

Abstract
The renin–angiotensin–aldosterone system (RAAS) is a key regulator of systemic blood pressure and renal function and a key player in renal and cardiovascular disease. However, its (patho)physiological roles and its architecture are more complex than initially anticipated. Novel RAAS components that may add to our understanding have been discovered in recent years. In particular, the human homologue of ACE (ACE2) has added a higher level of complexity to the RAAS. In a short period of time, ACE2 has been cloned, purified, knocked‐out, knocked‐in; inhibitors have been developed; its 3D structure determined; and new functions have been identified. ACE2 is now implicated in cardiovascular and renal (patho)physiology, diabetes, pregnancy, lung disease and, remarkably, ACE2 serves as a receptor for SARS and NL63 coronaviruses. This review covers available information on the genetic, structural and functional properties of ACE2. Its role in a variety of (patho)physiological conditions and therapeutic options of modulation are discussed. Copyright © 2007 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: ACE2, RAAS, ACE, renin, angiotensin
Introduction
The renin–angiotensin–aldosterone system (RAAS) is cardinal in renal and cardiovascular physiology and pathophysiology. Its architecture and functions are more complex than previously assumed. In the classical RAAS, the protease renin, which is secreted from renal juxtaglomerular cells, acts on the circulating precursor angiotensinogen to generate angiotensin (Ang) I (Figure 1). Ang I is converted by the dipeptidyl carboxypeptidase angiotensin‐converting enzyme (ACE) to Ang II, the main effector substance of the RAAS, with potent vasoconstrictive, pro‐inflammatory, and pro‐fibrotic properties. Consequently, ACE inhibitors (ACEi) and Ang II receptor blockers (ARBs) are effective in hypertension, heart failure, and progressive renal damage.
Figure 1.

Schematic diagram of the renin–angiotensin–aldosterone system which shows the role of ACE and ACE2 in the metabolism of the various angiotensin peptides. Modified from Warner et al 101, with permission
Recently, angiotensin fragments other than Ang II were also proposed to be relevant, in particular Ang(1–7), which mediates vasodilatation, anti‐proliferation, and apoptosis, thereby opposing the effects of Ang II 1. Further complexity was introduced by the discovery of an ACE homologue, ACE2. This enzyme cleaves Ang I into Ang (1–9), which can be converted to Ang(1–7) by ACE (Figure 1). Furthermore, ACE2 degrades Ang II to Ang(1–7). It has therefore been suggested that ACE2 acts in a counter‐regulatory manner to ACE by shifting the balance between Ang II and Ang(1–7), thus acting as a functional clearance mechanism for Ang II.
High ACE2 gene expression was initially reported in the testis, kidney, and heart 2, 3. Later studies showed widespread distribution of both rodent and human ACE2 in the lung, liver, small intestine, and brain, albeit much lower than in the kidneys 4, 5, 6, 7. Diverse roles have emerged for ACE2 since its identification in 2000 2, 3. Some 200 papers have subsequently addressed its structure, functions, and role in cardiovascular and renal disease, diabetes, SARS coronavirus infection, and lung injury.
This review covers available information on the genetic, structural, and functional properties of ACE2. Its role in a variety of (patho)physiological conditions and therapeutic options of modulation will be discussed.
The ACE2 gene and protein
The 40 kb ACE2 gene is located on chromosome Xp22 and contains 18 exons, many of which closely resemble exons in the ACE gene 3. Two alternative transcripts of the mouse ACE2 gene have been identified which probably arise by alternative splicing 8. Recently, an alternative 5′‐untranslated exon of human ACE2 and new polymorphisms have been reported 9.
The human ACE2 protein is a typical zinc metallopeptidase, which comprises 805 amino acids and is 40% identical in sequence with ACE, although it only contains a single catalytic domain. Critical active site residues, including the His‐Glu‐Met‐Gly‐His zinc‐binding motif, are highly conserved. ACE2 is a type I integral membrane glycoprotein orientated with the N‐terminus and the catalytic site facing the extracellular space (an ectoenzyme), where it can metabolize circulating peptides. The small C‐terminal, cytoplasmic domain has a number of potential regulatory sites. The similarity with ACE relates only to its topology and much of the extracellular domain (Figure 2); the juxtamembrane, transmembrane, and cytoplasmic domains of ACE2 share similarity with the renal, transmembrane protein collectrin 10 (Figure 2), which stimulates insulin exocytosis and pancreatic beta‐cell proliferation 11, 12. Targeted deletion of the collectrin gene in mice suggests that it also plays a major role in regulating renal amino acid transport 13.
