

Abstract
Porcine diarrhea and gastroenteritis are major causes of piglet mortality that result in devastating economic losses to the industry. A plethora of pathogens can cause these diseases, with the transmissible gastroenteritis virus (TGEV) and enterotoxigenic Escherichia coli K88 (ETEC) being two of the most salient. In the December 2017 issue of Proteomics Clinical Aplications, Xia and colleagues used comparative proteomics to shed light on how these microbes interact to cause severe disease 1. The authors discovered that TGEV induces an epithelial‐mesenchymal transition‐like phenotype that augments cell adhesion proteins mediating the attachment of ETEC to intestinal epithelial cells. Moreover, coinfection was found to modulate several host proteins that could bolster pathogen persistence. Importantly, the authors observed that ETEC suppresses the production of inflammatory cytokines induced by TGEV, which may in turn promote the long‐term survival of both microbes.
Keywords: cytokines, diarrhea, epithelial‐mesenchymal transition, host‐pathogen interactions, proteomics, TGEV–ETEC coinfection
Enteric infections affecting pigs are widespread, and cause a decrease in feed conversion and performance that ultimately result in high financial losses. Among these ailments, diarrhea and gastroenteritis are some of the most important as they cause high morbidity and mortality.2 The small intestine of a pig is a major site of nutrient absorption. Similar to the colon, this organ harbors a diverse microbiota that is pivotal to digestion and nutrient absorption. Although most of these microbes have a symbiotic relationship with the host, some can cause extensive harm.2, 3
Escherichia coli is a Gram‐negative, facultatively anaerobic bacterium that is usually harmless and aids digestion.3, 4 However, several E. coli strains have evolved toxins that cause extensive pathology in the gut. Indeed, E. coli is the most important cause of diarrhea in young swine.4 Of particular interest is enterotoxigenic E. coli K88 (ETEC), a noninvasive type that adheres to the microvilli of intestinal epithelial cells. These bacteria release toxins that evoke gastrointestinal hypersecretion of electrolytes and water. The diarrhea and vomiting that ensues causes dehydration, stunted growth, and death. Profuse diarrhea and gastroenteritis are also caused by the transmissible gastroenteritis virus (TGEV), which is a coronavirus that survives the acidic pH of the stomach and the proteolytic enzymes of the duodenum.5 It multiplies in the cell lining of the small intestine resulting in the loss of absorptive cells and villous atrophy.5, 6 TGEV spreads rapidly, causes outbreaks involving large numbers of pigs, and often leads to 100% mortality in young piglets. Diarrhea is often caused by coinfecting pathogens that synergize to cause severe disease. For instance, humans with diarrhea have been found to be coinfected with rotaviruses and E. coli or Giardia.7 Furthermore, infection by multiple microbes has also been reported to cause purulent diarrhea in swine.8 This raises many questions concerning the pathogenesis of coinfections. In a recent issue of Proteomics Clinical Applications, Xia and colleagues1 unveiled that TGEV infection augments the attachment of ETEC to intestinal cells, thence altering host cell homeostasis and the production of inflammatory cytokines (Figure 1).
Figure 1.
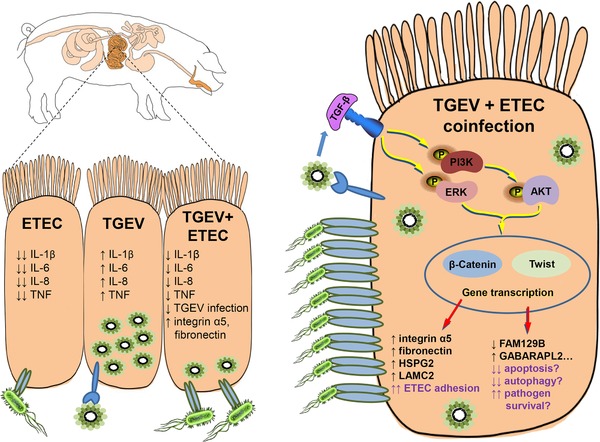
TGEV and ETEC elicit context‐dependent host cell responses. Infection of intestinal epithelial cells by TGEV induces a PI3K/Akt‐dependent EMT phenotype that elicits plasmalemmal expression of proteins such as integrin‐α5. Contrary to ETEC, TGEV augments the production of inflammatory molecules. Increased levels of integrin‐α5 promote the attachment of ETEC to epithelial cells, which quells the TGEV‐induced production of inflammatory cytokines. Coinfection by TGEV and ETEC also modulates the expression of many proteins involved in homeostasis and host defence, which could ultimately worsen disease severity by enhancing long‐term pathogen survival.
