

Abstract
Patients with rheumatoid arthritis (RA) may display atypical CD21−/lo B cells in their blood but the implications of this observation remains unclear. We report here that the group of patients with RA and elevated frequencies of CD21−/lo B cells shows decreased ataxia-telangiectasia mutated (ATM) expression and activation in B cells compared with other patients with RA and healthy donor controls. In agreement with ATM involvement in the regulation of V(D)J recombination, patients with RA who show defective ATM function displayed a skewed B cell receptor (BCR) Igκ repertoire, which resembled that of patients with ataxia-telangiectasia (AT). This repertoire was characterized by increased Jκ1 and decreased upstream Vκ gene segment usage, suggesting improper secondary recombination processes and selection. In addition, altered ATM function in B cells was associated with decreased osteoprotegerin (OPG) and increased RANKL production. These changes favor bone loss and correlated with a higher prevalence of erosive disease in patients with RA who show impaired ATM function. Using a humanized mouse model, we also show that ATM inhibition in vivo induces an altered Igκ repertoire and RANKL production by immature B cells in the bone marrow, leading to decreased bone density. We conclude that dysregulated ATM function in B cells promotes bone erosion and the emergence of circulating CD21−/lo B cells, thereby contributing to RA pathophysiology.
One Sentence Summary:
Defective ATM activation skews the B cell repertoire and increases RANKL production, promoting joint erosion in patients with rheumatoid arthritis.
Introduction
Rheumatoid arthritis (RA) is a debilitating autoimmune condition that causes irreversible bone erosions and affects millions of individuals worldwide (1). B cell-depleting therapy has shown efficacy in the treatment of RA, thereby demonstrating an important role for B cells in RA physiopathology (2). Autoantibodies including rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP) in patients with RA are associated with bone erosion and may arise from the activation of autoreactive naïve B cells that accumulate in the blood of patients with RA due to defective early B cell tolerance checkpoints that fail to counterselect developing autoreactive clones (3, 4). However, the clinical efficacy of anti-B cell therapy in RA does not correlate with a decrease in serum autoantibodies, suggesting that B cells may contribute to RA via additional mechanisms. Accordingly, it is becoming more appreciated that B cells have other pathological functions in RA besides generating autoantibodies, and osteoimmune interactions position B cells as key players in bone homeostasis (5–7). B cells are found near erosions in inflamed joints and produce pro-inflammatory cytokines such as TNFα, IL-6, and receptor activator of NFκB ligand (RANKL), which stimulate differentiation of bone-resorbing osteoclasts responsible for the clinical complications of osteoporosis and erosive joint disease (7–11). However, the potential mechanisms leading to increased osteoclastogenic cytokine production by B cells in RA remain unclear.
Premature immune system senescence is associated with many autoimmune diseases, including RA (12). Additionally, an increased frequency of circulating atypical CD21−/lo B cells accumulates prematurely in these autoimmune settings (13–17). An important mechanism implicated in cellular senescence is failure to repair damaged DNA (18). The ataxia-telangiectasia mutated (ATM) kinase is a key regulator of the DNA double-strand break (DSB) damage response (19, 20). Patients with biallelic mutations resulting in non-functional ATM suffer from ataxia-telangiectasia (AT), which is a neurodegenerative syndrome that is also marked by immune dysregulation and premature aging (20). In B cells, ATM plays a key role in regulating BCR gene rearrangements by recognizing, stabilizing, and mediating non-homologous end joining repair of DSB between newly cleaved Vκ and Jκ DNA segments (21, 22). In addition, ATM also enforces allelic exclusion at the Igκ locus (23, 24). Whereas ATM function was reported to be decreased in T cells from patients with RA compared to counterparts in healthy donor controls (HD) (25), dysregulated ATM expression in B cells and its putative impact in inflammatory joint disease and patients with RA have not been evaluated.
Here we identify a subgroup of patients with RA who display erosive disease and decreased ATM function in B cells, which was associated with reduced Igκ gene repertoire breadth. In addition, we found that ATM inhibition favors pro-osteoclastogenic cytokine production by B cells and decreased bone density in vivo, which further characterizes the important role played by B cells in RA.
Results
Decreased ATM expression in B cells identifies a subgroup of patients with RA
Since lower expression of DNA damage repair regulator ATM and associated factors was found in T cells from patients with RA (25, 26), we sought to determine if the expression of these DSB repair molecules was also altered in their B cells. To avoid potential differences related to different stages of B cell development, gene expression profiling was performed on naïve B cells isolated from PBMCs of consecutively enrolled patients with RA meeting American College of Rheumatology (ACR) diagnostic criteria (27). Transcripts for ATM and genes encoding key sensors and signaling adaptors in the ATM-related DNA DSB repair cascade, such as MRE11A and NBS1, were downregulated in a subgroup of patients with RA, referred henceforth as group I, relative to other patients (group II) (Fig. 1A). Decreased expression of genes encoding key mediators of the DNA DSB repair cascade regulated by ATM in group I patient B cells was further demonstrated by gene set comparison with the Molecular Signatures Database (Broad Institute), which revealed a highly significant overlap (P = 2.02 × 10−69, FDR = 1.94 × 10−66) in several hundred genes whose expression is directly correlated with that of ATM (Fig. 1B). In support of dysregulation of ATM and its related pathways in B cells from group I patients, we also found highly significant overlap with gene signatures in the database both up-regulated (P = 1.05 × 10−46, FDR = 1.72 × 10−43) and down-regulated (P = 8.85 × 10−96, FDR = 1.51 × 10−92) from experiments in which cells were incubated with doxorubicin, a DNA DSB-inducing chemotherapeutic agent that is used experimentally to activate ATM and ATM-dependent pathways (Fig. 1B) (28). Dysregulation of the DNA damage signaling and response cascade was further evidenced by the significant overlap in genes upregulated in B cells from group I patients with > 100 genes upregulated by p53 (P = 1 × 10−40), genes whose expression correlate directly with BRCA1 expression (P = 1 × 10−74), and genes involved in the response to DNA damage from UV radiation (P = 1 × 10−60) (Fig. 1B). Pathway analysis also revealed that the most significantly differentially expressed genes in B cells from group I versus group II patients with RA were involved in cell cycle checkpoint control, cellular senescence, cell stress, apoptosis, and the DNA DSB damage response, which are all pathways regulated by ATM (Fig. 1C) (P < 0.05 for all pathways in Fig. 1C) (19, 20). Thus, a subgroup of patients with RA can be identified by decreased expression of ATM and ATM-related genes in naïve B cells.
Fig. 1. Relative ATM insufficiency identifies a subgroup of patients with RA.
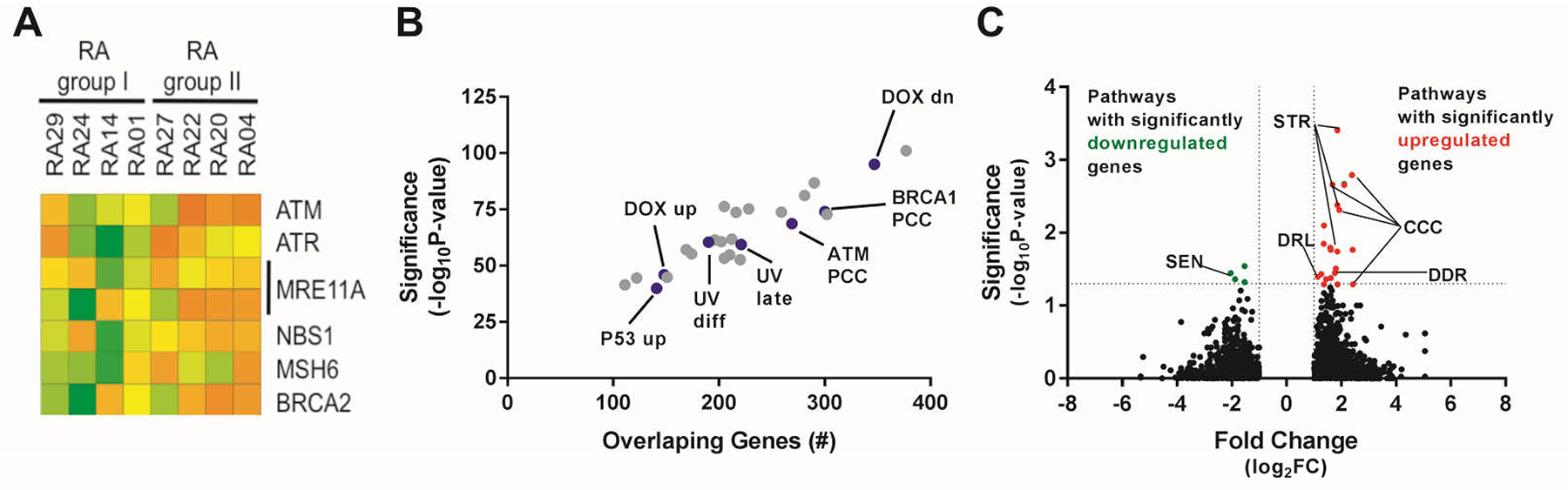
(A) Two groups of patients with RA: group I and group II identified based on differential expression of ATM and ATM-related genes amplified from 1 × 105 to 3 × 105 bulk-sorted CD19+CD21+CD27− naïve B cells. Gene array of genes in the ATM-regulated response to DNA DSB with at least 2-fold difference in expression between group I and group II patients. Lower expression (green); higher expression (orange). IDs of patients with RA are indicated. (B) Overlap between genes differentially regulated in group I vs group II patients with RA. Curated gene sets (blue dots) from the Molecular Signatures Database whose transcripts correlate significantly with ATM expression by Pearson correlation coefficient (ATM PCC), BRCA1 expression (BRCA1 PCC), genes induced or repressed by the DNA DSB-inducing agent doxorubicin (DOX up, DOX dn), genes induced in response to UV DNA damage (UV diff, UV late), and genes induced by overexpression of p53 (P53 up). Gray dots represent other gene sets with significant overlaps that do not relate to the ATM cascade. (C) Pathway analysis of genes that are differentially regulated in B cells from group I relative to group II patients with RA. Senescence pathway (SEN), cell stress pathway (STR), cell cycle checkpoint pathway (CCC), death receptors and ligands pathway (DRL), and DNA damage sensing and response pathway (DDR).
Altered Igκ gene segment usage in patients with RA and decreased ATM activation
ATM regulates V(D)J recombination processes that generate BCRs by recognizing stabilizing, and mediating repair of DSB between Vκ and Jκ DNA segments, and also by preventing simultaneous V-J recombination on both Igκ alleles (21–24). Since we found decreased ATM expression in a subgroup of patients with RA, we analyzed the Igκ repertoires of CD19+CD10+CD27‒CD21loIgMhi new emigrant/transitional B cells isolated from the blood of patients with RA and HD controls. These B cells display antibody repertoire and reactivity similar to immature B cells in the bone marrow (29). V(D)J rearrangements among the more than 40 Vκ and 5 Jκ gene segments on the Igκ locus can occur by deletion in which an 5’ upstream Vκ segment rearranges with a 3’ downstream Jκ segment thereby removing intervening Vκ and Jκ segments. Initial V(D)J rearrangements are biased toward the Jκ1 gene segment, allowing other Jκ genes to remain available for subsequent secondary rearrangements (30, 31). We found that the proportion of upstream Vκ gene segments in new emigrant/transitional B cells varied between the groups. The frequency in the four group I patients ranged between 3%−21% (average = 11%) compared to 19%−27% (average = 23.5%) in group II patients, similar to HDs in which Vκ gene segment usage averaged 22.3% (fig. S1A and table S1). This dearth in upstream Vκ gene segments in group I patients was associated with an increased usage of Jκ1, the most upstream of all human Jκs, and decreased downstream Jκ3/4/5 gene segment usage, which indicates that new emigrant/transitional B cells from group I patients express antibodies less likely encoded by secondary recombination events (fig. S1B and table S1). A similar bias in Vκ and Jκ gene segment usage was observed in new emigrant/transitional B cells from AT patients, suggesting that impaired ATM expression or function is responsible for this skewed Igκ repertoire (fig. S1 and table S1). The analysis of unfractionated patients with RA enrolled in the study cohort confirmed these findings in that their new emigrant/transitional B cells showed significantly increased Jκ1 gene segment usage relative to HDs, although this bias was driven by group I patients, whereas group II patients with RA displayed Jκ1 frequencies similar to HDs (Fig. 2A). In agreement with initial data from a smaller cohort of 9 patients with RA (4), the correlated analysis of the frequencies of upstream Vκ and downstream Jκ gene segment usage in new emigrant/transitional B cells revealed that both group I and AT patients exhibited a restricted utilization of Vκ and Jκ gene segments as demonstrated by decreased usage of upstream Vκ and downstream Jκ3/4/5 gene segments and segregated away from group II patients with RA who showed broader Vκ-Jκ gene segment usage as seen in HDs (Fig. 2B). Differences in Igκ repertoire between group I and group II patients did not appear to correlate with any biometrics including age and disease duration (average within 2–3 years of disease onset) or differences in disease-modifying anti-rheumatic drugs (DMARDs) since the types of RA therapy were similar between groups I and II patients (Table 1, table S2, and fig. S2, A–C). In addition, many patients with RA were enrolled before being medicated, suggesting that Igκ repertoire differences and ATM defects in group I patients were not induced by therapeutic regimen (Table 1, table S2, and fig. S2C). Moreover, group I and group II patients with RA displayed similar serum RF and anti-CCP autoantibody titers (Table 1, table S2, and fig. S2, D–G).