Figure 2.

The domain structure of somatic and testis ACE, ACE2, and collectrin. Each protein is a type I integral membrane protein with a cleaved signal peptide (black), an N‐terminal ectodomain, a transmembrane domain (blue in ACE; red in ACE2 and collectrin), and a C‐terminal cytoplasmic domain. Somatic ACE contains two HEMGH zinc binding active sites, while testis ACE and ACE2 contain a single HEMGH motif. Collectrin lacks a HEMGH motif. The ectodomain of ACE2 (yellow) is more similar to the N‐terminal domain of somatic ACE, while its juxtamembrane stalk, and transmembrane and cytoplasmic domains are more similar to collectrin. The numbers indicate the amino acid residues in each protein. Modified from Turner and Hooper 102, with permission
Substrate specificity of ACE2
ACE2, a strict carboxypeptidase, hydrolyses its substrates by removing a single amino acid from their respective C‐termini, rather than a dipeptide, as does ACE. ACE2 therefore has the ability to convert the decapeptide Ang I to Ang(1–9) and the octapeptide Ang II to Ang(1–7). A kinetic study evaluating the comparative roles of ACE and ACE2 in angiotensin metabolism 14 established that Ang II is hydrolysed two orders of magnitude more efficiently by ACE2 than Ang I. Hence, the major role of ACE2 in angiotensin metabolism seems to be the production of Ang(1–7), whose actions oppose those of Ang II. Different in vivo studies strongly support the concept that a major role of ACE2 is indeed the generation of Ang(1–7) from Ang II and that its conversion of Ang I to Ang(1–9) is not normally of physiological importance 15, 16, 17, 18, 19, 20, except possibly under conditions that raise Ang II levels, eg ACEi or ARB treatment 21.
Although most studies have focused on the role of ACE2 in angiotensin metabolism, the enzyme has broad substrate specificity. In addition to Ang I and Ang II, ACE2 hydrolyses apelin‐13, neurotensin‐ (1–11), dynorphin A‐(1–13), β‐casomorphin‐(1–7), and ghrelin 22. Although the ACE substrate brady‐ kinin is not hydrolysed by ACE2, its metabolite des‐Arg 9‐[bradykinin], which is an agonist for the B1 bradykinin receptor, is hydrolysed and a role for ACE2 in bradykinin metabolism cannot yet be dismissed. It is likely that other potential physiological substrates for ACE2 will emerge.
Cell biology and shedding of ACE2
Knowledge of the basic cell biology of ACE2 still remains limited, partly because few cell models expressing ACE2 at high levels are available 23. ACE2 is expressed as a cell‐surface non‐raft protein with little intracellular localization, and the protein does not readily internalize. However, binding of the SARS viral spike protein to ACE2 does trigger enzyme internalization, down‐regulating activity from the cell surface. In polarized cells, ACE2 is exclusively targeted to the apical surface 23, 24, in contrast to ACE which distributes equally between apical and basolateral surfaces 23. Another mechanism for down‐regulating ACE2 at the cell‐surface is by proteolytic shedding of its extracellular domain. This shedding, which is also undergone by ACE, is stimulated by phorbol esters and is blocked by inhibitors of the ADAMs family of zinc metalloproteinases. ADAM17 (TACE) is implicated as the primary enzyme involved in the regulated shedding of ACE2 25, resulting in detectable levels of ACE2 in plasma and urine 14, 23, 26.
ACE2 and the heart
The importance of ACE2 in cardiac physiology and disease was initially suggested by two independent groups, based on cardiac ACE2 expression, particularly in endothelial cells 2, 3. Subsequent studies revealed ACE2 not only in endothelial cells and smooth muscle cells from intra‐myocardial vessels, but also in cardiac myocytes 27 (Figure 3).
Figure 3.
