

Abstract
BACKGROUND&AIMS:
Chronic alcohol (EtOH) consumption is a leading risk factor for development of hepatocellular carcinoma (HCC), which is associated with marked increase of hepatic expression of pro-inflammatory IL-17A and its receptor IL-17RA.
METHODS:
Genetic deletion and pharmacological blocking was used to characterize the role of IL-17A/IL-17RA signaling in the pathogenesis of HCC.
RESULTS:
We demonstrate that global deletion of IL-17RA gene suppressed HCC in alcohol-fed DEN-challenged IL-17RA−/− and Mup-uPA/IL-17RA−/− mice compared to wild type mice. When the cell-specific role of IL-17RA signaling was examined, development of HCC was decreased in both alcohol-fed IL-17RAΔMΦ and IL-17RAΔHep mice devoid of IL-17RA in myeloid cells and hepatocytes, but not in IL-17RAΔHSCs mice (deficient of IL-17RA in hepatic stellate cells (HSCs)). Deletion of IL-17RA in myeloid cells ameliorated tumorigenesis via suppression of pro-tumorigenic/inflammatory and pro-fibrogenic responses in alcohol-fed IL-17RAΔMΦ mice. Remarkably, despite a normal inflammatory response, alcohol-fed IL-17RAΔHep mice developed the fewest tumors (compared to IL-17RAΔMΦ mice), with reduced steatosis and fibrosis. Steatotic IL-17RA-deficient hepatocytes downregulated expression of Cxcl1 and other chemokines, exhibited a striking defect in TNF-TNFR1-dependent Caspase-2-SREBP-1/2-DHCR7-mediated cholesterol synthesis, and upregulated production of anti-oxidant Vitamin D3. Pharmacological blocking of IL-17A/Th-17 cells using anti-IL-12/IL-23 Ab suppressed progression of HCC (by 70%) in alcohol-fed mice, indicating that targeting IL-17 signaling might provide novel strategies for treatment of alcohol-induced HCC.
CONCLUSIONS:
Overall, IL-17A is as a tumor promoting cytokine, which critically regulates alcohol-induced hepatic steatosis, inflammation, fibrosis, and HCC.
Keywords: Alcoholic liver disease (ALD), hepatocellular carcinoma (HCC), IL-17 signaling, inflammation, fibrosis, mutational signatures, cholesterol synthesis
Graphical Abstract
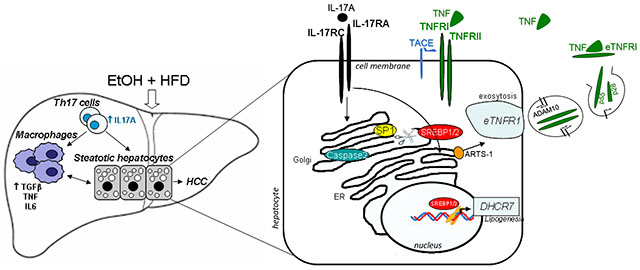
Lay summary:
IL-17A is a tumor-promoting cytokine, which critically regulates inflammatory responses in macrophages (Kupffer cells and bone marrow-derived monocytes), and cholesterol synthesis in steatotic hepatocytes in experimental model of alcohol-induced HCC. Therefore, IL-17A may be a potential therapeutic target for EtOH-induced HCC patients.
Introduction
Hepatocellular carcinoma (HCC) develops in response to chronic injury, such as HBV/HCV infections, and metabolic diseases including non-alcoholic steatohepatitis (NASH), and alcoholic liver disease (ALD). With the introduction of new therapies, the incidence of HBV/HCV-related HCC has declined1, while NASH- and ALD-induced HCC are rapidly rising, both of which typically progress from hepatic steatosis to steatohepatitis, fibrosis, cirrhosis, and cancer2. ALD remains a major risk factor of hepatic cirrhosis and aggressive HCC2. Consistent with the global epidemic of NASH, alcohol-induced HCC often occurs in patients with BMI > 253. Chronic alcohol consumption is believed to increase development of HCC in patients with NASH4, therefore, we used an experimental model of ALD and Western Diet which reflects the typical American patient with ALD.
Alcohol is metabolized in hepatocytes, leading to production of toxic metabolites (acetaldehyde and acetate), fatty acid and cholesterol synthesis2, and inhibition of DNA damage repair, causing accumulation of somatic mutations that drive HCC5–7.
Many cancers including HCC result from chronic inflammation. Previous studies have outlined the critical role of IL-17A and IL-17RA in the early development of colorectal adenomas8,9, lung fibrosis10, and toxic and NASH liver fibrosis11,12. IL-17A, a member of the IL-17 family of cytokines, was implicated in the pathogenesis of NASH- and ALD-induced fibrosis and HCC13. IL-6, TGF-β1, and IL-23 promote differentiation of IL-17A-producing Th17 cells from naïve Th0 cells14. In fibrotic liver, IL-17A is produced mainly by CD4+ Th17 cells11, and signals through its cognate receptor IL-17RA. Deletion of IL-17RA in Kupffer cells/macrophages was shown to prevent development of HCC in Diethylnitrosamine (DEN)-challenged mice with NASH12.
Meanwhile, the role of IL-17 signaling in steatotic hepatocytes and NASH- and ASH-induced HCC has not been previously explored. Hepatic lipogenesis is driven majorly by transcription factors SREBP 1 and 2 (sterol regulatory element binding proteins 1 and 2) that control fatty acid and cholesterol biosynthesis15. A novel Caspase-2-S1P-SREBP pathway has been recently identified in experimental models and patients with NASH and was shown to be activated in response to TNF signaling16. Similar to TNF/TNFR1-dependent activation of Caspase-2-S1P-SREBP signaling cascade in patients with NASH, here we demonstrate that caspase-2-S1P-SREBP mechanism is induced in patients with ASH, and activation of this pathway is regulated by IL-17 signaling.
Using DEN12- and MUP-uPA-17 models of HCC in mice, our study investigated the enhancing effect of alcohol on development of HCC in high fat diet (HFD)-fed mice. We demonstrate that alcohol enhances tumorigenesis, while global deletion of IL-17RA gene suppresses HCC. Furthermore, mice deficient of IL-17RA in macrophages or steatotic hepatocytes were protected from DEN-induced HCC due to reduced activation of inflammatory cells or suppression of cholesterol and fatty acid synthesis in steatotic hepatocytes, respectively. Therapeutic blocking of Th17 cells using anti-IL-12/23 Ab ameliorated development of HCC in EtOH+HFD-fed DEN-challenged mice, indicating that IL-17 signaling may become a novel target for treatment of ALD-associated HCC.