Fig. 2. Skewed Igκ repertoire in patients with RA who have decreased ATM function.
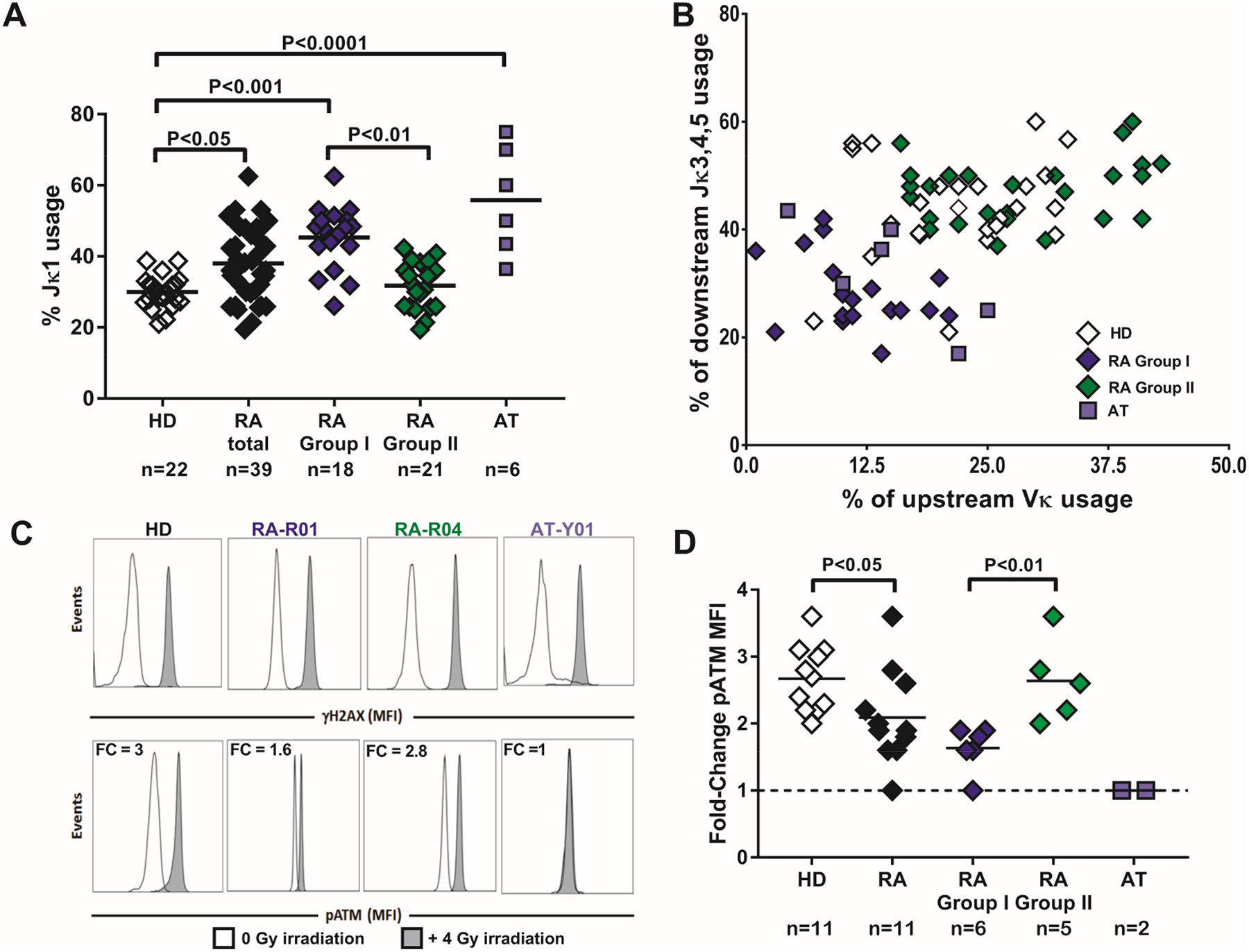
(A) Summary of Jκ1 gene usage frequency (HD = open diamond, combined RA = black diamonds, same subjects as shown in blue and green, group I RA= blue diamonds, group II RA= green diamonds, patients with AT = blue squares). (B) Upstream Vκ gene usage frequency was plotted against downstream Jκ 3-4-5 gene usage in transitional B cells from 25 HD controls, 6 patients with AT, and 43 patients with RA. (C) Flow cytometry performed on CD20+ B cells from a representative HD control, group I patient with RA (RA-R01), group II patient with RA (RA-R04), and a patient with AT (AT-Y01) for analysis of activated pATM and presence of DNA DSB (induction of γH2AX) with (gray histograms) or without (open histograms) X-ray irradiation. (D) Summary of pATM fold changes in B cells from HD (open diamonds), patients with RA (combined RA = black diamonds, group I = blue diamonds, group II = green diamonds), and patients with AT (blue squares). Each diamond/square represents an individual or patient, and bars represent mean value. Mann-Whitney U testing for difference between groups and P values are indicated when significant.
Table 1.
Characteristics of group I and group II patients with RA
| Group I (n = 18) | Group II (n = 25) | P value | |
|---|---|---|---|
| Age (yrs ± SD) | 55 ± 14.7 | 50 ± 11.7 | 0.55 |
| Disease Duration (yrs ± SD) | 3 ± 7.2 | 2 ± 6.9 | 0.45 |
| Prior DMARD use† (number of patients) | 9 | 9 | 0.36 |
| Rheumatoid factor titer ± SD | 387 ± 397.4 | 268 ± 307.8 | 0.35 |
| Anti-CCP antibody titer ± SD | 142 ± 86.5 | 178 ± 88.1 | 0.36 |
DMARD = disease modifying anti-rheumatic drug
includes conventional oral DMARDs and biologics/small molecule inhibitors with the exception of the anti-CD20+ B cell depleting biologic rituximab
SD = standard deviation
Mann-Whitney U testing for difference between groups except prior DMARD use which was compared by χ2 for equality in proportions. Significance at P<0.05.
We then tested ATM activation in total CD20+ B cells from patients with RA and HDs by inducing DSB with X-ray irradiation. Irradiation-induced DSB results in ATM activation by auto-phosphorylation at serine 1981, thereby generating phospho-ATM (pATM) when ATM is recruited to DSB sites, which are marked by γH2AX (32). We performed flow cytometry analysis on irradiated and non-irradiated cells to detect pATM and γH2AX as a readout of functional ATM activation and DSB formation, respectively. Whereas X-ray irradiation resulted in the generation of γH2AX in irradiated B cells from all patients with RA and HD, confirming the induction of DSB, there was a lower average induction of pATM in B cells from patients with RA, and pATM was not induced in AT patients (Fig. 2, C and D). Indeed, irradiated B cells from HD controls showed an average 2.7-fold induction of pATM relative to non-irradiated HD B cells compared with only 1.9 fold in B cells from patients with RA (Fig. 2D). In addition, B cells from group I patients with RA displayed the lowest fold-change in pATM following irradiation compared to B cells from group II patients (1.7 vs 2.6-fold induction, respectively, P<0.01, Fig. 2D). In agreement with a previous report (25), defects in pATM induction were also observed in T cells and in non-B and non-T cells from these patients, suggesting a global defect in ATM function affecting the hematopoietic lineage in RA (fig. S3 and S4). We conclude that decreased and impaired ATM activation in group I patients with RA and AT patients, respectively, alters the diversity of the BCR repertoire by affecting regulation of secondary V-J recombination events at the Igκ locus.
Elevated CD21−/lo B cell frequency and prevalence of joint erosions in group I patients with RA
Since decreased ATM and related DNA DSB repair factors are associated with premature cellular senescence, we analyzed the frequencies of atypical CD19+CD10−CD27−CD21−/lo (henceforth CD21−/lo) B cells, which often express T-bet and CD11c and accumulate in patients with autoimmune diseases including RA (13–17). We previously reported an increase in the frequency of CD21−/lo B cells in patients with RA (13) and the current larger cohort of patients not only confirmed this observation but also revealed that these B cells were specifically enriched in group I patients. Indeed, CD21−/lo B cells represented on average 8.9% of total circulating CD19+CD10−CD27− B cells in group I patients compared to only 1.8% and 0.8% in group II patients with RA and HDs, respectively (Fig. 3, A and B). In agreement with other reports, we found that the frequency of CD21−/lo B cells was also increased in AT patients, which suggests that impaired ATM function favors the emergence of these B cells (Fig. 3A and B) (33–35). CD21−/lo B cells expressed transcripts encoding CXCR3, important for homing to RA joints, and receptors for TNFα and IL-6, which promote B cell expression of the osteoclast differentiating factor RANKL (Fig. 3C), and may contribute to bone loss in both patients with RA and AT patients (9, 10, 36, 37). We therefore assessed the presence or absence of joint erosions in group I vs group II patients, and we found a significantly increased prevalence of erosive joint disease among group I patients with RA who show decreased pATM compared to group II patients (76.9% vs 31.6%, P<0.01) (Fig. 3D). As a corollary, the chance of finding a group I patient among patients without erosive disease was significantly lower, as 81% of the patients with RA without joint erosions were in group II (Fig. 3E). We conclude that decreased pATM induction is associated with the increased production of CD21−/lo B cells and high prevalence of bone erosion in group I patients with RA.
Fig. 3. Increased frequencies of circulating atypical CD21−/lo B cells correlate with high prevalence of joint erosion in group I patients with RA.
