

Abstract
A multiscale pharmacodynamic model was developed to characterize the receptor-mediated, transcriptomic, and proteomic determinants of corticosteroid (CS) effects on clinically relevant hepatic processes following a single dose of methylprednisolone (MPL) given to adrenalectomized (ADX) rats. The enhancement of tyrosine aminotransferase (TAT) mRNA, protein, and enzyme activity were simultaneously described. Mechanisms related to the effects of MPL on glucose homeostasis, including the regulation of CCAAT-enhancer binding protein-beta (C/EBPβ) and phosphoenolpyruvate carboxykinase (PEPCK) as well as insulin dynamics were evaluated. The MPL-induced suppression of circulating lymphocytes was modeled by coupling its effect on cell trafficking with pharmacogenomic effects on cell apoptosis via the hepatic (STAT3-regulated) acute phase response. Transcriptomic and proteomic time-course profiles measured in steroid-treated rat liver were utilized to model the dynamics of mechanistically relevant gene products, which were linked to associated systemic end-points. While time-courses of TAT mRNA, protein, and activity were well described by transcription-mediated changes, additional post-transcriptional processes were included to explain the lack of correlation between PEPCK mRNA and protein. The immune response model quantitatively discerned the relative roles of cell trafficking versus gene-mediated lymphocyte apoptosis by MPL. This systems pharmacodynamic model provides insights into the contributions of selected molecular events occurring in liver and explores mechanistic hypotheses for the multi-factorial control of clinically relevant pharmacodynamic outcomes.
Keywords: PK/PD systems modeling, Pharmacogenomics, Proteomics, mRNA–protein, Methylprednisolone, Liver
Introduction
The corticosteroids (CS) comprise an important class of anti-inflammatory agents widely used in the treatment of various immunological disorders such as rheumatoid arthritis [1], asthma [2], and systemic lupus erythematosus [3]. Despite a rise in the clinical use of targeted biologic therapies for autoimmune diseases, CS remain a mainstay due to their widespread anti-inflammatory and immunosuppressive effects. However, high dose or chronic CS therapy elicits various adverse metabolic effects in multiple tissues including bone, muscle, adipose, and liver, complicating their safety-to-efficacy profile [4].
While some actions of CS such as immune cell trafficking and adrenal suppression are rapid in onset [5], most effects of CS are mediated through delayed genomic mechanisms. These mechanisms include the steroid binding to its cytosolic receptor which leads to activation, dimerization, and translocation of the drug-receptor complex into the nucleus, thereby regulating message and protein expression by various mechanisms [6], including transactivation and transrepression. Mechanisms involved in CS action from molecular to systems physiology have been characterized. Pharmacokinetic/pharmacodynamic (PK/PD) models to quantitate the transcriptomic actions of CS in multiple tissues have resulted [7–13]. Although highly useful in understanding the mechanisms of pharmacogenomic regulation by CS, transcriptomic approaches are limited in that changes in mRNA expression may not correlate with, and hence fully reflect, drug effects [14, 15]. Since proteins are the direct mediators of drug-induced functional changes [16], incorporation of pharmacoproteomic information into mechanistic models can yield a better understanding of molecular drug actions. In addition, unique insights on regulatory networks can be gained from a ‘multi-omics’ integration [17].
The liver, one of the most sensitive organs to CS exposure, is involved in regulating essential physiological processes including amino acid metabolism, glucose regulation, and immune functioning [18, 19]. Tyrosine aminotransferase (TAT), the major rate-limiting enzyme involved in tyrosine metabolism, is transcriptionally regulated by CS in liver [20]. TAT and other aminotransferase enzymes break down amino acids, providing α-keto acid substrates for hepatic gluconeogenesis. Gluconeogenesis, which involves the rate-limiting enzyme, phosphoenolpyruvate carboxykinase (PEPCK) [21], stimulates hepatic glucose output and consequently, raises blood glucose concentrations. However, systemic glucose homeostasis is controlled not only by CS (via PEPCK) [22] but also other hormonal inputs such as insulin and glucagon [23, 24]. Separate models describing PEPCK mRNA regulation by CS [11] and the glucose-insulin system [23, 25] have been reported. In addition to its effects on energy metabolism, the CS also affect immune function through various mechanisms, including the production of acute phase proteins, which are associated with inflammation, tissue repair, and immune cell activity [26]. While models describing the ‘rapid’ effect of CS on inhibiting the trafficking of immune cells from tissues to the vascular space have been developed [5, 27], a mathematical basis for the ‘delayed’ genomic mechanism of CS action on immune cell apoptosis via the acute phase response (APR) has not been investigated. A model-based integration of both mechanisms is necessary to understand the immunopharmacodynamic actions of CS occurring in vivo.
We previously conducted a study wherein a population of animals received a single dose of the synthetic CS methylprednisolone (MPL), with multiple animals sacrificed at 11 time points following dosing, and livers harvested for proteomic analysis. The development of a robust and reproducible ion-current-based quantitative nano-LC/MS method that enabled assessment of drug-induced dynamic proteomic changes in vivo, and its application in examining the temporal proteomic response of liver from these animals was reported [28]. One of the major applications for this investigation was the development of mechanism-based PK/PD models that would provide a more wholistic depiction of CS actions occurring at the molecular level.
In this study, we developed a mechanism-based pharmacokinetic, pharmacodynamic, pharmacogenomic, and pharmacoproteomic (PK/PD/PG/PP) model, which integrates MPL pharmacokinetics, glucocorticoid receptor (GR) dynamics, expression of mRNA and protein biomarkers, and whole-body physiological measures using quantitative systems approaches to gain expanded insights into the dynamic effects of CS occurring in vivo. Temporal mRNA expression profiling using microarrays and high-throughput proteomic profiling using mass-spectroscopy strategies were integratively employed for developing mechanistic models describing the actions of CS and other intermediary controlling factors on: (i) TAT regulation at the levels of mRNA, protein, and enzyme activity, (ii) hepatic regulation of systemic glucose homeostasis, and (iii) blood lymphocyte modulation via direct (trafficking) and indirect (genomic) mechanisms.
Materials and methods
Experimental
Animal procedures
The data for this study comes from two sets of animal experiments performed in adrenalectomized male Wistar rats (Harlan Sprague–Dawley Inc., Indianapolis, IN), in which the endogenous glucocorticoid production is negligible. Our research protocol adheres to the ‘Principles of Laboratory Animal Care’ (NIH publication 85–23, revised in 1985) and was approved by the University at Buffalo Institutional Animal Care and Use Committee. The first set of experiments consists of 47 animals (group 1), which were given a single intravenous (IV) dose of 50 mg/kg of MPL succinate and sacrificed at different time points between 0 and 72 h. The second set of experiments consists of 55 animals (group 2) given a single intramuscular (IM) dose of 50 mg/kg of the same drug. The liver from these animals was perfused with cold heparinized saline (5 mL of heparin/1 L saline) before sacrifice to remove blood (as blood proteins can hinder accurate proteomic profiling in the tissue) and the animals were sacrificed at different time points between 0 and 66 h. Liver harvested from animals from both experiments were flash frozen in liquid nitrogen and stored at − 80 °C until further analysis. More information about the animal procedures can be obtained from our previous reports [28, 29].
Microarrays
Samples of 100 mg of powdered liver from each animal (group 1) were added to 1 mL of TRIzol Reagent (Invitrogen, Carlsbad, CA). Total RNA extractions were carried out according to manufacturer’s instructions and were further purified using RNeasy columns (QIAGEN, Valencia, CA). RNAs were quantified spectrophotometrically and purity and integrity were assessed by agarose gel electrophoresis. Isolated RNA from each individual liver was used to prepare target according to manufacturer’s protocols. The biotinylated cRNAs were hybridized to 47 individual Affymetrix GeneChips Rat Genome U34A (Affymetrix, Inc.), which contained 7000 probe sets. The high reproducibility of in situ synthesis of oligonucleotide chips allows accurate comparison of signals generated by samples hybridized to separate arrays. More information about this data set can be obtained from Gene Expression Omnibus (GEO) database (GSE490).
Proteomics
Proteins from perfused and flash frozen livers obtained from animals (group 2) were extracted, digested, and analyzed using a nano-LC/MS instrument. The Nano Flow Ultra-High Pressure LC system (nano-UPLC) consisted of a Spark Endurance autosampler (Emmen, Holland) and an ultra-high pressure Eksigent (Dublin, CA) Nano-2D Ultra capillary/nano-LC system, with a LTQ/Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA) used for detection. Protein quantification was based on the area under the curve (AUC) of the ion-current peaks. A more detailed description of our analytical methodology was published [28, 30].
Other measurements
The MPL concentrations were determined in both groups by normal phase high performance liquid chromatography (HPLC) with a lower limit of quantification of 10 ng/mL [31]. The TAT and PEPCK mRNA expression were measured in animals (group 1) using gene-specific TaqMan-based qRT-PCR assays as previously described [9, 11]. The TAT activity in freshly prepared liver homogenate (group 2) was determined by the Diamondstone colorimetric assay and is reported as the change in absorbance at 331 nm over time [32]. The values were normalized for protein concentrations of the crude liver supernatant measured using the Lowry assay [33]. Plasma insulin concentrations were quantified (group 1) using a rat-specific enzyme-linked immunosorbent assay kit (1-2-3 Rat Insulin ELISA, ALPCO Diagnostics, Windham, NH) [23]. Plasma glucose concentrations were measured in rats from group 2 using the glucose oxidase method (Sigma GAGO-20; Sigma-Aldrich, St. Louis, MO). Lymphocytes were counted in whole blood (group 2) using a Cell-Dyne 1700 instrument (Abbott Laboratories, Abbott Park, IL). Total lymphocyte counts were obtained by multiplying total number of leukocytes by the proportion of lymphocytes in total leukocytes.
Mechanism-based mathematical modeling
Figure 1 provides a general schematic for the entire systems pharmacodynamic model for MPL actions in liver as described by Eqs. (1) to (40).
Fig. 1.
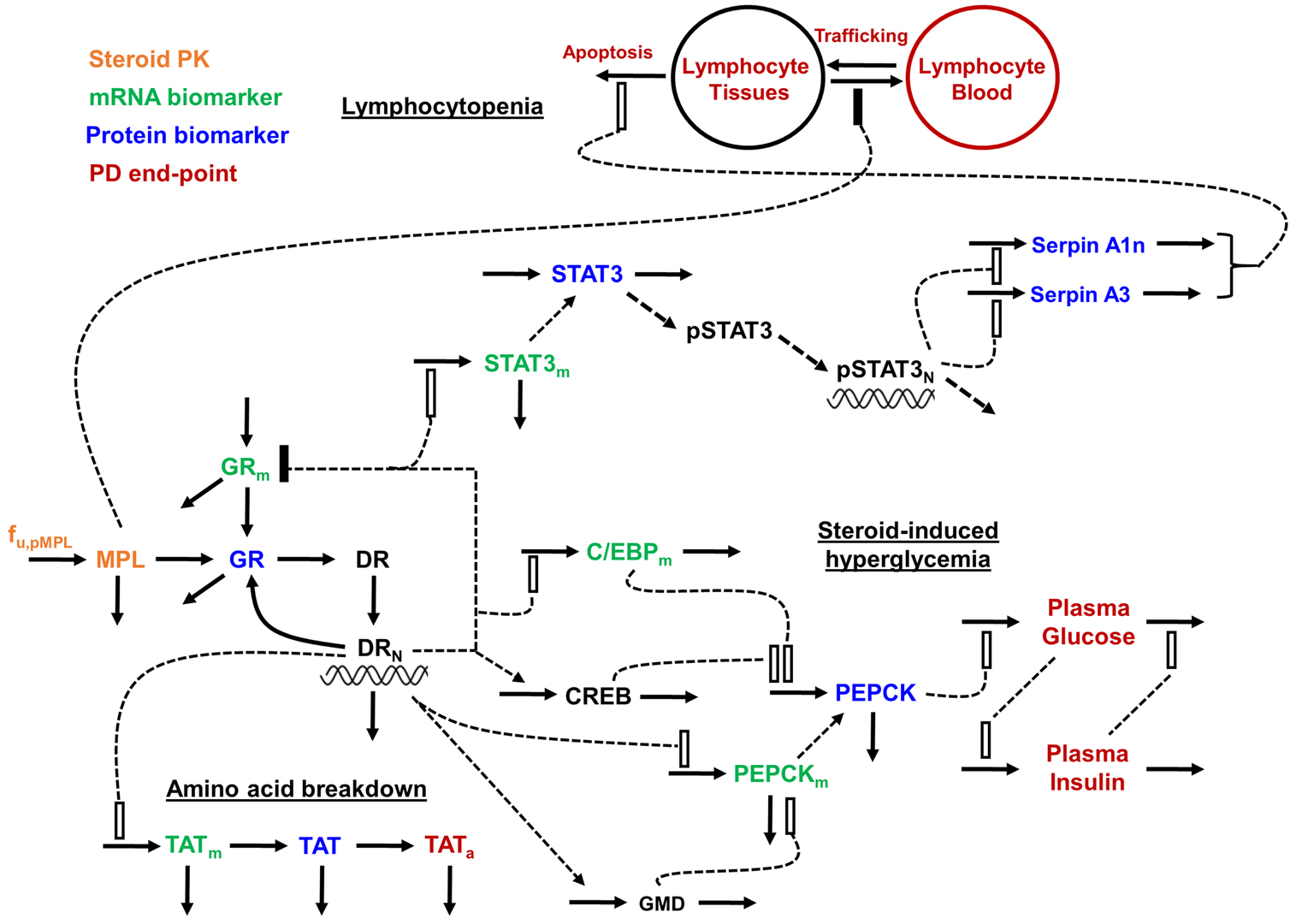
Schematic of systems pharmacodynamic model linking methylprednisolone pharmacokinetics, glucocorticoid receptor dynamics, and hepatic mRNA and protein biomarkers to select efficacy and toxicity end-points in rats. Stimulation is denoted as open rectangles and inhibition is denoted by closed rectangles. Heavy lines reflect turnover while broken lines reflect effects. Symbols are defined in the tables
MPL pharmacokinetics
MPL plasma concentrations following IV and IM dosing were modeled simultaneously. A two-compartment model with linear elimination was used to describe the plasma PK of MPL. In addition, two absorption components from the injection site was used to describe the absorption kinetics of MPL following IM dosing [34]. Equations and initial conditions (IC) describing the model are:
| (1) |
| (2) |
| (3) |
where C and D represent the concentration and dose of MPL in the corresponding plasma (P) and tissue (T) compartments, Fr and (1 – Fr) are fractions of dose absorbed through the absorption pathways described by first-order rate constants ka1 and ka2, CL is clearance from the central compartment, CLD is the distribution clearance, F is the overall bioavailability of MPL after IM injection, and Vc and VT are the central and peripheral volumes of distribution.
