

INTRODUCTION
A coronavirus, an RNA virus, has been identified in humans as the pathogen responsible for the recent worldwide Severe Acute Respiratory Syndrome (SARS) outbreak. Rapid study of the critical SARS enzymes will provide useful inhibitors for this disease as well as a methodology that may be applicable to fight emerging pathogens in the future. The necessity of this rapid approach is further justified by the fact that the SARS virus reservoir has not been identified and future viral outbreaks are still possible. Indeed, new cases have already been reported in 2004.1 The SARS coronavirus (SARS CoV) genome is a positive strand of RNA with 11 open reading frames and a genomic organization similar to other coronaviruses. However, phylogenic analysis and sequence alignments have shown that the SARS CoV has developed distinctly from other coronaviruses.2 The viral replication proceeds via synthesis of a complementary minus strand RNA using the genome as a template and the subsequent synthesis of genomic plus strand RNA from the minus strand RNA template. The key enzyme responsible for both steps is the RNA‐dependent RNA polymerase (RdRp), represented by nonstructural protein 12 (nsP12). Since RdRp activity is not essential for the host, its inhibition will not cause undesirable side effects during therapy. With the enzyme characterization in its early stages, a homology model based on the amino acid sequence can provide some clues concerning the residues critical for its function. We have built a homology model of the SARS polymerase using the 3D jury system.3 Recently, during preparation and revision of this study, Xu et al.4 reported an RdRp model using the program Modeller. Although their and our models have been built using a different approach, the r.m.s between the two models is about 1.6 Å (for Cα atoms). The similarity between the two models can be partially explained by the fact that the 3D‐jury system uses a set of prediction methods that includes the Pcons method on which the Modeller program is based on. The capability of the 3D‐jury in using consensus between Meta prediction methods makes it a very reliable tool for model building as proven by its highest ranking in the CASP5 (Critical Assessment of Techniques for Protein Structure Prediction) competition.5 This predictor has been used recently to successfully assign a methyl‐transferase function to nsP13 and to build a 3D model that identified residues critical for the enzyme function.6 Substrate and ligand docking in the active site were carried out with the Genetic Optimization Ligand Docking Program (Gold) giving us an estimate of ligand binding in the active site. This docking method, with a success rate of around 70% through its validation process, proved to be very reliable when used with caution.7
MATERIALS AND METHODS
In order to identify homologues for the nsP12 protein, the sequence (ID_NP828869) was initially submitted to the BLAST8 search engine. However, as this did not yield any significant results, we then used a more powerful consensus structure prediction method (meta‐predictor), the 3D‐jury system. We submitted the sequence to the 3D‐jury Meta predictor system. All models with a 3D‐jury score higher than 50 are considered to be significant. A three‐dimensional model for the SARS nsP12 comprising 474 residues was built based on the homologous rabbit Hemorrhagic Disease Virus RdR polymerase9 (pdb ID: 1KHW_B), which had a 3D‐jury score of 165. The final model was built using the GIGSAW program10 and its potential energy minimized using the GROMOS96 program.11 The model quality was evaluated with Procheck12 and then analyzed. The pdb ID for the model is 1SXF. Ligand docking in the active site was carried out with the Genetic Optimization Ligand Docking program (Gold).7 The ChemScore13 and ΔGbinding were evaluated to discriminate between ligands. The ChemScore value is obtained by adding a clash penalty and internal torsion terms to ΔGbinding, which militate against close contacts in docking and poor internal conformation.13
RESULTS AND DISCUSSION
As the sequence alignment search for SARS RdRp conducted with BLAST did not yield a homologous structure, we turned to the 3D jury Meta predictor to identify a homology model. The power of this method is illustrated by the fact that even with less than 10% sequence identity, the Rabbit Hemorrhagic Disease Virus (RHDV) RdR polymerase,9 was identified as a homologous structure, based on a combination of motif search, fold recognition, and structure‐based alignment orchestrated by the 3D‐Jury Meta server. The RdRp function was assigned to a C‐terminal segment of 537 residues out of the nsP12 932 residues with a highly significant score (3D‐jury score > 164 in our work). No characteristic motifs suggesting specific functions were found for the 396 N‐terminal residues. Subsequently, a three‐dimensional structure for the nsP12 SARS RdRp (residues 395 to 870) was built based on the homologue's structure. Our model has 78% of residues in the favored region of Ramachandran plot and the proportion reaches 98.8% when we consider residues in the allowed regions.
