

Abstract
Atopy is characterized by eosinophilic inflammation associated with recruitment of eosinophil/basophil (Eo/B) progenitors. We have previously shown that Eo/B progenitor phenotypes are altered in cord blood (CB) in infants at high risk of atopy/asthma, and respond to maternal dietary intervention during pregnancy. As respiratory tract viral infections have been shown to induce wheeze in infancy, we investigated the relationship between CB progenitor function and phenotype and acute respiratory illness (ARI), specifically wheeze and fever. CB from 39 high‐risk infants was studied by flow cytometry for CD34+ progenitor phenotype and by ex vivo Eo/B‐colony forming unit (CFU) responses to cytokine stimulation in relation to ARI in the first year of life. A consistent relationship was observed between increased numbers of granulocyte/macrophage (GM)‐colony‐stimulating factor (CSF)‐ and IL‐3‐responsive Eo/B‐CFU in CB and the frequency/characteristics of ARI during infancy. Comparable associations were found between ARI and CB IL‐3R+ and GM‐CSFR+CD34+ cell numbers. Conversely, a reciprocal decrease in the proportion of CB IL‐5R+ cells was found in relation to the clinical outcomes. The elevation of IL‐3/GM‐CSF‐responsive Eo/B progenitors in high‐risk infants in relation to ARI outcomes suggests a mechanism for the increased severity of inflammatory responses in these subjects following viral infection.
Keywords: progenitors, eosinophils, cytokine receptors, infant wheeze
Atopic diseases such as asthma have been increasing globally, and this appears to predominate in ‘westernized’ countries (1); factors favoring the development of atopy include small family size (2), high antibiotic use (3), good sanitation, low oral–fecal burden (4) and low rates of helminthic infections (5); large family size and living on a farm (6) appear to afford protection. These findings have been widely interpreted in the context of the ‘hygiene hypothesis’ which, in its broadest form, posits that natural exposure to microbial stimulation (both commensals and pathogens) provides obligatory signals required to drive the postnatal maturation of adaptive immune functions (7). The cellular targets for these microbial stimuli remain to be defined, but are likely to include populations within the innate immune system (7). Failure in receipt or transduction of these signals is hypothesized to result in postnatal prolongation of fetal Th2‐polarization within the immune system, thus increasing risk for atopic sensitization and atopic asthma (7). However, it is also apparent that acute respiratory illness (ARI) can serve as a double‐edged sword in this context, as intense wheezing‐associated ARI during infancy can synergize with inhalant allergy to drive asthma pathogenesis (8), presumably by disrupting sensitive differentiation programs regulating the growth and development of airway tissues during this life phase (9).
We have previously suggested that the increased severity of ARI in infants at high risk for atopy may be due in large part to a developmental delay in postnatal maturation of their adaptive immune functions (9), in particular their diminished capacity to secrete both Th2 and (especially) Th1 cytokines (9), which are central to host defence. However, it is equally feasible that non‐cognate inflammatory effector cells such as mast cells, basophils, eosinophils (Eo/B), or their progenitors may also play a critical role in determining the degree of inflammation and hence frequency/severity of symptoms accompanying ARI during infancy, thus contributing to asthma development. In this context, studies we have recently performed demonstrate that deficient maturation of cord blood (CB) eosinophil progenitors, as evidenced by reduced expression of hemopoietic cytokine receptors, may predict the subsequent development of atopic disease (10), including wheeze and atopic dermatitis in the first year of life (11), a process which is reversed by dietary supplementation with n‐3 omega fatty acids during pregnancy (12).
In the current study, we have therefore further examined the phenotype and function of CB hemopoietic Eo/B progenitors as potential predictors of the development of inflammation‐associated clinical responses, in particular ARI accompanied by fever or wheeze, in infants at high risk of atopy and asthma. Our aim was to examine more critically the concept of progenitor skewing or immaturity as a non‐T‐cell mechanism leading to tissue inflammation in atopic at‐risk subjects, and thus to provide further evidence for the ‘bone marrow hypothesis’ in atopy and asthma (13).
Method
Cohort childhood asthma study
The infants were a sub‐sample from a birth cohort of 263 Australian subjects born between 1997 and 1998, characterized as high risk for atopy/asthma, and recruited to study early events of allergy in the immunologic timeline and the effects of infant ARI on asthma/atopy outcomes. At least one parent of each child had a medical diagnosis of eczema, hay fever or asthma; atopy was confirmed by skin prick test (SPT) in one or more parent of 96% of the children. Selection of infants for this sub‐study was based on availability of cryo‐banked CB cells; preservation and viability of cells has been high and has not been known to alter properties of progenitors (10, 11, 13). Parents were asked to keep a daily diary of their child's health and to notify the study centre within 48 h of their child developing a runny/blocked nose, cough or fever (>38°C). The presence of fever for at least 24 h was recorded but did not form part of the clinical definition of ARI. Home visits were made to take postnasal aspirate samples of mucous within 72 h. Samples were collected and frozen at −85°C for future polymerase chain reaction (PCR) analysis. Follow‐up telephone calls were made every 2 wk until resolution of all symptoms. Information collected included the presence, as well as duration, of symptoms of fever, runny/blocked nose, cough, and wheeze. This report utilizes data collected during the first year of life.
