

Abstract
The authors have previously shown that acute lung injury (ALI) produces a wide spectrum of pathological processes in patients who die of severe acute respiratory syndrome (SARS) and that the SARS coronavirus (SARS‐CoV) nucleoprotein is detectable in the lungs, and other organs and tissues, in these patients. In the present study, immunohistochemistry (IHC) and in situ hybridization (ISH) assays were used to analyse the expression of angiotensin‐converting enzyme 2 (ACE2), SARS‐CoV spike (S) protein, and some pro‐inflammatory cytokines (PICs) including MCP‐1, TGF‐β1, TNF‐α, IL‐1β, and IL‐6 in autopsy tissues from four patients who died of SARS. SARS‐CoV S protein and its RNA were only detected in ACE2+ cells in the lungs and other organs, indicating that ACE2‐expressing cells are the primary targets for SARS‐CoV infection in vivo in humans. High levels of PICs were expressed in the SARS‐CoV‐infected ACE2+ cells, but not in the uninfected cells. These results suggest that cells infected by SARS‐CoV produce elevated levels of PICs which may cause immuno‐mediated damage to the lungs and other organs, resulting in ALI and, subsequently, multi‐organ dysfunction. Therefore application of PIC antagonists may reduce the severity and mortality of SARS. Copyright © 2006 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: SARS, SARS‐CoV, ACE2, acute lung injury, pro‐inflammatory cytokines
Introduction
Severe acute respiratory syndrome (SARS), an emerging infectious disease with significant morbidity and mortality, is caused by a novel coronavirus (SARS‐CoV) which mainly infects lung and bronchial tissues, leading to acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) 1, 2, 3, 4, 5. In vitro studies have shown that angiotensin‐converting enzyme 2 (ACE2) is the functional receptor for SARS‐CoV 6, 7, 8, 9. Human 293T or murine 3T3 cells transfected with human ACE2, but not those transfected with HIV‐1 receptors (CD4 and CCR5), permit SARS‐CoV entry. The soluble and cell‐associated ACE2 can specifically bind to the receptor‐binding domain (RBD) in the spike (S) protein of SARS‐CoV 6, 7, 8, 9. Most recently, Kuba et al demonstrated that ACE2‐knockout mice become unsusceptible to SARS‐CoV challenge, providing the first genetic proof that ACE2 plays a critical role in SARS‐CoV infection in vivo in animals 10. However, it is unclear whether ACE2‐expressing cells in humans can be infected in vivo by SARS‐CoV. The mechanisms of ALI and the pathogenesis of SARS have not been well defined.
In this study, we systematically analysed the expression of ACE2, SARS‐CoV spike (S) protein and pro‐inflammatory cytokines (PICs), and their RNAs, in autopsy specimens from SARS patients.
Materials and methods
Clinical materials
Autopsy samples of different organs and tissues were obtained from four patients who died of SARS in the Eighth People's Hospital of Guangzhou City, the Second Affiliated Hospital of Zhongshan University, and the Guangzhou Institute of Respiratory Diseases, with documented permissions for autopsies. All four cases met the diagnostic criteria for SARS defined by WHO 11. Detailed clinical data and pathological findings of these SARS victims have been reported previously 12, 13. Corresponding autopsy specimens were also collected from a young male who died suddenly in a traffic accident and had no detectable infection by SARS‐CoV or other pathogens, as a control subject. The ethical issues related to this study were reviewed and approved by the Research Administration Committees of the Southern Medical University (the former First Military Medical University) and the hospitals involved.
Histopathological examination
Lung specimens were fixed with 4% neutral formaldehyde, embedded in paraffin wax, and 4 µm sections were cut. Sections were stained with haematoxylin and eosin (H&E). Some of the sections of bronchial serous glands were also stained with Alcian blue (pH 2.5) to reveal acid mucosubstances.
Immunohistochemistry (IHC)
A monoclonal antibody (MAb) directed against a recombinant SARS‐CoV S protein was generated and characterized as previously described 14. MAbs specific for ACE2 and PICs, including TGF‐β1, and MCP‐1, IL‐1β, IL‐6, and TNF‐α were purchased from R&D (Minneapolis, MN, USA). MAbs against cell markers—Mac387, CD4, CD8, CD25, CD45RO, CD68, CK, and EMA—and the reaction kits for horseradish peroxidase (HRP) and alkaline phosphatase (AP) were purchased from DAKO (Carpinteria, CA, USA). Antibodies to Fas and Fas‐L were obtained from Maixin, Inc (Fuzhou, China). IHC was performed as previously described 15. Briefly, the autopsy samples were sectioned at 4 µm and pretreated with 3% hydrogen peroxide to quench endogenous peroxidase activity and with microwave‐induced heat for epitope retrieval. Slides were incubated with antibodies in a humid chamber at 4 °C overnight. Antibody binding to the cells in sections was detected with HRP or AP reaction kits according to the manufacturers' instructions.
In situ hybridization (ISH)
Details of the in situ hybridization methodology are given in the supplementary material, available at http://www.interscience.wiley.com/jpages/0022-3417/suppmat/path.2067.html.
Detection of DNA fragments in apoptotic cells
The Klenow‐FragEL DNA fragmentation kit (Oncogene Research Products, Germany) was used to evaluate apoptosis according to the manufacturer's instructions. Briefly, deparaffinized and rehydrated slides were rinsed in TBS and incubated in proteinase K for 20 min at room temperature. Endogenous peroxidases were blocked by 5 min incubation in 3% H2O2 in methanol. The slides were incubated with equilibration buffer for 30 min and then with the Klenow Enzyme for 2 h at room temperature. The entire specimens were covered with blocking buffer and incubated at room temperature for 10 min, followed by the immediate addition of 1× conjugate. After incubation in the stop solution for 5 min, the slides were counterstained for examination.