Materials and Methods
Mice:
IL-17RA−/− mice18, IL-17RAF/F mice9, MUP-uPA mice19, Lrat-cre mice20 are gifts of Drs. Rolls, Karin, Sandgren, Schwabe. Rag2−/− γc−/−, AlbCre and LyzMCre mice were obtained from JAX labs. Male age-matched littermates (C57BL/6, the number of mice/group/experiment is listed in the Figure legend) were used in this study, and were maintained under specific pathogen free conditions in filter-topped cages on autoclaved food, water, or liquid diet, and under 12 light/12 dark cycle, according to IACUC protocol S07088 (UCSD).
Human specimens:
Archived liver specimens were obtained in accordance with the ethical guidelines for epidemiological research in China (approved by Ethics Committee of Nanjing Medical University) from ALD patients with liver fibrosis and HCC (Metavir Score F3-4, n=3), and patients undergoing liver resection for reasons unrelated to liver fibrosis (normal liver, n=3) at Jiangsu People’s University Hospital (see Suppl. Materials).
Experimental models of alcohol-induced HCC:
IL-17RA−/−, IL-17RAΔMΦ, IL-17RAΔHep IL-17RAΔHSCs, and IL-17RAF/F and WT mice (male littermates, C57BL/6, 2 w old) were injected i.p. with DEN (25mg/kg body weight, Sigma). DEN-challenged mice, and MUP-uPA|IL-17RA−/− and MUP-uPA mice (male littermates, C57BL/6, 3 months of age) were subjected to isocaloric liquid EtOH feeding (EtOH+HFD: ethanol diet #710362, Dyets Inc.), or pair-fed (calorie-matched HFD: high fat diet #710142, Dyets Inc.), or chow-fed (Diet #5010, LAB) for 18 weeks or 24 weeks. Concentration of alcohol was gradually increased from 1%, to 2% and 3% (v/v, throughout 3 weeks) and maintained at 3.5% (v/v) in EtOH-fed mice for the rest of feeding21, 22 See Supplemental Methods for diet compositions.
HCC analysis:
AFP+YAP+ HCC (≈4 mm) and non-tumor tissues were microdissected from EtOH+HFD−, pair-, and chow-fed IL-17RA−/− and IL-17RAΔHep mice and littermates, and used for Western blotting, qRT-PCR, or RNA-Seq, see Suppl. Material.
Lipidomic analysis:
Snap-frozen non-tumor liver samples (~100mg) were homogenized in 1ml phosphotate-buffered saline containing 10% methanol and analyzed using HPLC/MS method to detect free and ester bonded cholesterol23.
Statistical Analysis:
All data are shown as mean ± standard error of deviation (S.D.). Comparisons of the two groups were analyzed using the unpaired two-tailed Student’s t test. Comparisons of three or more groups were analyzed using ANOVA. ANOVA with Dunnett’s test was used for comparing multiple mouse groups or treatments to a control. ANOVA with a Bonferroni test was used when making multiple pair-wise comparisons between different groups. p<0.05 was considered statistically significant. The analyses were performed by GraphPad Prism software (Graph Pad, La Jolla). De novo analysis of mutational signatures was performed using all mouse samples by applying our previously developed computational framework5.
Results
Chronic EtOH feeding accelerates HCC development in HFD-fed DEN-challenged WT mice.
WT male mice (C57BL/6, 14 days old) were challenged with a single dose of DEN (25 mg/kg, i.p.), and at the age of 3 months were fed with liquid diet containing either EtOH+HFD (EtOH-fed mice) or calorie matched HFD (pair-fed mice), or normal chow diet (chow-fed mice). HCC was assessed in all groups of mice after 18 or 24 weeks of feeding. EtOH strongly accelerated and enhanced tumorigenesis in HFD-fed DEN-challenged mice (vs pair-fed mice, Figure 1A–B). Since tumors reached >80% of liver mass after 24 weeks of EtOH feeding, the time point of 18 weeks was chosen to examine in further detail.
Figure 1. IL-17 signaling is activated in experimental model of EtOH/DEN-induced HCC in mice and patients with ALD.

(A) Gross liver images from DEN-challenged WT mice: chow-(n=3, 9 months), and pair-fed (HFD n=4) for 18 weeks, and EtOH-fed (EtOH+HFD) for 18 weeks (n= 8) and 24 weeks (n= 7).
(B) Serum ALT, tumor burden, and liver/body weight (BW) ratio were calculated.
(C) Livers were stained with H&E, Sirius Red, anti-Desmin, anti-F4/80, and anti-IL-17A Abs. Positive area was calculated as percent (micrographs were taken using x10, x20 and x40 objectives).
(D) Hepatic expression of IL-17A and IL-17RA mRNA was analyzed by qRT-PCR
(E-F) Livers from HCC patients with ALD (Alc. HCC, n=3, stage 3-4), or patients with no fibrosis (Normal, n=3) were (E) analyzed for expression of IL-17A and IL-17RA mRNA or (F) stained with H&E, Sirius Red, and anti-IL-17A Abs (x4, x20, and x60 objectives). Positive Sirius Red staining was calculated as percent.
Statistical analysis was performed using 2-tailed Student’s t test or ANOVA for comparisons between 2 groups or more than 2 groups. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001.
Liver injury, inflammation, and fibrosis were increased in EtOH+HFD-fed WT mice in comparison with the pair-fed WT mice (18 weeks), as shown by increased ALT (2 fold), positive staining for Sirius red (4 fold), F4/80 (2 fold), and IL-17A (12 fold, Figure 1B–C), and correlated with upregulation of IL-17A and IL-17RA mRNA in the livers of these mice (Figure 1D). High expression of IL-17A and IL-17RA mRNA was also found in patients with ALD-associated liver fibrosis and HCC (Figure 1E–F).
EtOH+HFD-fed IL-17RA−/− mice are protected from DEN-induced HCC.
The role of IL-17 signaling in the pathogenesis of alcohol-induced HCC was studied in EtOH+HFD-fed DEN-challenged WT and IL-17RA−/− littermates (males, C57BL/6). Despite similar food intake and blood alcohol content, tumorigenesis was strongly suppressed in IL-17RA−/− mice after 18 and 24 weeks of EtOH+HFD feeding compared to pair-fed WT mice (Figure 2A, Figures S1–2), and was associated with reduced steatosis, inflammation (≈2 fold, IL-6, IL-1β, TNF mRNA), fibrosis (2 fold, Col1a1, TIMP1 mRNA), and downregulation of cholesterol and fatty acid amounts (Figure 2B–C, E). IL-17RA-deficient tumors exhibited a less malignant phenotype, as shown by reduced expression of HCC markers AFP, YAP, Sox9, and phospho-STAT3 (Figure 2B, D and E). Unlike WT HCC, IL-17RA-deficient HCC showed a growth defect, and failed to form tumor nodules when adoptively transferred into the livers of Rag2−/− γc−/− mice (Figure 2F), suggesting that IL-17 signaling regulates DEN-induced tumorigenesis.
