screening (even though there are various repurposing subsets available from vendors). Notwithstanding, an approximation to a definitive approved set can be selected in PubChem as the intersect between curated drug resources such as DrugBank, DrugCentral and the IUPHAR/BPS Guide to PHARMACOLOGY, with totals of 9135, 4012 and 7674 molecules, respectively. The three-way compounds-in-common total is 1119 chemical identifiers (CID). PubChem indicates that 1197 molecules are available from chemical vendors. Relaxing the stringency of the repurposing provides triage steps that can certainly be useful for docking and/or machine learning or other modeling efforts. In this case the union of all three drug curation sources is 16\,468. Other drug-centric solutions include the selection of 9527 international non-proprietary names (INN) CID. The majority of these will have either been progressed to Phase I testing or are close to it.
Computational approaches to accelerate SARS-CoV-2 drug discovery
An alternative to screening every approved drug is to take one class of compounds. For example we have now taken the opportunity to dock FDA-approved HIV-1 protease inhibitors (amprenavir, atazanavir, darunavir, fosamprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and tipranavir) and hepatitis C NS3/4A protease inhibitors (asunaprevir, boceprevir, grazoprevir, paritaprevir, simeprevir and telaprevir) into the Mpro structure of SARS-CoV-2. The docking was performed with two different commercial docking software products: Biovia Discovery Studio and Schrödinger Glide 70, 71, 72, 73, and the best docking results were compared (see Supplementary material Table S2 online). After medicinal chemistry visual inspection, the results of both docking approaches indicated that azatanavir and lopinavir may be of most interest for in vitro/and/or in vivo testing (Fig. 4 ). Lopinavir (Fig. 4a) (LigandFit score of 191.196) may interact with Mpro by hydrogen bonding with Cys145 residue of the S1’ pocket. The aromatic ring of lopinavir also formed hydrophobic interactions with residues Cys44, Met49 (S2 pocket), Leu50, Pro52, Tyr54, and Cys145 (S1’ pocket). In relation to atazanavir (Fig. 4b) (LigandFit score of 175.497), hydrogen bonds were established between residues Cys145 and Glu166 both from S1’ pocket and nitrogen atoms of carbamate groups. Similar hydrophobic interactions observed with lopinavir occurred also with atazanavir, with residues Met49 (S2 pocket), Pro52 and Tyr54 (Fig. 4). In China and South Korea lopinavir 200 mg/ritonavir 50 mg has already been used for the treatment of COVID-19. From the day after lopinavir/ritonavir administration, the viral load started to decrease and no detectable or little coronavirus titers have been observed since then [11]. Lopinavir, co-formulated with ritonavir is commercially available and known as Kaletra® around the world–it is on formularies for HIV. By contrast, atazanavir is less commonly used. Kaletra® can be widely produced and distributed quickly through existing channels but it will be important to ensure HIV patients do not lose access to the medications they need. It is also a medication that clinicians are familiar with in terms of side effects, unlike other less commonly used medications. A broader analysis of all FDA-approved drugs docked in this protease structure could point to further drugs worth testing.
Figure 4.

Intermolecular 3D and 2D interactions of (a) lopinavir and (b) atazanavir with the main protease structure of SARS-CoV-2 (PDB ID 6LU7) obtained by docking. In the 3D representation (left), H-bonds are presented as magenta dashed lines and the color code of carbon, oxygen, nitrogen and hydrogen atoms are grey, red, blue and white, respectively. The S1, S1’, S2, and S4 pockets are presented as green, magenta, yellow, and blue surfaces, respectively. The PyMol software [79] was used for the visual inspection of 3D docking poses and to render the 3D molecular images.
