

Abstract
Severe acute respiratory syndrome (SARS) is a contagious and deadly disease caused by a new coronavirus. The protein sequence of the chymotrypsin‐like cysteine proteinase (CCP) responsible for SARS viral replication has been identified as a target for developing anti‐SARS drugs. Here, I report the ATVRLQp1Ap1'‐bound CCP 3D model predicted by 420 different molecular dynamics simulations (2.0 ns for each simulation with a 1.0‐fs time step). This theoretical model was released at the Protein Data Bank (PDB; code: 1P76) before the release of the first X‐ray structure of CCP (PDB code: 1Q2W). In contrast to the catalytic dyad observed in X‐ray structures of CCP and other coronavirus cysteine proteinases, a catalytic triad comprising Asp187, His41, and Cys145 is found in the theoretical model of the substrate‐bound CCP. The simulations of the CCP complex suggest that substrate binding leads to the displacement of a water molecule entrapped by Asp187 and His41, thus converting the dyad to a more efficient catalytic triad. The CCP complex structure has an expanded active‐site pocket that is useful for anti‐SARS drug design. In addition, this work demonstrates that multiple molecular dynamics simulations are effective in correcting errors that result from low‐sequence‐identity homology modeling. Proteins 2004. © 2004 Wiley‐Liss, Inc.
Keywords: anti‐SARS‐CoV drugs, coronavirus, 3CL‐PRO, main proteinase, protein modeling
INTRODUCTION
Severe acute respiratory syndrome (SARS) is a contagious and deadly disease in humans caused by a new coronavirus whose genome encodes a chymotrypsin‐like cysteine proteinase (CCP).1, 2, 3 This proteinase shares 40% and 44% sequence identity to human coronavirus strain 229E main proteinase and transmissible gastroenteritis coronavirus main proteinase (TGEVMP), respectively,3 and it processes the replicative polyproteins of the SARS coronavirus.2 Given the success of using viral proteinase inhibitors to treat viral infections, selective inhibitors of the CCP can be used to treat the SARS infection.
Two homology models of the apo‐CCP have been reported.3, 4 Three other homology models of the apo‐CCP were made available at the Protein Data Bank (PDB) (codes: 1PUK, 1P9T, and 1PA5) before the release of X‐ray structures of the apo‐CCP (PDB codes: 1Q2W, 1UK1, and 1UK2) and the inhibitor‐bound CCP (PDB code: 1UK4).5 Some of these models have reportedly been used in search of anti‐SARS drugs.3, 4, 6
It has been reported that the active site of an enzyme in the unbound state is slightly contracted and thus unsuitable for rigid‐body‐docking‐based search of anti‐SARS drugs.7 For this reason, protein modeling of the substrate‐bound CCP was carried out in this study using multiple molecular dynamics simulations (MMDSs)8, 9, 10, 11, 12 performed on teraflops computers. Additional reasons to carry out this study were to obtain information on the molecular flexibility of the active site of the CCP and to evaluate the effectiveness of MMDSs in protein modeling.
Here, I report the substrate‐bound CCP model that provides information regarding the molecular flexibility of the active site of the CCP. This model is useful for anti‐SARS drug design. The simulations of the CCP complex in water help explain why a catalytic dyad is observed in X‐ray structures of the CCP, TGEVMP, human coronavirus strain 229E main proteinase, and related 3C proteinases.13, 14 The results suggest that the catalytic efficiency of the CCP is regulated by substrate binding; the results also demonstrate that the SWISS‐MODEL‐based homology modeling followed by a refinement with MMDSs is effective in predicting 3D structures of proteins that have >40% sequence identities to known 3D protein structures.
METHODS
Homology Modeling
The homology model of the apo CCP used in this study was automatically generated by the SWISS‐MODEL program using the X‐ray structure of TGEVMP15 as a template, after submitting the protein sequence of the apo CCP to the website of SWISS‐MODEL.16
Multiple Molecular Dynamics Simulations
All MMDSs8, 9, 10, 11, 12 were performed according to a published protocol17 using the SANDER module of the AMBER 7.0 program18 with the Cornell et al. force field (parm96.dat).19 The topology and coordinate files used in the MMDSs were generated by the LINK, EDIT, and PARM modules of the AMBER 5.0 program.18 All simulations used (1) a dielectric constant of 1.0; (2) the Berendsen coupling algorithm;20 (3) a periodic boundary condition at a constant temperature of 300 K and a constant pressure of 1 atm with isotropic molecule‐based scaling; (4) the Particle Mesh Ewald method to calculate long‐range electrostatic interactions;21 (5) iwrap = 1 to generate the trajectories for interaction energy calculations using the EUDOC program;22 (6) a time step of 1.0 fs; (7) the SHAKE‐bond‐length constraints applied to all the bonds involving the H atom; (8) default values of all other inputs of the SANDER module. The initial structure of the substrate‐bound CCP used in the first set of simulations had no structural water molecules, whereas the corresponding structure used in the second set of simulations carried 218 structural water molecules. These structural water molecules were obtained from an instantaneous structure of the CCP complex from the first set of simulations (vide infra). The energy‐minimized initial CCP complexes used in the first and second sets of simulations were solvated with 8,274 and 8,713 TIP3P water molecules,23 respectively (EDIT input: NCUBE = 20, QH = 0.4170, DISO = 2.20, DISH = 2.00, CUTX = 7.8, CUTY = 8.0, and CUTZ = 8.0). The solvated CCP complex (29,550 atoms for the first set or 31,521 atoms for the second set) was first energy‐minimized for 100 steps to remove close van der Waals contacts in the system, then slowly heated to 300 K (10 K/ps) and equilibrated for 1.5 ns. All energy minimizations used the default method of AMBER 7.0 (10 cycles of the steepest descent method followed by the conjugate gradient method). The CARNAL module was used for geometric analysis and for obtaining the time‐average structure.