Figure 2. Global deletion of IL-17RA protects EtOH+HFD-fed DEN-challenged IL-17RA−/− mice from HCC.

(A). Gross liver images from DEN-challenged IL-17RA−/− and WT mice: chow-(n=4/group, 9 months, Figure S2A), pair-fed (HFD, n≥4/group) and EtOH-fed (EtOH+HFD, n≥8/group) for 18 weeks. Serum ALT, EtOH level, tumor burden, and liver/body weight (BW) ratio are shown.
(B) Livers were stained with H&E, Sirius Red, and anti-F4/80, anti-AFP, anti-YAP, anti-phospho-STAT3 Abs, positive area was calculated as percent (x10, x20, and x40 objectives).
(C) Cholesterol, fatty acids, and triglycerides were measured in livers of EtOH-fed IL-17RA−/− and wt mice.
(D) Expression of phospho-STAT3 in tumors was assessed by Western blot.
(E) Hepatic expression of fibrogenic, inflammatory genes and HCC markers was analyzed using qRT-PCR.
(F) Primary WT or IL-17RA-deficient AFP+YAP+ HCC from EtOH-fed mice were injected (1 x 106 cells) into the livers of Rag2−/−γc−/− pups (3 days old, n≥6/group). Mice were sacrificed at 1 month of age, livers were stained for AFP and YAP, representative macrographs are taken x60 objective; the number of engrafted AFP+ and YAP+ cells/field was calculated.
Statistical analysis was performed using 2-tailed Student’s t test. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001. (see Figures S1, S3).
MUP-uPA|IL-17RA−/− mice are protected from EtOH+HFD-induced HCC.
Similar results were obtained using another model of alcohol-induced HCC in Mup-uPA mice, in which urokinase-type plasminogen activator (uPA) is overexpressed in hepatocytes from the major urinary protein (MUP) promotor. EtOH+HFD feeding strongly accelerated tumorigenesis in MUP-uPA mice (vs pair-fed MUP-uPA mice). Global deletion of IL-17RA suppressed development of HCC (2 fold, Figure S3A–B), steatosis, inflammation (3 fold, IL-1β, TNF mRNA), and fibrosis (3 fold, Col1a1, α-SMA, TIMP1 mRNA) in EtOH+HFD-fed MUP-uPA|IL-17RA−/− mice (Figure S3C–F), suggesting that IL-17A is a common mediator of alcohol-induced HCC.
Pharmacological targeting of Th17 cells suppresses HCC in EtOH+HFD-fed DEN-challenged WT mice.
Of available IL-17A blocking agents, anti-IL-12/IL-23 Ab were shown to produce fewest adverse effects in patients (Phase III clinical trial24). To test if blocking of IL-17 signaling can attenuate development of alcohol-induced HCC, DEN-challenged WT mice were therapeutically administered with anti-IL-12/IL-23 Ab (vs IgG, 40mg/kg, i.p., once a week, Figure 3A). Indeed, treatment with anti-IL-12/IL-23 Ab significantly reduced serum and hepatic levels of IL-17A and IL-17RA in EtOH+HFD-fed DEN-challenged WT mice, and suppressed tumorigenesis (3 fold, Figure 3B–C), liver fibrosis (2 fold), inflammation (2 fold), and cholesterol accumulation (Figure 3D–F). We concluded that IL-17 signaling might become a novel target for treatment of alcohol-induced HCC.
Figure 3. Therapeutic blocking of IL-12/IL-23/IL-17 ameliorates development of HCC in EtOH+HFD-fed DEN-challenged mice.

(A) After 9 weeks of EtOH+HFD-feeding, DEN-injured WT male mice were administered with anti-IL-12/IL-23 Ab (40mg/kg, once a week, for 9 weeks) or IgG (n≥8/group).
(B) Gross liver images from EtOH+HFD-fed DEN-injured wt mice ± anti-IL-12/IL-23 Ab. Serum ALT, tumor burden, liver/body weight (BW) ratio are shown.
(C) Serum and hepatic levels of IL-17A and IL-17RA were measured by ELISA and qRT-PCR.
(D) Livers were stained with H&E, Sirius Red, anti-Desmin, anti-αSMA, and anti-F4/80, anti-AFP, anti-YAP, anti-phospho-STAT3 Abs, positive area was calculated as percent (x10, x20, and x40 objectives).
(E) Cholesterol, fatty acids, and triglycerides were measured in livers of EtOH-fed wt mice ± anti-IL-12/IL-23 Ab.
(F) Expression of inflammatory and fibrogenic genes was analyzed using qRT-PCR. Statistical analysis was performed using 2-tailed Student’s t test. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001.
AFP+YAP+ HCC from EtOH+HFD-fed DEN-challenged WT and IL-17RA−/− mice have similar mutational landscapes.
To investigate the mechanism by which IL-17 signaling promotes HCC, first, DEN-induced mutagenesis was compared in IL-17RA-deficient and WT HCC. Mutagenesis was accessed using paired-end RNA-Seq analysis, and identified somatic coding mutations within transcribed genes in HCC (vs non-tumor tissues). The patterns of both WT and IL-17RA−/− HCC (murine signature 1) indicated the activity of the mutational processes underlying COSMIC signatures 1, 5, 12, and 17, characteristic for HCC patients with NASH and ALD with an accuracy of 0.85 (Figure 4A)6. COSMIC signature 12, which has been found exclusively in human HCC6 was also enriched in HCC from EtOH+HFD-fed DEN-challenged mice. The same mutational signatures were identified in HCC from EtOH+HFD-fed MUP-uPA and MUP-uPA/IL-17RA−/− mice (Figure S4A–B). Our data demonstrate that murine HCC exhibited a mutational landscape similar to that observed in patients with NASH- and ALD-associated HCC.
Figure 4. Mutational landscapes of HCC from EtOH+HFD/DEN-injured WT and IL-17RA−/− mice are similar to that in human HCC.

(A-C) Paired-end RNA-Seq analysis was used to identify coding somatic gene mutations and gene expression profile of AFP+YAP+ HCC (4 mm, 1 tumor/mouse) and non-tumor tissue from DEN-challenged WT and IL-17RA−/− mice: chow- (n=3/group), pair-fed (n=3/group), and EtOH-fed (n=8/group). The data are normalized vs recurrent somatic mutations identified in livers of non-injured C57BL/6 mice.