Receptor dynamics
The molecular receptor-mediated mechanisms governing CS actions were used for developing the PK/PD/PG/PP model. The dynamics of drug-receptor complex and feedback inhibition of receptor production at the mRNA level was used as previously described [35]. The equations describing the receptor dynamics are:
| (4) |
| (5) |
| (6) |
| (7) |
where symbols represent the free cytosolic glucocorticoid receptor (R), cytosolic drug-receptor complex (DR), nuclear translocated drug-receptor complex (DRn) and receptor mRNA (GRm) concentrations. The ks,GR and kd,GR are first-order rate constants for the production of free receptor from the translation of GR mRNA and the degradation of the free receptor, kon is the second-order rate constant for formation of drug-receptor complex (DR) by the binding of free ligand and receptor in the cytosol, and kt is the first-order rate constant for translocation of the drug-receptor complex from cytosol (DR) into the nucleus (DRn). Part of DRn may recycle back to the cytosol controlled by the rate constant Rf·kre with the remainder degraded by rate constant (1 – Rf)·kre, and ks,GRm and kd,GRm are rate constants for the production and degradation of the receptor mRNA. The IC50,GRm is the concentration of DRn at which the synthesis rate of GR mRNA is reduced to 50% of its baseline.
Equations (4) and (7) yield the following baselines:
| (8) |
| (9) |
where GRm(0) and R(0) are the baseline values of receptor mRNA and free cytosolic GR density. These baseline values were fixed as the mean values obtained in liver from the control animals [9]. Parameters from our previous report [36] were used to simulate receptor dynamics and produce the driving force for genomic CS actions in the present study (Table 1).
Table 1.
Pharmacokinetic parameters for methylprednisolone (MPL) and glucocorticoid receptor (GR) dynamics
| Parameter (units) | Definition | Estimate (% CV) |
|---|---|---|
| MPL pharmacokinetics | ||
| CL (L/h/kg) | Clearance | 2.93 (0.89) |
| CLD (L/h/kg) | Distribution clearance | 2.51 (1.94) |
| Vc (L/kg) | Central volume of distribution | 0.803 (0.97) |
| VT (L/kg) | Peripheral volume of distribution | 0.974 (1.51) |
| F | Bioavailability | 0.2 (0.94) |
| Fr | Fraction absorbed by ka1 | 0.725 (fixed) |
| ka1 (h−1) | Absorption rate constant | 1.82 (2.8) |
| ka2 (h−1) | Absorption rate constant | 0.54 (4.1) |
| Glucocorticoid receptor dynamicsa | ||
| ks,GRm (fmol/g/h) | Synthesis rate constant for GR mRNA | 3.2 |
| kd,GRm (h−1) | Degradation rate constant for GR mRNA | 0.12 |
| ks,GR (nM/h) (fmol/g)−1 | Synthesis rate constant for receptor | 0.84 |
| IC50,GRm (nM) | DRn for 50% inhibition of GR mRNA synthesis | 123.7 |
| kd,GR (h−1) | Degradation rate constant for receptor | 0.04 |
| kon (nM−1 h−1) | Association rate constant | 0.019 |
| fmpl | Unbound fraction of MPL | 0.23 |
| kre (h−1) | DRn loss rate constant | 0.402 |
| Rf | Fraction recycled | 0.69 |
| kT (h−1) | Translocation rate constant | 58.2 |
| GRm (0) (fmol/g) | GR mRNA initial concentration | 25.8 |
| GR(0) (nM) | Free cytosolic receptor initial concentration | 540.7 |
| DR(0) (nM) | Drug-receptor complex initial concentration | 0 |
| DRn(0) (nM) | Nuclear complex initial concentration | 0 |
Parameter values obtained from Hazra et al. [36]
TAT dynamics
TAT is a highly studied biomarker of CS in liver. The CS are known to regulate hepatic TAT expression at the transcriptional level via transactivation, where the nuclear drug-receptor complex binds multiple glucocorticoid-response elements (GREs) located in the 5′-promoter region of the TAT gene, and alters the rate of synthesis of TAT mRNA. In the proposed model, drug-receptor complex in the nucleus (DRn) stimulates TAT mRNA expression (TATm), with the mRNA production and degradation controlled by rate constants ks,TATm and kd,TATm and stimulation by constant . The changes in TAT mRNA expression are translated to changes in TAT protein (TAT) and subsequently to TAT activity (TATa). The ks,TAT and kd,TAT and ks,TATa and kd,TATa represent production and degradation rate constants of TAT protein and activity. The γ1 parameter is an amplification factor representing the translational efficiency of TAT mRNA to protein. Similarly, γ2 represents the post-translational efficiency of TAT protein to TAT activity.
| (10) |
| (11) |
| (12) |
At time 0, the TAT pathway was assumed to be at its physiological steady-state, and Eqs. (10) to (12) yield the following baselines:
| (13) |
| (14) |
| (15) |
where TATm(0), TAT(0); and TATa(0) are the baseline values TAT mRNA, protein, and activity. These baseline values were estimated during model fittings.
PEPCK–insulin–glucose dynamics
The liver is a primary metabolic target of CS, where hepatic glucose output (HGO) is governed by glycogenolysis and gluconeogenesis. The CS strongly influence gluconeogenesis by affecting the availability of gluconeogenic precursors and by enhancing the expression of key gluconeogenic enzymes, including phosphoenolpyruvate carboxykinase (PEPCK), which in turn increases HGO. The molecular mechanisms of PEPCK regulation are, however, complex and only partially resolved. In addition to direct transcriptional regulation of PEPCK by CS via two complex GREs located within the PCK promoter region [37], the CCAAT/enhancer-binding protein-beta (C/EBPβ) as well as the cyclic AMP (cAMP) response element binding protein (CREB) regulate PEPCK expression [38–41]. Of interest, CS stimulate the expression of CREB and the C/EBP isoforms in vitro [42, 43], suggesting intertwined mechanisms of PEPCK regulation [44]. Added complexities in glucose control stem from the glucose-insulin feedback system [25], where increased insulin production from pancreatic β-cells in response to hyperglycemia counterbalances elevated plasma glucose, by promoting its utilization in peripheral tissues such as skeletal muscle. Based upon these biological mechanisms, a model consisting of single and tandem indirect responses, simple transduction, and feedback processes was developed to describe the receptor-mediated, genomic, and proteomic determinants of MPL (and insulin) effects on hepatic glucose regulation.
Enhancement of C/EBPβ mRNA
The CS causes an enhancement of C/EBPβ mRNA by stimulating its synthesis rate driven by the nuclear complex. The following equation describes the action of MPL on C/EBPβ mRNA synthesis:
| (16) |
The C/EBPβm is the C/EBPβ message level in liver expressed as fold-change compared to control animals. The DRn stimulates the expression of C/EBPβ mRNA, with the mRNA production and degradation controlled by rate constants ks,C/EBPβm and kd,C/EBPβm and stimulation by constant .
The message level was assumed to be at steady-state at time 0 (control animals), with Eq. (16) yielding the baseline:
| (17) |
where C/EBPβm(0) is the baseline value of normalized C/EBPβ mRNA. The obtained parameters were fixed to produce C/EBPβ mRNA dynamics in the following data analysis.
Enhancement of PEPCK mRNA and protein
The regulation of PEPCK mRNA by CS was modeled as described by Jin et al. [11] using Eqs. (18) and (19). The synthesis rate of PEPCK protein was assumed to be under the control of its message expression (PEPCKm), C/EBPβ (C/EBPβm), as well as a DRn—induced biosignal representing CREB. The equations are:
| (18) |
| (19) |
| (20) |
| (21) |
where GMD is the concentration of a presumed secondary biosignal mediated by the glucocorticoid-mediated mRNA decay (GMD) mechanism [45, 46], responsible for the stimulation of PEPCK mRNA degradation (kd,PEPCKm) by constant . A linear transduction model [47] was used to describe this biosignal, which was generated from DRn at the first-order rate kGMD. The DRn stimulates the PEPCK message synthesis rate (ks,PEPCKm) by constant . The PEPCKm is expressed as femtomoles per gram of liver. The assumed time-profile of CREB was driven by DRn at the first-order rate kCREB, which was fixed to a value of 2.77 h−1 to denote the in vitro half-life of activated CREB, which is reported to be 10–20 min [48]. The PEPCK protein was translated from its mRNA at the first-order rate ks,PEPCKm with efficiency factor γ and degraded at the first-order rate kd,PEPCK. The γ indicates that a variable number of copies of protein could be synthesized from a single mRNA transcript. The stimulation of translation by C/EBPβ and the CREB signal at constants and were assumed to be the major modes of molecular regulation of PEPCK at the level of protein expression. The fold-change of C/EBPβ mRNA from C/EBPβm (0) is used to drive its stimulation effect.
At time zero, the system was assumed to be at steady-state, with Eqs. (19) and (21) yielding the following baselines:
| (22) |
| (23) |
where PEPCKm(0) is the baseline value of PEPCK mRNA, which was fixed to 209 fmol/g liver as measured in control animals in our previous report [11], and PEPCK(0) is the baseline-normalized value of PEPCK protein. The initial conditions of Eqs. (18) and (20) were fixed as 0. Equations (18) to (21) were modeled simultaneously and the obtained parameters were fixed in the following data analysis.
Glucose–insulin dynamics
Based on the mechanisms described above for systemic glucose control, the gluconeogenic effect of MPL (i.e. PEPCK enhancement) was linked to the observed elevation in plasma glucose concentrations. In addition, the hyper-glycemic effect was assumed to affect the rate of insulin synthesis, which would in turn alter the dynamics of glucose by stimulating its degradation rate. The model structure describing glucose-insulin interplay was adapted from an established disease progression model [23]. The following equations describe MPL/glucose/insulin inter-regulation:
| (24) |
| (25) |
where Gluc and Ins are plasma glucose and insulin concentrations, which are controlled by their zero-order production (ks,Gluc and ks,Ins) and first-order removal rates (kd,Gluc and kd,Ins). The fold-change in PEPCK protein expression from its baseline (PEPCK – PEPCK(0)) is assumed to stimulate ks,Gluc by the constant . The change in plasma glucose concentrations from baseline (Gluc – Gluc(0)) in turn stimulates ks,Ins by the constant . Finally, the change in plasma insulin concentrations from baseline (Ins – Ins(0)) stimulates kd,Gluc by the constant .