Ninety percent of the simulations were performed on a cluster of 800 Intel Xeon P4 processors (2.2/2.4 GHz with hyperthreading) dedicated to the Computer‐Aided Molecular Design Laboratory of the Mayo Clinic College of Medicine, 9% on an HP SC45 with 512 Alpha EV6.8 processors (1.0 GHz) at the Aeronautical Systems Center of the High Performance Computing Modernization Program of the Department of Defense, and 1% on a Blue Horizon with 1,152 power3 processors (375 MHz) at the San Diego Supercomputing Center.
Interaction Energy
The intermolecular interaction energy and the average intermolecular interaction energy were calculated by the EUDOC program22 using the CCP complex that was refined only by energy minimization and the trajectories of the CCP complex that were obtained at 1.0‐ps intervals over a period of 3.5–4.0 ns in the second set of simulations, respectively. The interaction energy was calculated from the potential energy of the ligand–receptor complex relative to the potential energies of the two partners in their free state, according to equations 1 and 2, with the additive, all‐atom force field by Cornell et al.19 A distance‐dependent dielectric function is used for calculating the electrostatic interactions.24 No cutoff for steric and electrostatic interactions was used in the calculation of the intermolecular interaction energy.
| (1) |
| (2) |
RESULTS AND DISCUSSION
Model Construction
The homology model of the apo‐CCP was generated by the SWISS‐MODEL program16 using the CCP protein sequence that was obtained from GenBank (NP_828863) and aligned over the sequence of the X‐ray structure of TGEVMP15.1 There is only one residue insertion in the CCP sequence aligned with that of TGEVMP (Fig. 1). The model of the CCP bound with a peptide substrate (Alap6Thrp5Valp4Argp3Leup2Glnp1Alap1') was then built by manually docking the peptide into the active site of the homology model of the apo‐CCP. The sequence of the peptide substrate (residues 4224–4231) was identified from the SARS genome deposited at GenBank (NC_004718) according to the reported substrate specificities among coronavirus cysteine proteinases.25 The resulting complex was optimized by a 5,000‐step energy minimization using a positional constraint applied to the CCP, followed by a 50,000‐step energy minimization without the use of any positional constraint. Different orientations of the peptide relative to the active site were examined by repeating the energy‐minimization procedure.
Figure 1.

Sequence alignment of transmissible gastroenteritis coronavirus main proteinase (Protein Data Bank [PDB] code: 1LVO) with the chymotrypsin‐like cysteine proteinase (CCP) (PDB code: 1P76) generated by the SWISS‐MODEL program. The most varied sequence region of the active site of the CCP (residues 44–65), the inserted residue of the CCP, and the conserved Asp187 of the CCP are highlighted in yellow, green, and red, respectively.
The most energetically stable orientation of the peptide used in the subsequent MMDS refinement was consistent with the orientations of the peptide substrates in TGEVMP, alpha‐chymotrypsin, and gamma‐chymotrypsin complexes.15, 26, 27
The X‐ray structures of CCP were unavailable when the CCP complex model was built. The complex model was not rebuilt after the X‐ray structures were made available for the following reasons: (1) Part of the active‐site structure of the CCP (residues 46–51) is either not determined or determined with high B factors in the X‐ray structures of the CCP. (2) The homology model generated by the SWISS‐MODEL program is structurally close to the X‐ray structures (Fig. 2). The mass‐weighted root mean square deviations (RMSDs) of the backbone and all heavy atoms between the truncated X‐ray structure of the apo‐CCP (residues 8–45 and 52–197) and the corresponding segments of the homology model are 0.99 Å (backbone) and 1.71 Å (backbone and side chains), respectively (Table I). (3) The mass‐weighted RMSDs of the backbone between the truncated X‐ray structure of the apo‐CCP (residues 8–45 and 52–197) and the truncated X‐ray structure of the bound CCP or the corresponding segments of the X‐ray structure of TGEVMP is 0.40 Å or 1.01 Å (Table I). (4) One of the aims of this study was to evaluate the effectiveness of MMDSs in predicting protein 3D structures.
Figure 2.

Overlay of the backbone structures of the truncated chymotrypsin‐like cysteine proteinase (residues 8–45 and 52–197) generated by X‐ray (yellow), SWISS‐MODEL (red) and multiple molecular dynamics simulations (green).
Table I.