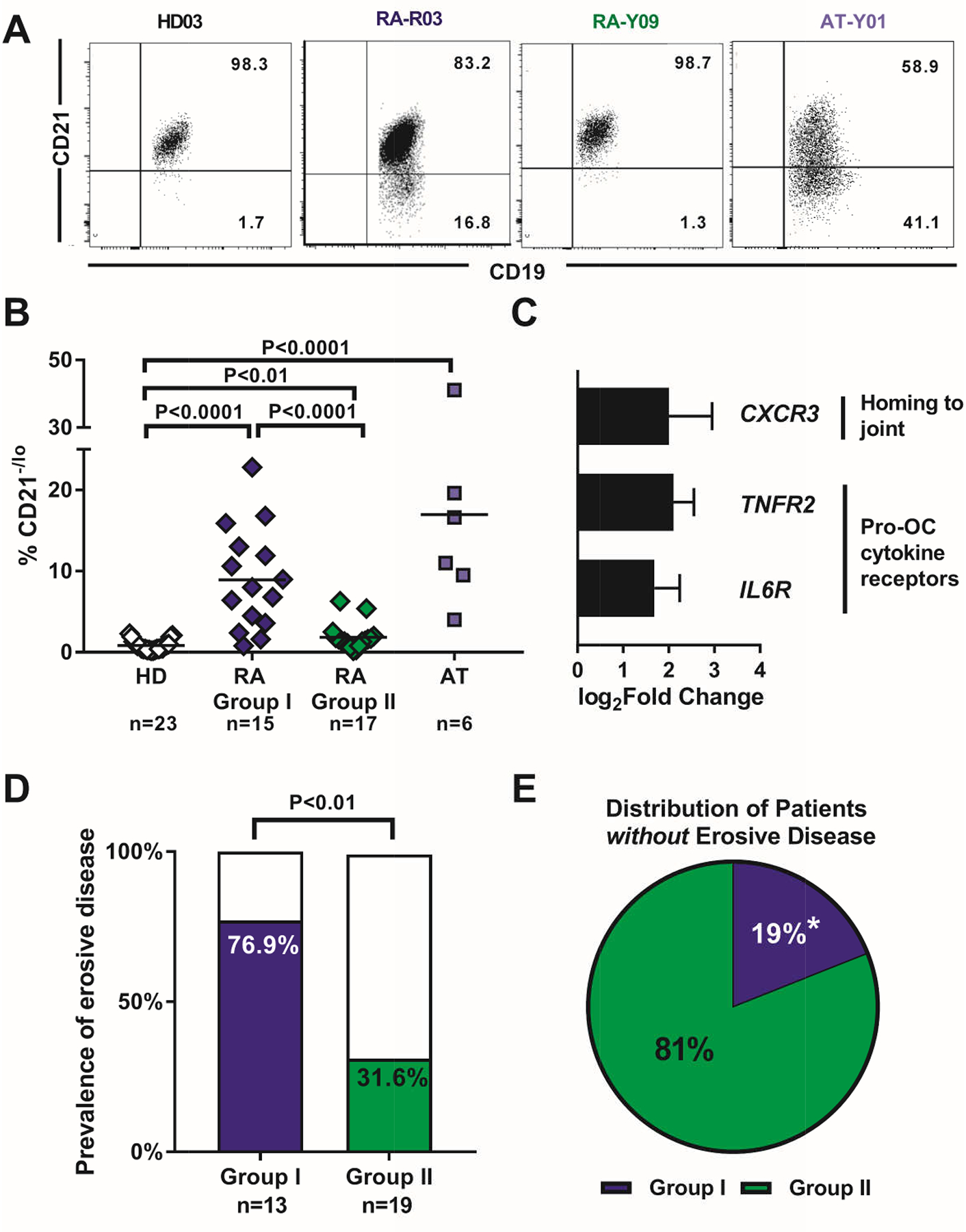
(A) Representative flow cytometry histograms on gated CD19+CD10−CD27− B cells from peripheral blood of a HD, group I and group II patients with RA, and a patient with AT for evaluating CD21−/lo B cell frequency. (B) Summary of frequencies of circulating CD21−/lo B cells among CD19+CD10−CD27− B cells in HD (open diamonds), group I (blue diamonds), group II (green diamonds), and AT (light blue squares). Each symbol represents an individual. Mann-Whitney U testing for difference between groups and P values are indicated when significant. (C) Relative fold change in gene expression for chemokine and cytokine receptors in CD19+CD10−CD27−CD21−/lo relative to conventional CD19+CD10−CD27−CD21+ naïve B cells from group I patients with RA. (D) Prevalence of erosive joint disease among group I and group II patients. Erosive disease was assessed by the patient’s rheumatologist and indicated present or absent. (E) Frequency of patients without joint erosions in group I vs group II. Prevalence of erosive joint disease compared with χ2 testing for difference in proportions between groups. * indicates significance with P<0.05.
ATM inhibition induces a pro-osteoclastogenic B cell phenotype
Bone resorption is mediated by osteoclasts, which play an important role in osteopenia and joint erosions in inflammatory arthritis (38, 39). Osteoclastogenesis is regulated by the balance of RANKL and its natural antagonist osteoprotegerin (OPG), with a high RANKL:OPG ratio being pro-osteoclastogenic (38). Although B cells are a major source of OPG in the bone marrow, recent data suggests that activated B cells can also support osteoclastogenesis and bone erosion in RA via RANKL production (9–11). To investigate the effect of ATM inhibition on B cell production of RANKL and other pro-osteoclastogenic cytokines independent of potentially confounding influences of RA disease factors, we cultured peripheral CD20+ B cells from healthy donors with or without ATM inhibitor KU55933 (ATMi) (40). ATMi abrogated the induction of pATM after B cells were irradiated, despite induction of DSB, which validated the efficacy of ATM inhibition in this in vitro system (mean fold change pATM = 1.2 vs 2.8 in controls, P<0.001; fig. S5A). Quantitative PCR revealed that ATM inhibition in B cells isolated from the blood of healthy donors significantly enhanced the transcription of IL6 and RANKL (P<0.05; Fig. 4A), both of which encode pro-inflammatory cytokines favoring osteoclastogenesis (5, 41). TNF expression and IL10 transcription were unaffected by ATMi (Fig. 4A). Transcription differences induced by ATMi were then validated at the protein level by measuring cytokines in supernatants of the B cells incubated with ATMi. We found that ATM inhibition significantly increased IL-6 and RANKL secretion by B cells (P<0.05; Fig. 4B). Secretion of OPG by B cells was also elevated by ATMi, but to a lesser degree than RANKL (Fig. 4B). When B cells were cultivated with ATMi in the presence of CD40L + anti-IgM for 2 days, there was still induction of RANKL but now decreased induction of OPG (Fig. 4C, fig. S5B). To better assess the relative contribution of B cell RANKL and OPG, we examined the RANKL:OPG ratio, which increased in favor of RANKL following ATM inhibition (Fig. 4D). The increase in RANKL:OPG ratio with ATMi was similar to the increased RANKL:OPG ratio following B cell activation with CD40L + anti-IgM, which are known to induce RANKL and OPG production (Fig. 4D). In addition, ATM inhibition in combination with BCR and CD40 triggering resulted in a further enhanced RANKL:OPG ratio compared to either ATMi or CD40L + anti-IgM stimulation alone (Fig. 4D). Thus, defects in ATM activation render human B cells pro-osteoclastogenic through increased RANKL and IL-6 secretion.
Fig. 4. ATM inhibition induces a pro-osteoclastogenic phenotype in human B cells.
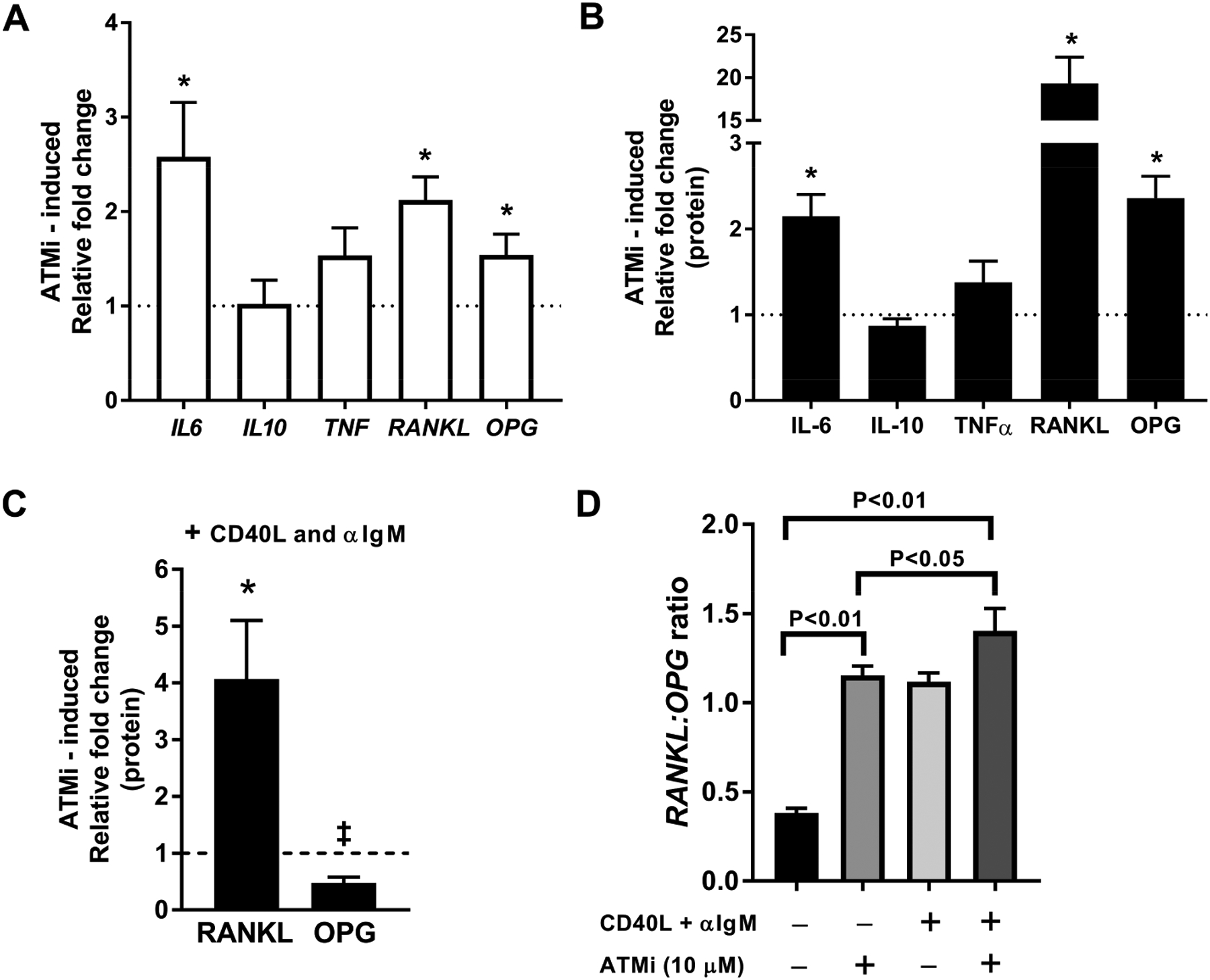
Cytokine gene transcripts assessed by qPCR (A) and secreted protein assessed by ELISA (B) in CD20+ B cells from HD cultured in the presence or absence of 10 μM KU55933 (ATMi). Dashed line represents unstimulated baseline. * = significant fold-induction over unstimulated baseline at P<0.05 with two-tailed t-testing. (C) Effect of ATM inhibition on the ability of CD40L and αIgM stimulation to induce RANKL and OPG protein expression. Comparison by two-tailed t-testing relative to culture with CD40L and αIgM alone (dashed line), * and ‡ represent significant difference relative to cultures with CD40L and αIgM alone. Error bars = SEM for all graphs (D) RANKL:OPG ratio in different culture conditions as noted. Bars represent mean ratios of the aggregated expression from 5 individual HD. Comparisons between conditions tested by ANOVA with multiple comparisons and P values are indicated when significant.