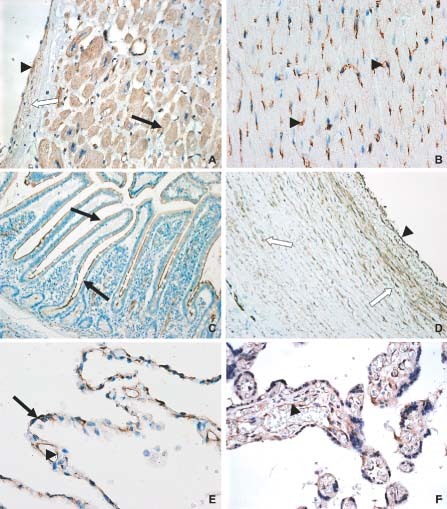
Immunohistochemical staining pattern of ACE2 in several organs. In the healthy human heart (A), ACE2 is expressed in cardiomyocytes (closed arrow), vascular endothelium (arrowhead), and smooth muscle cells (open arrow). In rat heart (B), ACE2 is predominantly expressed in vascular endothelium (arrowhead). (C) ACE2 expression in human small intestine (jejunum); abundant staining can be found in the brush border of enterocytes (arrow). In human aorta (D), ACE2 is expressed in the endothelium (arrowhead) and vascular smooth muscle cells (open arrow). In healthy human lung (E), ACE2 is present in alveolar epithelial cells (arrow) and in capillary endothelial cells (arrowhead). In human placenta (F), positive staining for ACE2 is found in the placental villi [syncytiotrophoblast, cytotrophoblast, vascular endothelium (arrowhead), and smooth muscle cells]
The importance of ACE2 in cardiac function was strengthened by Crackower et al, who described cardiac dysfunction in Ace2 knock‐out (KO) mice 28. Specifically, there was a 40% decrease in fractional shortening with slight ventricular dilatation. Interestingly, there was thinning of the left ventricular (LV) wall rather than LV hypertrophy and/or cardiac fibrosis. These changes progressed with age and were more prominent in male mice 28. The hearts of these Ace2 KO mice showed increased Ang II levels and up‐regulation of hypoxia‐inducible genes. The authors suggest that cardiac function is modulated by the balance between ACE and ACE2, and that the increase in local cardiac Ang II was involved in these abnormalities. This is supported by the fact that the cardiac phenotype and increased Ang II levels were completely reversible by concomitant deletion of the ACE gene in Ace2 KO mice. It remains unclear, however, why, despite elevated Ang II levels, the hearts of these Ace2 KO mice did not show any cardiac hypertrophy or fibrosis. This may partly be related to Ang II‐independent effects of ACE2, such as effects of the other substrates described above 22, which also can influence cardiac contractility.
Gurley et al generated an Ace2 KO mouse 29, deleting the same exon as the Crackower group 28, and although demonstrating changes in blood pressure regulation, particularly in response to Ang II, these investigators could not detect a specific cardiac phenotype. In another study, Yamamoto et al 30 also failed to identify cardiac abnormalities in their Ace2 KO mice. However, they did demonstrate reduced cardiac contractility after transverse aortic constriction (TAC), a model of pressure overload. TAC was associated, when compared with wild‐type mice subjected to the same procedure, with a marked increase in cardiac Ang II levels and increased fibrosis, LV dilation, and myofibrillar disarray. Indeed, in Ace2 KO mice followed for a longer period, this reduction in myocardial contractility led to pulmonary congestion and death 30.
Recent studies suggest that ACE2 possibly influences the electrical pathways of the heart. In ACE2 transgenic mice, cardiac conduction disturbances were present and some animals developed lethal ventricular fibrillation 31. The level of ACE2 up‐regulation correlated with the severity of the conduction disturbance.