Results
Co‐expression of ACE2 and SARS‐CoV S protein in cells in the lungs and other organs in SARS patients
Our previous studies have shown that SARS‐CoV N protein and its RNA can be detected in epithelial cells, pneumocytes, and macrophages in lung and bronchial tissues, indicating that these cells may be infected by SARS‐CoV 15. However, it is unclear whether these SARS‐CoV‐infected cells express ACE2, the functional receptor for SARS‐CoV demonstrated in the in vitro and animal in vivo studies 6, 7, 10. Using MAbs specific for the human ACE2 protein and the SARS‐CoV S protein by IHC, as previously described 15, we systematically analysed the expression of both ACE2 and SARS‐CoV S protein in the autopsy specimens that were randomly collected from the lungs and other organs of four patients who died of SARS and one control subject who died suddenly in a traffic accident. In the lung and bronchial tissues of the SARS patients and control subject, ACE2 protein was detected in the epithelial cells of alveoli, trachea/bronchi, and bronchial serous glands, and in the pneumocytes and monocytes/macrophages in alveolar cavities (Figure 1 and Table 1). The anti‐ACE2 MAb was also reactive with cells in other organs and tissues of the SARS patients and control subject, including gastric parietal cells, small intestinal epithelial cells, liver cells, myocardial cells, distal convoluted renal tubular cells, adrenal cortical cells, pancreatic islet cells, sweat gland epithelial cells, and acidophilic cells of the pituitary (Figure 2 and Table 1). Interestingly, all of the ACE2‐expressing cells in the SARS patients were reactive with the MAb directed against the SARS‐CoV S protein (Figures 1 and 2 and Table 1). However, the anti‐S MAb did not react with the ACE2− cells (eg bronchial mucous gland epithelial cells, gastric chief cells, thyroid follicular epithelial cells) in the SARS patients, nor did it react with ACE2+ and ACE2− cells in the control subject (Figures 1 and 2 and Table 1). We also detected the RNAs encoding the human ACE2 protein and the SARS‐CoV S protein in the autopsy specimens using the ISH assay as previously described 15. The distribution of ACE2 and S RNAs detected by ISH in the autopsy tissues is fully consistent with that of ACE2 and S protein measured by IHC (see Supplementary Figure 1 at http://www.interscience.wiley.com/jpages/0022-3417/suppmat/path.2067.html).
Figure 1.
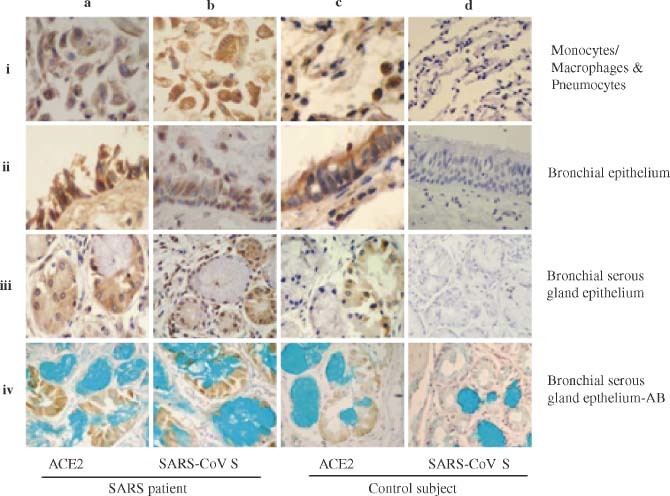
Detection of ACE2 and SARS‐CoV S protein in the lung and bronchial tissues by IHC. The MAbs against human ACE2 and the SARS‐CoV S protein were used to detect the expression of ACE2 (a–i to a–iv and c–i to c–iv) and SARS‐CoV S protein (b–i to b–iv and d–i to d–iv) in monocytes/macrophages and pneumocytes (a–i to d–i), bronchial epithelial cells (a–ii to d–ii), bronchial serous gland epithelium (a–iii to d–iii), and bronchial serous gland epithelium‐AB (a–iv to d–iv) in the SARS patients (a–i to b–iv) and control subject (c–i to d–iv). All sections were counter‐stained with haematoxylin, except the sections of bronchial serous gland epithelium‐AB, which were counter‐stained with haematoxylin and Alcian blue
Table 1.
Expression of ACE2, SARS‐CoV S protein, and pro‐inflammatory cytokines in specific tissues and cells in the SARS patients and control subject
| Tissues or cells | SARS patients | Control subject | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACE2 | SARS‐CoV S | MCP‐1 | TGF‐β | IL‐1β | IL‐6 | TNF‐α | ACE2 | SARS‐CoV S | MCP‐1 | TGF‐β | IL‐1β | IL‐6 | TNF‐α | |
| Alveolar epithelial cells | + + + | + + + | + + + | + + + | + + | + + | + + | + + | − | − | − | − | − | − |
| Bronchial epithelial cells | + + + | + + + | + + + | + + + | + + | + + | + + | + + | − | − | − | − | − | − |
| Bronchial serous gland epithelial cells | + + + | + + + | + + + | + + + | + + | + + | + + | + + + | − | − | − | − | − | − |
| Monocytes/macrophages | + + + | + + + | + + + | + + + | + + | + + | + + | + + | − | − | − | − | − | − |
| Bronchial mucous gland epithelial cells | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Gastric parietal cells | + + + | + + + | + + + | + + + | + + | + + | + + | + + + | − | − | − | − | − | − |
| Small intestinal epithelial cells | + + + | + + + | + + + | + + | − | − | − | + + | − | − | − | − | − | − |
| Hepatocytes | + | + | + + + | + + + | + | + | + + | + | − | − | − | − | − | − |
| Gastric chief cells | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Myocardial cells | + + + | + + + | + + + | + + + | + + | + + | + + | + + + | − | − | − | − | − | − |
| Distal convoluted renal tubules | + + + | + + + | + + + | + + + | + + + | + + + | + + | + + + | − | − | + | − | − | + |
| Adrenal cortical cells | + + + | + + + | + + + | + + | + + + | + | + | + + + | − | − | − | − | − | − |
| Pancreatic islet cells | + + + | + + + | + + + | + + + | + + + | + + + | + + | + + + | − | − | − | − | − | − |
| Acidophilic cells of the parathyroid | + + | + + | − | − | − | − | − | + + | − | − | − | − | − | − |
| Sweat gland epithelial cells | + + + | + + + | + + + | + + + | + + + | + + + | + + + | + + + | − | − | − | − | − | − |
| Acidophilic cells of the pituitary | + + + | + + + | + + + | + + + | + | + | + + + | + + + | − | − | − | − | − | − |
| Follicular epithelial cells of the thyroid | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
The number of positive cells and the total number of cells in the specified tissues were counted in ten high‐power views. The percentage of positive cells was calculated. 0%, 1–24%, 25–49%, 50–74%, and ≥ 75% positive cells were recorded as −, +, + +, + + +, and + + + +, respectively.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 2.