In vivo ATM inhibition results in a skewed Igκ repertoire
We sought to determine whether defective activation of ATM in vivo is sufficient to induce a restricted human Igκ repertoire as well as the resorptive bone changes seen to be more prevalent in group I patients with RA and the premature osteoporosis reported in AT patients (42). Since V(D)J recombination occurs during early B cell development in the bone marrow, we used NOD-scid-common gamma chain (γc) knockout (NSG) humanized mice engrafted with CD34+ hematopoietic stem cells (HSCs) isolated from human fetal livers as a dynamic model for human B cell development (43, 44). Reconstitution of human B and T cells was assessed by flow cytometry of mouse peripheral blood around weeks 8–12 after transplant. Absence of T cell activation reflecting potential ongoing graft-versus-host responses was confirmed by low expression of PD-1 activation marker as previously described (fig. S6A–C) (44). To examine the effect of functional ATM inhibition on engrafted human B cells, we injected NSG mice with 10 mg/kg of ATMi intraperitoneally for 10 days as previously reported (45). We compared bone marrow and splenic B cells to those from non-injected littermate controls transplanted with HSCs from the same fetal donors and treated with DMSO (vehicle). X-ray irradiation resulted in DSB as evidenced by the detection of γH2AX in CD45+CD19+ human B cells isolated from the spleen of NSG humanized mice both with and without ATMi treatment, but pATM failed to be induced in B cells from ATMi-injected animals, thereby validating our in vivo model for ATM inhibition (Fig. 5, A and B). Indeed, B cells from NSG humanized mice injected with ATMi showed a significantly lower mean fold induction of pATM after irradiation (fold change = 1.2) compared to a mean of 2.8-fold induction in human B cells isolated from DMSO-injected humanized littermate controls (P<0.001, Fig. 5B). In agreement with ATM inhibition, transcriptome analyses revealed that the gene expression signature of B cells from NSG humanized mice injected with ATMi displayed highly significant overlap with gene expression signatures that directly correlate with ATM and BRCA1 expression (fig. S6D). In contrast, the top three overlapping gene signatures for control NSG mice did not contain gene sets involving ATM or related DNA damage response genes (fig. S6D). Inhibition of pATM following irradiation was also observed in T cells from NSG humanized mice injected with ATMi (fig. S6E). Of note, ATM inhibition did not alter the proportion of new emigrant/transitional CD19+CD27−CD21loCD10+IgMhi that recently exited the bone marrow or mature naïve CD19+CD27−CD21+CD10−IgM+ B cells in the spleen of humanized mice and (fig. S6F). In addition, ATM inhibition did not alter the high proportion of human CD19+ B cell precursors relative to CD3+ T cell precursors in the bone marrow of humanized mice (fig. S7A–C). Altogether, these data show that ATMi injections in NSG humanized mice result in impaired ATM function in T cells and developing B cells.
Fig. 5. In vivo ATM inhibition recapitulates skewed Igκ repertoire.
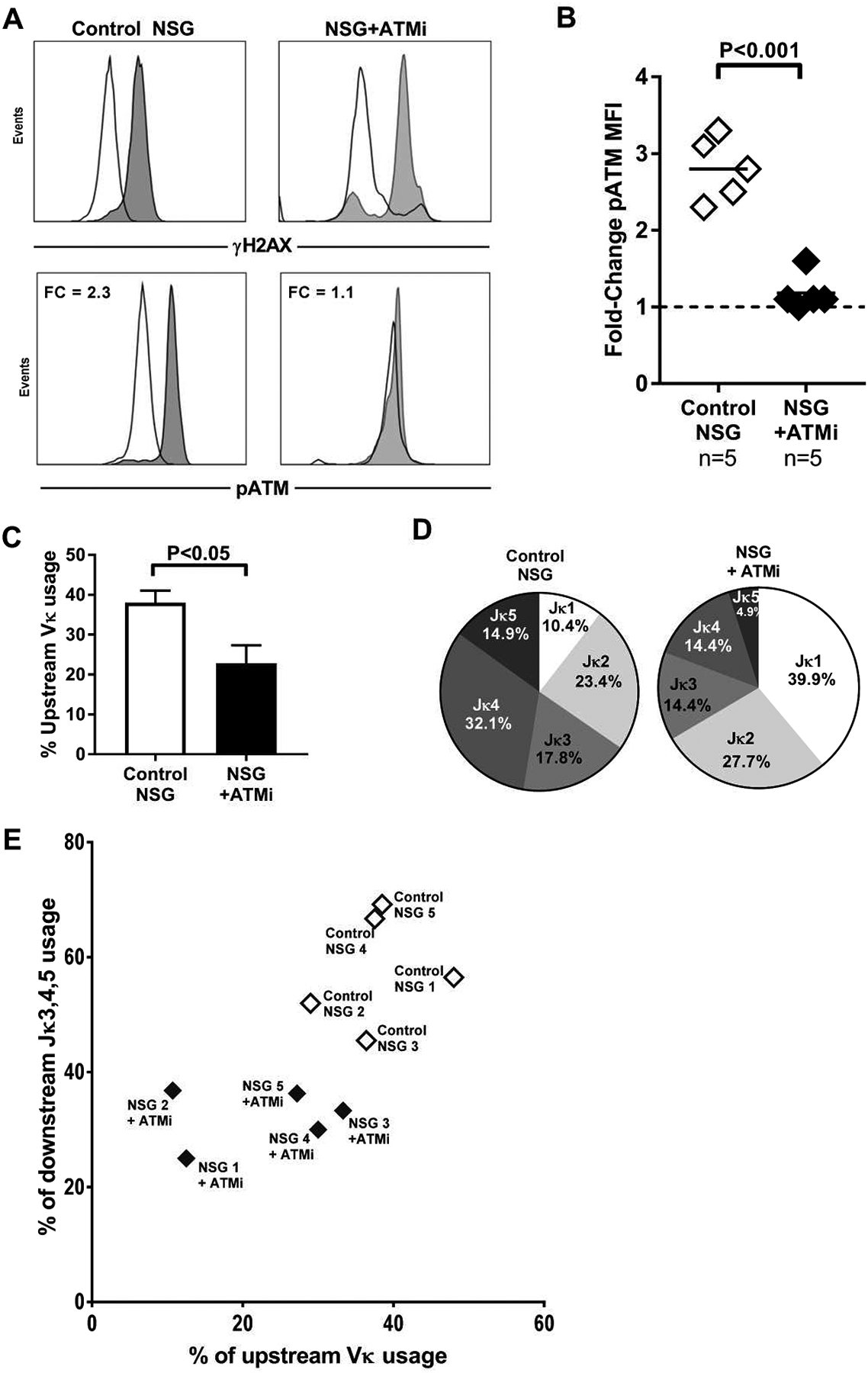
(A) Representative flow cytometry histograms for γH2AX induction on the top panels for human transitional B cells from a control (top left) and ATMi-injected NSG humanized mice (top right). Representative flow cytometry histograms for activated pATM on the bottom panels. (B) Summary of pATM fold changes. Each diamond represents a NSG humanized mouse (open = control, filled = injected with ATMi), bars represent mean value. Mann-Whitney U testing for differences between groups. (C) Summary of upstream Vκ gene segment usage in human transitional B cells isolated from NSG humanized mice, bars represent mean value. Mann-Whitney U testing for differences between groups. (D) Representative graphs of individual Jκ gene segment usage proportions in human transitional B cells from control and ATMi-injected NSG humanized mice. (E) Upstream Vκ gene usage frequency was plotted against that of downstream Jκ 3-4-5 gene usage in human transitional B cells from NSG humanized mice with or without ATMi injections. Data was obtained by analyzing human B cells isolated from 5 control and 5 NSG humanized mice injected with ATMi. Each diamond represents a mouse (open = control, filled = injected with ATMi). Human HSCs were obtained from 5 different human fetal donors and transplanted into 5 different sibling pairs of mice. Each murine sibling pair was born at the same time from the same mother and co-housed for life. Data represent experiments on 5 separate murine sibling pairs from 5 separate litters from 5 separate murine mothers. P values indicated when significant.
We then examined the Igκ repertoire of human new emigrant/transitional B cells isolated from the spleen of ATMi-injected and control NSG humanized mice (table S3). Upstream Vκ gene segment frequency accounted for 38% of total Vκs in control NSG humanized mice compared to 22% in NSG humanized mice injected with ATMi (Fig. 5, C and D). In vivo ATM inhibition also led to an overrepresentation of Jκ1 usage in new emigrant/transitional human B cells, which resembled patient counterparts in group I RA and AT patients (Fig. 5, C and D). In addition, upstream Vκ and downstream Jκ3/4/5 gene segment usage were both decreased in NSG humanized mice injected with ATMi, suggesting that ATM inhibition prevented the production of new emigrant/transitional B cells expressing BCRs resulting from secondary Vκ-Jκ recombination events that favor the utilization of these Igκ gene segments (Fig. 5E). The restricted BCR repertoire induced by ATMi is similar to those of group I patients with RA and AT patients who are both characterized by defective ATM function and differ from those of group II patients and HD in which ATM regulation appears normal (Fig. 1, 2 and 5). Thus, defective ATM activation decreases the diversity of the BCR repertoire characterized by a dearth of secondary recombination events on the Igκ loci.
ATM inhibition does not alter Igκ secondary recombination but affects immature B cell survival
We then tested whether secondary recombination events fail to occur when ATM regulation is compromised or, alternatively, if secondary rearrangements still proceed, but may be detrimental to B cell precursor survival. To distinguish between these two potential scenarios, we first assessed the Igκ: Igλ light chain ratio in newly emigrated transitional B cells from NSG humanized mice with or without ATMi injections. Since the Igκ loci rearrange before the Igλ loci, human B cells display a preferred Igκ usage and Igλ+ B cells have often inactivated their Igκ loci via recombination with the downstream κ deleting element (κDE) (31, 46). We found that ATM inhibition resulted in a significantly decreased percentage of Igκ+ relative to Igλ+ new emigrant/transitional B cells in ATMi-treated NSG humanized mice compared to DMSO-injected controls (κ:λ ratio 0.688 in NSG + ATMi vs 1.024 in NSG controls, P<0.001; Fig. 6A). Of note, ATM inhibition did not appear to alter light chain allelic exclusion since B cells only expressed either surface Igκ or Igλ but not both (Fig. 6A). We then tested if Igλ+ B cells produced when ATM was inhibited were generated in the absence of Igκ recombination by amplifying κDEs and kappa-deleting recombination excision circles (KRECs) by qPCR from sorted surface Igλ+ transitional B cells from control and ATMi-injected NSG humanized mice (Fig. 6B) (47). PCR products for κDEs and KRECs were amplified from Igλ+ clones of both NSG humanized mice injected or not with ATMi, thereby revealing that ATM inhibition did not prevent recombination on the Igκ loci and that Igλ+ transitional B cells developed after inactivating their Igκ loci (Fig. 6B).
Fig. 6. ATM inhibition does not impair secondary recombination.