At time zero, the system was assumed to be at its physiological steady-state, with Eqs. (24) and (25) yielding:
| (26) |
| (27) |
where Gluc(0) was fixed to a mean value of 148 mg/dL as measured in control animals (group 2), and Ins(0) was fixed to a mean value of 0.24 ng/mL as reported from measurements in untreated control rats [23].
Acute phase response and lymphocyte dynamics
The CS have profound effects on circulating immunocompetent cells and are therefore employed for their immune-modulating properties. A well-studied mechanism causing a suppression in circulating leukocytes is the redistribution of these cells out of the vasculature and into the extravascular tissue space [49–51]. Another important mechanism contributing to the transient lymphopenia by CS in rodents involves an induction of the apoptotic machinery in thymocytes, T cells, B cells, macrophages, mature dendritic cells, eosinophils, and natural killer cells [52], which requires functional GR and gene expression [53]. The hepatic APR plays a crucial role in the protection of tissues in response to potentially stressful or toxic stimuli, including CS exposure [26]. The CS stimulate the expression of acute phase proteins, including the serine anti-proteinases (Serpins), alpha-2-macroglobulin, and signal transducer and activator of transcription-3 (STAT3) [16]. Serpina1 (α1-antitrypsin) and Serpina3n (α1-antichymotrypsin) expression is up-regulated in response to hepatic stress in a STAT3-dependent manner [54, 55], and are correlated with the APR-induced apoptotic response. A model consisting of indirect responses, linear transduction, irreversible cell loss, and cell distribution processes was developed to recapitulate these mechanisms, which describe the direct (rapid) and the indirect (delayed) determinants of MPL on lymphocyte suppression.
Dynamics of STAT3
The model assumes that CS stimulate the synthesis rate of STAT3 mRNA (STAT3m). The synthesis rate of STAT3 protein was assumed to be under the control of STAT3m. The de novo synthesized STAT3 protein is then phosphorylated and translocates to the nucleus before exerting its PD effects, which were represented as rapid, linear transduction processes. The model equations are:
| (28) |
| (29) |
| (30) |
| (31) |
where the STAT3 mRNA synthesis and degradation are controlled by rate constants ks,STAT3m and kd,STAT3m and stimulation by constant . The ks,STAT3 and kd,STAT3 are production and degradation rate constants of STAT3 protein (STAT3). The γ3 parameter is an amplification factor representing the translational efficiency of STAT3 mRNA to protein. The presumed dynamics of phosphorylated STAT3 (pSTAT3) and nuclear pSTAT3 (pSTAT3N) are driven by STAT3 at the first-order rate kSTAT3, which was fixed to a value of 4.16 h−1 to denote the degradation half-life of pSTAT3, which is reported as approximately 10 min in vitro [56].
At time zero, the system was assumed to be at steady-state, with Eqs. (28) and (29) yielding:
| (32) |
| (33) |
where STAT3m(0) and STAT3(0) are baseline-normalized values of STAT3 mRNA and protein. The initial conditions of Eqs. (30) and (31) were fixed as 0.
Dynamics of serpins A1 and A3n
The enhancement of Serpins was assumed to be driven by pSTAT3N as follows:
| (34) |
| (35) |
where Serpina1 and Serpina3n synthesis (ks,Serpina1 and ks,Serpina3n) and degradation (kd,Serpina1 and kd,Serpina3n) are controlled by rate constants, and λ1 and λ2 are power coefficients for the effects of pSTAT3N on Sepina1 and Serpina3n.
At time zero, the system was assumed to be at steady-state, with Eqs. (34) and (35) yielding:
| (36) |
| (37) |
where Serpina1(0) and Serpina3n(0) are baseline-normalized values of protein.
Lymphocytopenia
The following equations were applied in the modeling of trafficking and tissue apoptosis of lymphocytes:
| (38) |
| (39) |
where movement of lymphocytes between blood (LYMB) and tissues (LYMT) are governed by first-order rate constants kin (from tissue to blood) and kout (from blood to tissue). The lymphocyte baselines in blood (LYMB (0)) and tissues (LYMT (0)) were fixed as 4.8 and 475.2 K/μL. The IC50 is the drug concentration that inhibits kin by 50% and kapoptosis is a second-order rate constant controlling LYMT apoptosis. The variable ∑Serpin is the influence of the Serpins on lymphocyte apoptosis defined as:
| (40) |
where β1 and β2 are the intrinsic activities of each individual Serpin protein on the apoptosis process.
Data analysis
Model fitting and nonlinear regression analysis
The ADAPT 5 software was used for all data fitting and simulation of model equations [57]. Parameter values employed in the final model were either fixed based on literature sources or estimated from the experimental data. The model was developed in three sequential phases: (i) assembly of the mechanistic model, (ii) fitting of molecular biomarkers, and (iii) fitting of systemic end-points. The maximum likelihood estimation (MLE) algorithm was applied for fitting the data. Replicate data at each time point from animals in each experiment were naïve-pooled. Separate variance models were specified for PK and PD outputs using:
| (41) |
| (42) |
where Vi is the variance of the ith data point, σ1 and σ2 are the variance parameters, and Yi is the model predicted concentration or response. Variance parameters σ1 and σ2 were estimated along with model parameters during fittings. The goodness-of-fit was assessed by system convergence, visual inspection of the fitted curves, objective function values such as akaike information criterion (AIC), improved likelihood, examination of residuals, and precision (CV%) of the estimated parameters.
Model simulation and evaluation
The developed PK/PD/PG/PP model in Fig. 1 was applied to simulate and predict the dynamics of select pharmacodynamic responses under different scenarios, including different doses and dosing regimens of MPL in ADX rats. The final parameter estimates obtained from fitting experimental data in the current study were fixed and applied in the simulations. All simulations were performed using ADAPT 5. The simulated responses were compared to previously published data from the literature [23, 58]. In order to understand the relative contribution of a particular pathway and/or parameter to its associated response, the parameter value was either set to zero (i.e. process “turned-off”) or varied 0.1×, 2×, 5×, and 10× from the estimated value. The extent of change in the associated response was simulated.
Results
Pharmacokinetics of MPL and glucocorticoid receptor dynamics
Plasma MPL shows a bi-exponential decline after IV administration which was modeled with a two-compartment model with linear elimination. However, with IM administration, the PK profile showed peak plasma concentrations at the earliest time point measured and an increased half-life. To capture this behavior, two first-order absorption rate constants were used to describe the absorption of the drug from the site of injection. The PK profiles of both the IV and IM doses were modeled simultaneously and the model was able to adequately describe the profiles (Fig. 2). The CL (3.0 L/h/kg) and Vc (0.803 L/kg) values were similar to previously reported values (CL 3.48 L/h/kg and Vc 0.73 L/kg) in ADX rats after single doses of MPL [59]. Parameter estimates for MPL PK are provided in Table 1. The receptor mRNA, free cytosolic receptors, and nuclear drug-receptor complex concentrations were simulated following IM dosing of MPL using parameter values obtained from a previous report [36] (Fig. S1). Parameter values for receptor dynamics are provided in Table 1.
Fig. 2.
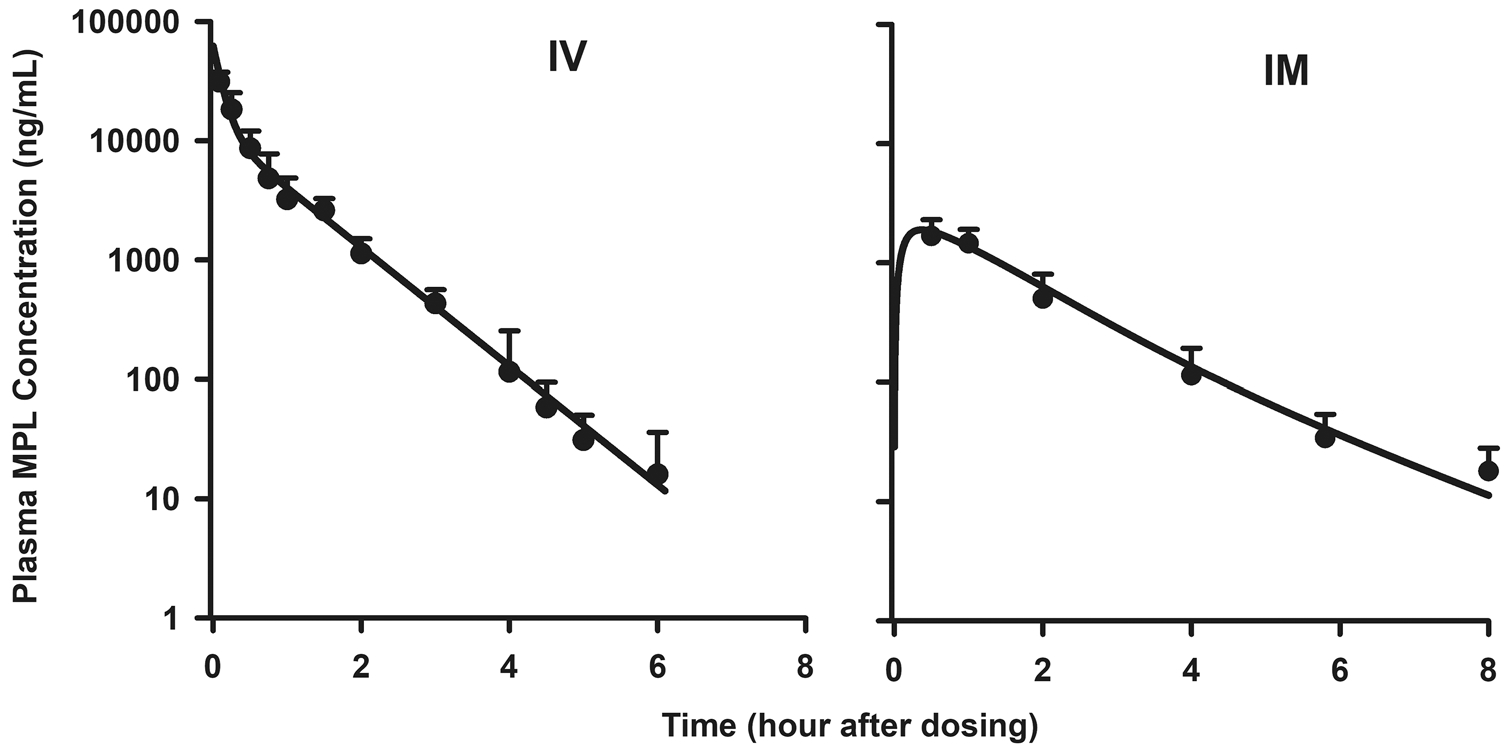
Pharmacokinetics of MPL in adrenalectomized male rats. Simultaneous fitting results of MPL pharmacokinetics in plasma (Eqs. 1 to 3) upon administration of 50 mg/kg intravenous (IV) injection of MPL (left panel) and following 50 mg/kg intramuscular (IM) injection (right panel). Solid lines represent model fittings, circles are means, and error bars are 1 standard deviation (SD) (n = 3–6 rats per point)
TAT dynamics
The temporal profiles of TAT mRNA, protein, and activity were well-captured by the model as shown in Fig. 3. Parameter estimates are listed in Table 2. TAT message expression began to increase about 1 h after MPL, peaked at 5 h showing an approximate two-fold increase, and returned to baseline between 18 and 30 h after dosing. The temporal profile of TAT mRNA was well-captured by the model, with an estimated SDRn,TATm of 0.002 nM−1 (48.1% CV) and kdeg,TATm of 0.22 h−1 (66.6% CV). The estimated degradation rate-constant for TAT mRNA was in close agreement with the value reported from normal rats (0.232 h−1) [9]. The expression of TAT protein followed a similar enhancement profile as its mRNA with a further onset delay of 0.5–1 h. Furthermore, TAT protein showed a six- to seven-fold increase at peak that occurred around 8 h, which was delayed compared to time of peak TAT mRNA. The estimated kdeg,TAT value of 0.29 h−1 (73.6% CV) translates to an in vivo half-life of 2.4 h, which is within the typical range reported in the literature (1.5–5 h) [60]. TAT activity showed an onset delay of about two hours, followed by a sharp rise to about 0.9/mg protein, and a decline to baseline by 24 h which paralleled the profile of TAT protein.