Accumulating evidence indicates that over‐activity of cardiac RAAS and myocardial Ang II production contributes to the progression of heart failure. Several studies characterized ACE2 expression and activity in heart failure. In experimental myocardial infarction, increased cardiac ACE2 expression was found in the infarct zone and the surrounding ischaemic zone 27. Moreover, local up‐regulation of ACE2 was also found in explanted human hearts with ischaemic cardiomyopathy 20, 27, 32 and idiopathic dilated cardiomyopathy 20, 32. These findings may imply that the up‐regulation of ACE2 is a compensatory response to the ischaemic insult and that the consequent increase in the vasodilatory Ang(1–7) may confer cardio‐protective effects in an attempt to counterbalance the effects of Ang II. Other groups did not observe up‐regulation of ACE2 33 or demonstrated down‐regulation of cardiac ACE2 in experimental heart failure 21, 34, albeit in different rat strains.
Several studies noted a marked increase in cardiac ACE2 expression in response to RAAS blockade by ACEi 21, 35, ARB 33, 34, 35, or an aldosterone antagonist 34, 36 (a diuretic with specific benefits in heart failure 37). This was interpreted as Ang II receptor blockade conferring cardio‐protection not only by reducing harmful Ang II‐mediated effects, but also by locally increasing Ang(1–7), which, through binding to a putative Ang(1–7) receptor, Mas 1 38, is postulated to have beneficial effects on the heart 1.
The potential for ACE2 to modulate cardiac function and remodelling is additionally suggested by the finding that lenti‐viral vector encoding mouse ACE2 injected intracardially in Sprague–Dawley rats significantly attenuated cardiac hypertrophy and myocardial fibrosis induced by Ang II infusion 39. Moreover, ACE2 overexpression after neonatal development provides protection from high blood pressure and cardiac pathophysiology in the SHR rat 40.
Altogether, these studies suggest that ACE2 is important in cardiac function and that ACE2‐related effects contribute to cardio‐protective effects of ACE inhibitors and ARBs. The increased ACE2 gene expression and activity during RAAS blockade suggest that at least part of their mode of action results from hydrolysis of the vasoconstrictor mitogenic Ang II to the vasodilator anti‐proliferative Ang(1–7) through a feed‐forward mechanism within the RAAS. Understanding the roles of ACE2 in heart failure may optimize current therapies and ultimately guide the development of new therapeutic strategies.
ACE2 and hypertension
It was initially hypothesized that disruption of the delicate balance between ACE and ACE2 would result in abnormal blood pressure control 41; ACE2 might protect against increases in blood pressure and, conversely, ACE2 deficiency might lead to hypertension. The localization of ACE2 in vascular endothelial cells and smooth muscle cells 6 (Figure 3) supports this.
Since hypertension was linked to loci on the X chromosome 42, 43 and ACE2 was mapped to the X chromosome 3, ACE2 became a candidate gene underlying the loci linked to hypertension. Crackower et al 28 were the first to test ACE2 as the gene underlying the blood pressure locus on the X chromosome. They showed reduced expression of renal ACE2 in the salt‐sensitive Sabra hypertensive rat compared with the normotensive rat. Both hypertensive SHR and SHRSP rats showed reduced renal ACE2 protein levels compared with the normotensive Sabra and WKY strains. Two other groups confirmed some of these findings by showing that SHR rats have lower renal ACE2 mRNA, protein, and activity compared with WKY rats 44, 45. However, others were unable to detect any differences in renal ACE2 mRNA, protein, and activity between adult hypertensive rats and their normotensive controls 46. On close scrutiny, in SHRSP rats the allele of the previously identified blood pressure locus on rat chromosome X contributes to a blood pressure‐lowering effect 42, while in the salt‐sensitive Sabra hypertensive rat this allele contributes to a blood pressure‐increasing effect 43, suggesting that it is not the same gene that underlies the blood pressure locus. Surprisingly, while the allelic effects of the blood pressure locus are discordant between these hypertensive strains, a similar, ie concordant, reduction in renal ACE2 expression was reported 28, which reduces the possibility that ACE2 is the candidate gene underlying the blood pressure locus.
Additional arguments came from studies in Ace2 KO mice. Ace2 KO mice on a mixed B6/129 background had normal blood pressure compared with the wild type 28. However, a recent study which also studied Ace2 KO animals on homozygous B6 and 129 backgrounds showed that Ace2 KO animals with a B6 background have a significantly lower blood pressure compared with wild‐type animals 29, suggesting that the genetic background with modifier genes present in the 129 strain may counteract the absence of Ace2, or that the 129 allele of another gene involved in blood pressure regulation moved with the KO into the B6 background and caused the difference in blood pressure in these animals.