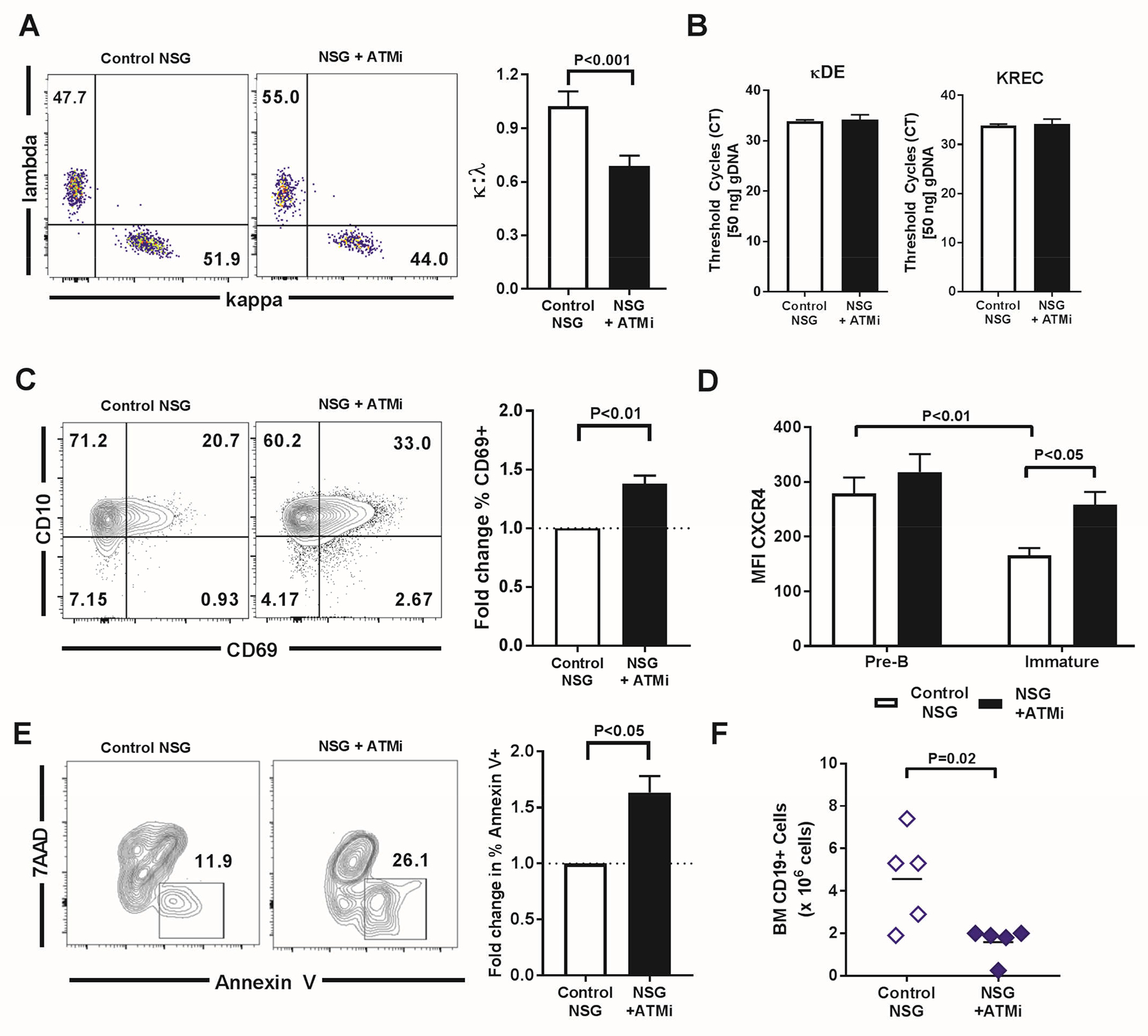
(A) Representative flow cytometry plots of κ:λ staining on peripheral transitional B cells from control NSG and ATMi injected (NSG + ATMi injection) humanized mice. Summary of κ:λ ratio compared between NSG control and NSG + ATMi injection. (B) κ-deletion elements (κDEs) and κ recombination excision circles (KRECs) PCRs in Igλ+ transitional human B cells from NSG humanized mice without (control) or with ATMi injection. Cycle threshold (CT) value to reach threshold is depicted. (C) Representative flow cytometry plots of CD69 and CD10 in bone marrow B cells from control NSG and NSG + ATMi injection humanized mice. Bar graph depicts fold change in frequency of CD10+CD69+ B cells. (D) Comparison of average MFI of CXCR4 in CD69+ pre-B cells and immature B cells in NSG control and NSG mice injected with ATMi. (E) Representative flow cytometry plots of Annexin V versus 7AAD staining in CD10+CD34−IgM+ B cells. Bar graph represents average fold change in frequency of Annexin V+ among CD10+CD34−IgM+ B cells. (F) CD19+ B cell precursors in the bone marrow of ATMi-treated (NSG+ATMi) compared to matched control NSG humanized mice. All experiments represent n = 4 or 5 mice per group. Error bars = SEM. Paired two-tailed t-testing and P values are indicated when significant.
Secondary rearrangements that mediate receptor editing and silence autoreactive BCRs occur at the immature B cell stage. As a consequence, immature B cell undergoing secondary rearrangements have delayed egress from the bone marrow, which requires CD69 and CXCR4 downregulation (46, 48, 49). We detected an increased frequency of CD69+ immature B cells in NSG humanized mice injected with ATMi compared to un-injected controls, suggesting that immature B cells remain longer in the bone marrow after ATM inhibition (Fig. 6C). Although CXCR4 expression was high in pre-B cells regardless of ATMi injection, its surface detection declined as B cells developed into IgM+ immature B cells in the absence of ATMi injection (Fig. 6D). In contrast, CXCR4 expression on IgM+ immature B cells was retained at pre-B cell intensity in mice receiving ATMi, thereby promoting immature B cell retention in the bone marrow (immature B cell CXCR4 MFI = 258 in NSG + ATMi compared to 166 in control NSG, P<0.05; Fig. 6D). Hence, ATM inhibition may allow immature B cells to remain in the bone marrow and undergo secondary recombination events by increasing both their CD69 and CXCR4 expression, leading to the production of many Igλ+ B cells. Moreover, ATM inhibition also increased the proportion of annexin V+ apoptotic immature B cells compared to controls and resulted in significantly decreased numbers of CD19+ B cell precursors in the bone marrow of ATMi-treated NSG humanized mice (Fig. 6, E and F). Immature B cells retained in the bone marrow after ATM inhibition may therefore be prone to cell death by apoptosis, which likely results from the genomic instability of unrepaired DNA DSB induced by ongoing secondary V(D)J recombination in the absence of proper ATM function (19).
In vivo ATM inhibition decreases bone density in NSG humanized mice
Since ATMi injections induced an altered Igκ repertoire in developing human B cells in NSG humanized mice that was similar to that of B cells from group I patients with RA, we next sought to determine if defective ATM activation could also promote bone loss in our humanized mouse model. We therefore assessed using X-ray micro-computed tomography (μCT) analysis the bone tissue density of tibiae from aged-matched and sex-matched NSG non-humanized and humanized mice with and without ATMi injections. We found that ATM inhibition did not alter either cortical (1098 mmHA/cm3 vs 1084 mmHA/cm3, control vs ATMi-injected) or trabecular (959 mmHA/cm3 vs 949 mmHA/cm3, control vs ATMi-injected) bone tissue density in NSG mice that were not engrafted with human HSCs (Fig. 7A). In contrast, interfering with ATM function induced a statistically significant decrease in cortical bone tissue density when human lymphocytes were present in NSG humanized mice (1103 mmHA/cm3 vs 1047 mmHA/cm3, P=0.014, Fig. 7A). Similarly, density of trabecular bone tissue from NSG humanized mice was significantly decreased by ATMi injections (944 mmHA/cm3 vs 904 mmHA/cm3, control vs ATMi-injected, respectively, P=0.001; Fig. 7B). In addition, whereas there was no difference in cortical thickness between ATMi-injected or control NSG untransplanted mice, ATM inhibition also led to a significant decrease in cortical thickness in NSG humanized mice (0.215 mm vs 0.197 mm, control vs ATMi-injected, respectively, P=0.016; Fig. 7C). In terms of the ratio of bone volume to tissue volume (BV/TV) in cortical bone, we also observed a significant decrease induced by ATM inhibition in engrafted NSG humanized mice (0.931 vs 0.926, control vs ATMi-injected, respectively, P=0.008; Fig. 7D). The differences in trabecular thickness in the NSG humanized mice did not reach statistical significance (fig. S8A). There was also no difference in trabecular BV/TV in control or ATMi-injected humanized or non-humanized mice (fig. S8B). We conclude that ATM inhibition leads to decreased bone tissue density, thickness, and volume, especially at the cortex, by impacting human cells engrafted in NSG humanized mice.
Fig. 7. Decreased bone density following ATM inhibition in humanized mice.
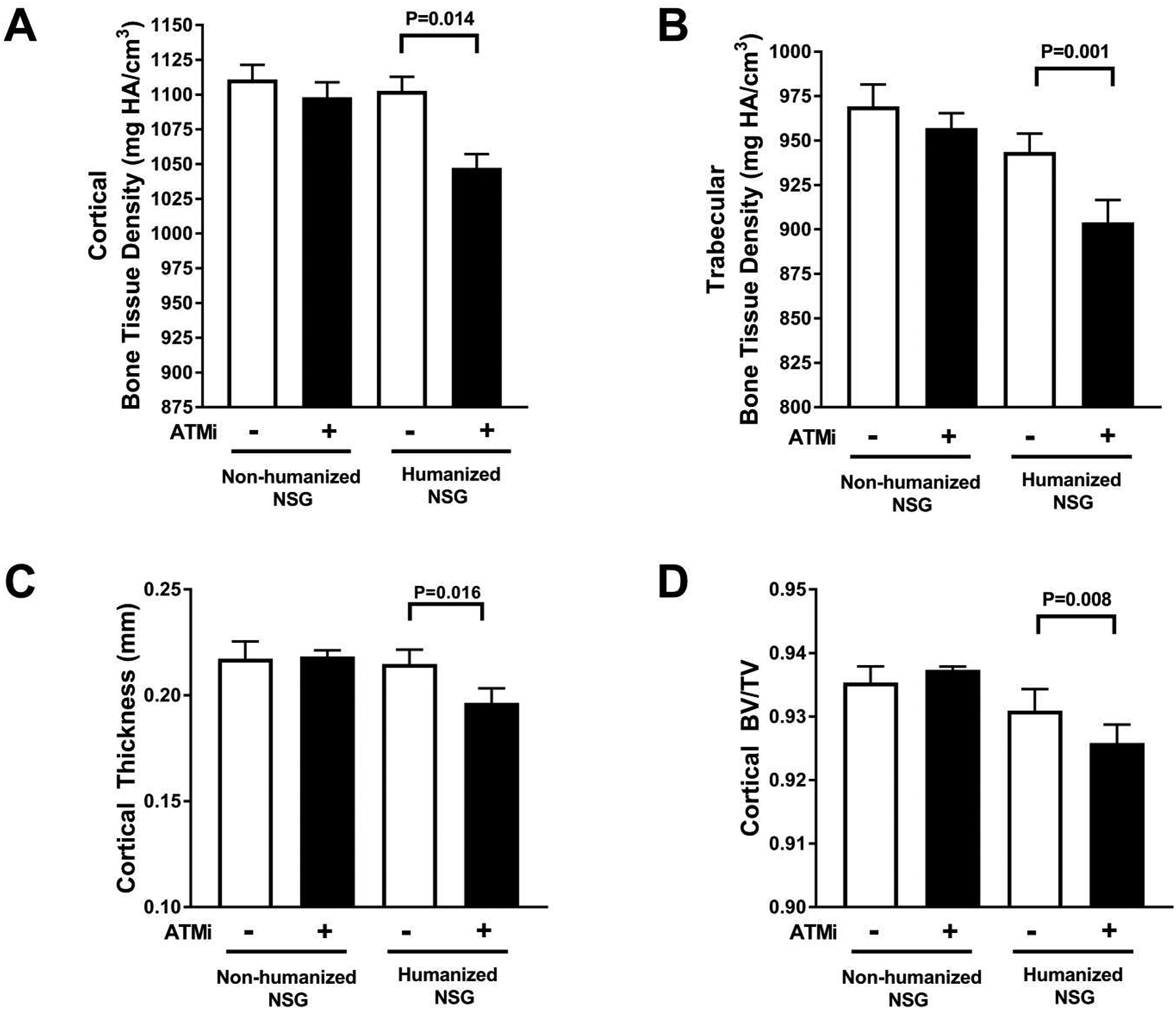
Average bone tissue density in (A) cortex and (B) trabeculae. (C) Average cortical thickness and (D) ratios of cortical bone volume to tissue volume (BV/TV) obtained by μCT performed on tibiae from non-humanized control and ATMi-injected NSG mice (open bars) and control and ATMi-injected NSG humanized mice (filled bars). Comparisons were made via paired t-testing with error bars = SEM and P values are indicated when significant.
In vivo ATM inhibition induces RANKL expression in B cell precursors of NSG humanized mice
We investigated the mechanisms by which in vivo ATM inhibition induced decreased bone density and cortical thickness in engrafted NSG humanized mice (Fig. 8). The analysis of human CD45+ cells in NSG humanized mice revealed that CD19+ B cell precursors represented the majority of human cells in the bone marrow and were 10–20 times more abundant than CD3+ T cells (fig. S7A–C). Since impaired or decreased CD40 expression in CD40−/− mice and AT patients, respectively, is associated with decreased bone density (6, 35, 50), we tested CD40 expression on B cells from NSG humanized mice. We found that ATM inhibition decreased CD40 expression on IgM+ immature B cells in the bone marrow of NSG humanized mice injected with ATMi compared to control NSG humanized mice (Fig. 8A). In addition, ATMi injections also induced higher frequencies of RANKL+ B cell precursors in the bone marrow (Fig. 8B). The analysis of OPG secretion by bone marrow B cells in vitro revealed no appreciable difference between ATMi-injected and control NSG humanized mice (Fig. 8C). However, RANKL secretion by bone marrow CD19+ cells in vitro was significantly enhanced by ATM inhibition as illustrated by a greater increase in RANKL relative to OPG produced by B cell precursors of NSG humanized mice that received ATMi compared to uninjected controls (Fig. 8D and E). Hence, bone loss induced by in vivo ATM inhibition correlates with decreased CD40 and increased RANKL expression by B cell precursors in the bone marrow of NSG humanized mice injected with ATMi.