Since both TGEV and ETEC infect and attach to a pig's intestinal epithelium, a suitable enterocytic cell line of porcine origin is required to study coinfection in the laboratory. In this regard, the IPEC‐J2 cell line is an appropriate model since they are morphologically differentiated cells derived from the small intestine of young piglets.9 Using these cells, Xia and colleagues investigated the dynamics of coinfection by TGEV and ETEC.1, 10 The authors showed that TGEV is able to grow1 and persist10 in IPEC‐J2 cells. Importantly, the authors found that a preexisting TGEV infection augmented ETEC attachment to intestinal cells. This observation raised the possibility that there might be a survival advantage for either pathogen during coinfection. To that effect, Xia and colleagues examined viral mRNA and protein expression and found that both decrease when ETEC is present. Why is coinfection beneficial for the bacterium but seemingly unfavorable for the virus? To gain important insight into how TGEV promotes ETEC attachment, Xia et al. undertook a comparative proteomics approach where they employed LC–MS/MS coupled to iTRAQ to study cells infected with TGEV, ETEC, or both.1 Relative to noninfected cells, TGEV infection modulated the expression of 77 proteins. Of particular interest was integrin‐α5, a matrix macromolecule known for binding fibronectin and stimulating angiogenesis. Adhesion molecules such as integrins, cadherins, and selectins have been found to facilitate bacterial attachment and invasion.11 Xia and colleagues demonstrated that integrin‐α5 mRNA and protein expression increased upon TGEV infection, and even more saliently so upon coinfection with ETEC. These findings were validated by flow cytometry, which revealed increased cell surface expression of integrin‐α5 on infected cells. In order to show a causal link between integrin‐α5 and increased ETEC adhesion to TGEV‐infected cells, the authors employed peptide agonists or inhibitors of integrin‐α5. When integrin‐α5 is inhibited on TGEV‐infected IPEC‐J2 cells, ETEC attachment decreases significantly; the opposite is observed upon treatment with an integrin‐α5 agonist.1 In an accompanying study, Xia and colleagues further elucidated how TGEV induces integrin‐α5.10 There, the authors established that TGEV evoked the production of TGF‐β, which in turn induced EMT through the PI3K/Akt pathway. This TGEV‐induced EMT was found to augment the expression of vimentin, fibronectin, and integrin‐α5 in IPEC‐J2 cells and pig intestines10 (Figure 1). Additionally, the proteome of TGEV–ETEC coinfected cells revealed higher expression of ECM‐related proteins HSPG2 and LAMC2.
TGEV causes severe intestinal inflammation.5, 6 Xia and colleagues expanded upon this by reporting that this viral infection induces production of proinflammatory cytokines TNF, IL‐1β, IL‐6, and IL‐8 in IPEC‐J2 cells1, 10 (Figure 1). Those cytokines contribute to immune cell infiltration at the infection site12 and may lead in viral clearance. Upon coinfection with ETEC, the authors found that the bacterium lowered cytokine levels by at least 40%.1 This finding can be explained by the fact that ETEC lowers TGEV replication,1 and may—by itself—induce apoptosis.13 As observed with other pathogens,12 one can hypothesize that a decreased inflammatory response might prevent removal of TGEV and ETEC from the gut. The question still remains on whether ETEC promotes the long‐term survival of TGEV. From the proteome of TGEV–ETEC coinfected cells reported by the authors, one can see an increase in FAM129B,1 a negative regulator of apoptosis that is overexpressed in cancer cells.14 Also exclusive to TGEV–ETEC coinfection was a decrease in GABARAPL2,1 a protein that is essential to autophagy.15 Decreased autophagy could prevent the elimination of TGEV from the intestine. Since increased viral persistence could translate into augmented transmission and greater financial losses, future experiments could investigate whether the presence of ETEC improves the long‐term fitness of TGEV in vitro and in vivo. Future investigations may also evaluate the roles of FAM129B and GABARAPL2 in TGEV–ETEC infection; their knockdown could hamper the survival of TGEV and ETEC in the porcine gut.