So far, three human association studies of ACE2 polymorphisms with hypertension have been performed 47, 48, 49. Two of these studies, using Chinese cohorts, reported an association between a SNP in intron 3 of ACE2 and blood pressure in females with metabolic syndrome 49 and females with essential hypertension 48. However, the use of only one SNP cannot rule out the possibility of this SNP being in linkage disequilibrium with SNPs in neighbouring genes that could cause the difference in blood pressure.
Altogether, the role of ACE2 in hypertension is not conclusive. Functional studies that show blood pressure effects after the administration of ACE2 inhibitors or stimulators are needed to further elucidate its significance in hypertension.
ACE2 and the kidney
ACE2 is highly expressed in the kidney 7; however, its role in the kidney has not been fully elucidated. In the human kidney, ACE2 is predominantly found in the proximal tubular brush border 50, 51, where it co‐localizes with ACE 23, 24. Moreover, ACE2 is found in endothelial and smooth muscle cells of renal vessels 50, 51 and in glomerular visceral (in podocyte/slit diaphragm complex) and parietal epithelial cells 51, 52 (Figure 4). The distribution of ACE2 is species‐specific. In human kidneys, the ACE2 expression pattern is comparable to that of mouse kidneys 5, 53, whereas in rat kidneys, ACE2 is predominantly found in glomeruli and to a lesser extent in tubules (Figure 4).
Figure 4.

Immunohistochemical staining pattern of ACE2 in the kidney. In the healthy human kidney (A), ACE2 is predominantly present in the brush border of proximal tubular cells (arrow) and to a lesser extent in the glomerular visceral and parietal epithelium. In rat kidney (B), ACE2 is predominantly found in glomeruli and to a lesser extent in distal tubules (open arrow). In mouse kidney (C), the distribution pattern of ACE2 is comparable to human kidney with predominant expression in proximal tubules (arrow). ACE2 is also expressed during human nephrogenesis (gestational age 16 weeks) (D)
Several lines of evidence support a role for renal ACE in renal damage 54. Individual differences in renal ACE activity predict the susceptibility for proteinuria‐associated renal damage in experimental conditions 55, 56. Furthermore, Ang II is increased in damaged tubules as a possible mediator of further renal damage in experimental and human renal disorders 57, 58. A disrupted balance between intrarenal ACE and ACE2 with consequent high levels of Ang II might therefore contribute to progressive renal damage. Indeed, in experimental hypertension and diabetes, renal ACE2 expression is decreased 28, 59. Moreover, Tikellis et al showed that in the kidneys of SHR rats ACE2 expression follows a developmental pattern with declining expression during development and onset of hypertension 45. In line with the beneficial effects of RAAS blockade on cardiac ACE2 as mentioned earlier, renal ACE2 activity is also increased in response to ACEi and ARB 59, 60. In contrast, further enhancement of the therapeutic efficacy of ACEi resulted in an unexpected reduction in renal ACE2 expression compared with ACEi alone 61. It looks like despite extensive research, the mechanisms of the effects of ACEi are still not completely understood 62. Further studies on the regulation of renal ACE2 during RAAS blockade might help to elucidate this.
Initial reports on Ace2 KO mice did not show any renal structural and functional abnormalities 28, 29, but recent studies showed that male Ace 2 −/y mice develop age‐dependent glomerulosclerosis and albuminuria, whereas the renal vasculature and interstitium were relatively protected 63. The authors hypothesized that the glomerular abnormalities were caused by chronic exposure to increased circulating and tissue Ang II, as these abnormalities were abolished by ARB treatment. The glomerulo‐protective role of ACE2 is supported by studies in which chronic infusion of the ACE2 inhibitor MLN4760 increased albuminuria in db/db mice, resulting in increased deposition of glomerular fibronectin 52. Albuminuria could be prevented by ARB, indicating Ang II dependency. Moreover, these diabetic female db/db mice have decreased glomerular ACE2 staining compared with db/m heterozygous littermates 52. The authors suggest that glomerular ACE2, present in the podocyte/slit diaphragm complex, could normally be reno‐protective by favouring rapid degradation of Ang peptides and thereby preventing exposure to high levels of Ang II.