Comparison of the X‐Ray Structure of the Bound or Unbound Chymotrypsin‐Like Cysteine Proteinase (CCP) with Different Theoretical Models of the CCP or the X‐Ray Structure of Transmissible Gastroenteritis Coronavirus Main Proteinase
| Protein | Method | PDB code | Release date | RMSDa (Å) | |
|---|---|---|---|---|---|
| Backbone | Backbone and side chains | ||||
| Unbound CCP | X‐ray | 1Q2W | 29‐JUL‐03 | 0 | 0 |
| Bound CCP | X‐ray | 1UK4 | 18‐Nov‐03 | 0.40 | 1.01 |
| Unbound CCP | Computational | 1PA5 | 27‐MAY‐03 | 1.01 | 1.68 |
| Unbound CCP | Computational | 1P9T | 24‐JUN‐03 | 2.14 | 2.61 |
| Bound CCP | Homologyb and MMDSc | 1P76 | 02‐JUL‐03 | 1.34/1.40d | 1.87/1.89d |
| Unbound CCP | Computational | 1PUK | 08‐JUL‐03 | 1.07 | 1.89 |
| Unbound CCP | Homologyb | — | NA | 0.99 | 1.71 |
| Unbound TGEVMP | X‐ray | 1LVO | 17‐JUL‐02 | 1.01 | NA |
RMSD: The mass‐weighted root‐mean‐square deviation between the truncated X‐ray structure of the unbound CCP (residues 8–45 and 52–197) and the corresponding segments of the theoretical model of CCPs or X‐ray structure of the bound CCP or transmissible gastroenteritis coronavirus main proteinase (residues 8–45 and 51–196). The coordinates of residues 46–51 are either not determined or determined with high B factors in the X‐ray structures of the CCP.
Homology: Homology modeling using the SWISS‐MODEL program.
MMDS = Multiple‐molecular‐dynamics‐simulation refinement.
The mass‐weighted RMSD between the truncated X‐ray structure of the bound CCP (residues 8–45 and 52–197) and the corresponding segments of the theoretical model.
Refinement
To correct potential errors resulting from low‐sequence‐identity homology modeling and the manual docking of the peptide substrate, MMDSs8, 9, 10, 11, 12 were used to simulate the substrate‐bound CCP solvated with explicit water molecules. Two hundred different simulations (2.0 ns for each simulation with a 1.0‐fs time step and different initial velocities) were performed according to a published protocol.17 In a previously reported single molecular dynamics simulation (2.0 ns with a 1.0‐fs time step) of a crystal structure of phosphotriesterase at 2.1‐Å resolution, this protocol was successful in generating a time‐average structure that fit into the difference electron density map.17
The sampling in the conformational space of the CCP complex achieved by the MMDSs was assessed by intermolecular distances calculated from the trajectories of the CCP complex used in the 200 simulations. The distance of the S atom of Cys145 to the C atom of the scissile bond (DS‐C) in the last instantaneous structure was calculated for each simulation, as was the distance of the main‐chain O atom of Glu47 to the main‐chain H atom of the P2 residue (DH‐O). The last instantaneous structure could be misleading, but it is reasonable to expect that 200 such structures resulting from different simulations represent the sampling of the MMDSs.
The two calculated distances (DS‐C and DH‐O) of the 200 simulations range continuously from 3.1 to 5.9 Å and 2.1 to 17.5 Å, respectively and demonstrate the conformational diversities of the substrate and the active site of the CCP in the 200 simulations. Furthermore, 99% of the DS‐C and DH‐O values can be found in multiple simulations, indicating that there was sufficient sampling of the conformations of the CCP and its substrate.
Of the 200 simulations, 188 resulted in partial unfolding in the active‐site region (residues 44–65); only 12 managed to adjust main‐ and side‐chain conformations and maintain the folded state in complex with the substrate. The active‐site region was considered partially unfolded if the distance between the two alpha carbon atoms of Glu47 and Leup2 was greater than 6.0 Å. In those partially unfolded structures, residues 44–50 and/or residues 61–65 unfolded in such a way that their side chains interacted mainly with water molecules.
The partial unfolding observed in the simulations may reflect the flexible nature of residues 44–65. This observation is consistent with all known X‐ray structures of the CCP (PDB codes: 1Q2W, 1UJ1, 1UK2, and 1UK4) in which residues 46–51 are either not determined or determined with high B factors. The CCP may uniquely have flexible residues of 44–65, because corresponding residues 44–64 are not disordered in the X‐ray structure of TGEVMP. It is possible, however, that the partial unfolding was caused by incorrect conformations of residues 44–65 that resulted from the lowest sequence identity in the region of residues 44–65 (Fig. 1). The partial unfolding could also have been caused by limited sampling of the bound conformations of the peptide substrate used in the manual docking.
Because the partial unfolding may be caused by limited sampling of the manual docking, the first set of simulations was considered more for re‐docking the peptide into the active site of the CCP than for refining the peptide‐bound CCP complex, namely, the first set of simulations was to “relax” the peptide substrate in the “restrained” CCP structure.
To test and refine the “MMDS‐docked” CCP complex, a second set of simulations was carried out using a water‐bound CCP complex structure obtained from one of the 12 simulations that kept the CCP active site in the folded state. Such simulations would result in unfolding or partial unfolding if the “MMDS‐docked” structure were energetically unstable.
For each of the 12 simulations, an average structure was obtained from 500 instantaneous structures at 1.0‐ps intervals over the last 0.5‐ns simulation. The average structure that had the smallest RMSD in the backbone structure of the CCP relative to that of the initial CCP complex used in the first set of simulations was selected as the initial structure for the second set of simulations. The use of the smallest‐RMSD‐based selection was for “relaxing” the substrate while “restraining” the enzyme. It is known that most water molecules converged to the center of the water box in the average structure of a long simulation. To retrieve the water molecules bound to the selected average structure, 500 instantaneous structures were compared to the selected average structure; the instantaneous structure that had bound water molecules and the smallest RMSD of all heavy atoms of the CCP relative to the selected average structure was actually used in the second set of simulations.
The second set of simulations was carried out using the same setup as the first set except that 220 simulations were carried out because additional computing resources were available. No unfolding in the active‐site region of the CCP complex was observed in any of the 220 simulations (2.0 ns for each simulation with a 1.0‐fs time step and different initial velocities). Otherwise, the simulations would have been repeated with an average structure of one of the rest of the 12 simulations until no unfolding was observed in the second set of simulations.