Detection of ACE2 and SARS‐CoV S protein in other organs and tissues by IHC. The MAbs against human ACE2 and the SARS‐CoV S protein were used to detect the expression of ACE2 (a–i to a–v and c–i to c–v) and SARS‐CoV S protein (b–i to b–v and d–i to d–v) in myocardial cells (a–i to d–i), gastric parietal cells (a–ii to d–ii), distal convoluted renal tubules (a–iii to d–iii), pancreatic islet (a–iv to d–iv), and sweat gland (a–v to d–v) in the SARS patients (a–i to b–v) and control subject (c–i to d–v). All sections were counter‐stained with haematoxylin
Expression of elevated levels of the pro‐inflammatory cytokines (PICs) in cells expressing both ACE2 and SARS‐CoV S proteins in the lungs and other organs in SARS patients
Subsequently, we examined the expression of PICs, including macrophage chemoattractant protein‐1 (MCP‐1), transforming growth factor‐β1 (TGF‐β1), tumour necrosis factor‐α (TNF‐α), interleukin‐1β (IL‐1β) and interleukin‐6 (IL‐6), in the autopsy specimens of the lungs and other organs of the SARS patients and control subject. This was accomplished by IHC using MAbs against the PICs. As shown in Figure 3 and Table 1, most of the cells expressing both ACE2 and SARS‐CoV S protein were strongly reactive with the MAbs against MCP‐1 and TGF‐β1 and reacted significantly with the MAbs to TNF‐α, IL‐1β, and IL‐6, respectively. However, these MAbs exhibited weak or no reactivity with the ACE2− cells in the SARS patients, and with the ACE2+ and ACE2− cells in the control subject. Similar results were obtained from the ISH assay for detecting the RNAs of MCP‐1, TGF‐β1, TNF‐α, IL‐1β, and IL‐6 in the corresponding cells and tissues (see Supplementary Figure 2 at http://www.interscience.wiley.com/jpages/0022-3417/suppmat/path.2067.html). These data suggest that PICs are overproduced in the SARS‐CoV‐infected ACE2+ cells in SARS patients.
Figure 3.
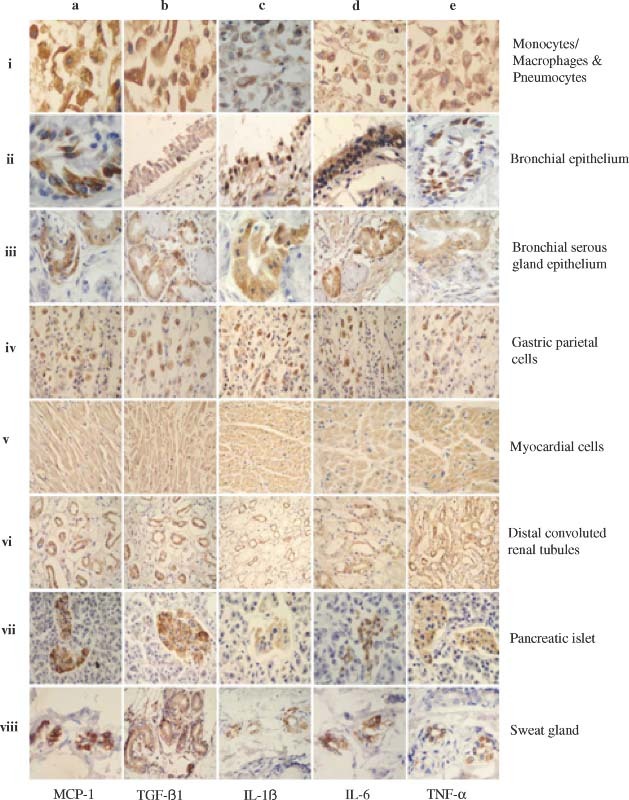
Detection of pro‐inflammatory cytokines in the lungs and other organs and tissues by IHC. The MAbs against human MCP‐1, TGF‐β1, IL‐1β, IL‐6, and TNF‐α, respectively, were used to detect the expression of MCP‐1 (a–i to a–viii), TGF‐β1 (b–i to b–viii), IL‐1β (c–i to c–viii), IL‐6 (d–i to d–viii), and TNF‐α (e–i to e–viii) in monocytes/macrophages and pneumocytes (a–i to e–i), bronchial epithelial cells (a–ii to e–ii), bronchial serous gland epithelium (a–iii to e–iii), gastric parietal cells (a–iv to e–iv), myocardial cells (a–v to e–v), distal convoluted renal tubules (a–vi to e–vi), pancreatic islet (a–vii to e–vii), and sweat gland cells (a–viii to e–viii) in the SARS patients. All sections were counter‐stained with haematoxylin
Pathological changes in the lungs of the SARS patients
Histopathological studies conducted in our laboratories and by other groups on biopsy tissues of SARS patients have shown that the most severe damage caused by SARS‐CoV infection occurs in the respiratory and immune systems 12, 16, 17. SARS‐CoV may also infect and cause pathological changes in other organs, including the digestive tract, liver, kidneys, adrenal glands, heart, brain, etc 12, 15, 17. Here we further examined the pathological changes in the lungs of the SARS patients. All the SARS patients exhibited diffuse alveolar damage (DAD). The alveolar cavities were filled with desquamated epithelial cells (Figure 4A), some of which were clearly enlarged and contained multiple nuclei (Figure 4B). This suggests that these cells had undergone fusion to form syncytia, which is the cytopathic effect (CPE) caused by virus replication in cells 3, 12. T‐lymphocyte infiltrates as shown by CD45RO staining (Figure 4C), the formation of hyaline membranes resulting from fibrin deposition (Figure 4D), organization and fibrinosis of alveolar exudates (Figure 4D), and local infiltration