We have also performed docking calculations of SARS-CoV-2 spike glycoprotein complexed with the host ACE2 protein against all FDA- approved drugs (∼2400 molecules) using Schrödinger Glide software 70, 71, 72, 73 . Because there was only one Cryo-EM structure of SARS-CoV-2 Spike protein with low resolution (3.46 Å) without the human ACE2 protein [48] at the time of doing this analysis, we preferred to build a model by homology modeling, because lower-resolution structures (3 Å or higher) show the basic contours of the protein chain and the atomic structure must be inferred. We built the SARS-CoV-2 Spike subunit S1 model using the SwissModel server [74]. The template used was the SARS-CoV Spike S1 protein (PDB ID 2GHV) [75], which has resolution of 2.2 Å and 73.22% of sequence identity with SARS-CoV-2 Spike protein. After the protein refinement, we added the human ACE2 protein to the model, based on the SARS-CoV Spike–ACE2 structure (PDB ID 2AJF) [76], and built the full SARS-CoV-2 Spike S1–ACE2 model (see Fig. S5 in Supplementary material online). The quality statistics of the modeled SARS-CoV-2 Spike–ACE2 were good: 96.53% of the residues lie in the most favorable regions of the Ramachandran plot and the Molprobity score was 1.66. Our SARS-CoV-2 Spike–ACE2 model is available (see Supplemental material PDB file online). Subsequently, we performed docking-based virtual screening with a grid centered at the Spike–ACE2 interface. As can be seen in Table 3 , docking scores indicated that rizatriptan (an antimigraine agent, Glide score = −7.49 kcal mol−1), dasabuvir (an antiviral for hepatitis C, Glide score = −7.09 kcal mol−1), pravastatin (a lipid-lowering agent, Glide score = −6.17 kcal mol−1) and empagliflozin (a hypoglycemic agent, Glide score = −6.37 kcal mol−1) may be of interest for in vitro testing. Details of intermolecular interactions of these drugs with the interface of Spike–ACE2 are shown in Fig. 5 . In summary, these drugs form π-cation interactions with Lys31a (ACE2’s residue), π–π interactions with Phe170b (Spike’s residue), and H-bonds with Glu35a and Asp38a residues of ACE2, Gly176b and Ser174b (residues of Spike). In good agreement with our results, a previous study suggests that an early and high dose of a statin (such as pravastatin) could be crucial to surviving MERS-CoV infection [77]. These suggested drugs could be validated in future in vitro experiments with SARS-CoV-2 cell-based assays and with Spike-ACE2 enzymatic assays.
Table 3.
Docking results for SARS-CoV-2 Spike–ACE2 model against an FDA-approved drug database
| Drug name | Drug original class | Docking score (kcal mol−1) |
|---|---|---|
| Rizatriptan | Antimigraine agent | −7.49 |
| Dasabuvir | Antiviral for hepatitis C | −7.09 |
| Pravastatin | Lipid-lowering agent | −6.17 |
| Empagliflozin | Hypoglycemic agent | −6.37 |
Figure 5.

3D and 2D intermolecular interactions of rizatriptan (a), dasabuvir (b) , pravastatin (c) and empagliflozin (d) with the spike–ACE2 interface. In 3D representation (left), the SARS-CoV-2 Spike (S1 subunit) is colored in light blue and human ACE2, in green. Hydrogen bonds are presented as magenta dashed lines and the color code of carbon, oxygen, nitrogen and hydrogen atoms is grey, red, blue and white, respectively. The PyMol software 71, 73, 79 was used for the visual inspection of 3D docking poses and to render the 3D molecular images.
Modern drug discovery efforts try to eliminate inefficiency by increasing knowledge, through extensive utilization of generated data to guide new experiments. Therefore, we have also taken a drug repurposing approach based on polypharmacology using a method called the ‘molecular and biological signature’ [78]. This is a profiling approach, guided by artificial intelligence, capable of presenting the complex relationships between the chemical substance with molecular targets, cells and/or organs. This could create a more complete understanding of the ‘multi-biological profile’ caused by the drug analyzed in response to a disease. As an example, we have applied the drugs ritonavir [9], favipiravir, ribavirin and chloroquine as scaffolds to find new potential molecular and biological signature profiles, to search for non-obvious, mechanism-of-action-based associations between drugs, molecular targets and diseases (Fig. 6 ; and Supplementary material Fig. S6 and Table S3 online). Comparing the molecular and biological signature of ritonavir with other drugs, we found that the drugs cobicistat, carfilzomib and ombitasvir presented the highest matching scores (81–71%), suggesting that those drugs could be of further interest for SARS-CoV-2.