The average structure of the second set of simulations during the period of 1.5–2.0 ns was selected as the refined substrate‐bound CCP complex model. Seven instantaneous structures with RMSDs less than 2.1 Å relative to all the heavy atoms of the refined substrate‐bound CCP complex were also selected as instantaneous structures of the CCP that provided information on protein dynamics and structural water molecules. The refined model folds into two antiparallel β‐barrel domains and one domain containing five α‐helices and one parallel β‐sheet (Fig. 3). The active site of the CCP has a heavy‐footprint‐like cavity, and is located at the interface of the two β‐barrel domains (Figs. 3, 4). The coordinates of the refined structure and the seven instantaneous structures bound with structural water molecules were released at the PDB (code: 1P76) on July 2, 2003.
Figure 3.
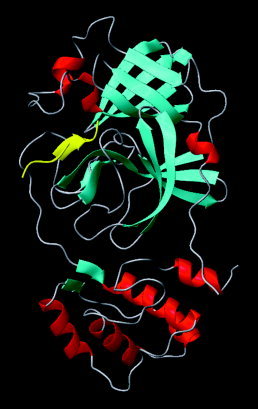
3D model of the substrate‐bound chymotrypsin‐like cysteine proteinase (red: α‐helix; cyan: β‐strand; yellow: substrate in the β‐strand conformation).
Figure 4.
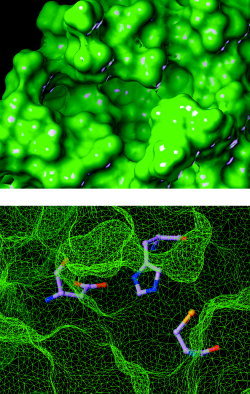
Top view of the active site of the chymotrypsin‐like cysteine proteinase showing the catalytic triad.
After the release of the coordinates of the structures obtained from the 220 simulations (2.0 ns for each simulation) at the PDB, each of these simulations was extended to 3.0 ns. The X‐ray structure of the apo‐CCP was released on July 29, 2003, before the completion of the 3.0‐ns simulations. For this reason, structures obtained from the 2.5‐ to 3.0‐ns or 1.0‐ to 3.0‐ns time frame were not submitted to the PDB. Reassuringly, no unfolding in the active‐site region of the CCP complex was observed in any of the 220 simulations that were extended to 3.0 ns.
The average structure of the 110,000 instantaneous structures obtained at 1.0‐ps intervals over the period of 1.5–2.0 ns in the second set of simulations was compared to the initial structure used in the second set of simulations. The RMSD of all the heavy atoms of the CCP complex (backbone and side chains) between the two structures is 2.6 Å. The average structure during the 1.5‐ to 2.0‐ns time frame was also compared to the average structure over the period of 2.5–3.0 ns. The RMSD of all the heavy atoms (backbone and side chains) of the CCP complex between the two average structures is 0.6 Å. These results indicate that the average structure over the period of 1.5–2.0 ns is converged.
Self‐Consistency Test
It has been reported that coronavirus cysteine proteinases have multiple cleavage sites.1, 2, 25 To computationally test the self‐consistency of the refined substrate‐bound CCP model, the substrate of the CCP complex was mutated to Asnp6Argp5Alap4Thrp3Leup2Glnp1Alap1'. The mutant contains an alternative cleavage site (residues 3919–3920) identified from the SARS viral genome.1, 2, 25 The refined CCP structure would be invalid if unfolding were observed in any of the MMDSs of the mutant complex or if the intermolecular interaction energies of the two complexes differed greatly.
Only 49 simulations (4.0 ns for each simulation with a 1.0‐fs time step and different initial velocities) were carried out for the mutant complex before the release of the first X‐ray structure of the CCP. Reassuringly, no unfolding in the active‐site region (residues 44–65) was observed in any of the simulations. The average intermolecular interaction energies of AsnArgAlaThrLeuGlnAla and AlaThrValArgLeuGlnAla in the refined substrate‐bound CCP complexes are −222.4 (Evdw = −71.3, Eele = −151.1) kcal/mol and −222.1 (Evdw = −74.3, Eele = −147.8) kcal/mol, respectively. The intermolecular interaction energy of AlaThrValArgLeuGlnAla in the substrate‐bound CCP complex that was refined only with energy minimization is −123.0 (Evdw = −59.0, Eele = −64.0) kcal/mol. Clearly, the MMDS refinement is significant and necessary in protein complex structure modeling, as it improved the interaction energy from −123.0 kcal/mol to −222.1 kcal/mol.
Protein Dynamics
The trajectories obtained from the first set of simulations reveal that the side chain of Cys145 adopts two distinct conformations, both of which enable a hydrogen bond with His41 (Fig. 5); the trajectories also demonstrate that the side chains of Asp187 and His41 have one conformation throughout the simulations. This observation explains why the S‐C bond of Cys145/144 is orthogonal to the imidazole ring of His41 in the crystal structures of the CCP and TGEVMP.3, 15 Interestingly, the side chain of Cys145 of the CCP in the second set of simulations adopted only one of the two conformations found in the first set of simulations; the other side‐chain conformation of Cys145 was disabled by the binding of Alap1'.
Figure 5.