Episodes of ARI that were associated with wheeze, reported by the parent or family doctor were considered to be a wheezy ARI. Wheeze was defined as a high‐pitched whistling sound heard coming from the chest on expiration. To reduce recall bias, symptoms reported during telephone calls were verified with the daily diary record.
To ensure accuracy and consistency, all families were given a digital thermometer at the commencement of the study and instructed how to use it to take temperatures orally. ‘Fever greater than 38°C for at least 24 h’ was defined as at least two separate temperature readings greater than 38°C taken at least 24 h apart. The temperature of 38°C and time frame of 24 h were chosen in consultation with three pediatricians and a family physician as these criteria would reduce bias by spurious fever episodes, which are often seen in young children and may not be associated with respiratory infections. Twenty‐seven percent of the episodes of ARI were associated with fever. Antipyretics such as Paracetamol were used in 44% of episodes of ARI, including non‐febrile episodes.
The ethics committees of King Edward Memorial, Princess Margaret Hospitals and the Research Ethics Board of McMaster University approved this study and subjects signed consent forms to participate.
Viral detection and analysis
The presence of rhinovirus (RV) RNA was detected using modifications of previously described real time (RT)‐PCR reactions (14). Polymerase chain reaction was utilized for diagnosis of Picornavirus ARI in subjects with and without respiratory symptoms. RV was identified by Bgl I digestion of picornavirus RT‐PCR amplicons (14). Positive and negative controls were included with each batch of RT‐PCR samples. Samples from symptomatic and asymptomatic subjects were analyzed blindly. ARI associated with positive RV PCR were classed as RV ARI. The presence of Respiratory Syncytial Virus (RSV) was also examined with positive and negative controls through RT‐PCR, similar to previous studies (15). Briefly, the samples were analyzed by a panel of RT‐PCR for rhinoviruses, other picornaviruses (coxsackie, echo, and enteroviruses), coronaviruses 229E and OC43, RSV, influenza A and B, parainfluenza viruses 1–3, adenoviruses, human metapneumovirus (HMPV), Chlamydia pneumoniae and Mycoplasma pneumoniae. Details of the PCR panel have been previously reported (16).
Preparation of CB cells
Thirty‐nine frozen samples were obtained from the Australian atopic at‐risk cohort. Samples were frozen in 1–1.5 ml aliquots containing 18–22 million cells. The samples were cooled to −80°C at 1°C per minute in a Nalgene cryo‐container with isopropyl alcohol (BDH, Victoria, Australia). They were then removed, placed in liquid nitrogen and shipped from Australia to Canada. Immune responses of these cells were assumed to be intact, as previously demonstrated by several other laboratories, including ours (10, 17). The cells were thawed by immersion in a warm water bath and slow drop‐wise application of 10% fetal calf serum (FCS), 1% penicillin/streptomycin, 2% HEPES buffer in RPMI medium 1640 was performed. Following this, they were resuspended in McCoy's 3+ medium and incubated for 2 h in plastic flasks (37°C, 5% CO2) in order to obtain the non‐adherent mononuclear cells (NAMNC). NAMNC were resuspended in Iscove's 2+ after a second cell count was performed. The viability counts for this cohort averaged to 98%. The recovery after incubation averaged to 67%, which is consistent with this procedure performed in other studies (10, 17). CD34+ cells are retained without loss after incubation; flasks are checked regularly under the inverted light microscope and counts performed before and after incubation to verify cell recovery (which is standardly >95%). Progenitor cells are also easily phenotypically distinguished in the hemocytometer.
Antibodies
Phycoerythrin (PE)‐conjugated CD34 (IgG1 anti‐HPCA‐2) and fluorescein isothiocyanate (FITC)‐conjugated CD45R (IgG1anti‐HLE‐1), PE‐conjugated isotype control antibody for IL‐3R, IL‐5R, PE‐conjugated isotype control antibody for GM‐CSFR, Streptavidin PerCP, biotin antihuman GM‐CSF receptor alpha chain (CD116), biotin antihuman IL‐3 receptor alpha chain (CDw123), were all obtained from BD Pharmingen (Mississauga, ON, Canada). IL‐5 receptor antibody was graciously given as a gift from Roche Laboratories (Ghent, Belgium) and biotinylated for use in our laboratory (18).