Fig. 8. Increased B cell RANKL relative to OPG in humanized mice with decreased bone density.
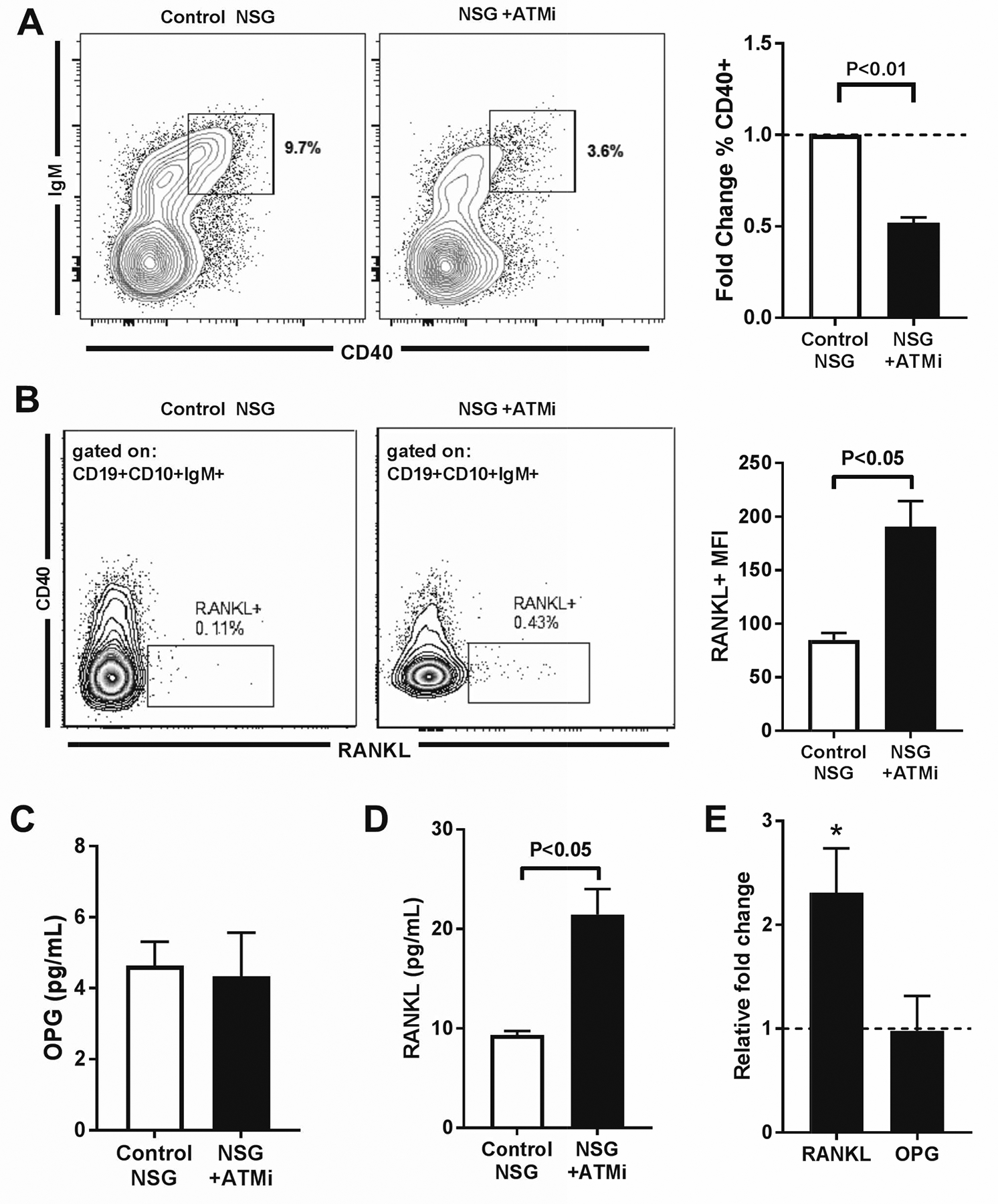
(A) Representative flow cytometry plot showing CD40 expression in human CD19+CD10+CD34− bone marrow B cells developing in control and ATMi-injected NSG humanized mice (left panel). Right panel summarizes fold changes in % CD40+ cells from control (open bar) and ATMi-injected (filled bar) mice. (B) Representative flow cytometry plot showing RANKL expression on gated bone marrow CD19+CD10+CD34−IgM+ B cells in control and ATMi-injected NSG humanized mice (left panel). Right panel shows RANKL MFI in RANKL+ B cells from control (open bar) and ATMi-injected (filled bar) NSG humanized mice. (C) OPG production measured by ELISA from in vitro cultures of human bone marrow B cells isolated from control and ATMi-injected NSG humanized mice. (D) Comparison of fold change in RANKL and OPG in bone marrow B cells from ATMi-injected relative to control NSG humanized mice. * represents significant fold change (P<0.05). Comparisons were made via paired t-testing with error bars = SEM and P values are indicated when significant
Discussion
We reported herein that decreased ATM function in B cells identified in some patients with RA is associated with a restricted BCR repertoire, increased frequency of circulating atypical CD21−/lo B cells, and the secretion of pro-osteoclastogenic cytokines that likely contribute to the increased prevalence of erosive disease that we observed in this subgroup of patients with RA. Decreased ATM expression and function was previously reported in T cells from patients with RA and associated with increased RANKL production, but neither presence of erosive joint disease nor ATM defects were reported as more prevalent in a specific patient subgroup in those studies (25, 51). The proportion of anti-CCP+ individuals and serum anti-CCP autoantibody titers were not different between group I and group II patients with RA and did not appear to correlate with erosive disease in our cohort. Our data suggest that the increased prevalence of erosive disease reported in anti-CCP+ compared to anti-CCP– patients may also be influenced by group I patients with ATM defects and elevated proportions of CD21−/lo B cells rather than the presence of anti-CCP autoantibodies alone, which are found at a similar frequency in group II patients with RA who rarely show bone erosion (3, 52). Although the limitation of our study may lie on our current cohort size, we did not identify a potential correlation between age, disease duration, DMARD use, or autoantibody titers and ATM insufficiency in group I patients with RA. A genetic or environmental origin of the impaired ATM expression in B and T cells from group I patients is currently unknown. The decreased ATM function in group I patients with RA does not result in the neurodegenerative or cutaneous features of AT patients in which ATM function is abrogated, revealing that loss of ATM function in RA is less severe than in AT. Support for a genetically-driven ATM insufficiency in some patients with RA may be suggested by both sparse case reports of inflammatory arthritis in AT (53) and the identification of a SNP tagged to the ATM locus in an RA genome-wide association study (54). However, none of the patients with RA in our study were positive for this SNP but our microarray data also showed decreased expression of other DSB repair factors and suggest a global defect in DSB repair in group I patients. Thus, other SNPs or epigenetic modifications are possible alternative origins of pathogenic ATM defects in RA.
Our in vivo NSG humanized mouse model demonstrated that ATM inhibition results in decreased bone tissue density and thereby reproduces the osteopenic phenotype seen in Atm−/− mice and in both AT patients and patients with RA (42, 55). Bone loss in humanized mice injected with ATMi depended on the presence of human cells, which demonstrates that ATM inhibition did not directly and significantly impact mouse osteoclasts or other mouse cells such as those from the myeloid compartment present in NSG mice. Since ATM inhibition in B cells in the absence of RA disease factors leads to increased production of RANKL, IL-6, and TNFα, which are known to induce osteoclastogenesis (39), we postulate that defective ATM function in B cells may induce bone loss. Indeed, B cells and their precursors expressed RANKL in vivo when ATMi was injected in humanized mice and were 10–20 times more abundant in the bone marrow than T cells, which can also produce pro-osteoclastogenic cytokines when ATM and its associated DNA DSB repair factor MRE11A are inhibited (26, 51). Regardless, OPG production by B cells normally antagonizes RANKL-mediated bone resorption but OPGi production is blunted following CD40 activation in the presence of ATMi (6). Indeed, CD40 plays an important role in the induction of OPG secretion by B cells and our data show that in vivo ATM inhibition decreases CD40 expression on B cells (6, 56). An important role for CD40 expression and proper bone metabolism is also supported by the report of decreased bone density when CD40/CD40L interactions are disrupted in Cd40−/− and Cd40l−/− mice (6). In addition, CD40 expression was also reported to be decreased on B cells from AT patients who also show low bone density (35). Moreover, we also previously showed decreased CD40 expression in CD21−/lo B cells, which we now show are more frequent in group I patients with defective ATM activation and correlate with high prevalence of erosive joint disease in these patients (13). Although circulating peripheral CD21−/lo B cells do not express high amounts of RANKL, these cells may be the precursors of the RANKL-expressing CD21−/lo B cells that are enriched in the synovial fluid from patients with RA (14, 57). In agreement with this hypothesis, peripheral CD21−/lo B cells have higher expression of CXCR3 important for lymphocyte trafficking to the joint in RA and enhanced expression of receptors for the RANKL-inducing cytokines IL-6 and TNFα. The mechanisms responsible for the generation of these B cells are currently not well understood and may involve dysregulated IL-21/IL-4/IFNγ production during B cell responses in germinal centers (16, 58, 59). Regardless, elevated circulating frequencies of CD21−/lo B cells in newly diagnosed patients with RA may represent an indicator for ATM insufficiency and identify early on in disease a subpopulation of patients with high risk for developing erosive joint damage. The presence of CD21−/lo B cells was also recently reported to be associated with more severe autoimmune disease in patients with SLE (16). Altogether, these data suggest that the efficacy of rituximab in RA may rely on the elimination of CD20-expressing CD21−/lo and other B cells that produce osteoclastogenic cytokines and favor local and generalized bone resorption in this disease (2).
Decreased or impaired ATM function also impacted the development of B cells by altering the BCR repertoire. The lack of breadth of Vκ-Jκ rearrangements in transitional B cells from group I RA and AT patients and in ATMi-injected NSG humanized mice may originate from the multiple important roles that ATM plays in V(D)J recombination. Since ATM controls the repression of Rag1 and Rag2 via down-regulation of Gadd45α, ATMi inhibition results in increased Rag activity that would favor secondary recombination on Ig light chain loci (24). In agreement with these observations, we found that ATM inhibition favored Igλ usage that occurred after the Igκ alleles were inactivated by rearrangements using the recombining κDE deleting element. Alternatively, ATM inhibition can result in loss of large regions of the Igκ locus by increased hybrid joint formation during Vκ to Jκ rearrangements by inversion, thereby potentially favoring the initiation of rearrangements on the Igλ locus (30, 31, 60). In addition, increased frequencies of CD69+CXCR4hi immature B cells that are retained in the bone marrow of NSG humanized mice injected with ATMi suggest ongoing secondary recombination when ATM is inhibited (48). Surprisingly, the Igκ repertoire of new emigrant/transitional B cells from group I RA and AT patients and ATMi-injected NSG humanized mice was characterized by a dearth of secondary Vκ-Jκ recombination events as illustrated by decreased upstream Vκ and downstream Jκ3/4/5 gene segment usage. However, this apparently paradoxical observation may be explained by ATM involvement in repairing RAG-mediated DNA DSB and regulating cell-cycle checkpoints. Indeed, secondary recombination will produce novel RAG-mediated DSBs in immature B cells that may fail to properly repair these DNA lesions when ATM function is defective, leading to genomic instability and cell loss (21). This scenario is supported by the increased frequency of annexin V+ immature B cells that are likely undergoing apoptosis in the bone marrow of NSG humanized mice injected with ATMi. Furthermore, delayed bone marrow and thymus egress combined to increased susceptibility to apoptosis of developing B and T lymphocytes provides a mechanism accounting for decreased numbers of new emigrant/transitional B cells and decreased thymic T cell output observed in AT patients (34, 61). It remains to be determined if the altered B cell repertoire induced by decreased ATM function in RA may contribute to disease pathogenesis or is solely a byproduct of improper ATM expression in developing B cells.