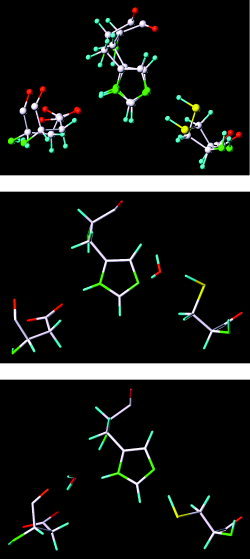
Different conformations of the catalytic triad in the chymotrypsin‐like cysteine proteinase.
The distance between the S atom of Cys145 and the Nϵ atom of His41 fluctuates between 1.8 and 3.3 Å in both sets of simulations. The trajectories of the two sets of simulations suggest that residues 46–51, Cys44, His163, Met165, and Leu167 are flexible and that these flexible residues should be modeled with caution in structure‐based design of anti‐SARS drugs.
Substrate Binding
The peptide substrate in the CCP complex adopts an antiparallel β‐strand conformation with four main‐chain hydrogen bonds conferred by Glnp1, Argp3, Valp4, His164, Glu166, and Thr190 (Fig. 6). The main‐chain H atom of Leup2 interacts with the main‐chain O atom of Glu47 via a hydrogen bond relay of a water molecule. An oxyanion hole comprises Gly143, Ser144, and Cys145 in the active site, and the scissile bond O atom of the substrate anchors at the oxyanion hole (Fig. 6). The carbonyl group of the scissile bond is best oriented for the nucleophilic attack by the thiolate of Cys145. This structural feature suggests that the Michaelis‐Menten state of the CCP complex is close to the transition state, and therefore the catalytic efficiency of the CCP is expected to be high. The methyl group of Alap1' is rigidly packed in the active site, preventing His41 from forming hydrogen bonds with water molecules. The side chain of Glnp1 adopts at least five different conformations that enable one or two hydrogen bonds with the main‐chain O atom of Argp3, the side‐chain O atom of Glu166, the main‐chain O atom of Leup2 via the hydrogen bond relay of a water molecule, the guanidinium H atoms of Argp3, and the Nϵ atom of His163 (Fig. 6). Both side chains of Leup2 and Valp4 are rigidly packed in the hydrophobic regions of the active site. The Argp3 residue is flexible in the active site as well.
Figure 6.
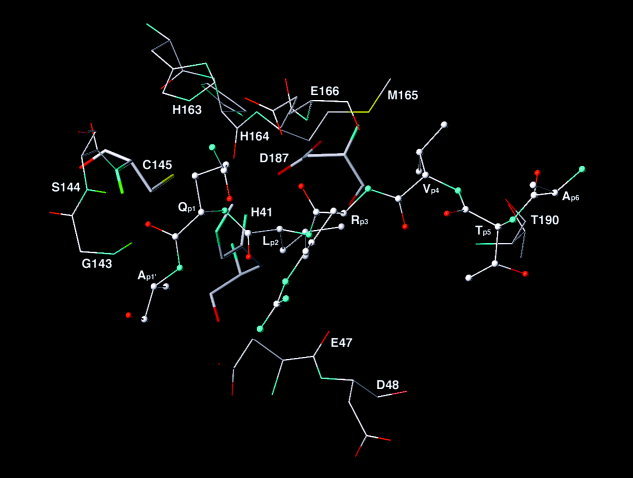
Residues that confer key intermolecular interactions in the multiple‐molecular‐dynamics‐simulations‐refined substrate‐bound chymotrypsin‐like cysteine proteinase (CCP) complex. Substrate is represented with the ball‐and‐stick model. Residues of the CCP are represented with the stick model. The catalytic triad is shown with the thick stick model. Hydrogen atoms are undisplayed except for those consisting of the oxyanion hole. C, O, N, S, and H atoms are shown in white, red, cyan, yellow, and green, respectively.
In the average structure of the CCP complex, the distances of the guanidinium C atom to the carboxylate C atoms of Glu47, Asp48, and Glu166 are 7.0 Å, 8.0 Å, and 7.0 Å, respectively (Fig. 6), although instantaneous structures reveal that the guanidinium group forms one or two hydrogen bonds with only one of the three residues at a time. The average structure of the CCP complex therefore indicates that the guanidinium group interacts electrostatically with all three residues in time and confers the entropic energy in binding.
Comparison with Crystal Structures
The MMDS‐refined model is essentially consistent with all the X‐ray structures of CCP (Fig. 2). The mass‐weighted RMSDs of the backbone and all heavy atoms between the truncated X‐ray structure of the bound/unbound CCP (residues 8–45 and 52–197) and the corresponding segments of the MMDS model are 1.40/1.34 Å and 1.89/1.87 Å, respectively; the corresponding RMSDs of the apo‐CCP models relative to the X‐ray structure are listed in Table I.
The backbone conformations of residues 184 and 151–158 in the X‐ray structures are identical to those in the MMDS model, but they are markedly different from those in all other theoretical models available at the PDB (Fig. 7). Residue 184 is a Pro that controls the active‐site loop conformation. The side‐chain of Gln192 is important to the function of the CCP, as it constitutes in part the active site of the CCP; its conformation in the MMDS model is identical to those in the X‐ray structures. However, the conformations of Gln192 in all other theoretical models of the CCP are different from those in the X‐ray structures (Fig. 8). These results demonstrate that the errors of side‐chain, main‐chain, and loop conformations that result from low‐sequence‐identity homology modeling can be corrected by MMDSs.
Figure 7.