of monocytes/macrophages and lymphocytes in pulmonary interstitial tissues (Figure 4F) were observed. Obvious exudation of proliferative and/or activated monocytes/macrophages in alveoli (Figures 4G–4I) was revealed. There was significant apoptosis in epithelial cells, monocytes/macrophages, lymphocytes, and pneumocytes as revealed by Klenow‐FragEL staining (Figure 4J), and the detection of Fas (Figure 4K) and FasL (Figure 4L). These data suggest that the main histological changes, including infiltration of macrophages and desquamated epithelial cells, and significant apoptosis of epithelial cells and pneumocytes in the lungs, which may be the major causes of ALI, may be associated with up‐regulation of PICs in the SARS‐CoV‐infected cells and the elevated levels of PICs in the blood of SARS patients.
Figure 4.
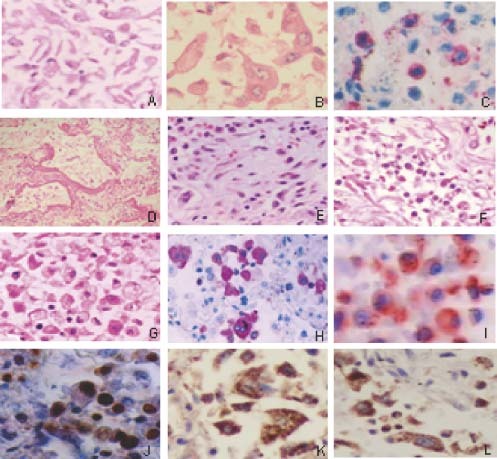
Pathological changes in the lungs of SARS patients. (A) Alveoli filled with desquamated epithelial cells (H&E); (B) mononuclear and multinucleate epithelial giant cells in alveoli (H&E); (C) T‐lymphocyte infiltrates (CD45RO staining); (D) formation of hyaline membranes (H&E); (E) organization and fibrinosis of alveolar exudates; (F) exudation of monocytes/macrophages and lymphocytes in pulmonary interstitial tissues (H&E); (G) exudation of monocytes/macrophages and lymphocyte in alveoli (H&E); (H) obvious proliferation of macrophages in the alveoli cavities (Mac387 staining positive); (I) active macrophages in the alveoli (CD25 staining positive); and apoptosis in the pneumocytes as shown by Klenow‐FragEL (J), Fas (K), and FasL (L) staining
Discussion
Although in vitro and animal in vivo studies have shown that ACE2 is the functional receptor for SARS‐CoV 6, 7, 8, 9, 10 and ACE2 is abundantly present in humans in the epithelia of the lungs, small intestine, and other organs 18, there is no direct evidence to show that ACE2‐expressing cells are the targets for SARS‐CoV infection in vivo in humans. Here we have demonstrated that the SARS‐CoV S protein, which is responsible for virus binding and entry, is detected in the ACE2‐expressing cells in the lungs and other organs of SARS patients. However, it was undetectable in the ACE2− cells in the SARS patients and in both ACE2+ and ACE2− cells in the control subject. ACE2 and S RNA expression was also detected in the cells expressing ACE2 and S proteins. The co‐detection of the SARS‐CoV S protein and its RNA in the ACE2‐expressing cells in the lungs and other organs of SARS patients suggests that only the ACE2+ cells in these tissues are susceptible to SARS‐CoV infection.
It has been reported that SARS‐CoV can infect and replicate in human lung epithelial cells 19. Macrophages and monocyte‐derived dentritic. cells (DCs) are not permissive for SARS‐CoV replication, but interaction of SARS‐CoV with these cells leads to up‐regulation of the expression of pro‐inflammatory cytokines (PICs), which may play a role in the pathogenesis of SARS 20, 21. However, it is unclear whether SARS‐CoV infection induces increased expression of PICs in the infected cells in SARS patients.
We identified the proteins and RNAs of five selected PICs using IHC and ISH methods, respectively. Strikingly, high levels of MCP‐1 and TGF‐β1, and intermediate levels of TNF‐α, IL‐1β, and IL‐6 were detected in the SARS‐CoV‐infected ACE2‐expressing cells, especially in the epithelial cells, pneumocytes, and macrophages in the lung and bronchial tissues. However, these PICs were either weakly detected in the ACE2− cells in the SARS patients and in both ACE2+ and ACE2− cells in the control subject, or not at all (Table 1). These data suggest that the expression of PICs is up‐regulated in the SARS‐CoV‐infected ACE2+ cells in SARS patients, which may lead to ALI and multi‐organ dysfunction.