Other studies support the assumption that increased ACE2 activity tied with decreased ACE activity may reflect a protective mechanism by limiting the renal accumulation of Ang II and favouring Ang(1–7) formation. Increased ACE2 expression is indeed coupled with profound reduction of ACE expression in renal tubules of young db/db mice 53. The authors speculate that this might be an early reno‐protective response. The kidneys of streptozotocin‐induced diabetic mice also show increased ACE2 expression at the post‐transcriptional level 64. In humans, de novo expression of ACE2 is found in glomerular and peritubular endothelium in biopsies in patients with primary and secondary renal disease, as well as in renal transplants 51. In renal biopsies of non‐diabetic and diabetic patients, significant up‐regulation of the ACE gene, and not the ACE2 gene, was found in diabetic nephropathy 65. Moreover, no associations between polymorphisms in the ACE2 gene and diabetic nephropathy could be established 66.
Altogether, ACE2 appears to be involved in the pathogenesis of renal damage, but its precise role is unclear and further studies are needed, in particular during renal disease.
ACE2 and pregnancy
The placenta is an organ with major Ang(1–7) and ACE2 expression 67 (Figure 3), suggesting that ACE2 may be involved in mother–fetus interactions, which is interesting regarding a potential role for ACE2 in fetal programming and pregnancy. In pregnant rats, renal expression of ACE2 is increased compared with virgin controls 15. A recent study showed no differences in ACE2 expression between normotensive and pre‐eclamptic placentas in the third trimester 67. However, pre‐eclampsia is determined early in pregnancy; thus, the role of ACE2 in pre‐eclampsia should be further studied, for example, in animal models.
ACE2 and lung disease
There is abundant expression of RAS components in the lung, including ACE and ACE2. Activation of the intrapulmonary RAS could influence the pathogenesis of lung injury 68. Indeed, increased levels of ACE have been associated with pulmonary hypertension 69, 70, sarcoidosis 71, 72, idiopathic pulmonary fibrosis 73, and the acute respiratory distress syndrome 74, 75. The alleged role for ACE2 as a counter‐regulatory mechanism of ACE may therefore be crucial in the lung. ACE2 is present in type I and type II alveolar epithelial cells and to a lesser extent in bronchiolar epithelial cells (Figure 3) 6, 76. Furthermore, as in other organs, ACE2 is present in endothelial cells and in arterial smooth muscle cells. With respect to the role of ACE2 in pulmonary hypertension, it can be envisaged that in particular, the presence and function in smooth muscle cells of small arterioles are of relevance, although the actual role in this disease is unknown 77.
Ace2 KO mice do not have lung abnormalities when compared with their wild‐type littermates 28. However, it was recently shown that loss of ACE2 expression precipitates severe acute lung failure 76. Moreover, injection of recombinant human ACE2 attenuates acute lung failure in Ace2 KO as well as in wild‐type mice 76. With respect to the possible role of ACE2 in the lung in relation to acute lung injury in particular, the relationship to the ACE2 expression at the alveolar capillary interface is of interest. It is tempting to speculate that increased ACE2 may play a role in reducing the initial leakage over the alveolar capillary interface. This would then slow down the vicious circle that often occurs after a damaging effect to this interface and leads to the clinical pathological picture of diffuse alveolar damage with intra‐alveolar oedema and fibrin deposits. These data support a critical role for the intrapulmonary RAS in the pathogenesis of acute lung injury and show that ACE2 is a key molecule involved in the development and progression of acute lung failure.
ACE2 and human coronaviruses
ACE2 acts as a receptor for two coronaviruses (CoV): severe acute respiratory syndrome (SARS)‐CoV and human CoV‐NL63 78, 79, positive stranded RNA viruses with a ‘corona’‐like appearance 80. Their genome is packaged together with several membrane proteins, the RNA binding nucleoprotein and the receptor binding spike protein that protrudes through the virion membrane. HCoV‐NL63 infection causes clinical respiratory symptoms resembling those observed in children infected with common cold viruses. SARS‐CoV causes severe lower respiratory tract disease including fever, non‐productive cough, myalgia, and dyspnoea 81. A specific region within the SARS‐CoV spike protein (S1) interacts with ACE2 79, 82, 83. The crystal structure at 2.9 angstrom resolution of this receptor binding domain bound with the peptidase domain of human ACE2 shows that it presents a gently concave surface, which cradles the N‐terminal lobe of the peptidase 82. After engagement with ACE2, SARS‐CoV fuses with host cell membranes, by which the conformational changes of the two heptad regions located in the S2 region, HR‐1 and HR‐2, cause the formation of an oligomeric structure, leading to fusion between the viral and target‐cell membranes.