Fig. 3.
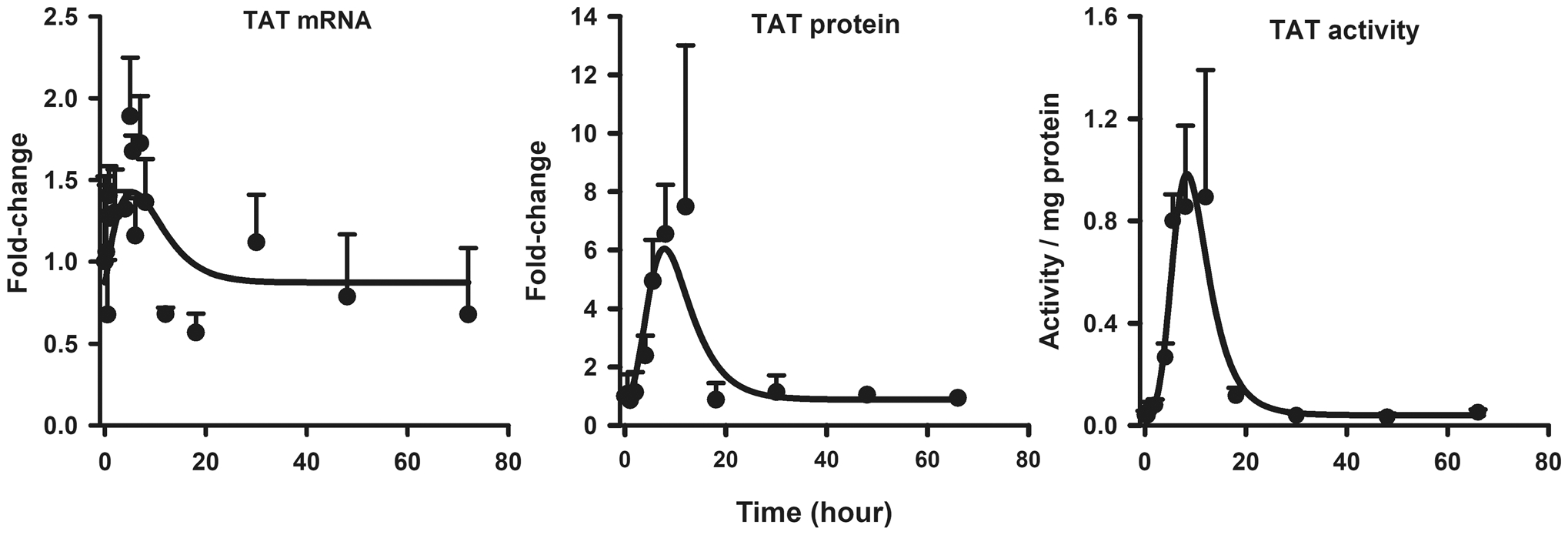
Hepatic TAT mRNA (left panel), TAT protein (center panel), and TAT activity (right panel) dynamics after 50 mg/kg MPL. The circles and bars are means + 1 SD and solid lines depict model (Fig. 1) fitting results based on the parameters shown in Tables 1 and 2
Table 2.
Parameter estimates for tyrosine aminotransferase (TAT) dynamics
| Parameter (units) | Definition | Estimate (% CV) |
|---|---|---|
| Tyrosine aminotransferase dynamics | ||
| kd,TATm (h−1) | Degradation rate constant for TAT mRNA | 0.22 (66.6) |
| Stimulation constant for TAT mRNA synthesis | 0.002 (48.1) | |
| kd,TAT (h−1) | Degradation rate constant for TAT protein | 0.29 (73.6) |
| γ1 | Amplification factor for TAT protein | 4.5 (34.9) |
| kd,TAT (h−1) | Degradation rate constant for TAT activity | 2.0 (96.0) |
| γ2 | Amplification factor for TAT activity | 1.67 (11.8) |
| TATm (0) | Baseline TAT mRNA | 0.87 (12.0) |
| TAT (0) | Baseline TAT protein | 0.88 (12.8) |
| TATa (0) (activity/mg protein) | Baseline TAT activity | 0.041 (7.3) |
PEPCK-insulin-glucose dynamics
The temporal profiles of C/EBPβ mRNA, PEPCK mRNA and protein, glucose, and insulin were well-described by the model as shown in Fig. 4. Parameter estimates are provided in Table 3. The mRNA expression for the transcription factor C/EBPβ showed a sharp rise following MPL, peaking at 1–2 h after dosing (~ 3.5-fold) followed by a return to baseline by 18 h. The sharp enhancement of C/EBPβ by MPL can be attributed to its rapid loss-rate constant, which was estimated as 3.2 h−1 (35.0% CV). PEPCK mRNA also showed a rapid increase by 0.75 h. This early peak of 794 ± 112 fmol/g liver was followed by an immediate decline below baseline after 4 h. PEPCK protein, however, showed a rather different profile—expression increased about three-fold from baseline by 5.5 h and returned to baseline by 12 h. The model equations for the PEPCK regulatory network (Eq. 18 to 21), as fitted simultaneously to the PEPCK mRNA and protein time-courses, satisfactorily described the complex dynamics of PEPCK. The estimated kdeg,pck value of 0.3 h−1 (41.9% CV) translating to a protein half-life of about 2.3 h, is shorter compared to a previously reported value (5–6 h) [61]. The stimulatory efficiency of PEPCK protein via the biosignal representing CREB (SCREB,pck) was estimated as 0.004 nM−1 (32.9% CV) whereas SC/EBP,pck was fixed as 0.001. Plasma glucose concentrations rose from a baseline of about 148 ± 28 to 208 ± 34 mg/dL by 8 h after MPL, followed by a return to baseline by 18 h. The profile of PEPCK protein was employed to stimulate the synthesis rate of plasma glucose concentrations with an estimated SPCK,glu of 0.33 (17.8% CV). Glucose-insulin interplay was adapted from an established disease progression model [23]. The change in plasma glucose from baseline stimulated the synthesis rate of insulin with an Sglu,Ins value of 0.5 (47.5% CV), while the change of insulin concentrations from its baseline stimulated the removal rate of glucose with an SIns,glu value of 0.043 (77.2% CV).
Fig. 4.
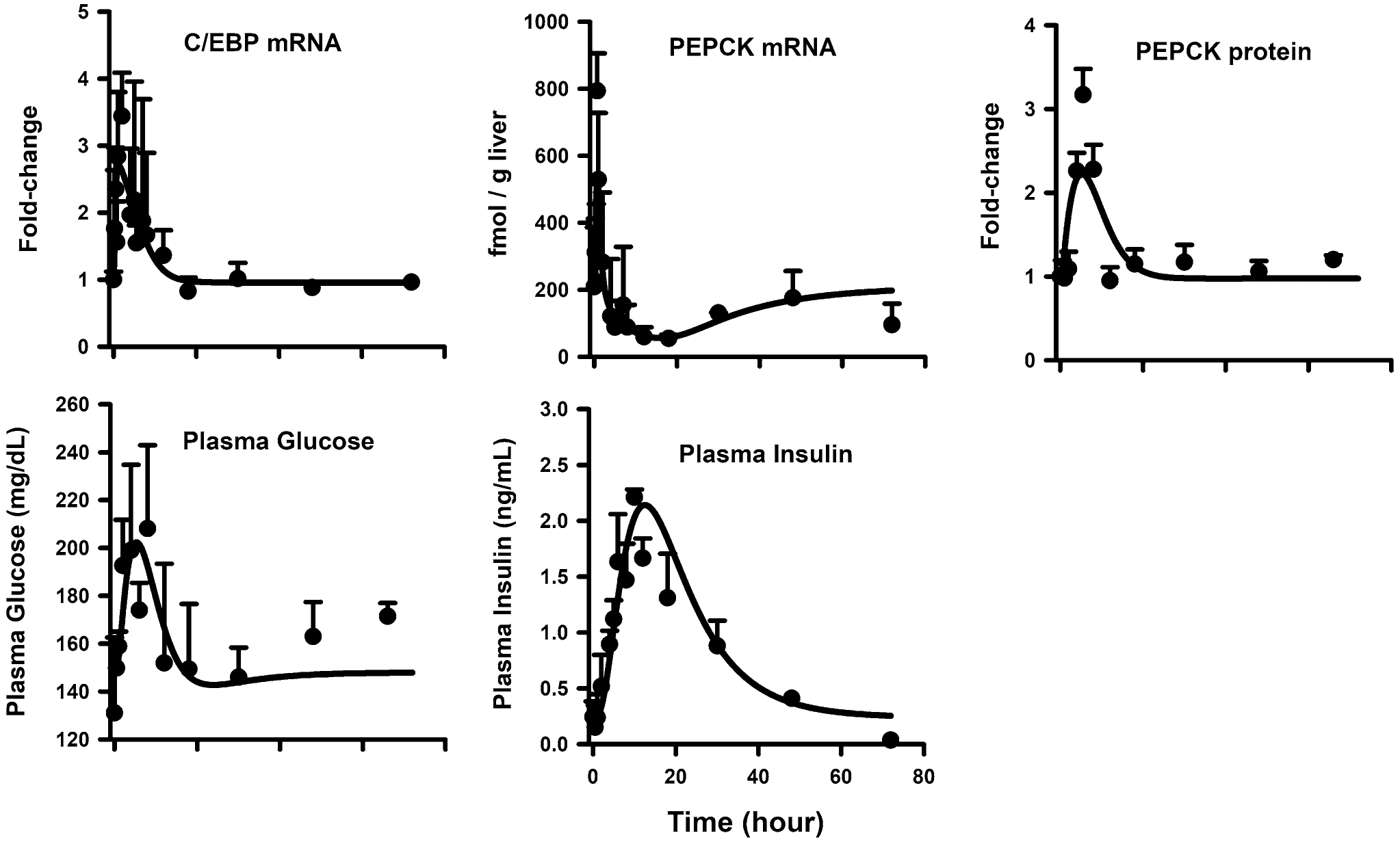
Hepatic C/EBP-β mRNA (top left panel), PEPCK mRNA (middle left panel), PEPCK protein (middle right panel), plasma glucose (bottom left panel), and plasma insulin (bottom right panel) dynamics after 50 mg/kg MPL. The circles and bars are means + 1 SD and solid lines depict model (Fig. 1) fitting results based on the parameters shown in Tables 1 and 3
Table 3.