(A) HCC mutational signature deciphered from EtOH+HFD-fed WT and IL-17RA−/− mice, and COSMIC signatures 1, 5, 12, and 17, characteristic for HCC patients with NASH and ALD6, and are displayed according to the 96 substitution classification defined by the substitution class and the sequence context immediately 3′ and 5′ to the mutated base, (see Figure S4).
(B) Gene set enrichment analysis (GSEA) identified the gene/pathway-specific mutations enriched in WT and IL-17RA-deficient HCC.
(C) Mutational frequency in HCC from WT and IL-17RA−/− mice is shown. Enrichment of variant counts per sample was observed between feeding groups, but not between different genotypes of mice.
Associations were assessed using Chi-square tests for categorical variables and ANOVA for quantitative features. The strength of association among gene mutation events was modeled using a binomial logistic regression. Statistical analysis was performed using 2-tailed Student’s t test. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001. (see Figure S5)
Specifically, exposure to HFD or EtOH+HFD progressively increased HCC mutational frequency in DEN-challenged WT and IL-17RA−/− mice (Figure S4C), especially in genes regulating cellular responses to DNA damage, RNA processing, and metabolic processes (Figure 4B). Meanwhile, WT and IL-17RA−/− HCC developed similar mutational signatures (Figure 4C), indicating that EtOH/DEN-driven mutagenesis occurs independently of IL-17 signaling.
WT HCC and IL-17-deficient HCC have different gene expression profiles.
Despite similarities in mutational patterns, the gene expression profile of tumor and non-tumor tissue in DEN-challenged IL-17RA−/− mice differed dramatically from that in WT mice (Figure S5). “Alcohol specific” signature genes were identified for EtOH+HFD-fed WT and IL-17RA−/− mice. In comparison with WT HCC, IL-17RA-deficient HCC downregulated expression of tumor-associated genes (IL-6, p53, NF-κB, and ERK-MAPK pathways, Figure S5A). Moreover, expression of inflammatory, fibrogenic, and lipogenic genes was suppressed in the non-tumor tissue of EtOH+HFD-fed IL-17RA−/− mice (Figure S5B–C), suggesting that IL-17 signaling in multiple cell types in the liver regulates responses to EtOH/HFD.
IL-17 signaling in Kupffer cells/macrophages (but not in HSCs) promotes tumorigenesis in EtOH+HFD-fed DEN-challenged mice.
To further delineate the role of IL-17 signaling in the pathogenesis of alcohol-induced HCC, cell-specific knockout mice lacking IL-17RA in hepatocytes (IL-17RAΔHep), myeloid cells (IL-17RAΔMΦ), HSCs (IL-17RAΔHSCs), or control IL-17RAF/F littermates were generated and subjected to DEN treatment and EtOH+HFD-feeding (18 weeks).
Similar to the NASH model of HCC12, EtOH+HFD-fed IL-17RAΔMΦ mice were protected from DEN-induced HCC (vs IL-17RAF/F mice, Figure 5A–B), and developed less steatosis (2 fold), fibrosis (2 fold), and inflammation (≈2 fold, TNF, IL-6, IL-1β, Cxcl1, Col1a1 mRNAs), and downregulated cholesterol and fatty acid synthesis (Figure 5C–D, Figure S6A), suggesting that IL-17RA signaling in myeloid cells is critical for cancer-related pathologies and inflammation.
Figure 5. IL-17 signaling in myeloid cells and steatotic hepatocytes is critical for HCC growth in EtOH+HFD-fed DEN-challenged mice.

(A) Gross liver images from EtOH+HFD-fed (18 weeks) DEN-injured IL-17RAΔMΦ mice (n=4, IL-17RAF/F mice x LyzMCre mice), IL-17RAΔHSC mice (n=5, IL-17RAF/F mice x LratCre mice), IL-17RAΔHep mice (n=7, generated by crossing IL-17RAF/F mice x AlbCre mice), and WT IL-17RAF/Flittermates (n=7). The efficiency of Cre-LoxP recombination for each Cre-mouse line has been tested39. Mice were chow-fed (n=3/group, 9 months), pair-fed (HFD, n≥4/group) and EtOH-fed (EtOH+HFD) for 18 weeks.
(B) Serum ALT, tumor burden, liver body weight (BW) ratio is shown.
(C) Livers were stained with H&E, Sirius Red, anti-αSMA, anti-Desmin, and anti-F4/80 Abs, positive area was calculated as percent (x20 objectives).
(D) Cholesterol, fatty acids were measured in livers of uninjured (black bar); chow-, pair-, EtOH+HFD-fed mice.
Statistical analysis was performed using 2-tailed Student’s t test. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001. (see Figure S2C–E, S6).
By contrast, EtOH+HFD-fed IL-17RAΔHSCs mice showed nearly as much tumor development, liver damage, inflammation, and steatosis as IL-17RAF/F mice (Figure 5A–D, Figure S6B), indicating that IL-17 signaling in aHSCs might not be critical for alcohol-induced HCC.
Deletion of IL-17RA in steatotic hepatocytes suppressed tumorigenesis in EtOH+HFD-fed DEN-challenged mice.
EtOH+HFD-fed IL-17RAΔHep mice showed the largest decrease in hepatic tumorigenesis, steatosis, and fibrosis (3 fold, Figure 5A–C), and expression of HCC markers (2 fold, YAP and AFP, Figure 6A) relative to IL-17RAF/F mice. Meanwhile, the macrophage numbers (Figure 5C, S6C) and expression of hepatic cytokines (IL-6, TGF-β1, TNF mRNA, Figure 6B) were not altered in EtOH+HFD-fed IL-17RAΔHep mice, with the exception of Cxcl1 mRNA, which expression was strongly downregulated in IL-17RAΔHep mice (4 fold, Figure 6B).
Figure 6. EtOH+HFD-fed DEN-challenged IL-17RAΔHep mice exhibit a defect in cholesterol synthesis.

(A) Expression of HCC markers was analyzed in tumors of EtOH+HFD-fed DEN-challenged IL-17RAΔHep mice using immunostaining (x10 objective) and qRT-PCR.
(B) Expression of inflammatory and fibrogenic genes was assessed using qRT-PCR (see Figure S6C).
(C) Human primary steatotic (from patients with ALD, n=4) and normal hepatocytes (from patients with no liver injury, n=3) were in vitro stimulated with IL-17A (20 ng/ml, or vehicle). Gene expression was analyzed using qRT-PCR, the data are average of four independent experiments (see Figure S7).