The SARS‐CoV nsP12 folds in a classic “right hand” shape that contains motifs shared by all RdRps in which the characteristic fingers, palm, and thumb sub‐domains can be recognized [Fig. 1(A)]. The finger domain is composed of three helices (residues 462–477, 561–584, and 619–637), the RdRp characteristic helix‐loop‐helix (residues 619–658), and two stranded β‐sheets (residues 535–541and 551–560). The palm domain consists of two helices (residues 687–706 and 776–793) and the β‐hairpin (residues 755–769) that contain the catalytic aspartates 760 and 761 responsible for the nucleotide transfer reaction. This domain contains the conserved “SDD” sequence in the RdRPs.14 The thumb domain consists essentially of two α‐helices (residues 833–843 and 854–869) and a large loop between residues 798–818. The thumb and finger domains are interconnected with loops running between them, as is the case for the Hepatitis C virus15 and RHDV RdRps.6 The active site is buried in the center of the protein. It has an architecture very similar to other RdRps [Fig. 1(B)]. We can identify all of the important residues by sequence homology. Sequence alignment of the SARS‐CoV polymerase with other known coronavirus polymerases showed a complete conservation of the basic residues (K545, R553, R555, and K551) and aspartates 760 and 761.16 These aspartate residues are responsible for the coordination of the metal ion involved in the nucleotidyl transfer reaction.9 In addition, residue R836, which is partially conserved in RdRp, contributes to nucleoside binding by making a hydrogen bond with the O4 of the cytidine moiety.
Figure 1.
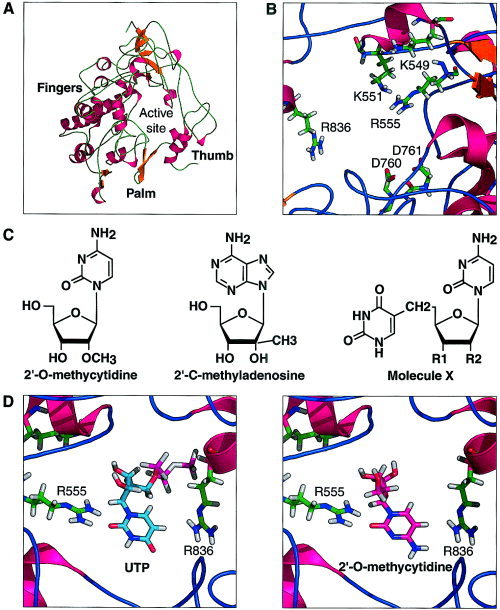
A: Ribbon diagram of SARS RNA‐dependent RNA polymerase. The characteristic fold of a “right hand” fingers, palm, and thumb can be recognized from residues 396 to 870 of nsP12. B: Structural detail of the active site of SARS RNA‐dependent RNA polymerase. The catalytic residues critical for the nucleotidyl transfer reactions are R553 and R555 and K551. D760 and D761 are responsible for the coordination of the metal ion. C: Structure of nucleoside analogs: derivatives from molecule X are XA1: R1 = OH; R2 = CH2OH, XA2; R1 = CH2OH; R2 = CH2OH. D: Nucleoside analogue, 2′‐O‐methylcytidine and uridine triphosphate (UTP) docking in SARS RdR polymerase using GOLD.
The present RdRp 3D‐model highlighted by the domain structure and by the active site architecture can provide a rationale for the design of inhibitors for the synthesis of viral RNA. In fact, there are presently no data on SARS coronavirus polymerase inhibition. In this context, we used data on the inhibition of other related enzymes, most notably hepatitis C virus (HCV) polymerase, to help us to design anti‐SARS agents. It has been shown that nucleotides can bind to the HCV polymerase catalytic site even in the absence of the template17 and that nucleoside analogues such as 2′‐C‐methyladenosine and 2′‐O‐methylcytidine are potent competitive inhibitors.17, 18
Starting from our RdRp model, we carried out the docking of the substrate UTP and nucleoside analogues. CTP, UTP, and GTP have a similar Km as determined for the hepatitis C virus NS5B RNA‐dependent RNA polymerase.19 As the active site determinant residues are conserved in the SARS CoV enzyme, we should expect very similar binding pattern for the nucleosides. The ΔGbinding value shown in Table I demonstrates as expected a somewhat lower binding energy for the substrate UTP compared to 2′‐O‐methylcytidine and 2′‐C‐methyladenosine.
Table I.