Overlays of the X‐ray structure of the substrate‐bound chymotrypsin‐like cysteine proteinase (yellow) with the corresponding multiple‐molecular‐dynamics‐simulations model (green) and the corresponding SWISS‐MODEL model (red) (top: residues 183–185; bottom: residues 151–158).
Figure 8.
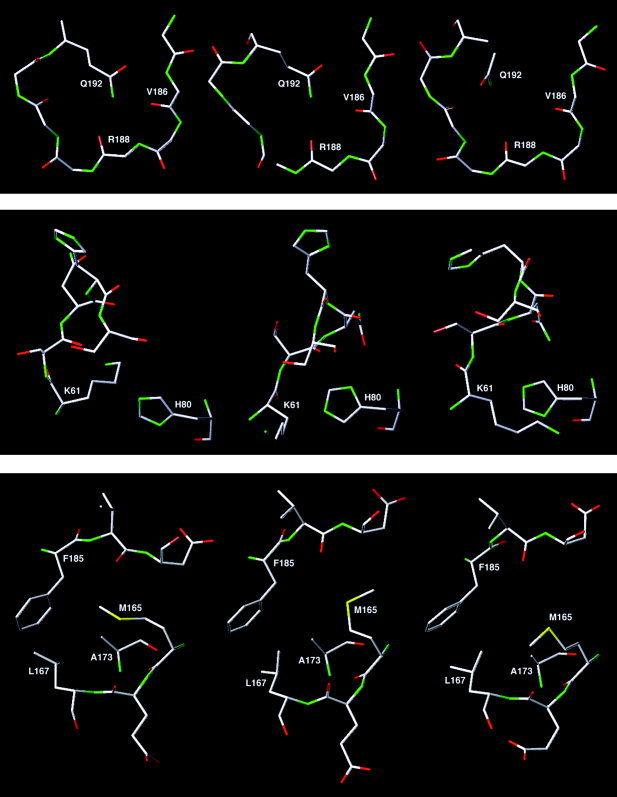
Comparison of the X‐ray structure of the substrate‐bound chymotrypsin‐like cysteine proteinase (middle) with corresponding the multiple‐molecular‐dynamics‐simulations model (left) and the corresponding SWISS‐MODEL model (right) (top: residues 185–192; middle: residues 61–66 and 80; bottom: residues 165–167, 173, and 185–187).
Significant differences between the MMDS model and the X‐ray structures are observed in the main‐chain conformation of residues 63–65 and the side‐chain conformations of Lys61 and Met165. Residues 63–65 of the MMDS model adopt a bend conformation, whereas the corresponding residues in the X‐ray structures all adopt the 310‐helix conformation. Related to this conformational difference, the ammonium group of Lys61 forms a hydrogen bond with His80 in the MMDS model, but it is away from His80 in the X‐ray structures (Fig. 8). The SWISS‐MODEL program incorrectly predicted the side‐chain conformation of Lys61 in which the ammonium group was surrounded by hydrophobic residues Ile43, Leu58, Phe66, Leu87, and Leu89. It has been reported that substrate binding in acetylcholinesterase can cause a large conformational change of a remote Arg residue (Fig. 9). Interestingly, the conformational difference of Arg in acetylcholinesterase between the bound and the unbound states is akin to the conformational difference of Lys61 in the CCP between the bound and the unbound states.28 Further computational and/or experimental studies are required to determine whether this difference is caused by substrate binding or by an error of the homology modeling that cannot be corrected by the MMDSs.
Figure 9.
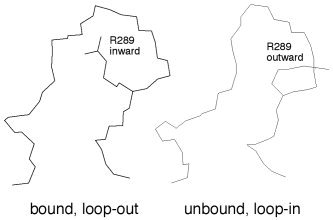
Different side‐chain conformations of Arg289 in the X‐ray structures of acetylcholinesterase in the bound and the unbound states.
The methyl group of Met165 in the active site of the MMDS model interacts with side chains of Leu167, Ala173, and Phe185. Therefore the methyl group of Met165 leaves more room to accommodate Valp4 of the substrate in the MMDS model than it does in the X‐ray structure of the apo‐CCP (Fig. 8). This reflects the difference of the CCP in the bound and the unbound states and suggests that the MMDS model with an expanded active‐site pocket is useful for virtual screening of chemical databases for the CCP inhibitor leads.29
Table II lists the torsions for the comparison of the backbone conformation of the substrate in the MMDS model with that of the inhibitor in the X‐ray structure of a CCP complex (PDB code: 1UK4).5 In the crystal structure, the inhibitor, AsnSerThrLeuGlnChloromethylKetone, was soaked into the active site of the CCP.5 Residues at the p6, p1, and p1' positions in the MMDS model are excluded in the comparison because the corresponding residues are either missing or modified in the X‐ray structure.5 The side‐chain conformations are excluded in the comparison too, because the sequences of the two peptides are different except for the p2 residue. For Leup2, the RMSD of all heavy atoms of Leu between the theoretical and experimental structures is 0.72 Å, indicating that the two conformations of the p2 residue in the two structures are almost the same. As apparent from Table II, the two backbone conformations of the peptides in the theoretical and experimental structures are nearly the same; on the contrary, the two backbone conformations of the same peptide before and after the two‐stage MMDS refinement are radically different as indicated by the large differences of torsions in the p3 and p5 residues. Again, the MMDS refinement is significant and necessary in protein complex structure modeling as demonstrated by the torsions in Table II.
Table II.