(D) Lipidomic analysis of free and bound lipids in the livers of EtOH+HFD-fed IL-17RAF/Fand IL-17RAΔHep mice.
(E) Schematic representation of cholesterol and fatty acid synthesis pathways. mRNA expression of FASN, DHCR7 and DHCR24 was assessed using qRT-PCR (and RNA-Seq, see Figure S8). Serum levels of Vitamin D3 were measured using ELISA.
Statistical analysis was performed using 2-tailed Student’s t test. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001.
Despite no obvious defect in macrophage activation, DEN-induced HCC was suppressed in EtOH+HFD-fed IL-17RAΔHep mice, but not in chow-fed IL-17RAΔHep mice (Figure S2C). These data are in concordance with our previous observation that normal hepatocytes lack responses to IL-17A11, indicating that IL-17 signaling regulates responses only in steatotic hepatocytes. We hypothesized that IL-17 signaling facilitates hepatic tumorigenesis via regulation of lipogenesis (cholesterol and fatty acid synthesis) and inflammatory responses in steatotic (vs normal) hepatocytes. In support, IL-17A-stimulated steatotic human and murine hepatocytes upregulated IL-17RA, IL-6, TNF, TNFR1, and Cxcl1, while normal hepatocytes did not (Figure 6C, S7A–B), and induced expression of phospho-NFκB and phospho-STAT3 (Figure S7C).
EtOH+HFD-fed DEN-challenged IL-17RAΔHep mice exhibit a defect in cholesterol and fatty acid accumulation.
To investigate the role of IL-17 signaling in cholesterol synthesis, lipidomics of EtOH+HFD-fed IL-17RAΔHep and IL-17RAF/F livers were compared. Intermediate lipid product 7α-hydroxy-4-cholesten-3-one (7a-OH-One, Figure 6D) were reduced in EtOH+HFD-fed IL-17RAΔHep mice vs IL-17RAF/F mice. Moreover, production of cholesterol precursors desmosterol and cholestanol was strongly suppressed in livers of these mice (2 fold, Figure 6D), indicating that IL-17 signaling might regulate 7-Dehydrocholesterol Reductase (DHCR7, that mediates conversion of 7-dehydrodesmosterol and 7-dehydrocholesterol into desmosterol and cholestanol, respectively). In support, expression of DHCR7 and DHCR24 (3β-Hydroxysterol Δ24-reductase) mRNA was downregulated in livers of EtOH+HFD-fed IL-17RAΔHep mice (Figure 6E), and in the absence of DHCR7, conversion of 7-dehydrocholesterol was shifted towards Vitamin D325, as shown by increased levels of Vitamin D3 in serum of EtOH+HFD-fed IL-17RAΔHep mice (Figure 6E). Since downregulation of cholesterol synthesis was linked to tumor suppression26, both impaired cholesterol synthesis and increased Vitamin D3 levels might contribute to a growth disadvantage for alcohol-induced IL-17RA-deficient HCC compared to WT HCC.
Furthermore, consistent with low levels of fatty acids, expression of FASN (fatty acid synthase) mRNA was reduced in the livers of EtOH+HFD DEN-challenged IL-17RAΔHep mice (Figure 6E), indicating that IL-17 signaling regulates lipogenesis in steatotic hepatocytes. In support, RNA-Seq analysis revealed that cholesterol and fatty acid synthesis pathways (including DHCR7 mRNA), p53, cytochrome P450, and cytokine/cytokine receptor interaction pathways (including Cxcl1, Cxcl4, Cxcl10, TGF-β3 mRNA) were suppressed in the livers of EtOH+HFD-fed IL-17RAΔHep mice (Figure S8), suggesting that DHCR7 and Cxcl1 might serve as IL-17A target genes.
IL-17RA-deficient steatotic hepatocytes exhibit a defect in Caspase-2-SREBP1/2 signaling.
Next, the potential mechanism by which IL-17 signaling regulates cholesterol and fatty acid synthesis was examined. A critical role of Caspase-2 in SREBP1 and 2 activation has been recently described in experimental models and in patients with NASH16. Here we demonstrate that the TNFR1-Caspase-2-S1P-SREBP1/2 pathway is also activated in livers of ALD patients (but not in healthy individuals, Figure 7A), and in livers of EtOH+HFD-fed DEN-challenged WT mice (Figure 7B, D). In turn, expression of Caspase-2 was completely blocked in EtOH+HFD-fed IL-17RAΔHep mice, and was associated with strong downregulation of SREBP1 and SREBP 2 proteins (Figure 7B, D). Our data indicate that IL-17 signaling may regulate cholesterol and fatty acid synthesis via Caspase-2-dependent activation of SREBP1/2.
Figure 7. EtOH+HFD-fed DEN-challenged IL-17RAΔHep mice exhibit a defect in Caspase 2-dependent activation of SREBP1/2.

(A) Expression of cleaved (C) or full length (F) SREBP-1, SREBP-2, Caspase2, S1P, and TNFR1 was analyzed by Western blotting in livers from patients with no liver injury (Normal), NASH, or ALD.
(B) Expression of cleaved (C) or full length (F) SREBP-1, SREBP-2, Caspase2, TNFR1, and TACE in livers of EtOH+HFD-fed IL-17RAF/F mice and IL-17RAΔHep littermates, representative images of ≥3 independent experiments are shown.
(C) Expression of full length (F) TNFR1 was analyzed by Western blot of livers of EtOH+HFD-fed IL-17RAF/F mice, IL-17RAΔMΦ and IL-17RAΔHSC littermates, representative images of ≥3 independent experiments are shown.
(D) Expression of SREBP-2, Caspase2, TNFR1 (Ab1: Santa Cruz, upper panel; Ab2, Abcam, lower panel), and TACE was analyzed in livers of EtOH+HFD-fed IL-17RAF/F mice and IL-17RAΔHep mice using immunohistochemistry (x60, x4, x10 objectives), positive area was calculated as a percent.
(E) Primary WT and IL-17RA-deficient steatotic hepatocytes from EtOH+HFD-fed IL-17RAF/F mice and IL-17RAΔHep mice were cultured ± Marimastat (10 μM), GW280264X (2 μM), or ± Brefeldin A (BFA, 1 or 5 μg/ml) for 6 h. Expression of full length (F) or cleaved (C) TNFR1 and ARTS-1 was analyzed using Western blotting of cell lysates and supernatants from WT mice and IL-17RAΔHep. BFA inhibited TNFR1 exocytosis in WT and IL-17RA-deficient hepatocytes in a dose-dependent manner.
Statistical analysis was performed using 2-tailed Student’s t test. Data are means ± SD, *p<0.05, **p<0.01, ***p<0.001.