This proposition is further supported by clinical observations. The levels of PICs, eg IL‐6, IL‐8, and TNF‐α, in the blood were rapidly elevated during the early phase of SARS and the timing of PIC elevations is associated with progression of pulmonary infiltrates and damage 22, 23, 24, 25. The elevated levels of PICs are sensitive correlates of disease severity and viral load in serum 26. Furthermore, dysregulation of cytokines may account for the haemophagocytosis and severity of SARS 16.
PICs are the mediators of hyper‐immune responses induced by many infectious pathogens 22, 23, 24, 25, 27, 28, 29 and can significantly damage host organs and tissues. In this study, we found that high concentrations of MCP‐1 and TGF‐β1 were produced in the SARS‐CoV‐infected ACE2‐expressing cells, suggesting that these two PICs may play major roles in the pathogenic mechanism of ALI. MCP‐1 can act as a chemotactic factor to induce the migration of monocytes/macrophages from the bloodstream to the affected sites to combat the invading pathogens. However, these cells can also attack uninfected bystander cells. Microscopic examination of the SARS patients' lungs revealed significant infiltration and aggregation of macrophages in the alveolar cavities. Under stimulation by some PICs (eg TGF‐β1 and TNF‐α, etc), macrophages become proliferative and/or activated 30, which in turn produces more PICs, leading to further immuno‐mediated damage of the tissues in the host 31. The majority of the macrophages in the alveolar cavities showed signs of proliferation (Mac387+) and/or activation (CD25+).
TGF‐β1 is a mediator that can enhance Fas‐mediated cell apoptosis, which may be the main cause of death of alveolar epithelial cells, lymphocytes, and platelets, leading to ALI, lymphopenia and thrombocytopenia in SARS patients 32, 33, 34. Expression of Fas and FasL and apoptosis were observed in epithelial cells and pneumocytes in the lungs of the SARS patients. TNF‐α may play critical roles in the process of pulmonary fibrosis since it can stimulate the proliferation of fibroblasts to synthesize a large amount of collagen fibres and extracellular matrix, resulting in subsequent fibrinogen exudates 35, 36, 37. The deposited fibrin, when mixed with remnants of necrotic epithelial cells, forms hyaline membranes and subsequent fibrinosis, both of which were observed in the lungs of all of the SARS patients. IL‐1β and IL‐6, an endogenous pyrogen, can stimulate the production of C‐reactive protein (CRP) that mediates inflammatory responses 38.
Based on the findings from this study, we have proposed a model to elucidate the possible relationship between the up‐regulation of PIC expression and ALI, as well as the pathogenesis of SARS (Figure 5). SARS‐CoV‐infected cells produce high levels of PICs in order to combat the invading virus. However, the overproduced PICs may also attack the host cells, including both the infected and the uninfected cells, resulting in significant ALI and ARDS. The elevated levels of PICs in blood released from the SARS‐CoV‐infected cells and the uninfected cells stimulated by viral antigens or some PIC‐regulatory factors, eg nuclear factor‐kappaB (NF‐κB) and p38 (a mitogen‐activated protein kinase) 39, 40, 41, may damage the cells in the lungs and other organs, eg small intestine, kidney, adrenal gland, liver, and brain, etc. In addition, blood hypo‐oxygenation due to ARDS and disseminated intravascular coagulation resulting from impairment of microvessel endothelial cells may further damage the structure and function of different organs in SARS patients, resulting in multi‐organ dysfunction.
Figure 5.
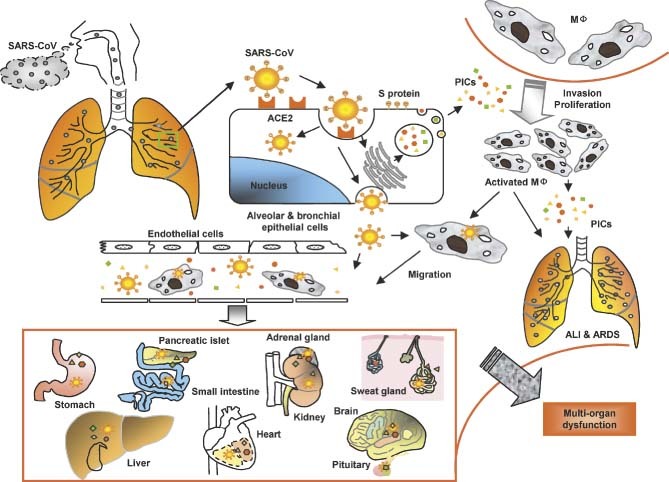
A model for the immunopathogenesis of SARS. SARS‐CoV in droplets enters into the lung, where the virus binds via its S protein to ACE2 on the alveolar or bronchial epithelial cells. The virus replicates in these cells, from which new virions are released into the blood. The infected cells under the stimulation of SARS‐CoV and some uninfected cells induced by viral antigens or PIC‐regulatory factors produce high levels of PICs to mediate inflammatory responses for combating the virus. However, these PICs also damage the host cells. Some of the PICs, eg monocyte chemoattractant protein‐1 (MCP‐1), attract monocytes in blood to migrate to the alveolar cavities, where the monocytes are stimulated by other PICs to become proliferative and/or activated macrophages (MΦ). The activated macrophages can produce more PICs and may transmit SARS‐CoV to other sites. Some of the PICs, including TGF‐β1 and TNF‐α, may induce apoptotic death of the epithelial cells, pneumocytes, and lymphocytes, or mediate pulmonary fibronosis, resulting in ALI and ARDS. The cell‐free and MΦ‐associated SARS‐CoV in the blood can be transmitted from the lung to other organs to infect the ACE2‐expressing cells in the local sites. More PICs are produced and the level of PICs in the blood is rapidly elevated, leading to multi‐organ dysfunction. MΦ = macrophages; ALI = acute lung injury; PICs = pro‐inflammatory cytokines
Most SARS patients have pronounced peripheral T‐cell lymphopenia and spleen and lymph node atrophy 12, 17, 42. However, depletion of T lymphocytes in the blood and lymph tissues may not be due to CPE‐mediated cell death because of SARS‐CoV infection and replication in these cells. Human T lymphocytes lack ACE2 expression 18, 43 and therefore they may not be permissive to SARS‐CoV replication. Furthermore, no significant CPE in T lymphocytes has been observed in the autopsy tissues of SARS patients 12. We believe that the rapid depletion of T lymphocytes may be caused by overproduced PICs because the increase of blood PIC levels correlates with the decrease of T lymphocytes in the blood of SARS patients 22, 44. As discussed above, some of the PICs may enhance T‐lymphocyte apoptosis 32, 33, 34. Some cytokines may mobilize the T lymphocytes in blood and immune organs to the local inflammatory sites 45, resulting in lymphopenia in the blood and atrophy of the spleen and lymph nodes.