Backbone Torsions (Degrees of Arc) of the Substrate or Inhibitor Bound in Different Chymotrypsin‐Like Cysteine Proteinase (CCP) Structures
| Torsion name | Theoretical structure | X‐ray Structure | Torsion difference | ||
|---|---|---|---|---|---|
| A1 | B2 | C3 | A–B | B–C | |
| p5: ψ | −119.7 | −49.8 | −93.1 | 69.9 | 43.3 |
| p5: ω | 4.8 | −2.2 | 0.2 | 7.0 | 2.4 |
| p4: ϕ | −75.4 | −66.2 | −37.2 | 9.2 | 29 |
| p4: ψ | −37.9 | −18 | −55.6 | 19.9 | 37.6 |
| p4: ω | 10.5 | 22 | 0.2 | 11.5 | 21.8 |
| p3: ϕ | −145.9 | −121.2 | −108.6 | 24.7 | 12.6 |
| p3: ψ | −116.7 | −62.9 | −36.4 | 53.8 | 26.5 |
| p3: ω | 143.3 | −5.5 | 1.8 | 148.8 | 7.3 |
| p2: ϕ | −66.3 | −72.1 | −113.2 | 5.8 | 41.1 |
| p2: ψ | −37 | −41.5 | 13.5 | 4.5 | 55 |
A1: Thrp5Valp4Argp3Leup2 substrate bound in the theoretical model of the CCP complex before the multiple‐molecular‐dynamics‐simulation (MMDS) refinement.
B2: Thrp5Valp4Argp3Leup2 substrate bound in the theoretical model of the CCP complex after the MMDS refinement.
C3: Asnp5Serp4Thrp3Leup2 inhibitor bound in the X‐ray structure of the CCP complex (PDB code: IUK4).
Dyad versus Triad
In stark contrast to the catalytic dyad observed in X‐ray structures of the CCP5 and other coronavirus cysteine proteinases,3, 13, 14, 15 a catalytic triad comprising Asp187, His41, and Cys145 is found in the active site of the refined substrate‐bound CCP complex model (Fig. 4). Sequence analysis reveals that Asp187 of the CCP corresponds to Asp186 of TGEVMP (Fig. 1). Asp186 is reportedly conserved among the main proteinases of feline infectious peritonitis coronavirus, human coronavirus 229E, bovine coronavirus, murine hepatitis coronavirus, avian infectious bronchitis coronavirus, and transmissible gastroenteritis coronavirus (GenBank accession codes: AF326575, X69721, AF391542, M55148, M95169, and AJ271965, respectively).15
In the first set of simulations, a water molecule was occasionally sandwiched by Cys145 and His41 or by His41 and Asp187 when the peptide substrate failed to shield the catalytic triad from water (Fig. 5). This observation is consistent with reports that a water molecule is entrapped by the catalytic His and Asp residues in X‐ray structures of coronavirus cysteine proteinases and a related 3C proteinase.3, 14, 15 Interestingly, no water molecule was entrapped at the catalytic triad in the second set of simulations because the tight binding of the peptide at the active site completely shields the triad from water. The present results suggest that substrate binding leads to the displacement of a water molecule entrapped by Asp187 and His41, thus converting the dyad to a more efficient catalytic triad in which the nucleophilicity of the thiol group of Cys145 is increased by the hydrogen bonding of His41 to the carboxylate of Asp187.
While experimental studies are certainly required to confirm the catalytic role of Asp187 in the CCP, the exchange between dyad and triad observed in the present simulations suggests that one might be able to experimentally determine the structure of a 3C proteinase cocrystallized with a peptide‐like inhibitor of appropriate length that carries the p1' residue and changes the Asp residue to a catalytically competent position. This experiment may settle the long‐standing dispute over the existence of a catalytic triad in coronavirus cysteine proteinases and related 3C proteinases.3, 13, 14, 15 The simulations suggest further that the catalytic efficiency of the CCP is regulated by substrate binding, which is insightful to protein design of enzymes with high substrate selectivity.
Conclusion
This work demonstrates that the SWISS‐MODEL‐based homology modeling followed by a refinement with MMDSs generates the 3D structure of the substrate‐bound CCP from its amino acid sequence with a mass‐weighted RMSD of 1.89 Å for all heavy atoms of the protein relative to the corresponding X‐ray structure. The simulations of the substrate‐bound CCP suggest that substrate binding results in a slight expansion of the active‐site pocket, formation of a more efficient catalytic triad, and conformational changes of residues 61–65. The complex model with an expanded active‐site pocket is useful for anti‐SARS drug design. The simulations also provide a mechanism of catalytic efficiency regulated by substrate binding and suggest new experiments that may settle the debate on the existence of a catalytic triad in coronavirus cysteine proteinases and related 3C proteinases. In addition, this work suggests that MMDSs are effective in correcting errors of side‐chain, main‐chain, and loop conformations that result from low‐sequence‐identity homology modeling.
Acknowledgements
Supported by the Aeronautical Systems Center of the High Performance Computing Modernization Program of the U.S. Department of Defense, the San Diego Supercomputing Center, the University of Minnesota Supercomputing Institute, and the Compaq Medical Sciences Group. The opinions or assertions contained herein belong to the author and are not necessarily the official views of the U.S. Army, the U.S. Department of Defense, or the National Institutes of Health.