Expression of TNFR1 is downregulated in IL-17RA-deficient steatotic hepatocytes.
TNF/TNFR1 signaling is a critical activator of Caspase 2-SREBP1/2 pathway in steatotic hepatocytes16. Expression of TNFR1 (55 kDa) was dramatically reduced in livers of EtOH+HFD-fed IL-17RAΔHep mice, but not IL-17RAΔMΦ, IL-17RAΔHSCs, or IL-17RAF/F mice, as shown by Western blotting and immunohistochemistry (Figure 7B–C). To gain insight into the underlying mechanism, primary WT and IL-17RA-deficient steatotic murine hepatocytes were further analyzed.
A TACE-independent mechanism is responsible for TNFR1 downregulation in IL-17RA-deficient steatotic hepatocytes.
The potential mechanism by which TNFR1 is downregulated in IL-17RA-deficient hepatocytes was studied in vitro. We confirmed that expression of surface TNFR1 is reduced in IL-17RA-deficient hepatocytes (vs WT hepatocytes), and soluble TNFR1 (28 kDa, proteolytically cleaved from the cell surface, C) and full length TNFR1 (55 kDa, F) are released into the supernatant (Figure 7E). The presence of both full length and soluble forms of TNFR1 in the supernatant of IL-17RA-deficient hepatocytes suggests that the mechanism of TNFR1 can be regulated either by shedding (via TACE-dependent proteolytic cleavage of 28-kDa ectodomain of TNFR1, sTNFR1)27 or via exocytosis (a proteolytic cleavage-independent pathway that enables constitutive release of exosome-like vesicles containing the full length 55-kDa TNFR1 (eTNFR1) into the extracellular compartment)28. The full length 55-kDa eTNFR1 appears to be a predominant form released into the supernatant of IL-17RA-deficient hepatocytes compared to sTNFR1 form. Moreover, release of sTNFR1 and eTNFR1 forms by IL-17RA-deficient hepatocytes was not affected by the TACE inhibitors (Marimastat, or GW280264X) but was diminished by Brefeldin A (BFA, 1 or 5 μg/ml for 6 h), an inhibitor of Golgi-ER traffic and exocytosis29 (Figure 7E). We conclude that exocytosis (rather than shedding) might be responsible for downregulation of surface TNFR1 in steatotic IL-17RA-deficient hepatocytes (vs WT hepatocytes), and release of TNFR1 into the extracellular space. In support, ARTS-1 (aminopeptidase regulator of TNFR1 shedding), that regulates constitutive release of eTNFR1 (55-kDa) via exocytosis30, was upregulated in IL-17RA-deficient steatotic hepatocytes (Figure 7E). Consistent with previous observations30, surface expression of TNFR1 negatively correlated with expression levels of ARTS-1 in WT and IL-17-deficient hepatocytes. We concluded that suppression of Caspase-2-S1P-SREBP1/2 pathway results from increased exocytosis of TNFR1 in IL-17RA-deficient steatotic hepatocytes.
Discussion
Using a model of EtOH+HFD feeding in DEN-challenged and MUP-uPA mice, we demonstrate that alcohol potentially accelerates and enhances tumorigenesis; IL-17 signaling is driving metabolic alterations in NASH- and ALD-injured hepatocytes and increasing the development of HCC. Cell specific deletion of IL-17RA in hepatocytes caused a profound defect in the Caspase-2-SREBP1/2 pathway and DHCR7-dependent cholesterol synthesis, and as a result, EtOH-fed IL-17RAΔHep mice were protected from alcohol-induced steatosis, fibrosis, and HCC. Similar to NASH12, deletion of IL-17RA in Kupffer cells/macrophages ameliorated development of HCC in alcohol-fed IL-17RAΔMΦ mice, indicating that IL-17 signaling regulates pro-tumorigenic/inflammatory and pro-fibrogenic responses in myeloid cells. Therapeutic administration of anti-IL-12/IL-23 Ab strongly suppressed HCC progression in alcohol-fed mice, suggesting that targeting IL-17 signaling may become a novel strategy to treat NASH- and ALD-induced liver injury and HCC.
Using experimental models of HCC, we demonstrate that EtOH+HFD increased HCC by 2 fold versus HFD alone (pair-fed). Development of NASH and ALD-induced HCC correlated with the increased levels of hepatic IL-17A and IL-17RA in patients and mice. Although a role of IL-17A in tumorigenesis has been suggested8,9,12, the insights into the mechanism(s) of IL-17 signaling in the pathogenesis of NASH and ALD-induced HCC are new.
Specifically, the critical role of somatic mutations as drivers of tumorigenesis has been described. Using two experimental models of HCC in EtOH+HFD DEN-challenged12 or MUP-uPA-transgenic17 mice, here we report that WT and IL-17RA-deficient HCC exhibit the same mutational signature of transcribed genes (mutational signatures of non-transcribed DNA was not assessed by this study). Despite the similarities of mutational landscapes5, development of HCC was strongly suppressed in alcohol-injured IL-17RA−/− mice compared to WT mice. Moreover, IL-17RA-deficient HCC exhibited a less malignant phenotype, suggesting that tumor microenvironment plays an important role in NASH- and ALD-induced HCC. In concordance, IL-17 signaling regulates steatosis, inflammation, and fibrosis in NASH- and ALD-injured mice.
Previous studies have outlined the critical role of IL-17 signaling in activation of Kupffer cells/macrophages11,12. In agreement, the current study demonstrates that deletion of IL-17RA in myeloid cells strongly attenuates development of alcohol-induced HCC due to a defect in expression of pro-tumorigenic/inflammatory cytokines in IL-17RAΔMΦ mice. Consistently, tumorigenesis was reduced in all groups of DEN-challenged IL-17RAΔMΦ mice independent of feeding (chow, HFD, EtOH+HFD). Consistently, tumorigenesis was reduced in all groups of DEN-challenged IL-17RAΔMΦ mice independent of feeding (chow, HFD, EtOH+HFD). Furthermore, alcohol-injured IL-17RAΔMΦ mice developed less steatosis due to reduced expression of TNF by IL-17RA-deficient macrophages, and as a result, reduced activation of TNFR1-Caspase 2-SREBP1/2-DHCR7-dependent lipogenesis in alcohol-damaged hepatocytes.
In comparison, IL-17RAΔHep mice (devoid of IL-17RA in hepatocytes) were protected from DEN-induced HCC only when fed with HFD or EtOH+HFD, but not with normal chow, implying that IL-17 signaling in myeloid cells and hepatocytes promotes HCC via distinct mechanisms. Furthermore, we demonstrate that IL-17 signaling in steatotic hepatocytes (but not in normal hepatocytes) promotes hepatocellular damage and HCC.