Immunofluorescent staining
A protocol for triple color staining, previously designed in our laboratory (19) was utilized to measure cytokine receptors IL‐3Rα, IL‐5Rα, and GM‐CSFRα on CD34+ enriched cells. Designated controls were used for the corresponding receptors: mouse IgG1 isotype for IL‐3R, IL‐5R and mouse IgM for GM‐CSFR. The cells were stained with biotin‐conjugated anti‐IL‐3R, anti‐IL‐5R and anti‐GM‐CSFR, or the corresponding controls, in PBS (0.1% NaN3 and 0.1% BSA) for 30 min. Following this, the cells were washed in 0.1% NaN3 and 0.1% BSA PBS and stained with Streptavidin‐conjugated PerCP, anti‐CD45 FITC and anti‐CD34 PE for 30 min. PBS 0.1% azide was used to wash the cells and they were fixed in PBS and 1% paraformaldehyde. They were then covered and stored in the refrigerator until FACS acquisition.
Flow cytometry
Acquisition was performed on the cells using FACScan flow cytometer (BD Biosciences, Mississauga, ON, Canada). Compensation settings were created by staining NAMNC with anti‐CD34 PE, anti‐CD45 FITC and anti‐IL‐3Rα PerCP. For each sample, 50,000–100,000 events were collected. Analysis was carried out with 50,000 events.
Flow cytometry data were analyzed by Winlist software (Verify Software House, Topsham, ME) using a multi‐parameter gating strategy. Primarily, a dot plot of CD45 vs. side scatter was created and denoted by region (R1), to classify nucleated leukocytes. CD34+ stained cells were extracted from these CD45 events and denoted region (R2). A dot plot of CD45 vs. SSC was created to distinguish true CD34+ cells, which have low side scatter and low CD45 expression, as shown by region (R3). Following this, CD34+ cells were analyzed for their lymphoblastoid characteristics on a dot plot of side scatter vs. forward scatter in region (R4), illustrating their small size and low granularity (10). The net percentage of cytokine receptor expression on CD34+/CD45+ cells was determined by subtracting the staining achieved by the designated isotype control from the staining of progenitor cells with the specific receptor (10). Mean fluorescence intensity (MFI) was not performed because of limited numbers of cells in samples. CB differential cell counts were not performed at the time of collection.
Methylcellulose cultures
Culture dishes (35 × 10 mm; Falcon, Oxnard, CA, USA) were used to culture 2.0 × 105 cells in the following media: semisolid methycellulose, Iscove's 2+ (from Iscove's Dulbecco medium with 1% penicillin/streptomycin and 1% two mercaptoethanol), and FCS. The materials for the cultures were obtained as follows: methylcellulose, paraformaldehyde, bovine serum albumin (grade V), sodium azide, and heparin from (Sigma Aldrich Ltd, Oakville, ON, Canada); Dulbecco McCoy's 5A medium, FCS, penicillin, HEPES buffer, RPMI medium 1640 and trypan blue from (Gibco, Burlington, ON, Canada) and 2‐mercaptoethanol from BDH. Recombinant human cytokines IL‐3 (1 ng/ml), IL‐5 (1 ng/ml) and GM‐CSF (10 ng/ml) (BD Biosciences) were utilized along with an equal volume of Iscove's 2+ as a control. Duplicate conditions were made for each cytokine and cells were cultured for 14 days in the Revco incubator at 37°C (5%CO2). On day 14, colonies were identified and counted, using a light inverted microscope (20). Eo/B‐colony forming units (CFU) were denoted as colonies if there was a minimum of 40 cells in a group, each of which was tight, compact and round, as described previously (20); GM colonies were also counted and identified.
Statistical analyses
Statistical analyses were performed using SPSS 12.0 software. Biological markers of atopy such as cytokine receptor percentages and cytokine stimulated Eo/B colonies were associated with ARI and ARI associated with fever or wheeze by using Spearman's correlation coefficient for ranked data. Mann–Whitney tests were performed for analyses of progenitor phenotypic responses in infants who acquired ARI in the first year of life. Additionally, non‐parametric tests, such as Kruskal–Wallis, were performed for progenitor functional responses and ratio analyses. Non‐parametric Kruskal–Wallis and Spearman's Rho tests were also performed to determine the significance of cytokine stimulated Eo/B colonies and ARI associated with wheeze or fever. Ratio analyses were performed using the median for enumeration of colonies and receptor expression for each cytokine per subject.
Results
A subset of 39 cohort infants at high risk for atopy and asthma was assessed for progenitor phenotypic and functional markers in response to ARI in the first year of life. All the infants acquired one or more ARI within the first year of life. Of these 39 infants, 21 acquired infection accompanied by febrile episodes, and of these 21, 10 had one documented episode of fever and 11 had two or more episodes. Additionally, 25 of the 39 infants acquired infection accompanied by wheeze in the first year of life, of which 10 had one episode of documented wheeze and 15 had two or more episodes. Bacterial contamination and non‐viable cells, due to transfer between liquid nitrogen containers, reduced the total number of analysable events related to cellular studies.