In conclusion, defective ATM function in human B cells induces pro-osteoclastogenic RANKL and IL-6 secretion, is associated with decreased bone density in humanized mice and a high prevalence of joint erosions in patients with RA, thereby providing additional support for the pathogenic role of B cells in RA bone processes. Thus, identifying patients with RA and decreased ATM function using their elevated frequencies of circulating atypical CD21−/lo B cells, which correlate with high prevalence of erosive disease, may have implications for improving our understanding, diagnosis, and treatment of RA with the growing arsenal of biologic monoclonal antibody and small molecule inhibitor therapies.
Materials and Methods
Study Design
This study explored the presence and implications of decreased ATM function in B cells on the Igκ repertoire and the potential for pathogenic bone resorption in a subset of patients with RA. We obtained peripheral blood from consecutively enrolled, RF and/or anti-CCP autoantibody positive, mostly female, American, patients with RA of diverse ethno-racial backgrounds, on no or minimal DMARD treatment at the time of blood draw. Patients were enrolled from 2003-present as part of an ongoing multi-center recruitment for the study of B cell tolerance in RA. None of the patients had been treated with the B cell-depleting monoclonal antibody rituximab. All patients met ACR diagnostic criteria for RA as determined by their treating rheumatologist (27). Presence of erosive disease was assessed as a binary value (present or absent) by the treating rheumatologist. Characteristics of patients with RA are summarized in Table 1 and table S2. In addition, we obtained peripheral blood from six AT patients who have naturally occurring biallelic loss-of-function mutations in ATM. Peripheral blood was also obtained from 25 healthy donor volunteers (HD) without RA or AT as controls. The study protocol was approved by the Institutional Review Board (HIC#0906005336), and informed consent was obtained from all patients or their legal surrogates before participation. With the help of the Yale Center for Analytical Sciences, we used the Power Analysis and Sample Size software (version 2008) to determine patient with RA (18–23 patients per group) and NSG mouse (5 per group) sample sizes for a power = 90% and α = 0.05 needed to detect a difference in erosive disease and human B cell Igκ gene segment usage. Primary data are reported in data file S1.
Mice
Two-day old NOD.Cg-PrkdcscidIl2rγtm1Wjl/SzJ (NSG) mice were engrafted after irradiation with 105 CD34+ human hematopoietic stem cells (HSCs) isolated from fetal tissues obtained from the Birth Defects Research Laboratory of the University of Washington, Seattle, as previously described (44). ATM function was inhibited in vivo by daily intraperitoneal injection of 10 mg/kg of KU55933 for 10 days. At 10–12 weeks of age, humanized mice displayed at least 20% of human CD45+ cells in their blood and bone marrow cells, splenocytes, and tibia bones were harvested. NSG littermates engrafted with HSCs from the same, non-RA, human fetal donor but only injected with DMSO served as controls for each biological replicate. All animals were treated, and experiments were conducted in accordance with the Yale institutional reviewed guidelines on treatment of experimental animals.
Flow Cytometry
Peripheral B cells were purified from the blood of patients and HD by Ficoll density gradient purification followed by positive selection using CD20 magnetic beads (Miltenyi Biotec). Bone marrow and splenic B cells from humanized mice were enriched using anti-human CD19 magnetic beads (Miltenyi Biotec). Purified B cells were then stained with the following antibodies: anti-CD10 (clone: HI10A), anti-CD19 (clone: HIB19), anti-CD21 (clone: B-LY4), anti-CD27 (clone: O323), anti-CD34 (clone: 581), anti-CD45 (clone: HI30), anti-CD69 (clone: FN50), anti-CD86 (clone: IT2.2), anti-CXCR4 (clone: 12G5), anti-IgM (clone: MHM88), anti-IgD (clone: IA6–2), anti-PD-1 (clone: EH12.2H7), anti-RANKL (clone: MIH24) (all from Biolegend), anti-CD3 (clone: OKT3) and 7AAD (eBioscience), and annexin V (AF488 conjugated) (ThermoFisher Scientific). Intracellular staining was performed with anti-p-ATM (clone: 10H11.E12) and anti-γ-H2AX (clone: 2F3) (Biolegend) after staining for surface markers using a fixation-permeabilization solution kit (eBioscience). Flow cytometry was performed using a BD LSRII, and the data were analyzed using Flow Jo software (Treestar).
Immunoglobulin Light Chain Gene Usage Sequencing
RNA from single CD19+CD10+CD27‒CD21−/loIgMhi new emigrant/transitional B cells was reverse transcribed in the original 96-well PCR plate into which single cells were sorted by FACS. Reverse transcription and PCR were performed as previously described (29). Immunoglobulin sequences were obtained by sequencing of the Igκ PCR products and analyzed by comparison with GenBank using the National Center for Biotechnology Information IgBlast server (NIH) (http://www.ncbi.nlm.nih.gov/igblast/).
DNA Double-Strand Break (DSB) Induction
PBMCs from human subjects or cells from NSG humanized mice were isolated into pre-warmed, 37 °C PBS and irradiated in Petri dishes at 4 Gy as previously reported (62). Immediately after irradiation, cells were incubated at 37 °C in PBS for 1 hour before surface staining for B or T cell phenotyping and intracellular staining to detect pATM and γH2AX.
Cell culture and in vitro ATM inhibition
B cells were plated at 3 × 105 cells per well in 96-well plates and were cultivated with RPMI media + 10% FBS and a combination of 2 μg/mL polyclonal F(ab′)2 rabbit anti-human IgM (Jackson ImmunoResearch) and 0.05 μg/mL of multimeric soluble recombinant-human CD40L (Alexis Biochemicals) for 2 days with or without the ATM inhibitor KU55933 (Sigma) used at the validated kinase inhibitory concentration of 10 μM (40).
Gene expression analyses
RNA extracted from 1 × 105 to 3 × 105 sorted CD19+CD21+CD27− naïve B cells from patients with RA, amplified and labeled to produce cDNA. Labeled cDNA was hybridized on chips containing the whole human genome (Human Genome U133 2.0 from Affymetrix). Differential gene expression for genes with at least 2-fold difference in expression between group I and group II patients was compared to curated gene sets in the Molecular Signatures Database (Broad Institute, http://software.broadinstitute.org/gsea/login.jsp). Pathway analysis was done by analyzing differentially up and down regulated genes in group I relative to group II with the REACTOME software (http://www.reactome.org). RNA Sequencing was performed on RNA extracted from 3 × 105 bone marrow B cells from NSG humanized mice injected or not with the ATMi. Fragment per kilobase million (FPKM) values were obtained and the top 500 genes were compared to curated gene sets in the Molecular Signatures Database. For quantitative PCR (qPCR), total RNA was isolated from 1.5 × 105 CD20+ B cells from healthy donor volunteers. Gene expression normalized to ACTB were determined by a ΔΔCT method. The presence of κ-deletion recombination excision circles (KRECs) and κ deletion elements (κDEs) was determined using qPCR as previously described from 50 ng of genomic DNA from λ+ sorted transitional splenic B cells (47).
ELISA
ELISA was performed on supernatants from healthy donor volunteer B cells cultured in either RPMI media alone or with CD40L and anti-human IgM with or without KU55933. Soluble IL-6, TNFα, IL-10, and OPG were measured using a multiplex platform (Millipore). Soluble RANKL was measured by ELISA (PeproTech).
Skeletal Analysis
Tibiae from the NSG mice engrafted or not with fetal human HSCs and injected or not with KU55933 ATMi were harvested for X-ray micro-computed tomography (μCT) analysis using a ScanCo MicroCT 35 machine with 3.5 μm resolution.
Statistical Analysis
Data were analyzed using Prism 7.01 software (GraphPad). All data are presented as means ± SEM except where specifically noted in the figure legend. Analysis of statistical differences was performed using tests indicated in the figure legends as appropriate for the type of comparison. D’Agostino and Pearson normality testing was done for values from HD and patients with RA. Shapiro-Wilk normality testing was done for values relating to NSG mice because of the smaller sample size. A P value of < 0.05 was considered significant.
Supplementary Material
Fig. S1. Skewed Igκ repertoire in patients with RA and decreased ATM function.
Fig. S2. Group I and group II patients with RA display similar biometric characteristics.
Fig. S3. Decreased pATM induction in T cells from patients with RA.
Fig. S4. Decreased pATM induction in non-T and non-B cells from patients with RA.
Fig. S5. In vitro inhibition of ATM activation in human B cells.
Fig. S6. Reconstitution of human lymphocytes in NSG humanized mice.
Fig. S7. B cell precursors are the major human CD45+ cell population in the bone marrow of NSG humanized mice.
Fig. S8. Trabecular thickness and ratios of bone volume to tissue volume in NSG humanized mice injected or not with ATMi.
Table S1. Igκ repertoire of antibodies expressed by new emigrant/transitional B cells from patients with RA, patients with AT and healthy donors.
Table S2. Additional characteristics of group I and group II patients with RA.
Table S3. Igκ repertoire of antibodies expressed by new emigrant/transitional B cells from NSG humanized mice.
Data file S1. Primary data
Acknowledgments:
We thank Dr. Lesley Devine and Chao Wang for cell sorting. We also thank the Yale Core Center for Musculoskeletal Disorders Micro CT facility. We thank Dr. Peter K. Gregersen for providing patient’s material and Drs. Roberta Pelanda, David G. Schatz and Richard Bucala for helpful discussions.
Funding: Supported by the Robert Leet Patterson and Clara Guthrie Patterson Trust Mentored Research Award, Bank of America, N.A., Trustee, and NIH (AR007107-41 to KAM, HHSN272201100019C to RRM, AR40072 to JEC, and AI071087 to EM).
Footnotes
Competing interests: E.M. is an advisor for and receives funding from AbbVie, Inc.
Data and materials availability: All data associated with this study are present in the paper or Supplementary Materials. Accession numbers for gene array and RNA-Seq data are GSE132832 and GSE132963, respectively.