Figure 6.

Molecular and biological signature profiles generated for ritonavir compared with other drugs, using artificial intelligence to create thousands of decision-makers that mimic a biological response through the interaction of a small molecule with multiple enzyme targets, off-targets and in vitro and in vivo endpoints. The drugs cobicistat, carfilzomib and ombitasvir presented the higher matching scores (81–71%) with ritonavir.
Concluding remarks
Although there are already several drugs being assessed clinically for SARS-CoV-2, there is still a need to identify additional treatments as alternatives (to offset drug shortages) and possibly discover molecules with increased efficacy, safety and cost-effectiveness. We have previously proposed and applied computational drug discovery approaches such as machine learning and docking to aid in the discovery of new candidate molecules for Ebola and Zika, respectively. What we have learnt in these efforts can be applied to SARS-CoV-2 as we have demonstrated here. However, the research in this case has moved at a much faster pace initially, delivering a first crystal structure in just a few weeks after first public notification of the virus. Companies have mobilized to bring their drugs to patients and clinicians have used prior in vitro research that proposed compounds active against MERS-CoV and SARS-CoV to suggest treatments for SARS-CoV-2. We currently do not clinically use any broad-spectrum antivirals in the USA but they would be invaluable in situations like this for SARS-CoV-2 and other viruses. Identifying medications with side-effect profiles known by clinicians that are already produced at scale and available at local pharmacies around the world will allow more patients to be treated, especially as travel restrictions and quarantines could reduce production and distribution of certain drugs. It will remain important that those who need these medications at baseline, such as HIV patients who take ritonavir/lopinavir, are able to continue to access the medications they need or else there is a risk of poor clinical outcomes and antiviral resistance in these patients. Although other viruses such as the flu might have killed more patients in the USA this season so far, the crippling financial impact of SARS-CoV-2 remains to be seen globally. If past Ebola, ZIKV, MERS-CoV and SARS-CoV outbreaks are anything to go by this SARS-CoV-2 pandemic will be far more significant. Having multiple effective treatments available for SARS-CoV-2 that can be rapidly delivered to patients could save lives. We can also supplement these treatments by a combination of computational screening with machine learning and docking to select additional drugs to test as proposed herein and this can be repeated for future virus outbreaks. The sense of déjà vu will probably be repeated again and again with the next virus outbreaks until we develop broader- spectrum antivirals. Each time we hope to be better prepared. Hope however does not save lives. Effective antiviral treatments do, and we need to have them ready in the future.
Conflict of interest
S.E. is founder and owner of Collaborations Pharmaceuticals. D.H.F., K.M.Z. and A.C.P. are employees of Collaborations Pharmaceuticals. R.C.B. is co-founder and CTO of InsilicAll. C.S. is owner of TW2Informatics. All other authors declare no conflicts of interest. Collaborations Pharmaceuticals owns the trademark for Assay Central™.
Acknowledgments
We kindly acknowledge NIH funding: R21TR001718 from NCATS and R44GM122196- 02A1 from NIGMS (PI–Sean Ekins). We also acknowledge Brazilian funding agencies, CNPq, FAPEG and CAPES (Finance Code 001) for the financial support and fellowships. M.M. and C.H.A. thank the support of the Brazilian CNPq/FAPEG (grant 300508/2017-4). C.H.A. also thanks the ‘L’Oréal-UNESCO-ABC Para Mulheres na Ciência’ and ‘L’Oréal-UNESCO International Rising Talents’ for the awards and fellowships received, which partially funded this work. C.H.A. is CNPq research fellow. We kindly acknowledge the support and encouragement of the IBM World Community Grid team, and the community of volunteers who donate their dormant computer power for our docking calculations. S.E. kindly thanks Biovia for providing Discovery Studio and the many collaborators on our Ebola and Zika projects. S.E. acknowledges Dr Alex Clark (Molecular Materials Informatics) for assistance with Assay Central™.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:https://doi.org/10.1016/j.drudis.2020.03.019.