CB CD34 cells
Infants with two or more fever‐associated ARI in the first year of life had a lower percentage of CD34+ cells at birth than those with fewer febrile episodes (p = 0.028, Kruskal–Wallis test): a dose‐response relationship was obtained, with the mean percentage of CD34+ cells being 0.96 ± 0.13 among infants with no fever‐associated ARI, 0.78 ± 0.18 among those with one episode, and 0.48 ± 0.07 among those with two or more (Fig. 1). Spearman's tests for correlation confirmed that the percentage of CD34+ cells at birth was negatively correlated with the frequency of ARI associated with fever (p = 0.008, Rs = −0.42). The percentage of CD34+ cells was also negatively correlated with the number of RV ARI in the first year of life (p = 0.047, Rs = −0.33).
Figure 1.
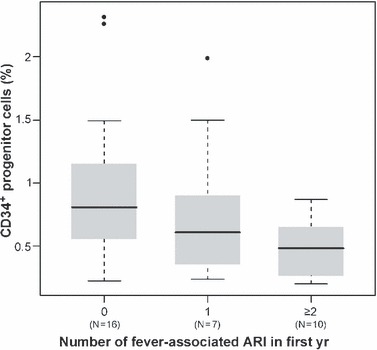
Boxplots representing the percentage of CD34+ progenitor cells among children with 0, 1, and 2 or more fever‐associated acute respiratory illnesses (ARI) in the first year of life. Indicated are the median value (bold line), interquartile range (grey box) and maximum and minimum values (extended lines). Outlying data points are shown as dots. The number of infants (N) in each group is indicated below the corresponding boxplot, and the mean percentage of CD34+ progenitor cells differs significantly between the groups (p = 0.028, Kruskal–Wallis test).
Cytokine receptors on CD34+ progenitors and infections in infancy
The expression levels of IL‐3 and GM‐CSF receptors were positively correlated with the number of ARI in the first year of life (p = 0.046, 0.026; Rs = 0.322, 0.350, respectively). In particular, expression of these receptors was highest among infants who had five or more ARI in the first year of life (p < 0.05, Mann–Whitney test). These groups were chosen on the basis of division by the midline. The highest number of ARI in a subject was eleven, while the lowest was one. For example, the distribution of GM‐CSF receptor expression within these groups is shown in Fig. 2.
Figure 2.
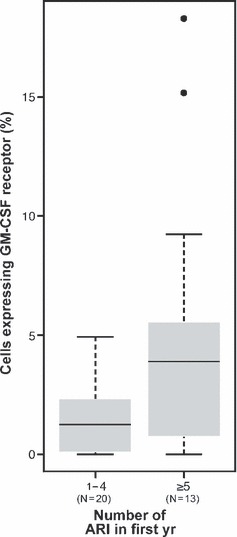
Boxplots representing the percentage of CD34+ progenitor cells expressing the GM‐CSF receptor, among children with one to four vs. five or more acute respiratory illnesses (ARI) in the first year of life. Indicated are the median value (bold line), interquartile range (grey box) and maximum and minimum values (extended lines). Outlying data points are shown as dots. The number of infants (N) in each group is indicated below the corresponding boxplot, and the mean percentage of CD34+ progenitor cells expressing the GM‐CSF receptor differs significantly between the groups (p = 0.048, Mann–Whitney test).
There were no significant correlations between CD34+ cells with RSV ARI. More specifically, RSV ARI correlated with IL‐3, IL‐5 and GM‐CSF phenotypically and functionally and resulted in p‐values of 0.261, 0.446, 0.227, and 0.056, 0.845, and 0.244, respectively. RSV ARI accompanied by wheeze did not correlate significantly with phenotypic or functional characteristics of CD34+ cells. RSV ARI accompanied by fever showed similar results with the exception of a trend towards significance of IL‐3 Eo/B CFU (p = 0.056).
CB Eo/B‐CFU
We examined functional progenitor responses and found positive correlations between GM‐CSF‐stimulated Eo/B CFU and the number of RV ARI in the first 6 months (p = 0.004, Rs = 0.35) and in the first year of life (p = 0.029, Rs = 0.37). Additionally, IL‐3‐stimulated Eo/B‐CFU correlated positively with ARI accompanied by fever during the first year of life (p = 0.013, Rs = 0.41; see Fig. 3a). Similarly, GM‐CSF‐stimulated CFU positively correlated with overall frequency of ARI accompanied by wheeze during the same period (p = 0.023, Rs = 0.38; see Fig. 3b).
Figure 3.