Parameter estimates for phosphoenolpyruvate carboxykinase (PEPCK) regulation and glucose homeostasis
| Parameter (units) | Definition | Estimate (% CV) |
|---|---|---|
| CCAAT enhancer binding protein-β mRNA dynamics | ||
| kd,C/EBP-βm (h−1) | Degradation rate constant for C/EBP-β mRNA | 3.2 (35.0) |
| Stimulation constant for C/EBP-β mRNA synthesis | 0.0038 (13.9) | |
| C/EBP-βm (0) | Baseline C/EBP-β mRNA | 0.96 (3.9) |
| Phosphoenolpyruvate carboxykinase mRNA and Protein Dynamics | ||
| kGMD (h−1) | First-order rate constant for GMD signal | 0.42 (20.3) |
| kd,PEPCKm (h−1) | Degradation rate constant for PEPCK mRNA | 0.053 (53.7) |
| Stimulation constant for PEPCK mRNA synthesis | 0.17 (63.1) | |
| Stimulation constant for PEPCK mRNA degradation | 0.27 (59.8) | |
| kd,PEPCK (h−1) | Degradation rate constant for PEPCK protein | 0.3 (41.9) |
| γ | Power coefficient for PEPCK translation from mRNA | 0.0048 (fixed) |
| kCREB (h−1) | First-order rate constant for CREB signal | 2.77 (fixed) |
| Stimulation constant for PEPCK protein - CREB | 0.004 (32.9) | |
| Stimulation constant for PEPCK protein - C/EBP-β | 0.001 (fixed) | |
| PEPCKm (0) (fmol/g) | Baseline PEPCK mRNA | 209 |
| PEPCK (0) | Baseline PEPCK protein | 0.98 (8.6) |
| Plasma glucose and insulin dynamics | ||
| kd,Gluc (h−1) | Degradation rate constant for glucose | 1.48 (fixed) |
| Stimulation constant for glucose synthesis by PEPCK | 0.33 (17.8) | |
| Insulin sensitivity | 0.043 (77.2) | |
| Kd,Ins (h−1) | Degradation rate constant for insulin | 0.049 (55.3) |
| Glucose sensitivity | 0.5 (47.5) | |
| Glucose (0) (mg/dL) | Baseline plasma glucose concentration | 148 |
| Insulin (0) (ng/mL) | Baseline plasma insulin concentration | 0.24 |
Acute phase response and lymphocyte dynamics
With their anti-inflammatory properties, MPL affects immune cell trafficking and also induces the apoptosis-mediated death of immune cells via the acute phase response. The STAT3 transcription factor plays a central role in initiating the downstream activation of acute phase response proteins such as the Serpins. The time-course profiles of STAT3 mRNA and protein, Serpina1, Serpina3n, and blood lymphocyte counts along with the model predictions are shown in Fig. 5. Parameter estimates are listed in Table 4. After MPL, both STAT3 mRNA and protein displayed robust time-dependent increases in expression. STAT3 mRNA was up-regulated (~ 2.5-fold) by 5 h and returned to baseline by 18 h, while STAT3 was enhanced to peak (~ 3.3-fold) by 8 h and showed a prolonged response before returning to baseline beyond 30 h. Consistent with the observed time profiles, the degradation rate-constants for STAT3 mRNA and protein were estimated as 0.33 h−1 (24.0% CV) and 0.12 h−1 (22.3% CV). The estimated in vivo STAT3 half-life of about 6 h (0.693/kdeg,STAT3) is in reasonable agreement with that obtained from pulse-chase experiments in vitro using COS-7 cells (8 h) [62]. While both Serpins peaked between 8 and 12 h, Serpina1 protein expression was enhanced to a lesser extent compared to Serpina3n (2.5-fold vs. 5-fold). The power coefficients λ1 and λ2 for pSTAT3N effects on Serpina3n and Serpina1 were 1.5 (17.5% CV) and 0.6 (20.9% CV), accordingly. The model tended to over-predict the peak and return of Serpina3n expression. Blood lymphocyte counts showed a baseline around 5.0 K/μL in control animals. The cell counts in blood showed a decrease after MPL dosing reaching a nadir of around 0.8 K/μL (five-fold decrease) at 8 h after dosing. The cell counts attained a new lower steady-state baseline value around 3.5 K/μL. The proposed cell trafficking-apoptosis model captured both the initial fall in blood lymphocyte counts and the return back to the new steady-state reasonably well. The kout of 0.23 h−1 (5.0% CV) suggests that the half-life of blood lymphocyte movement from blood to tissues is about 3 h. The value of kin is much lower than kout primarily due to the very large peripheral pool compared to blood. Due to overparameterization, the IC50,MPL inhibiting the movement of cells from tissues to blood was fixed to 6.15 ng/mL based on literature estimates [58]. For the same reason, the intrinsic activity parameters β1 and β2 controlling Serpin-mediated apoptosis of tissue lymphocytes were fixed to 1 (i.e. equal contributions assumed).
Fig. 5.
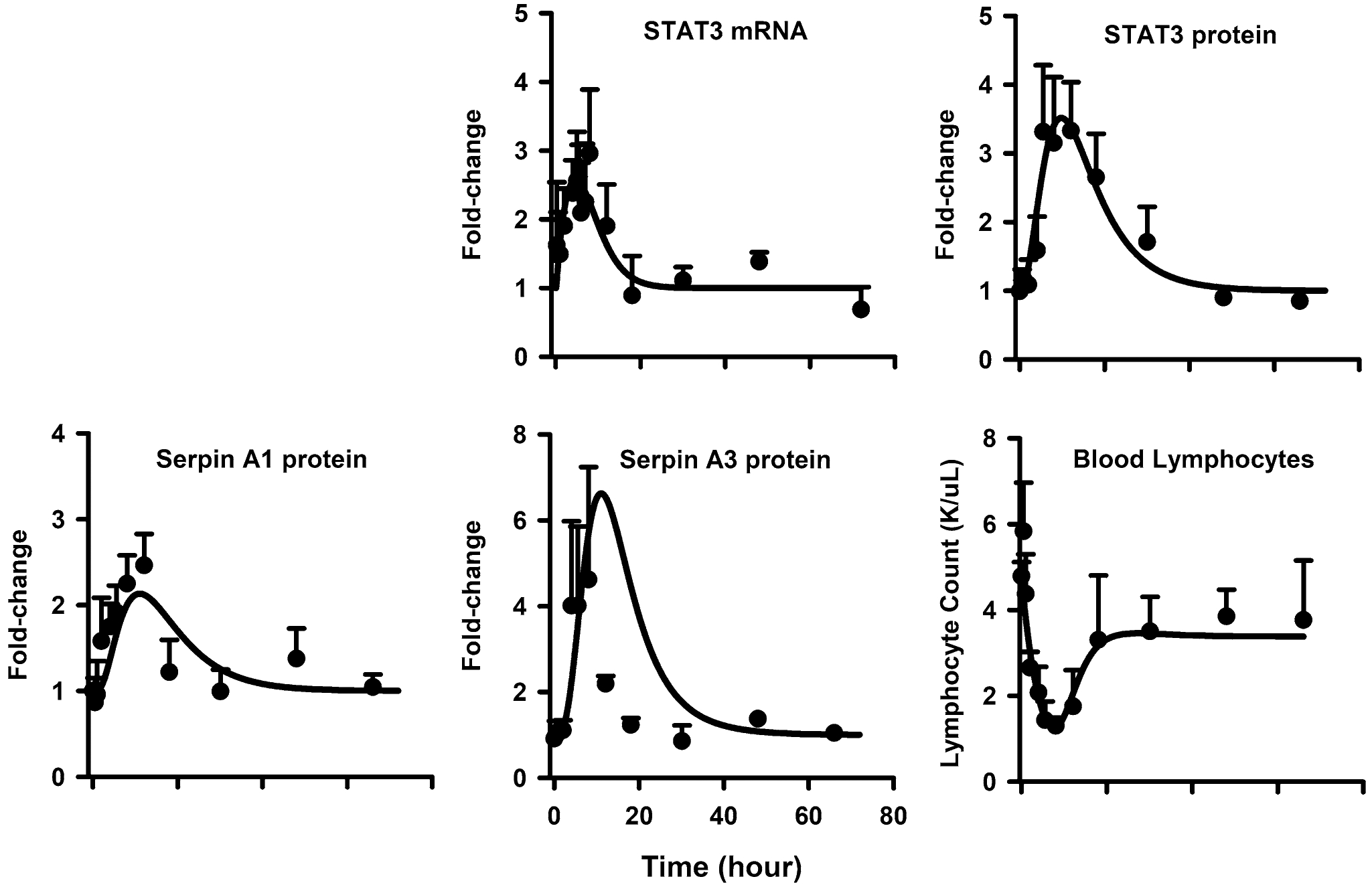
Hepatic STAT3 mRNA (top left panel), STAT3 protein (top right panel), Serpin A1 protein (middle left panel), Serpin A3n (middle right panel), and blood lymphocyte (bottom left panel) dynamics after 50 mg/kg MPL. The circles and bars are means + 1 SD and solid lines depict model (Fig. 1) fitting results based on the parameters shown in Tables 1 and 4
Table 4.
Parameter estimates for acute phase response dynamics and blood lymphocyte count
| Parameter (units) | Definition | Estimate (% CV) |
|---|---|---|
| Signal transducer and activator of transcription-3 mRNA and protein dynamics | ||
| kd,STAT3m (h−1) | Degradation rate constant for STAT3 mRNA | 0.33 (24.0) |
| Stimulation constant for STAT3 mRNA synthesis | 0.005 (10.9) | |
| kd,STAT3 (h−1) | Degradation rate constant for STAT3 protein | 0.12 (22.3) |
| γSTAT3 | Amplification factor for STAT3 protein | 1.9 (9.3) |
| kSTAT3 (h−1) | Phosphorylation/translocation rate constant | 4.16 (fixed) |
| STAT3m (0) | Baseline STAT3 mRNA | 1 (fixed) |
| STAT3 (0) | Baseline STAT3 protein | 1 (fixed) |
| Serpin A1n and serpin A3 protein dynamics | ||
| kd,SerpinA3n (h−1) | Degradation rate constant for serpin A3n | 1.4 (fixed) |
| λ1 | Power coefficient for pSTAT3N effect on serpin A3n | 1.5 (17.5) |
| kd,SerpinA1 (h−1) | Degradation rate constant for serpin A1 | 1.4 (fixed) |
| λ2 | Power coefficient for pSTAT3N effect on serpin A1 | 0.6 (20.9) |
| Serpin A3n (0) | Baseline SerpinA3n protein | 1 (fixed) |
| Serpin A1 (0) | Baseline SerpinA1 protein | 1 (fixed) |
| Blood lymphocyte dynamics | ||
| kin (h−1) | Trafficking rate constant for lymphocytes from tissue to blood | 0.002 (14.3) |
| kout (h−1) | Trafficking rate constant for lymphocytes from blood to tissue | 0.23 (5.0) |
| IC50, MPL (ng/mL) | Drug concentration inhibiting kin by 50% | 6.15 (fixed) |
| kapoptosis (nM−1h−1) | Second-order rate constant of LYMT apoptosis | 0.0019 (72.6) |
| β1 | Intrinsic efficiency of Serpin A1 | 1 (fixed) |
| β2 | Intrinsic efficiency of Serpin A3n | 1 (fixed) |
| LYMB (0) (K/uL) | Baseline blood lymphocyte count | 4.8 (fixed) |
| LYMT (0) (K/uL) | Baseline tissue lymphocyte count | 475.2 (fixed) |
Model simulation and validation
The dynamic profiles of TAT mRNA, protein, and activity were simulated using an MPL dosing regimen of 0.3 mg/kg/h SC infusion over a seven-day period. As shown in Fig. 6, the predicted profiles were compared with hepatic TAT mRNA and activity measures obtained from a previous chronic infusion study [58]. The MPL clearance value was adjusted to 5.61 L/h/kg based on a previous estimate at lower drug exposures [58]. While the model underestimated the peak of TAT mRNA, it was able to capture the general trend of the rather variable data. The simulated profile of TAT protein was linked to TAT activity, which was captured well by the model after adjusting for the baseline value of TAT activity in this cohort of animals. Similarly, MPL PK, PEPCK protein, and plasma glucose and insulin concentrations were simulated at an IV bolus dose of 10 mg/kg in ADX rats. As depicted in Fig. 7, the predicted profiles were superimposed on previously published profiles of plasma drug PK, glucose, and insulin concentrations [23]. The model was able to satisfactorily capture the extent of the glucose and insulin spikes following the 10 mg/kg IV dose of MPL. Simulations were performed to assess the relative influence of the serpin-driven kapoptosis parameter on lymphocyte apoptosis versus the trafficking process in contributing to the observed decreases in circulating lymphocyte counts. The kapoptosis parameter was set to zero and also varied 0.1×, 2×, 5×, and 10× from the estimated value to observe the associated change in blood lymphocyte profiles. Model simulations suggest that the initial drop in cell counts to the nadir is driven largely through the inhibition of cell trafficking whereas the extent of decrease in counts upon return to baseline is controlled by the rate of cell apoptosis (Fig. S2). The time taken to return back to a given steady baseline, however, is dependent upon the drug concentration producing 50% inhibition of movement of cells from tissues to blood (IC50, MPL).
Fig. 6.