In sum, Xia and colleagues convincingly showed that TGEV promotes ETEC adhesion to intestinal epithelial cells.1, 10 The coinfection that ensues lowers the production of proinflammatory molecules and may modulate—to the pathogens’ advantage—the expression of host proteins involved in homeostasis and microbial clearance (Figure 1).
Abbreviations
- EMT
epithelial‐mesenchymal transition
- ETEC
enterotoxigenic Escherichia coli K88
- FAM129B
family with sequence similarity 129 member B
- GABARAPL2
GABA(A) receptor‐associated protein like 2
- HSPG2
heparan sulfate proteoglycan 2
- IPEC‐J2
intestinal columnar epithelial cells
- iTRAQ
isobaric tags for relative and absolute quantification
- LAMC2
laminin gamma 2
- LC‐MS/MS
liquid chromatography coupled to tandem mass spectrometry
- TGEV
transmissible gastroenteritis virus
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
G.A.D. was partially supported by a Frederick Banting and Charles Best Doctoral Award from the Canadian Institutes of Health Research, and by bridging funds from the Centre for Host‐Parasite Interactions.
Arango Duque G., Acevedo Ospina H. A., Prot. Clin. Appl. 2018, 12, 1700143 10.1002/prca.201700143
See accompanying article by Lu Xia et al. https://doi.org/10.1002/prca.201600137
References
- 1. Xia L., Dai L., Zhu L., Hu W., Yang Q., Proteomics Clin. Appl. 2017, 10.1002/prca.201600137. [DOI] [PubMed] [Google Scholar]
- 2. Lee I. K., Kye Y. C., Kim G., Kim H. W., et al, Asian‐Australas. J. Anim. Sci. 2016, 29, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fouhse J. M., Zijlstra R. T., Willing B. P., Animal Frontiers 2016, 6, 30. [Google Scholar]
- 4. Fairbrother J. M., Nadeau E., Gyles C. L., Anim. Health Res. Rev. 2005, 6, 17. [DOI] [PubMed] [Google Scholar]
- 5. Kim L., Hayes J., Lewis P., Parwani A. V., Chang K. O., Saif L. J., Arch. Virol. 2000, 145, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morin M., Morehouse L. G., Can. J. Comp. Med. 1974, 38, 227. [PMC free article] [PubMed] [Google Scholar]
- 7. Bhavnani D., Goldstick J. E., Cevallos W., Trueba G., Eisenberg J. N. S., Am. J. Epidemiol. 2012, 176, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de la Fé Rodríguez P. Y., Martin L. O. M., Muñoz E. C., H. Imberechts, Butaye P., Goddeeris B. M., Cox E.,Trop. Anim. Health Prod. 2013, 45, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nossol C., Barta‐Böszörményi A., Kahlert S., Zuschratter W., Faber‐Zuschratter H., Reinhardt N., Ponsuksili S., Wimmers K., Diesing A.‐K., Rothkötter H.‐J., PLoS One. 2015, 10, e0132323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia L., Dai L., Yu Q., Yang Q., J. Virol. 2017, 91, e01256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boyle E. C., Finlay B. B., Curr. Opin. Cell Biol. 2003, 15, 633. [DOI] [PubMed] [Google Scholar]
- 12. Arango Duque G., Descoteaux A., Front. Immunol. 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson A. M., Kaushik R. S., Rotella N. J., Hardwidge P. R., Infect. Immun. 2009, 77, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito S., Fujii H., Matsumoto T., Abe M., Ikeda K., Hino O., Head Neck 2010, 32, 96. [DOI] [PubMed] [Google Scholar]
- 15. Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., Elazar Z., EMBO J. 2010, 29, 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]