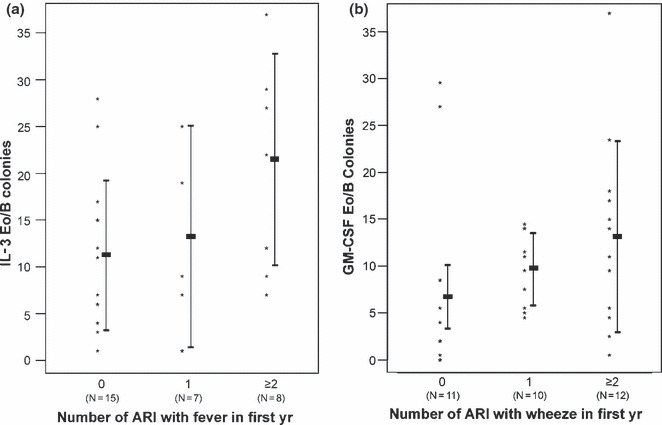
Counts of IL‐3 Eo/B colonies for children with 0, 1, and 2 or more (a) fever‐ and (b) wheeze‐associated acute respiratory illnesses (ARI) in the first year of life. Colony counts were correlated with the number of fever‐associated ARI (p = 0.013, Spearman's rho = 0.41). Rectangles represent the mean ± standard deviation for each group, and the number of infants (N) in each group is indicated below. Colony counts were correlated with the number of wheeze‐associated ARI (p = 0.023, Spearman's Rho = 0.38). Rectangles represent the mean ± standard deviation for each group, and the number of infants (N) in each group is indicated below.
In contrast to these findings relating to GM‐CSF and IL‐3‐responsive Eo/B CFU, no significant correlations were found between ARI frequency alone or ARI associated with fever or wheeze and IL‐5‐responsive CFU. These findings suggested that ARI susceptibility may be associated with skewing of the Eo/B progenitor population towards GM‐CSF/IL‐3‐responsive lineages. To further examine this possibility, ratio analyses were undertaken, re‐expressing cytokine‐stimulated CFU as GM‐CSF: IL‐5 and/or IL‐3: IL‐5. Positive correlations were found between GM‐CSF:IL‐5‐responsive CFU and frequency of ARI with fever in the first year of life (p = 0.012), and IL‐3:IL‐5 ratios and ARI frequency (p = 0.006). In agreement with the above functional (differentiation) data, phenotypic analyses of cytokine receptor expression on CB progenitors in this cohort revealed ratios of 5.0 each for IL‐3Rα: IL‐5Rα and GM‐CSFRα: IL‐5Rα; thus, IL‐5Rα expression appears to be substantially lower in high‐risk cohorts.
Discussion
Our current findings extend and qualify aspects of the hemopoietic progenitor (‘bone marrow’) hypothesis of atopy and asthma, by suggesting that markers detected in CB are used to predict clinical outcomes and establish how this might relate to T‐cell‐oriented models of disease etiology. Our laboratory has previously shown that the measurement of cytokine receptor expression on hemopoietic CD34+ progenitors in CB can be used as a marker related to risk and prediction of atopy and in this study, we attempt to demonstrate that progenitors can serve as markers of ARI and symptoms. Specifically, we have observed that there is a significant, inverse correlation between the numbers of undifferentiated progenitors (CD34+/45+ cells overall) and febrile responses to ARI (Fig. 1); this result is totally in agreement with our previous observation of ‘restoration’ of a normal, high ambient level of undifferentiated progenitors after polyunsaturated fatty acids (PUFA) treatment of the mother, which correlates with improved first year atopic outcomes (12, 21). PUFA has been thought to play in role in the prevention/reversal of allergy. Moreover, we have recently observed that CB CD34+ progenitors of infants at high risk for atopy and asthma (as evidenced by maternal SPT) have an increased expression of IL‐3R (p = 0.018, R = −0.887), and decreased IL‐5R expression (p = 0.021, R = −0.879) (Cyr et al, in preparation). Similarly, in the current study, the absence of IL‐5 responsiveness, the combined IL‐3R and GM‐CSFR expression on CD34+ CB cells, as well as corresponding functional (CFU) responses to these cytokines, correlated positively and significantly with ARI frequency and accompanying symptoms from three months through the first year of life. This was also reflected as an apparent skewing towards IL‐3 and GM‐CSF responsive myeloid progenitor phenotypes and functions, as shown by ratio analyses comparing CB from infants at high risk for atopy/asthma and those at low risk.
One interpretation of these findings is that the intensity of acute inflammatory responses triggered by ARI in infancy, which is determined to a large extent by the efficiency of recruitment of inflammatory effector cells such as neutrophils and eosinophils, may be directly related to the numbers or phenotype of available, lineage‐committed hemopoietic progenitors in the circulation. While IL‐3, GM‐CSF and IL‐5 are each involved in eosinophil differentiation, IL‐3‐ and GM‐CSF‐responsive progenitors represent earlier stages in lineage commitment than IL‐5‐responsive progenitors, and thus would be expected to be more ‘promiscuous’ in the sense that they could contribute to development of multi‐cellular (neutrophilic and eosinophilic) inflammatory responses. Situations in which the frequency of these progenitors exceeds a critical threshold results in significant pathological consequences, contrasting with the situation involving a more ‘mature’ compartment dominated by eosinophil progenitors which have differentiated further towards exclusive IL‐5‐responsiveness, in which eosinophil generation is tightly regulated by the availability of IL‐5 (22). It is thought that high‐risk infants are subject to delayed immune deviation (22). Therefore, initial lack of IL‐5 in infancy could result in compensation by an overshoot of the IL‐5 response later in childhood, causing increased deposition of tissue eosinophilia in response to environmental stimuli, deeming the child atopic. Alternatively, decreased IL‐5R expression may be a consequence of increased maternal T‐cell production of IL‐5, causing downregulation of receptor in utero (23). We do not have CB differential counts to accompany the progenitor data, so these explanations remain speculative.