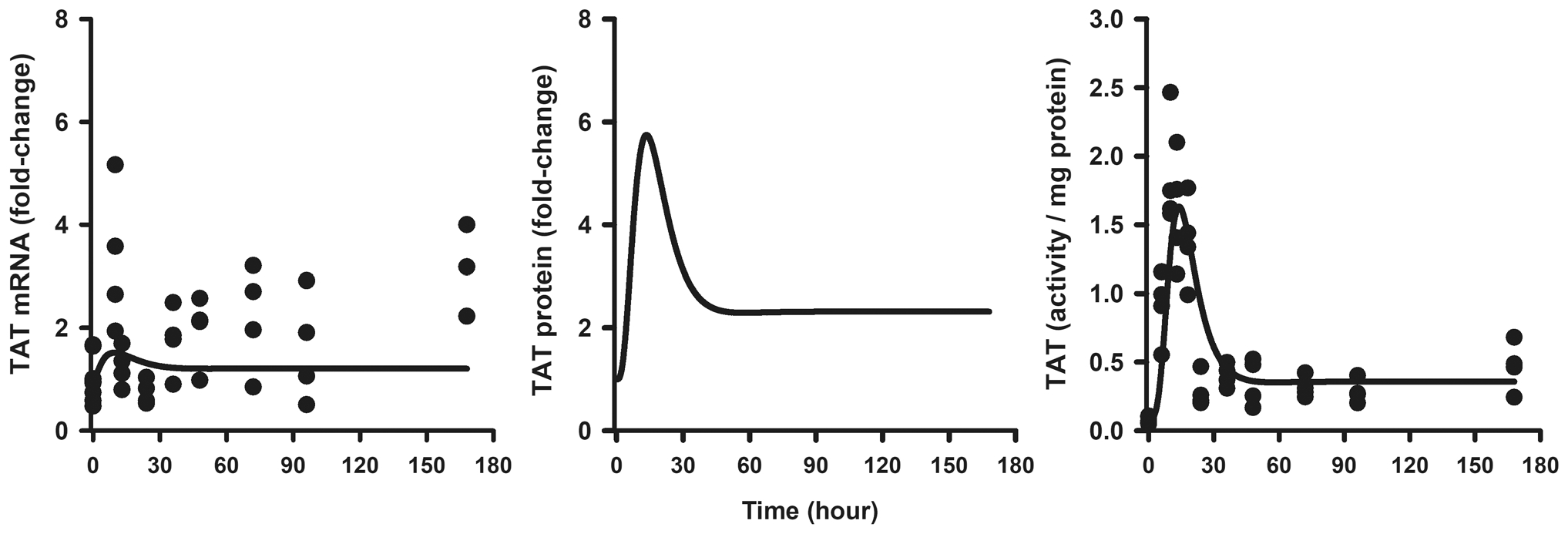
Hepatic TAT mRNA (left panel), TAT protein (center panel), and TAT activity (right panel) dynamics during a 0.3 mg/kg/h SC infusion of MPL for 7 days. Each circle is an individual measurement from one rat and solid lines depict simulated predictions performed based on the developed model (Fig. 1) using parameters shown in Tables 1 and 2. Data were from a previous report. Reproduced with Permission from Ramakrishnan et al. [35]
Fig. 7.
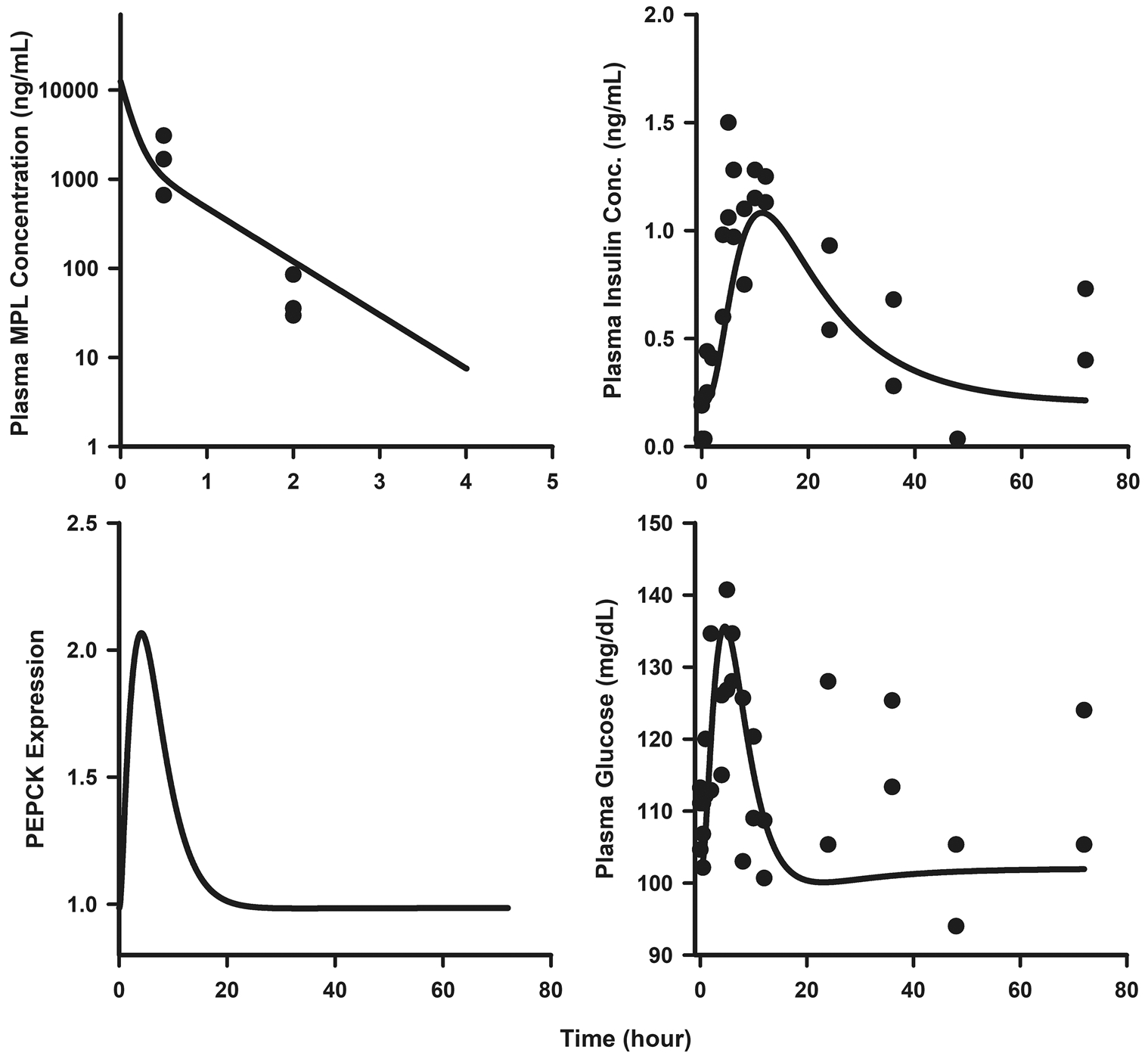
Plasma PK of MPL (top left panel), plasma insulin (top right panel), hepatic PEPCK protein (bottom left panel), and plasma glucose (bottom right panel) dynamics following a 10 mg/kg IV bolus of MPL. Circles are measurement from single rats and solid lines depict simulated predictions performed based on the developed model (Fig. 1) using parameters shown in Tables 1 and 3. Data were from a previous report. Reproduced with Permission from Jin and Jusko [23]
Discussion
The receptor-mediated pharmacodynamics of CS has been of interest in our laboratory for over three decades [35, 36, 59, 63, 64]. This report extends these studies with additional biomarkers to demonstrate that MPL elicits pharmacodynamic/toxicodynamic responses through down-stream effects controlled by receptor/gene/protein-mediated signaling. Although efficacious as anti-inflammatory agents, the CS evoke adverse metabolic effects in various organs, including liver, through coupled genomic mechanisms. We present a mechanistic model-based platform (Fig. 1) to evaluate the role of mRNA and protein biomarkers in regulating the exposure–response relationships of MPL to select efficacy and toxicity end-points measured in steroid-treated ADX rats.
Temporal responses of several mRNA and proteins revealed that biological cascades remained active well after the drug was cleared from the system (Figs. 2, 3, 4 and 5). Of interest was to examine the complementarity (or lack thereof) in mRNA and protein dynamics within the signaling cascades studied. A concordance of about 40–50% between mRNA and protein profiles was reported in a previous global analysis of these—omic datasets [65]. While direct transcription-mediated changes were readily established for TAT and STAT3, a disconnect between PEPCK mRNA and protein dynamics was evident. This disconnect was explained through secondary/post-transcriptional gene regulation mediated by the transcription factors CREB (presumed biosignal in the absence of data) and C/EBP-β. Since proteins are regarded as the mediators of functional changes, protein dynamics were linked to final pharmacodynamic outcomes.
TAT dynamics
As prototypic biomarkers of receptor/gene-mediated CS effects, TAT mRNA and enzyme activity have been frequently quantified in our experimental studies [9, 58, 59, 66]. The development and application of quantitative nano-LC/MS methods [28] enabled the measurement of TAT protein, allowing a complete characterization of the signal transduction pathway. Time-dependent increases in TAT mRNA, TAT protein, and associated activity proceeded as cascading events. Changes in mRNA expression caused by the activated drug-receptor complex were translated into changes in protein expression and subsequently changes in enzyme activity. Power coefficients were needed to optimally capture the profiles of TAT protein and TAT activity. The model was able to predict both hepatic TAT mRNA and activity in ADX rats given a different dosage regimen (0.3 mg/kg/h SC for 7 days), including their attainment of a new pharmacodynamic steady-state (Fig. 6). Interestingly, our previous fifth-generation model [58] showed a decoupling between prediction of TAT mRNA and activity upon chronic MPL. Among other mechanistic hypotheses such as secondary regulation by insulin or cAMP [58], protein translation of TAT from its mRNA may also serve as a rate-controlling step, as illustrated in this study.
PEPCK–insulin–glucose dynamics
Blood glucose homeostasis is sustained mainly through the balance between hepatic glucose output (HGO) and glucose uptake primarily by brain, muscle and adipose tissue [67]. Liver is the major site for gluconeogenesis and also a primary metabolic target of CS. The present model sought to quantitatively describe the signaling processes controlling the MPL-induced rise in blood glucose by understanding the regulation of the rate-limiting gluconeogenic enzyme, PEPCK, at the mRNA and protein levels along with its secondary regulatory mechanisms. The dynamics of C/EBP protein is an important transduction step and an appropriate driver of post-transcriptional PEPCK regulation. C/EBP protein abundance, however, was not identified within our proteomics dataset. Hence, C/EBP mRNA served as a surrogate marker. While an additional transit step could have been employed to represent C/EBP protein dynamics, the model would have become overparameterized, since a transit step representing CREB signal was already applied within this regulatory network. The current model is limited in that the roles of skeletal muscle and adipose tissue in the development of insulin resistance, which also contribute to increases blood glucose were not accounted for. While a multi-organ model of glucose regulation by transcriptomic CS actions has been reported [67], our goal in this analysis was to link protein-level measures to PD outcomes. While the present model could adequately predict glucose and insulin dynamics from a different acute dosing study (Fig. 7), caution must be taken in extrapolating the model to long-term dosing regimens, where insulin resistance becomes of particular importance. Additional experiments to assess protein-level changes in insulin resistance genes in adipose and muscle would allow for expansion of the present model to account for this pathophysiological process. Under homeostasis, glucose concentrations are controlled not only by PEPCK and insulin but also by other signaling factors such as cAMP, glucagon, and free fatty acids, which are linked to feeding [7]. This model assumes that in the presence of exogenous corticosteroid, the induction of gluconeogenesis, as evidenced by strong enhancement of PEPCK expression, is the primary driver of hyperglycemia. In our experimental study, animals were not fasted at any time, had free access to food and water during these studies, and food intake is gradual rather than rapid as with meals in humans. Food intake can also influence baseline glucose and insulin concentrations, which were not accounted for in this model. However, this current model can be expanded to include the effects of food intake on glucose homeostasis as well as the effect of MPL on food intake based upon previous work.
Acute phase response and lymphocyte dynamics
The response of blood lymphocytes to systemic administration of various CS including dexamethasone, MPL, and prednisolone have been assessed in humans [27, 68, 69]. In those studies, lymphocytopenia induced by CS was accounted for only by cell trafficking. The data revealed that human blood lymphocytes returned to a baseline circadian rhythm by 24 h. However, in this study and in normal male rats [5], blood lymphocyte numbers were significantly reduced even beyond 60 h after MPL dosing. This discrepancy may be attributed to two reasons. First, human lymphocytes are more resistant to the apoptotic machinery induced by CS. Second, the doses used in the human studies were relatively low compared to the dose used in these rodent studies. The CS elicit prominent acute phase protein responses, especially at high doses. Among these, up-regulation of STAT3 was for the first time observed as a result of CS. STAT3 is capable of increasing the expression of its corresponding down-stream acute phase proteins [70], e.g. Serpin proteins, which is consistent with our findings. Moreover, STAT3 has a GRE in its highly conserved promoter region as suggested from Gene2promoter (Genomatix, Munich, Germany), indicating that the elevation of STAT3 occurred via receptor/gene-mediated signaling induced by MPL. The concordant up-regulation of both STAT3 mRNA and protein supports the proposed mechanism for enhanced expression of STAT3. The present model assumes that the production and elimination of lymphocytes from the lymphoid system are balanced and do not contribute to the observed drug response within the study period. Reflected by the model parameters are the pooled responses of several types of lymphocytes, since this model does not account for differences in the sensitivity of lymphocyte subpopulations to CS.