Moreover, immunodeficiency due to T‐cell lymphopenia is unlikely to be the primary cause of acute syndrome and death. Unlike SARS‐CoV, HIV can directly infect and replicate in CD4+ T lymphocytes, resulting in rapid death of these cells 46. Even so, HIV infection does not cause acute disease. HIV‐infected individuals take several years to progress to AIDS and opportunistic infection is the main cause of death in patients with HIV/AIDS 47. Since the elevated levels of PICs may be the major mediators of the exaggerated immune responses that cause ALI and multi‐organ dysfunction in SARS patients, the use of immunosuppressive agents may effectively inhibit the production and function of PICs and reduce the severity of ALI. Corticosteroids were the treatment of choice of SARS during 2003–2004. However, several groups reported that early application of large doses of corticosteroids with Ribavirin, a non‐specific antiviral drug, for treating SARS patients did not show significant clinical benefits 48, 49 and even caused serious adverse effects, eg osteonecrosis 50 and aspergillosis 13, though the data were not obtained from well‐designed double‐blind, randomized, and placebo‐controlled clinical trials. One concern is that corticosteroids may non‐specifically suppress the function of most immuno‐modulators rather than specifically inhibit the activities of those causing harmful inflammation. Therefore, specific regimens may be designed to include antagonists specifically against the individual PICs, eg MCP‐1 and TGF‐β, that are highly expressed in the SARS‐CoV‐infected cells and in elevated levels in blood, plus antiviral drugs for the treatment of patients with SARS. Some drugs that selectively block pro‐inflammatory cytokines or their cell receptors may also be used for SARS treatment 51. The phenomenon of the rapidly increased levels of PICs in blood and their positive correlation with the severity of ALI has also been observed in patients who have been infected with other viruses, eg avian influenza virus subtype H5N1 27, 28, 52. This suggests that up‐regulation of PIC expression induced in the virus‐infected cells may be common mediators of ALI and ARDS in patients who have been infected with viruses via the respiratory pathway. Therefore the anti‐PIC strategy may also be applied, in combination with the specific antiviral therapeutics against the corresponding viruses, for the early treatment of these infectious diseases.
Supplementary material
Supplementary material may be found at the web address http://www.interscience.wiley.com/jpages/0022-3417/suppmat/path.2067.html
Supporting information
This article contains supplementary material available via the Internet from the Journal http://www.interscience.wiley.com/jpages/0022-3417/suppmat/path.2067.html
Supplementary Figure 1. Detection of ACE2 and SARS‐CoV S RNA in the lungs and other organs and tissues by ISH. ISH assay was used to detect ACE2 RNA (a‐i to a‐viii and c‐i to c‐viii) and SARS‐CoV S RNA (b‐i to b‐viii and d‐i to d‐viii) in pneumocytes (a‐i to d‐i), bronchial epithelial cells (a‐ii to d‐ii), bronchial serous gland epithelium (a‐iii to d‐iii), and myocardial cells (a‐iv to d‐iv), gastric parietal cells (a‐v to d‐v), distal convoluted renal tubules (a‐vi to d‐vi), pancreatic islet (a‐vii to d‐vii), sweat gland (a‐viii to d‐viii) in the SARS patient (a‐i to b‐viii) and the control subject (c‐i to d‐viii). All negatively stained samples were counterstained with nuclear Fast Red to identify cell nuclei
Supplementary Figure 2.Detection of proinflammatory cytokines in lung and other organs and tissues by ISH. ISH assay was used to detect the RNA of MCP‐1 (a‐i to a‐viii), TGF‐β1 (b‐i to b‐viii), IL‐1β (c‐i to c‐viii), IL‐6 (d‐i to d‐viii), and TNF‐α (e‐i to e‐viii) in monocytes/macrophages and pneumocytes (a‐i to e‐i), bronchial epithelial cells (a‐ii to e‐ii), bronchial serous gland epithelium (a‐iii to e‐iii), gastric parietal cells (a‐iv to e‐iv), myocardial cells (a‐v to e‐v), distal convoluted renal tubules (a‐vi to e‐vi), pancreatic islet (a‐vii to e‐vii), and sweat gland cells (a‐viii to e‐viii) in the SARS patients
Supplementary text
Acknowledgements
This work was supported by the Chinese National Foundation of Natural Sciences (No 30340015) and the SARS Foundation of China and Germany (No GZ228‐202‐1).