If the CD34+ progenitor pool is dominated by cells that can differentiate into eosinophils and/or neutrophils in response to the more ubiquitous cytokines IL‐3 and/or GM‐CSF, an expected consequence will be the development of a mixed inflammatory response at sites of microbial challenge in the absence of a local Th2 (IL‐5) response in infancy. However, as the individual progresses from infancy to childhood, viral modulation of the host response could result in modification from a multi‐cellular inflammatory profile to a purely eosinophilic response. It is the snapshot of the progenitor phenotype in infancy that captures host progenitor regulation processes and allows us to compare these with the picture of inflammation in childhood, thus serving as an indicator that mirrors deficiencies in all other immune compartments.
Further interpretation of elevated numbers of CB IL‐3/GM‐CSF‐responsive, ‘immature’ Eo/B progenitors would accordingly deem infants to be at increased risk of eosinophilic and neutrophilic airway inflammation in response to viral ARI, accompanied by symptoms. For instance, local generation of IL‐3 and/or GM‐CSF would be an integral component of host defence mechanisms, which in turn may result in fever. The immunological progenitor profiles of infants with ARI accompanied by fever differ from infants who acquired ARI without fever. An imbalance in these profiles is more evident in the symptomatic group, as demonstrated by significant correlations between febrile ARI and IL‐3 Eo/B CFU and an inverse significant correlation between febrile ARI and CD34+ cells. This indicates that decreased numbers, but increased ratios of undifferentiated CD34+ progenitors to mature immune cells exist during ARI with fever. The profile that occurs with ARI alone may have this imbalance to a lesser degree, as evidenced by significant correlations between IL‐3R and GM‐CSFR phenotypes with ARI in the first year of life. A similar imbalance can also be attributed to ARI with wheeze, as indicated by the findings that GM‐CSF‐stimulated CFU positively correlated with overall frequency of ARI accompanied by wheeze during the first year of life (p = 0.023). Therefore, imbalances in progenitor phenotypic and functional responses appear to be more evident in symptomatic ARI, as opposed to ARI alone.
The role of wheeze with respect to ARI and progenitors assessed in this age group is slightly more complex to interpret. Given the central role of eosinophils and neutrophils in viral wheeze in infancy (22), this finding provides a plausible mechanism for viral induced wheeze, often termed transient, in at‐risk children in this age group. Martinez et al. (24) found that the wheeze present in high‐risk infants may be transient or remain persistent through childhood, and have suggested that 15% of infants who wheeze progress to chronic asthma, mostly those associated with familial atopic disease . These principles have been extended by other groups to incorporate the bronchoalveolar lavage (BAL) profile of different subtypes of wheezers, showing that the total number of lymphocytes and eosinophils in the BAL is increased in wheezing infants compared with controls; this may parallel our findings of increased numbers of ‘immature progenitors’ in CB (25, 26). RSV may also contribute directly and indirectly to the factor of wheeze, given that 80% of infants experience at least one RSV ARI in the first year of life. Whereas severe RSV ARI are thought to be accompanied by high IL‐4/IFN‐γ production, both Th1 and Th2 type responses occur during this infection (27). Our results showed that RSV ARI at 1 yr of age were not significantly correlated with phenotypic or functional changes in CD34+ cells, with the exception of febrile RSV ARI, which tended towards significance with IL‐3 Eo/B CFU (p = 0.056), consistent with our positing an earlier, perhaps more ‘immature’ progenitor in these high‐risk infants.
Other groups have indicated that, in subjects with ARI (28) and in children under 3 yr who had severe recurrent wheeze (29), bronchial inflammation is mainly neutrophilic, consistent with our finding of an increased availability of progenitors committed to both eosinophilic and neutrophilic lineages in these subjects.
In our study, ARI could be triggers and/or propagators of CD34+ progenitor subset IL‐3/GM‐CSF‐responsiveness, leading to increased episodes of concomitant fever and wheeze. Nonetheless, it is also likely that many of these high‐risk children have parallel defects in other immune and inflammatory pathways that contribute towards enhanced susceptibility to ARI. In particular, we have established that a central element of genetic risk for atopy and asthma is transient developmental deficiency in adaptive immune function, which manifests as attenuated capacity to secrete both Th1 and Th2 cytokines (9), and this at‐risk immunophenotype is already discernible in the fetal compartment (17). Comparable atopic risk‐related developmental deficiencies have also been reported in the innate compartment in monocytic/dendritic cell lineages (30), and our recent studies (31) indicate that the latter also are associated with increased susceptibility to ARI.