Culminating from the modeling of the chosen biological systems were quantitative descriptions of three distinct modes of molecular CS actions. These mechanisms include: (i) simple, direct transcription-mediated signaling as employed for describing the TAT pathway, (ii) secondary and/or post-transcriptional regulation of mRNA-protein expression, as used to characterize PEPCK regulation by C/EBP and CREB, and (iii) downstream activation of proteins lacking direct GREs via upstream signaling molecules (i.e. serpin activation via STAT3-signaling). While the mechanisms characterized in this model apply to CS actions, such types of genomic control should be generalizable to other drugs acting through similar mechanisms. An extended PK/PD/PG/PP paradigm that builds upon a previous generalized model by Jin et al. [11] is introduced (Fig. 8). Upstream activation of receptors by drug at the sites of action(s) would promote DNA binding and consequent alterations in the production rate (kin) or the removal (kout) of target mRNAs. Changes in target mRNA expression is assumed to drive subsequent protein dynamics. Drugs producing large-scale perturbations such as CS may also affect other endogenous regulatory mechanisms such as hormones, cytokines, microRNA, and ligand-specific mRNA decay mechanisms [11, 45, 71] that secondarily regulate mRNA during transcription, and proteins at the post-transcriptional and translation stages. Such processes may be described by either incorporating “real” measurements of the endogenous regulator or more simply by linear transduction, with a rate-limiting first-order rate constant in the absence of data. Signal transduction steps occurring along the pathway to produce a final response can be modeled by linking the major proteins involved in that pathway, or represented empirically as transit steps, depending upon the data available and the extent of onset delay in final response.
Fig. 8.
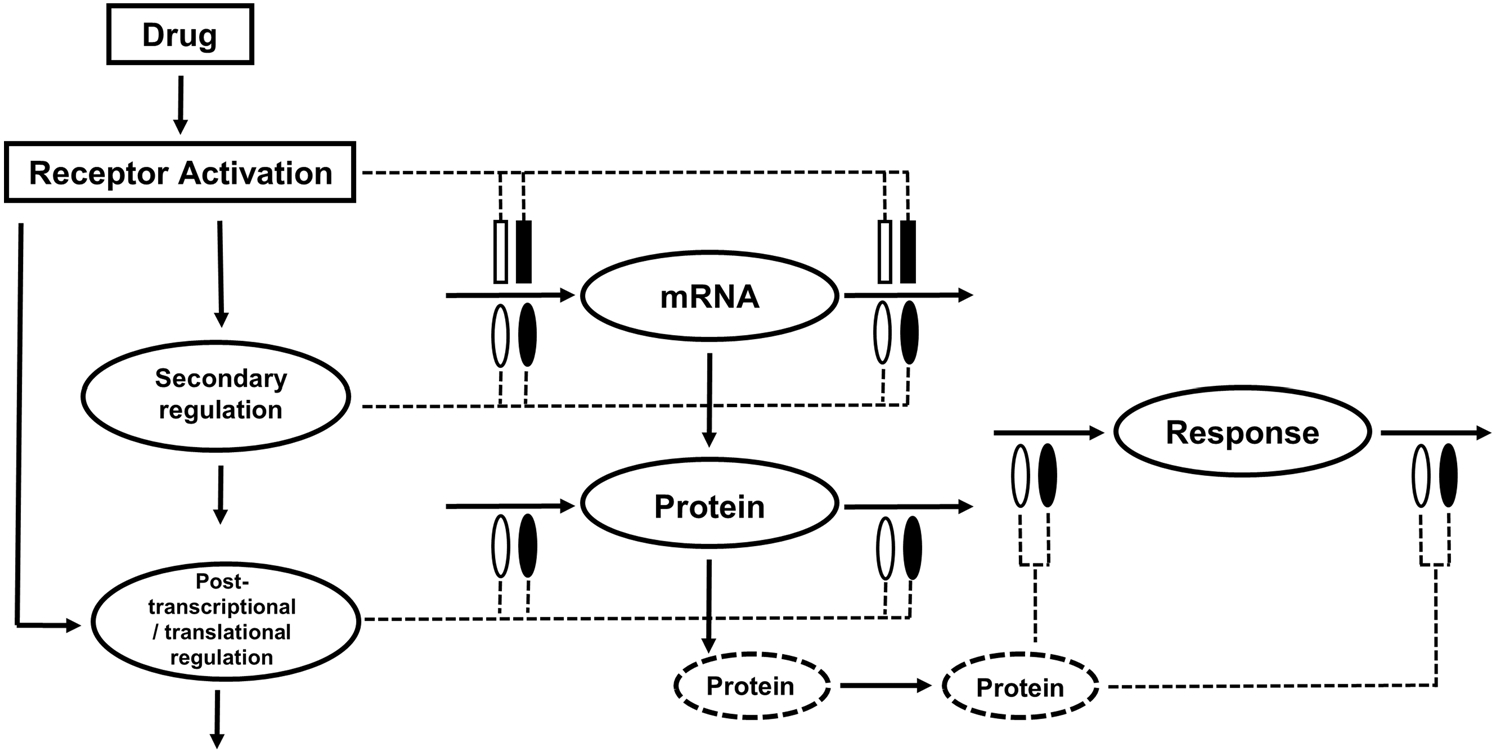
Generalized model of primary and secondary drug effects occurring at the scales of mRNA and protein regulation. The dashed circles represent downstream proteins involved in the signal transduction and mediation of pharmacodynamic responses. The dotted lines and symbols indicate stimulation (open symbol) and inhibition (solid symbol) of the various turnover processes (solid lines) via indirect mechanisms. The rectangles represent primary drug action. The ellipses represent secondary drug effects via secondary mechanisms
Basic “mechanism-based” PK/PD models tend to incorporate one or more of the major rate-limiting processes, including pharmacokinetics, receptor binding, and/or homeostatic mechanisms controlling drug responses [72]. Such models typically seek parsimony and robust statistical reliability of model parameters. Systems-based models, on the other hand, employ “bottom-up” approaches to describe in detail systems or network-based structures in order to generate biological/pharmacological hypotheses in silico. One of the challenges addressed in the development of our current CS systems PKPD model, which sought to combine systems structures with fundamental PK/PD principles [73] was finding a suitable ‘middle ground’ between model parsimony and a more incisive recapitulation of mechanistic complexity within the system. In this effort, some unidentifiable model parameters were fixed based on available experimental evidence, and some biochemical species were represented as empirical transit compartments where necessary, while the major rate-limiting steps were captured by appropriate model fitting to data.
Certain limitations exist within our study paradigm. Male ADX rats were used in our experiments to prevent the confounding effects of endogenous corticosterone and to enact stable pharmacodynamic baselines. In reality, several steroid-regulated hormones, mRNAs, and proteins display circadian rhythms [7, 13], which were not considered in this analysis. The microarray and proteomic data were obtained from two separate but very similar animal studies, with the only major difference being that the gene expression analysis were performed in rats given an IV dose (group 1) whereas the proteomic measurements were performed in rats given an IM injection of MPL (group 2). In our earlier studies, IV doses was administered through an implanted jugular vein cannula in order to precisely time the administration of drug. The route of administration was changed because subsequent studies demonstrated that the more convenient IM dosing can replace the IV route without causing marked differences in gene-mediated pharmacodynamics [34]. The IM dosing route is easier for time-controlled administration with no cannulation required. While the kinetics following IM and IV were modestly different, previous experiments combined with simulations showed that profiles of cytosolic free receptor density and drug receptor complex in the nucleus were similar following both routes. Simulated profiles of GR mRNA, cytosolic receptor density, and nuclear complex in group 2 animals (Fig. S1) were comparable with receptor dynamics from previous IV studies [36]. This phenomenon conceivably explains the nearly identical profiles seen for TAT mRNA dynamics following IM and IV dosing [34]. The nuclear complex profile that drives gene expression dynamics showed rapid rises following either dosing route. Therefore, estimates of transit parameters are unlikely to be strongly affected. In some cases, simultaneous estimation of several parameters using single-dose data led to generation of parameter estimates with high CV%. Therefore, these parameters were fixed to values obtained from more parsimonious PD models fitted to multiple-dose data [23, 58], thereby providing confidence in their values. In the case of the parameter, kstat3, the data at hand was not able to support its estimation. Therefore, in vitro information from the literature [56] was leveraged to identify a biologically plausible value. Although the model was developed based on a single dose using linear effect terms, the final model showed good predictivity at lower doses and different dosing regimens. Despite these limitations, the signaling model offers a mechanism- based description of CS effects, provides a biomarker driven molecular- to-whole body translational approach, and serves as a platform for further testing and exploration of mechanistic hypotheses.
In summary, we present a mechanistic model that integrates and evaluates the role of mRNA and protein biomarkers as well as secondary mediators as they relate to the exposure–response relationships of MPL with TAT dynamics, glucose homeostasis, and lymphocyte modulation. This study exemplifies the importance of capturing the dynamics of critical molecular biomarkers or mediators and their utility in the development of mechanistic pharmacodynamic models that link pharmacokinetics to various therapeutic and toxic responses. Finally, the model offers a mechanism-based platform to explore dosing schedules for enhancing immunosuppressive efficacy while minimizing adverse metabolic toxicities and immune cell apoptosis.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health—National Institute of General Medical Sciences (Grant GM24211).
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10928-018-9585-x) contains supplementary material, which is available to authorized users.