Docking of Substrate and Nucleoside Analogues to SARS‐CoV RdR Polymerase Active Site†
| Ligand | ChemScore | ΔGbinding (kJ · mol−1) |
|---|---|---|
| Uridine triphosphate | 19.77 | −20.3 |
| 2′‐C‐Methyladenosine | 18.27 | −18.8 |
| 2′‐O‐Methylcytidine | 15.27 | −15.4 |
| Molecule XA1 | 16.52 | −17.4 |
| Molecule XA2 | 19.77 | −21.0 |
ChemScore = ΔGbinding + Clash penalty + internal torsion terms.20
Based on our nucleoside analogue bound models, we have designed several compounds and determined their binding energy. Efforts were made to create more binding interactions between the protein and the nucleoside analogue through the modification of the latter's. The most promising result has been obtained with thymidine substituted 2′‐O‐methylcytidines. These are molecules X1A and XA2, as shown on Figure 1©, where in XA1: R1 = OH; R2 = CH2OH; and in XA2: R1 = CH2OH; R2 = CH2OH. The computed binding energy (ΔGbinding) of molecule XA2 is 40% lower than the lead compound and is even slightly better than substrate UTP (Table I).
This encouraging result prompts us to continue with the modeling and synthesis of this compound. Such attempts are important in advancing the design of potent drugs for the SARS therapy.
REFERENCES
- 1. Paterson R. SARS returns to China. Lancet Infect Dis 2004; 4: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marra MA et al. The genome sequence of the SARS‐associated coronavirus. Science 2003; 300: 1399–1404 [DOI] [PubMed] [Google Scholar]
- 3. Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D‐Jury: a simple approach to improve protein structure predictions. Bioinformatics 2003; 19: 1015–1018. [DOI] [PubMed] [Google Scholar]
- 4. Xu X, et al. Molecular model of SARS coronavirus polymerase: Implications for biochemical functions and drug design. Nucleic Acids Res 2003; 31: 7117–7130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. von Grotthuss M, Pas J, Wyrwicz L, Ginalski K, Rychlewski L. Application of 3D‐Jury, GRDB, and Verify3D in fold recognition. Proteins 2003; 53: 418–423 [DOI] [PubMed] [Google Scholar]
- 6. von Grotthuss M, Wyrwicz LS, Rychlewski L. mRNA cap‐1 methyltransferase in the SARS genome. Cell 2003; 113: 701–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones G, Willett P, Glen RC Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997; 267: 727–748 [DOI] [PubMed] [Google Scholar]
- 8. Altschul SF, et al. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ng KK, Cherney MM, Vazquez AL, Machin A, Alonso JM, Parra F, James MN. Crystal structure of active and inactive conformations of a caliciviral RNA‐dependent RNA polymerase. J Biol Chem 2002; 277: 1381–1387. [DOI] [PubMed] [Google Scholar]
- 10. Bates P, Kelley LA, MacCallum RM, Sternberg MJE. Enhancement of protein modeling by human intervention in applying the automatic programs 3D‐JIGSAW and 3D‐PSSM. Proteins 2001; 5: 39–46 [DOI] [PubMed] [Google Scholar]
- 11. Van Gunsteren WF, Berendsen HJC. Computer simulation of molecular dynamics: methodology, applications and perspectives in chemistry. Angew Chem Int Ed Engl 1990; 29: 992–1023 [Google Scholar]
- 12. Laskowski RA, McArthur MW, Moss DS, Thomson JD. PROCHECK: a program to check the stereochemistry of protein structure. J Appl Cryst 1993; 26: 183–291. [Google Scholar]
- 13. Baxter CA, Murray CW, Clark DE, Westhead DR, Eldridge MD. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins 1998; 33: 367–382. [PubMed] [Google Scholar]
- 14. Koonin EV, Dolja VV. Evolution and taxonomy of positive‐strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit Rev Biochem Mol Biol 1993; 28: 375–430 [DOI] [PubMed] [Google Scholar]
- 15. Bressanelli S, Tomei L, Rey FA, De Francesco R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J Virol 2002; 76: 3482–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. http://www.ncbi.nlm.nih.gov/genomes/SARS/nsP9.html
- 17. Bera S, et al. Synthesis and evaluation of optically pure dioxolanes as inhibitors of hepatitis C Virus RNA replication. Bioorg Med Chem Lett 2003; 13: 4455–4458. [DOI] [PubMed] [Google Scholar]
- 18. Carroll SS, et al. Inhibition of hepatitis C virus RNA replicon by 2′‐modified nucleoside analogs. J Biol Chem 2003; 278: 11979–11984. [DOI] [PubMed] [Google Scholar]
- 19. Lohmann V, Roos A, Korner F, Koch JO, Bartenschlager R. Biochemical and structural analysis of the NS5B RNA‐dependent RNA polymerase of the hepatitis C virus. J Viral Hepat 2000; 7: 167–174. [DOI] [PubMed] [Google Scholar]
- 20. Eldridge MD, Murray CW, Auton TR, Paolinine GV, Mee JRP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. Comput Aided Mol Des 1997; 11: 425–445. [DOI] [PubMed] [Google Scholar]