Contributor Information
Sean Ekins, Email: sean@collaborationspharma.com.
Carolina Horta Andrade, Email: carolina@ufg.br.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- 1.Furuyama W., Marzi A. Ebola virus: pathogenesis and countermeasure development. Annu. Rev. Virol. 2019;6:435–458. doi: 10.1146/annurev-virology-092818-015708. [DOI] [PubMed] [Google Scholar]
- 2.Musso D. Zika virus infection – after the pandemic. N. Engl. J. Med. 2019;381:1444–1457. doi: 10.1056/NEJMra1808246. [DOI] [PubMed] [Google Scholar]
- 3.Gorbalenya A.E. Severe acute respiratory syndrome-related coronavirus – the species and its viruses, a statement of the Coronavirus Study Group. BioRxiv. 2020 doi: 10.1101/2020.02.07.937862. [DOI] [Google Scholar]
- 4.Holshue M.L. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020 doi: 10.1056/nejmoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO Director-General's Opening Remarks at the Media Briefing on COVID-19. Available at: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020.
- 6.FDA First FDA-approved vaccine for the prevention of Ebola virus disease, marking a critical milestone in public health preparedness and response. Case Med. Res. 2019 doi: 10.31525/cmr-24ab0c1. [DOI] [Google Scholar]
- 7.Pan Y. Initial CT findings and temporal changes in patients with the novel coronavirus pneumonia (2019-nCoV): a study of 63 patients in Wuhan, China. Eur. Radiol. 2020 doi: 10.1007/s00330-020-06731-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J. Med. Virol. 2020 doi: 10.1002/jmv.25709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.AbbVie Statement on Coronavirus and Lopinavir/Ritonavir | AbbVie News Center. https://news.abbvie.com/news/media[HYPHEN]statements/abbvie-statement-on-coronavirus-and-lopinavirritonavir.htm
- 10.Momattin H. A systematic review of therapeutic agents for the treatment of the Middle East respiratory syndrome coronavirus (MERS-CoV) Travel. Med. Infect. Dis. 2019;30:9–18. doi: 10.1016/j.tmaid.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim J. Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: the application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. J. Korean Med. Sci. 2020;35:e79. doi: 10.3346/jkms.2020.35.e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulangu S. A randomized, controlled trial of Ebola virus disease therapeutics. N. Engl. J. Med. 2019;381:2293–2303. doi: 10.1056/NEJMoa1910993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Wit E. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. U.S.A. 2020 doi: 10.1073/pnas.1922083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019- nCoV) in vitro. Cell Res. 2020 doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV) Biosci. Trends. 2020 doi: 10.5582/bst.2020.01020. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L., Liu Y. Potential interventions for novel coronavirus in China: a systemic review. J. Med. Virol. 2020 doi: 10.1002/jmv.25707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H. Potential antiviral therapeutics for 2019 novel coronavirus. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:E002. doi: 10.3760/cma.j.issn.1001-0939.2020.0002. (in Chinese) [DOI] [PubMed] [Google Scholar]
- 18.Nishiura H. Initial cluster of novel coronavirus (2019-nCoV) infections in Wuhan, China is consistent with substantial human-to-human transmission. J. Clin. Med. 2020;9:488. doi: 10.3390/jcm9020488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bajema K.L. Persons evaluated for 2019 novel coronavirus – United States, January 2020. Morb. Mortal. Wkly. Rep. 2020;69:166–170. doi: 10.15585/mmwr.mm6906e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith N., Fraser M. Straining the system: novel coronavirus (COVID-19) and preparedness for concomitant disasters. Am. J. Public Health. 