References
- 1.Kirwan JR (1995) The effect of glucocorticoids on joint destruction in rheumatoid arthritis. N Engl J Med 333:142–147 [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ (1998) Efficacy of inhaled corticosteroids in asthma. J Allergy Clin Immunol 102:531–538 [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Irastorza G, Danza A, Khamashta M (2012) Glucocorticoid use and abuse in SLE. Rheumatology 51:1145–1153 [DOI] [PubMed] [Google Scholar]
- 4.Vegiopoulos A, Herzig S (2007) Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol 275:43–61 [DOI] [PubMed] [Google Scholar]
- 5.Yao Z, DuBois DC, Almon RR, Jusko WJ (2008) Pharmacokinetic/pharmacodynamic modeling of corticosterone suppression and lymphocytopenia by methylprednisolone in rats. J Pharm Sci 97:2820–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jusko WJ (1995) Pharmacokinetics and receptor-mediated pharmacodynamics of corticosteroids. Toxicology 102:189–196 [DOI] [PubMed] [Google Scholar]
- 7.Sukumaran S, Jusko WJ, DuBois DC, Almon RR (2011) Mechanistic modeling of the effects of glucocorticoids and circadian rhythms on adipokine expression. J Pharmacol Exp Ther 337:734–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hazra A, DuBois DC, Almon RR, Snyder GH, Jusko WJ (2008) Pharmacodynamic modeling of acute and chronic effects of methylprednisolone on hepatic urea cycle genes in rats. Gene Regul Syst Bio 2:1–19 [PMC free article] [PubMed] [Google Scholar]
- 9.Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ (2007) Modeling receptor/gene-mediated effects of corticosteroids on hepatic tyrosine aminotransferase dynamics in rats: dual regulation by endogenous and exogenous corticosteroids. J Pharmacokinet Pharmacodyn 34:643–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin JY, Almon RR, DuBois DC, Jusko WJ (2003) Modeling of corticosteroid pharmacogenomics in rat liver using gene microarrays. J Pharmacol Exp Ther 307:93–109 [DOI] [PubMed] [Google Scholar]
- 11.Jin JY, DuBois DC, Almon RR, Jusko WJ (2004) Receptor/gene-mediated pharmacodynamic effects of methylprednisolone on phosphoenolpyruvate carboxykinase regulation in rat liver. J Pharmacol Exp Ther 309:328–339 [DOI] [PubMed] [Google Scholar]
- 12.Yao Z, Hoffman EP, Ghimbovschi S, DuBois DC, Almon RR, Jusko WJ (2008) Mathematical modeling of corticosteroid pharmacogenomics in rat muscle following acute and chronic methylprednisolone dosing. Mol Pharm 5:328–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayyar VS, DuBois DC, Almon RR, Jusko WJ (2017) Mechanistic multi-tissue modeling of GILZ regulation: integrating circadian gene expression with receptor-mediated corticosteroid pharmacodynamics. J Pharmacol Exp Ther 363:45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payne SH (2015) The utility of protein and mRNA correlation. Trends Biochem Sci 40:1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maier T, Güell M, Serrano L (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett 583:3966–3973 [DOI] [PubMed] [Google Scholar]
- 16.Ayyar VS, Almon RR, DuBois DC, Sukumaran S, Qu J, Jusko WJ (2017) Functional proteomic analysis of corticosteroid pharmacodynamics in rat liver: relationship to hepatic stress, signaling, energy regulation, and drug metabolism. J Proteomics 160:84–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamisoglu K, Acevedo A, Almon RR, Coyle S, Corbett S, Dubois DC, Nguyen TT, Jusko WJ, Androulakis IP (2017) Understanding physiology in the continuum: integration of information from multiple -omics levels. Front Pharmacol 8:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kmiec Z (2001) Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol 161:1–151 [DOI] [PubMed] [Google Scholar]
- 19.Rui L (2014) Energy metabolism in the liver. Compr Physiol 4:177–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DuBois DC, Xu ZX, McKay L, Almon RR, Pyszczynski N, Jusko WJ (1995) Differential dynamics of receptor down-regulation and tyrosine aminotransferase induction following glucocorticoid treatment. J Steroid Biochem Mol Biol 54:237–243 [DOI] [PubMed] [Google Scholar]
- 21.Pilkis SJ, Granner DK (1992) Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 54:885–909 [DOI] [PubMed] [Google Scholar]
- 22.Kuo T, McQueen A, Chen TC, Wang JC (2015) Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol 872:99–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin JY, Jusko WJ (2009) Pharmacodynamics of glucose regulation by methylprednisolone. I. Adrenalectomized rats. Biopharm Drug Dispos 30:21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin JY, Jusko WJ (2009) Pharmacodynamics of glucose regulation by methylprednisolone. II. Normal rats. Biopharm Drug Dispos 30:35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palumbo P, Ditlevsen S, Bertuzzi A, De Gaetano A (2013) Mathematical modeling of the glucose–insulin system: a review. Math Biosci 244:69–81 [DOI] [PubMed] [Google Scholar]
- 26.Baumann H, Prowse KR, Marinković S, Won KA, Jahreis GP (1989) Stimulation of hepatic acute phase response by cytokines and glucocorticoids. Ann N Y Acad Sci 557:280–296 [DOI] [PubMed] [Google Scholar]
- 27.Wald JA, Jusko WJ (1994) Prednisolone pharmacodynamics: leukocyte trafficking in the rat. Life Sci 55:PL371–PL378 [DOI] [PubMed] [Google Scholar]
- 28.Nouri-Nigjeh E, Sukumaran S, Tu C, Li J, Shen X, Duan X, DuBois DC, Almon RR, Jusko WJ, Qu J (2014) Highly multiplexed and reproducible ion-current-based strategy for large-scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Anal Chem 86:8149–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almon RR, DuBois DC, Brandenburg EH, Shi W, Zhang S, Straubinger RM, Jusko WJ (2002) Pharmacodynamics and pharmacogenomics of diverse receptor-mediated effects of methylprednisolone in rats using microarray analysis. J Pharmacokinet Pharmacodyn 29:103–129 [DOI] [PubMed] [Google Scholar]
- 30.Tu C, Li J, Sheng Q, Zhang M, Qu J (2014) Systematic assessment of survey scan and MS2-based abundance strategies for label-free quantitative proteomics using high-resolution MS data. J Proteome Res 13:2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haughey DB, Jusko WJ (1988) Analysis of methylprednisolone, methylprednisone and corticosterone for assessment of methylprednisolone disposition in the rat. J Chromatogr 430:241–248 [DOI] [PubMed] [Google Scholar]
- 32.Diamondstone TI (1966) Assay of tyrosine transaminase activity by conversion of p-hydroxyphenylpyruvate to p-hydroxybenzaldehyde. Anal Biochem 16:395–401 [Google Scholar]
- 33.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275 [PubMed] [Google Scholar]
- 34.Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ (2007) Pharmacokinetics of methylprednisolone after intravenous and intramuscular administration in rats. Biopharm Drug Dispos 28:263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ (2002) Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn 29:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazra A, DuBois DC, Almon RR, Jusko WJ (2007) Assessing the dynamics of nuclear glucocorticoid-receptor complex: adding flexibility to gene expression modeling. J Pharmacokinet Pharmacodyn 34:333–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott DK, Stromstedt PE, Wang JC, Granner DK (1998) Further characterization of the glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. The role of the glucocorticoid receptor-binding sites. Mol Endocrinol 12:482–491 [DOI] [PubMed] [Google Scholar]
- 38.Jurado LA, Song S, Roesler WJ, Park EA (2002) Conserved amino acids within CCAAT enhancer-binding proteins (C/EBP(alpha) and beta) regulate phosphoenolpyruvate carboxykinase (PEPCK) gene expression. J Biol Chem 277:27606–27612 [DOI] [PubMed] [Google Scholar]
- 39.Park EA, Gurney AL, Nizielski SE, Hakimi P, Cao Z, Moorman A, Hanson RW (1993) Relative roles of CCAAT/enhancer-binding protein beta and cAMP regulatory element-binding protein in controlling transcription of the gene for phosphoenolpyruvate carboxykinase (GTP). J Biol Chem 268:613–619 [PubMed] [Google Scholar]
- 40.Park EA, Roesler WJ, Liu J, Klemm DJ, Gurney AL, Thatcher JD, Shuman J, Friedman A, Hanson RW (1990) The role of the CCAAT/enhancer-binding protein in the transcriptional regulation of the gene for phosphoenolpyruvate carboxykinase (GTP). Mol Cell Biol 10:6264–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M (2001) CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413:179–183 [DOI] [PubMed] [Google Scholar]
- 42.Jungmann RA, Wang XS, Milkowski DM, Short ML (1992) Glucocorticoid induction of CRE-binding protein isoform mRNAs in rat C6 glioma cells. Nucleic Acids Res 20:825–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woltje M, Tschoke B, von Bulow V, Westenfeld R, Denecke B, Graber S, Jahnen-Dechent W (2006) CCAAT enhancer binding protein beta and hepatocyte nuclear factor 3beta are necessary and sufficient to mediate dexamethasone-induced up-regulation of alpha2HS-glycoprotein/fetuin-A gene expression. J Mol Endocrinol 36:261–277 [DOI] [PubMed] [Google Scholar]
- 44.Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK (1993) Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem 268:5353–5356 [PubMed] [Google Scholar]
- 45.Cho H, Park OH, Park J, Ryu I, Kim J, Ko J, Kim YK (2015) Glucocorticoid receptor interacts with PNRC2 in a ligand-dependent manner to recruit UPF1 for rapid mRNA degradation. Proc Natl Acad Sci USA 112:E1540–E1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park OH, Park J, Yu M, An H-T, Ko J, Kim YK (2016) Identification and molecular characterization of cellular factors required for glucocorticoid receptor-mediated mRNA decay. Genes Dev 30:2093–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun YN, Jusko WJ (1998) Transit compartments versus gamma distribution function to model signal transduction processes in pharmacodynamics. J Pharm Sci 87:732–737 [DOI] [PubMed] [Google Scholar]
- 48.McPherson CS, Lawrence AJ (2007) The nuclear transcription factor CREB: involvement in addiction, deletion models and looking forward. Curr Neuropharmacol 5:202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisher LE, Ludwig EA, Jusko WJ (1992) Pharmacoimmunodynamics of methylprednisolone: trafficking of helper T lymphocytes. J Pharmacokinet Biopharm 20:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wald JA, Salazar DE, Chen HY, Jusko WJ (1991) Two-compartment basophil cell trafficking model for methylprednisolone pharmacodynamics. J Pharmacokinet Biopharm 19:521–536 [DOI] [PubMed] [Google Scholar]
- 51.Fauci AS, Dale DC (1975) The effect of hydrocortisone on the kinetics of normal human lymphocytes. Blood 46:235–243 [PubMed] [Google Scholar]
- 52.Gruver-Yates AL, Cidlowski JA (2013) Tissue-specific actions of glucocorticoids on apoptosis: a double-edged sword. Cells 2:202–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cidlowski JA, King KL, Evans-Storms RB, Montague JW, Bortner CD, Hughes FM Jr (1996) The biochemistry and molecular biology of glucocorticoid-induced apoptosis in the immune system. Recent Prog Horm Res 51:457–490 [PubMed] [Google Scholar]
- 54.Ahmed ST, Darnell JE (2009) Serpin B3/B4, activated by STAT3, promote survival of squamous carcinoma cells. Biochem Biophys Res Commun 378:821–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dasgupta M, Dermawan JKT, Willard B, Stark GR (2015) STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci USA 112:3985–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaefer TS, Sanders LK, Park OK, Nathans D (1997) Functional differences between Stat3a and Stat3b. Mol Cell Biol 17:5307–5316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.D’Argenio D, Schumitzky A, Wang X (2009) ADAPT 5 user’s guide: Pharmacokinetic/pharmacodynamic systems analysis software [Google Scholar]
- 58.Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ (2002) Pharmacodynamics and pharmacogenomics of methylprednisolone during 7-day infusions in rats. J Pharmacol Exp Ther 300:245–256 [DOI] [PubMed] [Google Scholar]
- 59.Sun YN, DuBois DC, Almon RR, Jusko WJ (1998) Fourth-generation model for corticosteroid pharmacodynamics: a model for methylprednisolone effects on receptor/gene-mediated glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm 26:289–317 [DOI] [PubMed] [Google Scholar]
- 60.Salinas M, Wallace R, Grisolia S (1974) Comparative studies in vivo and in vitro of rat-liver enzymes. Eur J Biochem 44:375–381 [DOI] [PubMed] [Google Scholar]
- 61.Hopgood MF, Ballard FJ (1973) Synthesis and degradation of phosphoenolpyruvate carboxylase in rat liver and adipose tissue. Changes during a starvation-re-feeding cycle. Biochem J 134:445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siewert E, Muller-Esterl W, Starr R, Heinrich PC, Schaper F (1999) Different protein turnover of interleukin-6-type cytokine signalling components. Eur J Biochem 265:251–257 [DOI] [PubMed] [Google Scholar]
- 63.Boudinot FD, D’Ambrosio R, Jusko WJ (1986) Receptor-mediated pharmacodynamics of prednisolone in the rat. J Pharmacokinet Biopharm 14:469–493 [DOI] [PubMed] [Google Scholar]
- 64.Nichols AI, Boudinot FD, Jusko WJ (1989) Second generation model for prednisolone pharmacodynamics in the rat. J Pharmacokinet Biopharm 17:209–227 [DOI] [PubMed] [Google Scholar]
- 65.Kamisoglu K, Sukumaran S, Nouri-Nigjeh E, Tu C, Li J, Shen X, Duan X, Qu J, Almon RR, DuBois DC, Jusko WJ, Androulakis IP (2015) Tandem analysis of transcriptome and proteome changes after a single dose of corticosteroid: a systems approach to liver function in pharmacogenomics. OMICS 19:80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun YN, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ (1998) Dose-dependence and repeated-dose studies for receptor/gene-mediated pharmacodynamics of methylprednisolone on glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm 26:619–648 [DOI] [PubMed] [Google Scholar]
- 67.Fang J, Sukumaran S, Dubois DC, Almon RR, Jusko WJ (2013) Meta-modeling of methylprednisolone effects on glucose regulation in rats. PLoS ONE 8:e81679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mager DE, Lin SX, Blum RA, Lates CD, Jusko WJ (2003) Dose equivalency evaluation of major corticosteroids: pharmacokinetics and cell trafficking and cortisol dynamics. J Clin Pharmacol 43:1216–1227 [DOI] [PubMed] [Google Scholar]
- 69.Stark JG, Werner S, Homrighausen S, Tang Y, Krieg M, Derendorf H, Moellmann H, Hochhaus G (2006) Pharmacokinetic/pharmacodynamic modeling of total lymphocytes and selected subtypes after oral budesonide. J Pharmacokinet Pharmacodyn 33:441–459 [DOI] [PubMed] [Google Scholar]
- 70.Kushner I, Rzewnicki DL (1994) The acute phase response: general aspects. Baillieres Clin Rheumatol 8:513–530 [DOI] [PubMed] [Google Scholar]
- 71.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R (2006) Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 20:515–524 [DOI] [PubMed] [Google Scholar]
- 72.Jusko WJ (2013) Moving from basic toward systems pharmacodynamic models. J Pharm Sci 102:2930–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mager DE, Kimko HHC (2016) Systems Pharmacology and Pharmacodynamics: An Introduction In: Mager DE, Kimko HHC (eds) Systems Pharmacology and Pharmacodynamics. Springer International Publishing, Cham, Switzerland, pp 3–14 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.