The role of IL-17 in hepatocytes has not been previously explored, mostly because healthy non-steatotic hepatocytes showed no responses to IL-17A11. Here we demonstrate for the first time that IL-17 signaling in steatotic hepatocytes is critical for the pathogenesis of NASH- and ALD-induced HCC. IL-17RAΔHep mice were protected from NASH- and ALD-induced HCC due to suppression of inflammatory responses and downregulation of cholesterol synthesis in steatotic hepatocytes.
Despite normal levels of IL-6, TNF, IL-1β, and TGF-β1, alcohol-fed IL-17RAΔHep mice developed less fibrosis. To dissect this phenomenon, the gene expression of WT and IL-17RAΔHep livers was profiled. Steatotic (vs normal) hepatocytes serve as a significant source of chemokines, Cxcl1, CCL2, CCL5, Cxcl10, and others. Moreover, both murine and human steatotic (but not normal) hepatocytes responded to IL-17A stimulation by upregulation of IL-6, TNF, TNFR1, and Cxcl1, and strong activation of IL-17 signaling cascades (phospho-NFκB, and phospho-STAT3), and NFκB- and STAT3-mediated pathways. These responses were suppressed in alcohol-fed IL-17RAΔHep mice, proposing that IL-17 signaling is an important activator of pro-inflammatory responses in steatotic, but not control, hepatocytes.
IL-17 signaling plays a critical role in hepatic lipogenesis. In agreement with a previous report31, steatosis was reduced in the whole body IL-17RA−/− mice, hepatocyte- and myeloid cell-specific IL-17RA knockout mice, and was associated with downregulation of hepatic levels of cholesterol, bile acids, and fatty acids. Our study revealed that HCC progression relies on cholesterol synthesis in steatotic hepatocytes, which is regulated by IL-17 signaling. Similar metabolic co-dependency was proposed for brain cancers28, suggesting that cholesterol is required for growth and proliferation of HCC and serves as a source of energy, nutrients, and structure26. On the other hand, excessive accumulation of free cholesterol might produce a lipotoxic effect on hepatocytes, causing their compensatory proliferation, release of tumor promoting cytokines IL-6, TNF, IL-1β1, constitutive activation of NF-κB and JAK2-STAT37 signaling, leading to hepatocyte de-differentiation and further facilitating HCC progression.
Investigation into the mechanism of IL-17A-dependent lipogenesis revealed that a defect in cholesterol and fatty acid synthesis in steatotic IL-17RA-deficient hepatocytes was associated with reduced expression of Caspase-2 and the lipogenic transcription factors SREBP1/2, and subsequent downregulation of cholesterol/fatty acid-producing enzymes DHCR7, DHCR24, and FASN. SREBP1/2 are key regulators of cholesterol and fatty acid synthesis15. Caspase-2-S1P has a critical role in SREBP1/2 activation, and Caspase-2 knockout mice were protected from NASH16. Moreover, ER stress and TNF/TNFR1 signaling trigger activation of Caspase-2-S1P-SREBP1/216. Here we demonstrate that IL-17 signaling regulates TNF/TNFR1-mediated Caspase-2-S1P-SREBP1/2 pathway in experimental model and patients with ALD-induced HCC. Mice deficient of IL-17 signaling in hepatocytes fail to activate Caspase-2-S1P-SREBP1/2, and are protected from alcohol-induced HCC. Moreover, IL-17RA-deficient steatotic hepatocytes exhibit a profound defect in TNFR1 expression. Our data suggest that IL-17 signaling mediates alcohol-induced Caspase 2-S1P-SREBP1/2-dependent lipogenesis via regulation of TNFR1 expression/turnover in steatotic hepatocytes. Hepatocyte-specific deletion of IL-17RA results in strong downregulation of surface TNFR1 expression due to increased exocytosis, an alternative pathway that enables constitutive release of TNFR1-containing exosome-like vesicles (eTNFR1) into the extracellular space.
Although it is unknown, IL-17 signaling might control Caspase 2-S1P-SREBP1/2 pathway directly or via its target genes. Suppression of SREBP1/2 results in downregulation of DHCR7 and FASN in alcohol-injured IL-17RA-deficient hepatocytes. In support, the dhcr7 promoter contains binding sites for SREBP-133. DHCR7 is the sole enzyme that converts 7-dehydrocholesterol and 7-dehydrodesmosterol into cholestanol and desmosterol34, suggesting that DHCR7 may serve as a IL-17A target in steatotic hepatocytes.
DHCR7 plays a critical role in physiological cholesterol maintenance and turnover. Ablation of dhcr7 gene in mice resulted in complete loss of endogenous cholesterol biosynthesis in the liver and brain causing early postnatal lethality35. Furthermore, due to the ability of DHCR7 to interact with DHCR24 and provide a substrate for channeling Vitamin D3 synthesis, the role of DHCR7 in the “cholesterol metabolon” was recently proposed36. Specifically, DHCR7 negatively regulates production of Vitamin D3, which possess strong anti-proliferative and anti-inflammatory properties25. A similar mechanism was observed in alcohol-fed IL-17RAΔHep mice. In the absence of DHCR7, the conversion of 7-dehydrocholesterol into cholesterol was suppressed in IL-17RA-deficient steatotic hepatocytes, thereby enabling its conversion into Vitamin D3 upon exposure to ultraviolet light25.
Vitamin D3 insufficiency is linked to various types of cancer, including patients with ALD-induced HCC37, perhaps through its antioxidant function. Therefore, Vitamin D3 and its analogs are potential therapeutic regimens for HCC. Pharmaceuticals were also being designed to inhibit DHCR7 as a suggested treatment for hepatitis C38. Our study demonstrates that pharmacological targeting of IL-17A/Th17 cells may provide a novel strategy for treatment of alcohol-induced liver fibrosis and HCC. Three classes of IL-17A/Th17 cell inhibitors (anti-IL-17A Ab, RORγt inhibitor, and anti-IL-12/IL-23 Ab) were used in clinical trials, but anti-IL-12/IL-23 Ab caused no/minimal adverse effects in patients24. Therapeutic administration of anti-IL-12/IL-23 Ab to EtOH/DEN-injured mice produced a strong anti-tumorigenic effect and suppressed HCC progression by 70%. Our study provides preclinical evidence for synergistic targeting of IL-12/IL-23/IL-17A for treatment of alcohol-induced liver fibrosis and HCC.