Thus, our findings of increased numbers of ‘immature’ CB progenitors in relation to more symptoms in infancy lead us to speculate that susceptibility to ARI and atopy later in life is complicated, challenging a simplistic rendition of the hygiene hypothesis which states that increased exposure to infections protects against atopy/asthma later in childhood (2). We postulate that atopic high‐risk infants have developmental deficiencies in many compartments, perhaps in conjunction with genetic lung deficiencies and environmental triggers. This has been shown in the Childhood Origins of ASThma (COAST) study: cytokine dysregulation accompanied by environmental insult could predispose high‐risk infants to atopic asthma (32) and predict immune and therapeutic profiles later in childhood (33). Current concepts of the immunological basis for risk of asthma/atopy must now include altered functional status of cell populations within the bone marrow as well as the cord blood. The common factor for risk of disease development appears to be postnatal persistence of functionally immature cell phenotypes in several (myeloid and lymphoid) compartments. It is yet to be determined how discrete, or lineage‐specific, genetic mechanisms and/or environmental factors are involved.
Acknowledgments
I would especially like to thank Dr. Malcolm Sears for kindly offering his advice on the manuscript and the statistics. Additionally, I would like to thank Holly Hatfield for her contributions to this study. I also extend my thanks to Lynn Crawford and Lynne Larocque for their technical assistance and support. This research was supported by grants from the Canadian Institutes of Health Research, the National Health and Medical Research Council of Australia, and AllerGen NCE Inc.
References
- 1. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee . Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. Lancet 1998: 351: 1225–32. [PubMed] [Google Scholar]
- 2. Strachan DP. Hay fever, hygiene, and household size. Br Med J 1989: 299: 1259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Farooqi IS, Hopkin JM. Early childhood infection and atopic disorder. Thorax 1998: 53: 927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matricardi PM, Rosmini F, Riondino S, et al. Exposure to foodborne and orofecal microbes versus airborne viruses in relation to atopy and allergic asthma: epidemiological study. Br Med J 2000: 320: 412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cooper PJ, Chico ME, Rodrigues LC, et al. Reduced risk of atopy among school‐age children infected with geohelminth parasites in a rural area of the tropics. J Allergy Clin Immunol 2003: 111: 995–1000. [DOI] [PubMed] [Google Scholar]
- 6. Von Ehrenstein OS, VonMutius E, Illi S, Baumann L, Bohm O, Von Kries R. Reduced risk of hay fever and asthma among children of farmers. Clin Exp Allergy 2000: 30: 187–93. [DOI] [PubMed] [Google Scholar]
- 7. Martinez FD, Holt PG. Role of microbial burden in aetiology of allergy and asthma. Lancet 1999: 354 (Suppl. II): S12–5. [DOI] [PubMed] [Google Scholar]
- 8. Holt PG, Sly PD. Interactions between respiratory tract infections and atopy in the aetiology of asthma. Eur Respir J 2002: 19: 538–45. [DOI] [PubMed] [Google Scholar]
- 9. Holt PG, Clough JB, Holt BJ, et al. Genetic ‘risk’ for atopy is associated with delayed postnatal maturation of T‐cell competence. Clin Exp Allergy 1992: 22: 1093–9. [DOI] [PubMed] [Google Scholar]
- 10. Upham JW, Hayes LM, Lundahl J, Sehmi R, Denburg JA. Reduced expression of hemopoietic cytokine receptors on cord blood progenitor cells in neonates at risk for atopy. J Allergy Clin Immunol 1999: 104: 370–5. [DOI] [PubMed] [Google Scholar]
- 11. Dunstan JA, Mori TA, Barden A, et al. Maternal fish oil supplementation in pregnancy reduces interleukin‐13 levels in cord blood of infants at high risk of atopy. Clin Exp Allergy 2003: 33: 442–8. [DOI] [PubMed] [Google Scholar]
- 12. Denburg JA, Hatfield HM, Cyr MM, et al. Fish oil supplementation in pregnancy modifies neonatal progenitors at birth in infants at risk of atopy. Pediatr Res 2005: 57: 276–81. [DOI] [PubMed] [Google Scholar]
- 13. Gauvreau GM, Denburg JA. Hemopoietic progenitors: the role of eosinophil/basophil progenitors in allergic airway inflammation. Expert Rev Clin Immunol 2005: 1: 87–101. [DOI] [PubMed] [Google Scholar]