References and Notes
- 1.Gibofsky A, Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis. The American journal of managed care 18, S295–302 (2012). [PubMed] [Google Scholar]
- 2.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T, Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. The New England journal of medicine 350, 2572–2581 (2004). [DOI] [PubMed] [Google Scholar]
- 3.van Gaalen FA, van Aken J, Huizinga TW, Schreuder GM, Breedveld FC, Zanelli E, van Venrooij WJ, Verweij CL, Toes RE, de Vries RR, Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis and rheumatism 50, 2113–2121 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E, Impaired early B cell tolerance in patients with rheumatoid arthritis. The Journal of experimental medicine 201, 1659–1667 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mensah KA, Li J, Schwarz EM, The emerging field of osteoimmunology. Immunologic research 45, 100–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, Weitzmann MN, B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109, 3839–3848 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horowitz MC, Fretz JA, Lorenzo JA, How B cells influence bone biology in health and disease. Bone 47, 472–479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duddy ME, Alter A, Bar-Or A, Distinct profiles of human B cell effector cytokines: a role in immune regulation? Journal of immunology 172, 3422–3427 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Ota Y, Niiro H, Ota S, Ueki N, Tsuzuki H, Nakayama T, Mishima K, Higashioka K, Jabbarzadeh-Tabrizi S, Mitoma H, Akahoshi M, Arinobu Y, Kukita A, Yamada H, Tsukamoto H, Akashi K, Generation mechanism of RANKL(+) effector memory B cells: relevance to the pathogenesis of rheumatoid arthritis. Arthritis research & therapy 18, 67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meednu N, Zhang H, Owen T, Sun W, Wang V, Cistrone C, Rangel-Moreno J, Xing L, Anolik JH, Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis & rheumatology 68, 805–816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeo L, Toellner KM, Salmon M, Filer A, Buckley CD, Raza K, Scheel-Toellner D, Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Annals of the rheumatic diseases 70, 2022–2028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goronzy JJ, Shao L, Weyand CM, Immune aging and rheumatoid arthritis. Rheumatic diseases clinics of North America 36, 297–310 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, Samuels J, Berman J, Buckner JH, Cunningham-Rundles C, Meffre E, Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood 115, 5026–5036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, van der Burg M, van Dongen JJ, Wiech E, Visentini M, Quinti I, Prasse A, Voelxen N, Salzer U, Goldacker S, Fisch P, Eibel H, Schwarz K, Peter HH, Warnatz K, Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proceedings of the National Academy of Sciences of the United States of America 106, 13451–13456 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, Joly F, Rosenzwajg M, Sene D, Benech P, Musset L, Klatzmann D, Meffre E, Cacoub P, Expansion of autoreactive unresponsive CD21-/low B cells in Sjogren’s syndrome-associated lymphoproliferation. Arthritis and rheumatism 65, 1085–1096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, Tomar D, Woodruff MC, Simon Z, Bugrovsky R, Blalock EL, Scharer CD, Tipton CM, Wei C, Lim SS, Petri M, Niewold TB, Anolik JH, Gibson G, Lee FE, Boss JM, Lund FE, Sanz I, Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 49, 725–739 e726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P, Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW, DNA repair, genome stability, and aging. Cell 120, 497–512 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Lavin MF, Kozlov S, ATM activation and DNA damage response. Cell cycle 6, 931–942 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Ambrose M, Gatti RA, Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 121, 4036–4045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, Sleckman BP, ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 442, 466–470 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Schatz DG, Ji Y, Recombination centres and the orchestration of V(D)J recombination. Nature reviews. Immunology 11, 251–263 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Hewitt SL, Yin B, Ji Y, Chaumeil J, Marszalek K, Tenthorey J, Salvagiotto G, Steinel N, Ramsey LB, Ghysdael J, Farrar MA, Sleckman BP, Schatz DG, Busslinger M, Bassing CH, Skok JA, RAG-1 and ATM coordinate monoallelic recombination and nuclear positioning of immunoglobulin loci. Nature immunology 10, 655–664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinel NC, Lee BS, Tubbs AT, Bednarski JJ, Schulte E, Yang-Iott KS, Schatz DG, Sleckman BP, Bassing CH, The ataxia telangiectasia mutated kinase controls Igkappa allelic exclusion by inhibiting secondary Vkappa-to-Jkappa rearrangements. The Journal of experimental medicine 210, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM, Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. The Journal of experimental medicine 206, 1435–1449 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, Zhang H, Goronzy JJ, Weyand CM, Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity 45, 903–916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G, 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis and rheumatism 62, 2569–2581 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Kurz EU, Douglas P, Lees-Miller SP, Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. The Journal of biological chemistry 279, 53272–53281 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC, Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003). [DOI] [PubMed] [Google Scholar]
- 30.van der Burg M, Tumkaya T, Boerma M, de Bruin-Versteeg S, Langerak AW, van Dongen JJ, Ordered recombination of immunoglobulin light chain genes occurs at the IGK locus but seems less strict at the IGL locus. Blood 97, 1001–1008 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Vettermann C, Schlissel MS, Allelic exclusion of immunoglobulin genes: models and mechanisms. Immunological reviews 237, 22–42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM, DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. The Journal of biological chemistry 273, 5858–5868 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM, Ataxia telangiectasia: a review. Orphanet journal of rare diseases 11, 159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson A, Trip M, Warris A, van der Flier M, Wulffraat N, Verhagen MM, Taylor MA, van Zelm MC, van Dongen JJ, van Deuren M, van der Burg M, Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. The Journal of allergy and clinical immunology 131, 1367–1375 e1369 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Pereira CTM, Bichuetti-Silva DC, da Mota NVF, Salomao R, Brunialti MKC, Costa-Carvalho BT, B-cell subsets imbalance and reduced expression of CD40 in ataxia-telangiectasia patients. Allergologia et immunopathologia, (2018). [DOI] [PubMed] [Google Scholar]
- 36.Ruth JH, Rottman JB, Katschke KJ Jr., Qin S, Wu L, LaRosa G, Ponath P, Pope RM, Koch AE, Selective lymphocyte chemokine receptor expression in the rheumatoid joint. Arthritis and rheumatism 44, 2750–2760 (2001). [DOI] [PubMed] [Google Scholar]
- 37.McInnes IB, Schett G, Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 389, 2328–2337 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Teitelbaum SL, Bone resorption by osteoclasts. Science 289, 1504–1508 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Schwarz EM, Looney RJ, Drissi MH, O’Keefe RJ, Boyce BF, Xing L, Ritchlin CT, Autoimmunity and bone. Annals of the New York Academy of Sciences 1068, 275–283 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC, Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer research 64, 9152–9159 (2004). [DOI] [PubMed] [Google Scholar]
- 41.O’Brien W, Fissel BM, Maeda Y, Yan J, Ge X, Gravallese EM, Aliprantis AO, Charles JF, RANK-Independent Osteoclast Formation and Bone Erosion in Inflammatory Arthritis. Arthritis & rheumatology 68, 2889–2900 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rasheed N, Wang X, Niu QT, Yeh J, Li B, Atm-deficient mice: an osteoporosis model with defective osteoblast differentiation and increased osteoclastogenesis. Human molecular genetics 15, 1938–1948 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R, Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. Journal of immunology 174, 6477–6489 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Schickel JN, Kuhny M, Baldo A, Bannock JM, Massad C, Wang H, Katz N, Oe T, Menard L, Soulas-Sprauel P, Strowig T, Flavell R, Meffre E, PTPN22 inhibition resets defective human central B cell tolerance. Science immunology 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stagni V, Manni I, Oropallo V, Mottolese M, Di Benedetto A, Piaggio G, Falcioni R, Giaccari D, Di Carlo S, Sperati F, Cencioni MT, Barila D, ATM kinase sustains HER2 tumorigenicity in breast cancer. Nature communications 6, 6886 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Luning Prak ET, Monestier M, Eisenberg RA, B cell receptor editing in tolerance and autoimmunity. Annals of the New York Academy of Sciences 1217, 96–121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ, Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. The Journal of experimental medicine 204, 645–655 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beck TC, Gomes AC, Cyster JG, Pereira JP, CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. The Journal of experimental medicine 211, 2567–2581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL, S1P1 receptor directs the release of immature B cells from bone marrow into blood. The Journal of experimental medicine 207, 1113–1124 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Panach L, Pineda B, Mifsut D, Tarin JJ, Cano A, Garcia-Perez MA, The role of CD40 and CD40L in bone mineral density and in osteoporosis risk: A genetic and functional study. Bone 83, 94–103 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, Weyand CM, Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Science translational medicine 8, 331ra338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, Jakobsson PJ, Baum W, Nimmerjahn F, Szarka E, Sarmay G, Krumbholz G, Neumann E, Toes R, Scherer HU, Catrina AI, Klareskog L, Jurdic P, Schett G, Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. The Journal of clinical investigation 122, 1791–1802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasini AM, Gagro A, Roic G, Vrdoljak O, Lujic L, Zutelija-Fattorini M, Ataxia Telangiectasia and Juvenile Idiopathic Arthritis. Pediatrics 139, (2017). [DOI] [PubMed] [Google Scholar]
- 54.Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, Graham RR, Manoharan A, Ortmann W, Bhangale T, Denny JC, Carroll RJ, Eyler AE, Greenberg JD, Kremer JM, Pappas DA, Jiang L, Yin J, Ye L, Su DF, Yang J, Xie G, Keystone E, Westra HJ, Esko T, Metspalu A, Zhou X, Gupta N, Mirel D, Stahl EA, Diogo D, Cui J, Liao K, Guo MH, Myouzen K, Kawaguchi T, Coenen MJ, van Riel PL, van de Laar MA, Guchelaar HJ, Huizinga TW, Dieude P, Mariette X, Bridges SL Jr., Zhernakova A, Toes RE, Tak PP, Miceli-Richard C, Bang SY, Lee HS, Martin J, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Rantapaa-Dahlqvist S, Arlestig L, Choi HK, Kamatani Y, Galan P, Lathrop M, consortium R, consortium G, Eyre S, Bowes J, Barton A, de Vries N, Moreland LW, Criswell LA, Karlson EW, Taniguchi A, Yamada R, Kubo M, Liu JS, Bae SC, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Raychaudhuri S, Stranger BE, De Jager PL, Franke L, Visscher PM, Brown MA, Yamanaka H, Mimori T, Takahashi A, Xu H, Behrens TW, Siminovitch KA, Momohara S, Matsuda F, Yamamoto K, Plenge RM, Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirozane T, Tohmonda T, Yoda M, Shimoda M, Kanai Y, Matsumoto M, Morioka H, Nakamura M, Horiuchi K, Conditional abrogation of Atm in osteoclasts extends osteoclast lifespan and results in reduced bone mass. Scientific reports 6, 34426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yun TJ, Chaudhary PM, Shu GL, Frazer JK, Ewings MK, Schwartz SM, Pascual V, Hood LE, Clark EA, OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. Journal of immunology 161, 6113–6121 (1998). [PubMed] [Google Scholar]
- 57.Amara K, Clay E, Yeo L, Ramskold D, Spengler J, Sippl N, Cameron JA, Israelsson L, Titcombe PJ, Gronwall C, Sahbudin I, Filer A, Raza K, Malmstrom V, Scheel-Toellner D, B cells expressing the IgA receptor FcRL4 participate in the autoimmune response in patients with rheumatoid arthritis. Journal of autoimmunity 81, 34–43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meffre E, Louie A, Bannock J, Kim LJ, Ho J, Frear CC, Kardava L, Wang W, Buckner CM, Wang Y, Fankuchen OR, Gittens KR, Chun TW, Li Y, Fauci AS, Moir S, Maturational characteristics of HIV-specific antibodies in viremic individuals. JCI insight 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, Bengsch B, Linderman SL, Stelekati E, Spolski R, Wherry EJ, Hunter C, Hensley SE, Leonard WJ, Cancro MP, Cutting Edge: IL-4, IL-21, and IFN-gamma Interact To Govern T-bet and CD11c Expression in TLR-Activated B Cells. Journal of immunology 197, 1023–1028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bredemeyer AL, Huang CY, Walker LM, Bassing CH, Sleckman BP, Aberrant V(D)J recombination in ataxia telangiectasia mutated-deficient lymphocytes is dependent on nonhomologous DNA end joining. Journal of immunology 181, 2620–2625 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giovannetti A, Mazzetta F, Caprini E, Aiuti A, Marziali M, Pierdominici M, Cossarizza A, Chessa L, Scala E, Quinti I, Russo G, Fiorilli M, Skewed T-cell receptor repertoire, decreased thymic output, and predominance of terminally differentiated T cells in ataxia telangiectasia. Blood 100, 4082–4089 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Hamasaki K, Imai K, Nakachi K, Takahashi N, Kodama Y, Kusunoki Y, Short-term culture and gammaH2AX flow cytometry determine differences in individual radiosensitivity in human peripheral T lymphocytes. Environmental and molecular mutagenesis 48, 38–47 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Skewed Igκ repertoire in patients with RA and decreased ATM function.
Fig. S2. Group I and group II patients with RA display similar biometric characteristics.
Fig. S3. Decreased pATM induction in T cells from patients with RA.
Fig. S4. Decreased pATM induction in non-T and non-B cells from patients with RA.
Fig. S5. In vitro inhibition of ATM activation in human B cells.
Fig. S6. Reconstitution of human lymphocytes in NSG humanized mice.
Fig. S7. B cell precursors are the major human CD45+ cell population in the bone marrow of NSG humanized mice.
Fig. S8. Trabecular thickness and ratios of bone volume to tissue volume in NSG humanized mice injected or not with ATMi.
Table S1. Igκ repertoire of antibodies expressed by new emigrant/transitional B cells from patients with RA, patients with AT and healthy donors.
Table S2. Additional characteristics of group I and group II patients with RA.
Table S3. Igκ repertoire of antibodies expressed by new emigrant/transitional B cells from NSG humanized mice.
Data file S1. Primary data