The authors of this article declared they have no financial conflicts of interest.
Contributor Information
Y Ding, Email: dyq@fimmu.com.
S Jiang, Email: sjiang@nybloodcenter.org.
REFERENCES
- 1. Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1953–1966. [DOI] [PubMed] [Google Scholar]
- 2. Drosten C, Gunther S, Preiser W, Van Der WS, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1967–1976. [DOI] [PubMed] [Google Scholar]
- 3. Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003; 361: 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marra MA, Jones SJM, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YSN, et al. The genome sequence of the SARS‐associated coronavirus. Science 2003; 300: 1399–1404. [DOI] [PubMed] [Google Scholar]
- 5. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003; 300: 1394–1399. [DOI] [PubMed] [Google Scholar]
- 6. Li WH, Moore MJ, Vasilieva NY, Sui JH, Wong SK, Berne AM, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426: 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS‐CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun 2003; 312: 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li W, Greenough TC, Moore MJ, Vasilieva N, Somasundaran M, Sullivan JL, et al. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin‐converting enzyme 2. J Virol 2004; 78: 11429–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He Y, Zhou Y, Liu S, Kou Z, Li W, Farzan M, et al. Receptor‐binding domain of SARS‐CoV spike protein induces highly potent neutralizing antibodies: implication for developing subunit vaccine. Biochem Biophys Res Commun 2004; 324: 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nature Med 2005; 11: 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. World Health Organization : Severe acute respiratory syndrome (SARS): multi‐country outbreak updated 34. http://www.who.int/csr/don/2003_04_02b/en/index.html.
- 12. Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 2003; 200: 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H, Ding Y, Li X, Yang L, Zhang W, Kang W. Fatal aspergillosis in a patient with SARS who was treated with corticosteroids. N Engl J Med 2003; 349: 507–508. [DOI] [PubMed] [Google Scholar]
- 14. Wen K, Mei YB, Qiu LW, Liao ZY, Yuen KY, Che XY. Preparation and characterization of monoclonal antibodies against S1 domain at N‐terminal residues 249 to 667 of SARS‐associated coronavirus S1 protein. Di Yi Jun Yi Da Xue Xue Bao 2004; 24: 1–6. [PubMed] [Google Scholar]
- 15. Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J, et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS‐CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 2004; 203: 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003; 361: 1773–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med 2005; 202: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004; 203: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ziegler T, Matikainen S, Ronkko E, Osterlund P, Sillanpaa M, Siren J, et al. Severe acute respiratory syndrome coronavirus fails to activate cytokine‐mediated innate immune responses in cultured human monocyte‐derived dendritic cells. J Virol 2005; 79: 13800–13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheung CY, Poon LL, Ng IH, Luk W, Sia SF, Wu MH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus‐infected macrophages in vitro: possible relevance to pathogenesis. J Virol 2005; 79: 7819–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up‐regulation in SARS‐coronavirus‐infected, monocyte‐derived human dendritic cells. Blood 2005; 106: 2366–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sheng WH, Chiang BL, Chang SC, Ho HN, Wang JT, Chen YC, et al. Clinical manifestations and inflammatory cytokine responses in patients with severe acute respiratory syndrome. J Formos Med Assoc 2005; 104: 715–723. [PubMed] [Google Scholar]
- 23. Theron M, Huang KJ, Chen YW, Liu CC, Lei HY. A probable role for IFN‐gamma in the development of a lung immunopathology in SARS. Cytokine 2005; 32: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang Y, Xu J, Zhou C, Wu Z, Zhong S, Liu J, et al. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med 2005; 171: 850–857. [DOI] [PubMed] [Google Scholar]
- 25. Tang NL, Chan PK, Wong CK, To KF, Wu AK, Sung YM, et al. Early enhanced expression of interferon‐inducible protein‐10 (CXCL‐10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin Chem 2005; 51: 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ward SE, Loutfy MR, Blatt LM, Siminovitch KA, Chen J, Hinek A, et al. Dynamic changes in clinical features and cytokine/chemokine responses in SARS patients treated with interferon alfacon‐1 plus corticosteroids. Antivir Ther 2005; 10: 263–275. [PubMed] [Google Scholar]
- 27. Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J Clin Invest 1998; 101: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 2002; 360: 1831–1837. [DOI] [PubMed] [Google Scholar]
- 29. Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 2006; 117: 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fox SW, Fuller K, Bayley KE, Lean JM, Chambers TJ. TGF‐beta 1 and IFN‐gamma direct macrophage activation by TNF‐alpha to osteoclastic or cytocidal phenotype. J Immunol 2000; 165: 4957–4963. [DOI] [PubMed] [Google Scholar]
- 31. Fujiwara N, Kobayashi K. Macrophages in inflammation. Curr Drug Targets Inflamm Allergy 2005; 4: 281–286. [DOI] [PubMed] [Google Scholar]
- 32. Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, et al. TGF‐beta 1 as an enhancer of Fas‐mediated apoptosis of lung epithelial cells. J Immunol 2002; 168: 6470–6478. [DOI] [PubMed] [Google Scholar]