2020 doi: 10.2105/ajph.2020.305618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekins S. Finding small molecules for the “next Ebola”. F1000Research. 2015;4:58. doi: 10.12688/f1000research.6181.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ekins S. A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000Research. 2014;3:277. doi: 10.12688/f1000research.5741.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Letterman N. Small molecules with antiviral activity against the Ebola virus. F1000Research. 2015;1:38. doi: 10.12688/f1000research.6120.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekins S., Coffee M. FDA approved drugs as potential Ebola treatments. F1000Research. 2015;4:48. doi: 10.12688/f1000research.6164.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madrid P.B. Evaluation of Ebola virus inhibitors for drug repurposing. ACS Infect. Dis. 2016;1:317–326. doi: 10.1021/acsinfecdis.5b00030. [DOI] [PubMed] [Google Scholar]
- 26.Madrid P.B. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLOS ONE. 2013;8:e60579. doi: 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gignoux E. Effect of artesunate-amodiaquine on mortality related to Ebola virus disease. N. Engl. J. Med. 2016;374:23–32. doi: 10.1056/NEJMoa1504605. [DOI] [PubMed] [Google Scholar]
- 28.Ekins S. Machine learning models identify molecules active against the Ebola virus in vitro. F1000Research. 2015 doi: 10.12688/f1000research.7217.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekins S. Efficacy of tilorone dihydrochloride against Ebola virus infection. Antimicrob. Agents Chemother. 2018;62 doi: 10.1128/AAC.01711-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lane T.R. Repurposing quinacrine against Ebola virus infection in vivo. Antimicrob. Agents Chemother. 2019;63 doi: 10.1128/AAC.01142-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lane T.R. Repurposing the antimalarial pyronaridine tetraphosphate to protect against Ebola virus infection. PLoS Negl. Trop. Dis. 2019;13:e0007890. doi: 10.1371/journal.pntd.0007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ekins S. Open drug discovery for the Zika virus. F1000Research. 2016 doi: 10.12688/f1000research.8013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekins S. Illustrating and homology modeling the proteins of the Zika virus. F1000Research. 2016;5:275. doi: 10.12688/f1000research.8213.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ekins S. OpenZika: an IBM World Community Grid project to accelerate Zika virus drug discovery. PLoS Negl. Trop. Dis. 2016;10:e0005023. doi: 10.1371/journal.pntd.0005023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mottin M. Molecular dynamics simulations of Zika virus NS3 helicase: insights into RNA binding site activity. Biochem. Biophys. Res. Commun. 2017;492:643–651. doi: 10.1016/j.bbrc.2017.03.070. [DOI] [PubMed] [Google Scholar]
- 36.Mottin M. The A–Z of Zika drug discovery. Drug Discov. Today. 2018;23:1833–1847. doi: 10.1016/j.drudis.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva S. A diarylamine derived from anthranilic acid inhibits ZIKV replication. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-54169-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chemdiv Database. Available at: https://www.chemdiv.com/cysteine-proteases-inhibitors-library/.
- 39.Asinex Database. Available at: http://www.asinex.com/protease/.
- 40.GISAID Initiative the Genome Sequences of the Newly Emerging Coronavirus. Available at: https://www.gisaid.org/epiflu-applications/next-sars-cov2-app/.
- 41.Jesus J.G. 2020. First Report of COVID-19 in South America. Available at: http://virological.org/t/first-report-of-covid-19-in-south-america/409. [Google Scholar]
- 42.Comparative Model of Novel Coronavirus 2019-nCoV Protease Mpro. Available at: https://figshare.com/articles/Comparative_model_of_novel_coronavirus_2019-nCoV_protease_Mpro/11752752.