REFERENCES
- 1. Marra MA, Jones SJM, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY, et al. The genome sequence of the SARS‐associated coronavirus. Science 2003; 300: 1399–1404. [DOI] [PubMed] [Google Scholar]
- 2. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003; 300: 1394–1399. [DOI] [PubMed] [Google Scholar]
- 3. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science 2003; 300: 1763–1767. [DOI] [PubMed] [Google Scholar]
- 4. Xiong B, Gui C‐S, Xu X‐Y, Luo C, Chen J, Luo H‐B, Chen L‐L, Li G‐W, Sun T, Yu C‐Y, et al. A 3D model of SARS‐CoV 3CL proteinse and its inhibitors design by virtual screening. Acta Pharmacol Sin 2003; 24: 497–504. [PubMed] [Google Scholar]
- 5. Yang HT, Yang MJ, Ding Y, Liu YW, Lou ZY, Zhou Z, Sun L, Mo LJ, Ye S, Pang H, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA 2003; 100: 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chou KC, Wei DQ, Zhong WZ. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem Bioph Res Commun 2003; 308: 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pang YP, Silva ND, Hydock C, Prendergast FG. Docking studies on the complexed and uncomplexed FKBP12 structures with bound and unbound ligands: an implication of conformational selection mechanism for binding. J Mol Model 1997; 3: 240–248. [Google Scholar]
- 8. Caves LSD, Evanseck JD, Karplus M. Locally accessible conformations of proteins— multiple molecular dynamics simulations of crambin. Protein Sci 1998; 7: 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith LJ, Daura X, van Gunsteren WF. Assessing equilibration and convergence in biomolecular simulations. Proteins 2002; 48: 487–496. [DOI] [PubMed] [Google Scholar]
- 10. Snow CD, Nguyen N, Pande VS, Gruebele M. Absolute comparison of simulated and experimental protein‐folding dynamics. Nature 2002; 420: 102–106. [DOI] [PubMed] [Google Scholar]
- 11. Zagrovic B, Snow CD, Shirts MR, Pande VS. Simulation of folding of a small alpha‐helical protein in atomistic detail using worldwide‐distributed computing. J Mol Biol 2002; 323: 927–937. [DOI] [PubMed] [Google Scholar]
- 12. Oelschlaeger P, Schmid RD, Pleiss J. Modeling domino effects in enzymes: Molecular basis of the substrate specificity of the bacterial metallo‐beta‐lactamases IMP‐1 and IMP‐6. Biochemistry 2003; 42: 8945–8956. [DOI] [PubMed] [Google Scholar]
- 13. Malcolm BA. The picornaviral 3C proteinases: cysteine nucleophiles in serine proteinase folds. Protein Sci 1995; 4: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bergmann EM, Mosimann SC, Chernaia MM, Malcolm BA, James MN. The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J Virol 1997; 71: 2436–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha‐helical domain. EMBO J 2002; 21: 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwede T, Diemand A, Guex N, Peitsch MC. Protein structure computing in the genomic era. Res Microbiol 2000; 151: 107–112. [DOI] [PubMed] [Google Scholar]
- 17. Pang Y‐P. Successful molecular dynamics simulation of two zinc complexes bridged by a hydroxide in phosphotriesterase using the cationic dummy atom method. Proteins 2001; 45: 183–189. [DOI] [PubMed] [Google Scholar]
- 18. Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE III, Debolt S, Ferguson D, Seibel G, Kollman PA. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun 1995; 91: 1–41. [Google Scholar]
- 19. Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 1995; 117: 5179–5197. [Google Scholar]
- 20. Berendsen HJC, Postma JPM, van Gunsteren WF, Di Nola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81: 3684–3690. [Google Scholar]
- 21. Darden TA, York DM, Pedersen LG. Particle Mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys 1993; 98: 10089–10092. [Google Scholar]
- 22. Pang YP, Perola E, Xu K, Prendergast FG. EUDOC: A computer program for identification of drug interaction sites in macromolecules and drug leads from chemical databases. J Comp Chem 2001; 22: 1750–1771. [DOI] [PubMed] [Google Scholar]
- 23. Jorgensen WL, Chandreskhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1982; 79: 926–935. [Google Scholar]
- 24. McCammon JA, Gelin BR, Karplus M. Dynamics of folded proteins. Nature 1977; 267: 585–590. [DOI] [PubMed] [Google Scholar]
- 25. Hegyi A, Ziebuhr J. Conservation of substrate specificities among coronavirus main proteases. J Gen Virol, Part 3. 2002; 83: 595–599. [DOI] [PubMed] [Google Scholar]
- 26. Fujinaga M, Sielecki AR, Read RJ, Ardelt W, Laskowski M, Jr. , James MN. Crystal and molecular structures of the complex of alpha‐chymotrypsin with its inhibitor turkey ovomucoid third domain at 1.8 Å resolution. J Mol Biol 1987; 195: 397–418. [DOI] [PubMed] [Google Scholar]
- 27. Yennawar NH, Yennawar HP, Farber GK. X‐ray crystal structure of gamma‐chymotrypsin in hexane. Biochemistry 1994; 33: 7326–7336. [DOI] [PubMed] [Google Scholar]
- 28. Millard CB, Kryger G, Ordentlich A, Greenblatt HM, Harel M, Raves ML, Segall Y, Barak D, Shafferman A, Silman I, Sussman JL. Crystal structures of aged phosphonylated acetylcholinesterase: Nerve agent reaction products at the atomic level. Biochemistry 1999; 38: 7032–7039. [DOI] [PubMed] [Google Scholar]
- 29. Perola E, Xu K, Kollmeyer TM, Kaufmann SH, Prendergast FG, Pang YP. Successful virtual screening of a chemical database for farnesyltransferase inhibitor leads. J Med Chem 2000; 43: 401–408. [DOI] [PubMed] [Google Scholar]