Overall, IL-17A is as a tumor promoting cytokine, which critically regulates alcohol-induced hepatic steatosis, inflammation, fibrosis, and HCC. Genetic deletion or pharmacological blockade of Th17 cells substantially reduces HCC incidences via inhibition of Kupffer cells/macrophage activation, and suppression of cholesterol synthesis in fatty hepatocytes.
Supplementary Material
Highlights.
IL-17A promotes alcohol-induced HCC
IL-17A regulates activation of macrophages
IL-17A facilitates TNF/TNFR-mediated lipogenesis in alcohol-damaged hepatocytes
IL-17A promotes lipogenesis via activation of Caspase2-SP1-SREBP1/2-DHCR7 pathway
IL-17 signaling prevents TNFR1 exocytosis in steatotic hepatocytes
Acknowledgments:
We want to thank Miss Karin Diggle for her excellent management; The Cancer Genome Atlas (TCGA) Research Network Group for sharing the human HCC sequencing data.
Grant support: Supported by the National Institutes of Health R01 DK101737-01A1, U01 AA022614-01A1, R01 DK099205-01A1, P50AA011999 (T.K. and D.A.B.); and Herman Lopata Memorial Hepatitis Postdoctoral ALF Fellowship (J.X.), AI043477; P42 ES010337; U01 AA027681; R01 CA211794; R01 DK120714 (M.K.).
Abbreviations:
- ALD
alcoholic liver disease
- Col1a1
collagen α1(I)
- α-SMA
α-smooth muscle actin
- EtOH
ethanol
- aHSCs
activated Hepatic Stellate Cells
- GSH
antioxidant glutathione
- Kupffer cells
are here referred as liver resident and bone marrow-derived myeloid cells
- DHCR7
7-dehydrocholesterol reductase
- HMGCS1
cytoplasmic hydroxymethylglutaryl-CoA synthase
- SREBP
sterol regulatory element-binding protein
- c-SREBP
cleaved sterol regulatory element-binding protein
- MUP-uPA
major urinary protein-urokinase-type plasminogen activator
Footnotes
Conflicts of Interest: The authors declare they have no conflicts of interest.
References
- 1.Morgan RL, Baack B, Smith BD, et al. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med 2013;158:329–37. [DOI] [PubMed] [Google Scholar]
- 2.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandl K, Hartmann P, Jih LJ, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goossens N, Hoshida Y. Is Hepatocellular Cancer the Same Disease in Alcoholic and Nonalcoholic Fatty Liver Diseases? Gastroenterology 2016;150:1710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB. Understanding the origins of human cancer. Science 2015;350:1175. [DOI] [PubMed] [Google Scholar]
- 6.Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006;6:674–87. [DOI] [PubMed] [Google Scholar]
- 8.Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012;491:254–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, Kim MK, Di Caro G, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014;41:1052–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson MS, Madala SK, Ramalingam TR, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 2010;207:535–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng F, Wang K, Aoyama T, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012;143:765–76 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomes AL, Teijeiro A, Buren S, et al. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2016;30:161–175. [DOI] [PubMed] [Google Scholar]
- 13.Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol 2011;2011:345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity 2004;21:467–76. [DOI] [PubMed] [Google Scholar]
- 15.Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes Dev 2009;23:2578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JY, Garcia-Carbonell R, Yamachika S, et al. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018;175:133–145 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shalapour S, Lin XJ, Bastian IN, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017;551:340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 2001;194:519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weglarz TC, Degen JL, Sandgren EP. Hepatocyte transplantation into diseased mouse liver. Kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. Am J Pathol 2000;157:1963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brandon-Warner E, Walling TL, Schrum LW, et al. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res 2012;36:641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazaro R, Wu R, Lee S, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015;61:129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quehenberger O, Armando AM, Brown AH, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J Lipid Res 2010;51:3299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reich K, Langley RG, Papp KA, et al. A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N Engl J Med 2011;365:1586–96. [DOI] [PubMed] [Google Scholar]
- 25.Boland MR, Tatonetti NP. Investigation of 7-dehydrocholesterol reductase pathway to elucidate off-target prenatal effects of pharmaceuticals: a systematic review. Pharmacogenomics J 2016;16:411–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikonen E Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 2008;9:125–38. [DOI] [PubMed] [Google Scholar]
- 27.Chanthaphavong RS, Loughran PA, Lee TY, et al. A role for cGMP in inducible nitric-oxide synthase (iNOS)-induced tumor necrosis factor (TNF) α-converting enzyme (TACE/ADAM17) activation, translocation, and TNF receptor 1 (TNFR1) shedding in hepatocytes. J Biol Chem 2012;287:35887–35898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawari FI, Rouhani FN, Cui X, et al. Release of full-length 55-kDa TNF receptor 1 in exosome-like vesicles: a mechanism for generation of soluble cytokine receptors. Proc Natl Acad Sci U S A 2004; 101: 1297–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lippincott-Schwartz J1, Yuan LC, Bonifacino JS, et al. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 1989;56:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xinle C, Feras H, Sura A, et al. Identification of ARTS-1 as a novel TNFR1 binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest 2002;110: 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giles DA, Moreno-Fernandez ME, Stankiewicz TE, et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat Med 2017;23:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villa GR, Hulce JJ, Zanca C, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016;30:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JH, Lee JN, Paik YK. Cholesterol biosynthesis from lanosterol. A concerted role for Sp1 and NF-Y-binding sites for sterol-mediated regulation of rat 7-dehydrocholesterol reductase gene expression. J Biol Chem 2001;276:18153–60. [DOI] [PubMed] [Google Scholar]
- 34.Moebius FF, Fitzky BU, Lee JN, et al. Molecular cloning and expression of the human delta7-sterol reductase. Proc Natl Acad Sci U S A 1998;95:1899–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitzky BU, Moebius FF, Asaoka H, et al. 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J Clin Invest 2001;108:905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luu W, Hart-Smith G, Sharpe LJ, et al. The terminal enzymes of cholesterol synthesis, DHCR24 and DHCR7, interact physically and functionally. J Lipid Res 2015;56:888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trepo E, Ouziel R, Pradat P, et al. Marked 25-hydroxyvitamin D deficiency is associated with poor prognosis in patients with alcoholic liver disease. J Hepatol 2013;59:344–50. [DOI] [PubMed] [Google Scholar]
- 38.Rodgers MA, Villareal VA, Schaefer EA, et al. Lipid metabolite profiling identifies desmosterol metabolism as a new antiviral target for hepatitis C virus. J Am Chem Soc 2012;134:6896–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang S, Ma HY, Zhong Z, et al. NADPH Oxidase 1 in liver macrophages promotes inflammation and tumor development in mice. Gastroenterology 2019;156:1156–1172 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.