- 14. Johnston SL, Sanderson G, Pattemore PK, et al. Use of polymerase chain reaction for diagnosis of picornavirus infection in subjects with and without respiratory symptoms. J Clin Microbiol 1993: 31: 111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papadopoulos NG, Moustaki M, Tsolia M, et al. Association of rhinovirus infection with increased disease severity in acute bronchiolitis. Am J Respir Crit Care Med 2002: 165: 1285–9. [DOI] [PubMed] [Google Scholar]
- 16. Seemungal T, Harper‐Owen R, Bhowmik A, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001: 164: 1618–23. [DOI] [PubMed] [Google Scholar]
- 17. Macaubas C, DeKlerk NH, Holt BJ, et al. Association between antenatal cytokine production and the development of atopy and asthma at age 6 years. Lancet 2003: 362: 1192–7. [DOI] [PubMed] [Google Scholar]
- 18. Sehmi R, Wood LJ, Watson R, et al. Allergen‐induced increases in IL‐5 receptor α‐subunit expression on bone marrow‐derived CD34+ cells from asthmatic subjects. A novel marker of progenitor cell commitment toward eosinophilic differentiation. J Clin Invest 1997: 100: 2466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sehmi R, Howie K, Sutherland DR, Schragge W, O'Byrne PM, Denburg JA. Increased levels of CD34+ hemopoietic progenitor cells in atopic subjects. Am J Respir Cell Mol Biol 1996: 15: 645–54. [DOI] [PubMed] [Google Scholar]
- 20. Denburg JA, Gauldie J, Dolovich J, Ohtoshi T, Cox G, Jordana M. Structural cell‐derived cytokines in allergic inflammation. Int Arch Allergy Appl Immunol 1991: 94: 127–32. [DOI] [PubMed] [Google Scholar]
- 21. Dunstan JA, Mori TA, Barden A, et al. Fish oil supplementation in pregnancy modifies neonatal allergen‐specific immune responses and clinical outcomes in infants at high risk of atopy: a randomised control trial. J Allergy Clin Immunol 2004: 112: 1178–84. [DOI] [PubMed] [Google Scholar]
- 22. Holt PG. Postnatal maturation of immune competence during infancy and childhood. Pediatr Allergy Immunol 1995: 6: 59–70. [DOI] [PubMed] [Google Scholar]
- 23. Tavernier J, Van der Heyden J, Verhee A, et al. Interleukin 5 regulates the isoform expression of its own receptor alpha‐subunit. Blood 2000: 95: 1600–7. [PubMed] [Google Scholar]
- 24. Martinez FD, Wright AL, Taussig LM, et al. Asthma and wheezing in the first six years of life. N Engl J Med 1995: 332: 133–8. [DOI] [PubMed] [Google Scholar]
- 25. Krawiec ME, Westcott JY, Chu HW, et al. Persistent wheezing in very young children is associated with lower respiratory inflammation. Am J Respir Crit Care Med 2001: 163: 1338–43. [DOI] [PubMed] [Google Scholar]
- 26. Stevenson EC, Turner G, Heaney LG, et al. Bronchoalveolar lavage findings suggest two different forms of childhood asthma. Clin Exp Allergy 1997: 27: 1027–35. [DOI] [PubMed] [Google Scholar]
- 27. Psarras S, Papadopoulos NG, Johnston SL. Pathogenesis of respiratory syncytial virus bronchiolitis‐related wheezing. Paediatr Respir Rev 2004: 5 (Suppl. A): S179–84. [DOI] [PubMed] [Google Scholar]
- 28. Jarjour NN, Gern JE, Kelly EA, Swenson CA, Dick CR, Busse WW. The effect of an experimental rhinovirus 16 infection on bronchial lavage neutrophils. J Allergy Clin Immunol 2000: 105: 1169–77. [DOI] [PubMed] [Google Scholar]
- 29. Le Bourgeois M, Goncalves M, Le Clainche L, et al. Bronchoalveolar cells in children <3 years old with severe recurrent wheezing. Chest 2002: 122: 791–7. [DOI] [PubMed] [Google Scholar]
- 30. Hagendorens MM, Ebo DG, Schuerwegh AJ, et al. Differences in circulating dendritic cell subtypes in cord blood and peripheral blood of healthy and allergic children. Clin Exp Allergy 2003: 33: 633–9. [DOI] [PubMed] [Google Scholar]
- 31. Upham JW, Rate A, Kusel M, Sly PD, Holt PG. The frequencies of plasmacytoid and myeloid dendrite cell subsets in infancy are differentially associated with risk for viral respiratory infections, allergic sensitization and asthma. Proc Am Thorac Soc 2006; 3: A827. [Google Scholar]
- 32. Lemanske RF, Jr. The childhood origins of asthma (COAST) study. Pediatr Allergy Immunol 2002: 13 (Suppl. 15): 38–43. [DOI] [PubMed] [Google Scholar]
- 33. Niggemann B. Functional symptoms confused with allergic disorders in children and adolescents. Pediatr Allergy Immunol 2002: 13: 312–8. [DOI] [PubMed] [Google Scholar]