- 43.Stoermer M. Homology models of Wuhan coronavirus 3CLpro protease. ChemRxiv. 2020 doi: 10.26434/chemrxiv.11637294.v1. [DOI] [Google Scholar]
- 44.Zhang C. 2020. Structure Models of All Mature Peptides in COVID-19 Genome by C-I-TASSER. Available at: https://zhanglab.ccmb.med.umich.edu/C-I-TASSER/2019-nCov/ [Google Scholar]
- 45.Zhang H. Deep learning based drug screening for novel coronavirus 2019-nCov. Preprints. 2020 doi: 10.20944/preprints202002.0061.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu X. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X. 2020. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3. Available at: http://www.rcsb.org/structure/6LU7. [Google Scholar]
- 48.Wrapp D. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020 doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Y., Sun F. 2020. Crystal Structure of HR2 Domain of 2019 -nCoV S2 Subunit. Available at: http://www.rcsb.org/structure/6LXT; http://www.rcsb.org/structure/6LVN. [Google Scholar]
- 50.Song H. Zika virus NS1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat. Struct. Mol. Biol. 2016;23:456–458. doi: 10.1038/nsmb.3213. [DOI] [PubMed] [Google Scholar]
- 51.Chou C.Y. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43:14958–14970. doi: 10.1021/bi0490237. [DOI] [PubMed] [Google Scholar]
- 52.Galasiti Kankanamalage A.C. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 2018;150:334–346. doi: 10.1016/j.ejmech.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ziebuhr J. The 3C-like proteinase of an invertebrate nidovirus links coronavirus and potyvirus homologs. J. Virol. 2003;77:1415–1426. doi: 10.1128/JVI.77.2.1415-1426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xue X. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82:2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andrade C.H. In silico chemogenomics drug repositioning strategies for neglected tropical diseases. Curr. Med. Chem. 2019;26 doi: 10.2174/0929867325666180309114824. [DOI] [PubMed] [Google Scholar]
- 58.Chong C.R., Sullivan D.J. New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 59.Ekins S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today. 2011;16:298–310. doi: 10.1016/j.drudis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 60.Sisk J.M., Frieman M.B. Screening of FDA-approved drugs for treatment of emerging pathogens. ACS Infect. Dis. 2016;1:401–402. doi: 10.1021/acsinfecdis.5b00089. [DOI] [PubMed] [Google Scholar]
- 61.Dyall J. Middle East respiratory syndrome and severe acute respiratory syndrome: current therapeutic options and potential targets for novel therapies. Drugs. 2017;77:1935–1966. doi: 10.1007/s40265-017-0830-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li C.C. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov. Today. 2019;24:726–736. doi: 10.1016/j.drudis.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Wilde A.H. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014;58:4875–4884. doi: 10.1128/AAC.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dyall J. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014;58:4885–4893. doi: 10.1128/AAC.03036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coleman C.M. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus fusion. J. Virol. 2016;90:8924–8933. doi: 10.1128/JVI.01429-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ekins S. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019;18:435–441. doi: 10.1038/s41563-019-0338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russo D.P. Comparing multiple machine learning algorithms and metrics for estrogen receptor binding prediction. Mol. Pharm. 2018;15:4361–4370. doi: 10.1021/acs.molpharmaceut.8b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zorn K.M. Multiple machine learning comparisons of HIV cell-based and reverse transcriptase data sets. Mol. Pharm. 2019;16:1620–1632. doi: 10.1021/acs.molpharmaceut.8b01297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anantpadma M. Ebola virus Bayesian machine learning models enable new in vitro leads. ACS Omega. 2019;4:2353–2361. doi: 10.1021/acsomega.8b02948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Friesner R.A. Extra Precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 71.Halgren T.A. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 72.Friesner R.A. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 73.Glide, Schrödinger, LLC, New York, NY, 2019. https://www.schrodinger.com/glide
- 74.Biasini M. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hwang W.C. Structural basis of neutralization by a human anti-severe acute respiratory syndrome Spike protein antibody, 80R. J. Biol. Chem. 2006;281:34610–34616. doi: 10.1074/jbc.M603275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li F. Structure of SARS coronavirus Spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 77.Yuan S. Statins may decrease the fatality rate of Middle East respiratory syndrome infection. MBio. 2015;6 doi: 10.1128/mBio.01120-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pushpakom S. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 79.Schrödinger. The PyMOL Molecular Graphics System, Version 1.8 2015.
- 80.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;(80-) doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.