- 33. Matute‐Bello G, Martin TR. Science review: apoptosis in acute lung injury. Crit Care 2003; 7: 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen RF, Chang JC, Yeh WT, Lee CH, Liu JW, Eng HL, et al. Role of vascular cell adhesion molecules and leukocyte apoptosis in the lymphopenia and thrombocytopenia of patients with severe acute respiratory syndrome (SARS). Microbes Infect 2005; 8: 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sime PJ, Marr RA, Gauldie D, Xing Z, Hewlett BR, Graham FL, et al. Transfer of tumor necrosis factor‐alpha to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor‐beta1 and myofibroblasts. Am J Pathol 1998; 153: 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peiris JSM, Chu CM, Cheng VCC, Chan KS, Hung IFN, Poon LLM, et al. Clinical progression and viral load in a community outbreak of coronavirus‐associated SARS pneumonia: a prospective study. Lancet 2003; 361: 1767–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martin M, Lefaix J, Delanian S. TGF‐beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys 2000; 47: 277–290. [DOI] [PubMed] [Google Scholar]
- 38. Vermeire S, Van Assche G, Rutgeerts P. C‐reactive protein as a marker for inflammatory bowel disease. Inflamm Bowel Dis 2004; 10: 661–665. [DOI] [PubMed] [Google Scholar]
- 39. Bernasconi D, Amici C, La Frazia S, Ianaro A, Santoro MG. The IkappaB kinase is a key factor in triggering influenza A virus‐induced inflammatory cytokine production in airway epithelial cells. J Biol Chem 2005; 280: 24127–24134. [DOI] [PubMed] [Google Scholar]
- 40. Lee DC, Cheung CY, Law AH, Mok CK, Peiris M, Lau AS. p38 mitogen‐activated protein kinase‐dependent hyperinduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. J Virol 2005; 79: 10147–10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 2006; 117: 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nature Med 2004; 10: S88–S97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Z, Xu D, Li Y, Jin L, Shi M, Wang M, et al. Longitudinal alteration of circulating dendritic cell subsets and its correlation with steroid treatment in patients with severe acute respiratory syndrome. Clin Immunol 2005; 116: 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Z, Guo X, Hao W, Wu Y, Ji Y, Zhao Y, et al. The relationship between serum interleukins and T‐lymphocyte subsets in patients with severe acute respiratory syndrome. Chin Med J (Engl) 2003; 116: 981–984. [PubMed] [Google Scholar]
- 45. Lisignoli G, Toneguzzi S, Piacentini A, Cristino S, Cattini L, Grassi F, et al. Recruitment and proliferation of T lymphocytes is supported by IFNgamma‐ and TNFalpha‐activated human osteoblasts: involvement of CD54 (ICAM‐1) and CD106 (VCAM‐1) adhesion molecules and CXCR3 chemokine receptor. J Cell Physiol 2004; 198: 388–398. [DOI] [PubMed] [Google Scholar]
- 46. Grossman Z, Meier‐Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nature Med 2006; 12: 289–295. [DOI] [PubMed] [Google Scholar]
- 47. Ioannidis J, Wilkinson D. HIV: prevention of opportunistic infections. Clin Evid 2005; 834–853. [PubMed] [Google Scholar]
- 48. Ho JC, Ooi GC, Mok TY, Chan JW, Hung I, Lam B, et al. High‐dose pulse versus nonpulse corticosteroid regimens in severe acute respiratory syndrome. Am J Respir Crit Care Med 2003; 168: 1449–1456. [DOI] [PubMed] [Google Scholar]
- 49. Auyeung TW, Lee JS, Lai WK, Choi CH, Lee HK, Lee JS, et al. The use of corticosteroid as treatment in SARS was associated with adverse outcomes: a retrospective cohort study. J Infect 2005; 51: 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffith JF, Antonio GE, Kumta SM, Hui DS, Wong JK, Joynt GM, et al. Osteonecrosis of hip and knee in patients with severe acute respiratory syndrome treated with steroids. Radiology 2005; 235: 168–175. [DOI] [PubMed] [Google Scholar]
- 51. Soldacki D, Mucha K, Foroncewicz B. Drugs selectively blocking proinflammatory cytokines and cell receptors. Pol Arch Med Wewn 2005; 113: 609–618. [PubMed] [Google Scholar]
- 52. Ng WF, To KF, Lam WW, Ng TK, Lee KC. The comparative pathology of severe acute respiratory syndrome and avian influenza A subtype H5N1—a review. Hum Pathol 2006; 37: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This article contains supplementary material available via the Internet from the Journal http://www.interscience.wiley.com/jpages/0022-3417/suppmat/path.2067.html
Supplementary Figure 1. Detection of ACE2 and SARS‐CoV S RNA in the lungs and other organs and tissues by ISH. ISH assay was used to detect ACE2 RNA (a‐i to a‐viii and c‐i to c‐viii) and SARS‐CoV S RNA (b‐i to b‐viii and d‐i to d‐viii) in pneumocytes (a‐i to d‐i), bronchial epithelial cells (a‐ii to d‐ii), bronchial serous gland epithelium (a‐iii to d‐iii), and myocardial cells (a‐iv to d‐iv), gastric parietal cells (a‐v to d‐v), distal convoluted renal tubules (a‐vi to d‐vi), pancreatic islet (a‐vii to d‐vii), sweat gland (a‐viii to d‐viii) in the SARS patient (a‐i to b‐viii) and the control subject (c‐i to d‐viii). All negatively stained samples were counterstained with nuclear Fast Red to identify cell nuclei
Supplementary Figure 2.Detection of proinflammatory cytokines in lung and other organs and tissues by ISH. ISH assay was used to detect the RNA of MCP‐1 (a‐i to a‐viii), TGF‐β1 (b‐i to b‐viii), IL‐1β (c‐i to c‐viii), IL‐6 (d‐i to d‐viii), and TNF‐α (e‐i to e‐viii) in monocytes/macrophages and pneumocytes (a‐i to e‐i), bronchial epithelial cells (a‐ii to e‐ii), bronchial serous gland epithelium (a‐iii to e‐iii), gastric parietal cells (a‐iv to e‐iv), myocardial cells (a‐v to e‐v), distal convoluted renal tubules (a‐vi to e‐vi), pancreatic islet (a‐vii to e‐vii), and sweat gland cells (a‐viii to e‐viii) in the SARS patients
Supplementary text