Using soluble ACE2 molecules, peptides derived thereof, and antibodies directed against ACE2, the SARS‐CoV infection can be blocked 79, 84. Conversely, expression of ACE2 in refractory cell lines resulted in SARS‐CoV replication 85. Experiments in Ace2 KO mice revealed the importance of ACE2 as a receptor for SARS‐CoV 86. Moreover, autopsy specimens of patients who died of SARS revealed that ACE2‐expressing cells are a direct target of SARS‐CoV 87.
Besides ACE2, lysosomal proteases such as cathepsin‐L are required for productive SARS‐CoV 88, 89, explaining discrepancies observed between ACE2 expression and absence of SARS‐CoV replication in endothelial cells 6, 85. Members of the DC‐SIGN family of proteins may enhance SARS‐CoV infection but are not sufficient to infect cells 90.
Murine and rat ACE2 less efficiently bound the S1 domain of late‐phase SARS‐CoV isolates and supported less efficient S‐protein‐mediated infection 91. In addition, spike proteins from isolates of palm civets utilized civet ACE2 more efficiently than human ACE2, whereas late‐phase isolates such as TOR2 utilized both receptors with equal efficiency 92. Subsequent studies demonstrated that sequence variation in the spike protein binding sites of ACE2 hindered efficient binding 92 (Figure 5). The lower affinity of some of these S proteins could be complemented by altering specific residues within the S‐protein‐binding site of human ACE2 to those of civet ACE2, or by altering S‐protein residues 479 and 487 to residues conserved during the 2002–2003 outbreak. This indicates that specific molecular interactions are important in the adaptation process of SARS‐CoV to human cells. Animal precursors of SARS‐CoV are thus likely to be less pathogenic to humans, and exposure to such viruses may have led to limited clinical symptoms but antigenic stimulation that results in a serological response. This was observed when SARS‐CoV re‐emerged in Guangdong in 2003, causing milder clinical disease 93. Similarly, animal traders had high seroprevalence for human and animal SARS coronavirus, without having a history of SARS.
Figure 5.

Alignment of amino acid sequences of ACE2 from human, macaque (Mf), cat (Felis), rat (Rn), and mouse (Mumu) critical to SARS‐CoV spike protein interactions
ACE2 is down‐regulated in the lungs of mice after acute lung injury, including SARS‐CoV infection 86. The cytokines IL‐4 and IFN‐γ down‐regulated cell‐surface expression of ACE2 (Figure 6), decreased ACE2 mRNA levels, and also inhibited SARS‐CoV replication in Vero E6 cells 94. Furthermore, experiments in vitro indicated that ACE2 expression is dependent on the differentiation state of epithelia 95, and a role for the GATA family of transcription factors in regulating the expression of ACE2 has been suggested 96. Down‐regulation of ACE2 expression may not only affect SARS‐CoV entry, but also hamper angiotensin II cleavage, causing pathological changes due to angiotensin II type 1a receptor activation 86. Therefore, intervention strategies using soluble recombinant ACE2 proteins may neutralize SARS‐CoV and dampen lung pathology. However, studies analysing the role of ACE2 gene polymorphisms in the progression of SARS have not found evidence that these affect outcome 9, 97.
Figure 6.

IFN‐gamma down‐regulates ACE2 expression in Vero E6 cells. ACE2 expression was determined by Facs analysis after treatment with TNF‐α (A), IFN‐γ (B), and IFN‐γ combined with TNF‐α (C), all at 48 h, or Vero E6 cells incubated for 96 h in the presence of IFN‐γ combined with TNF‐α (D). Dotted lines represent cytokine‐treated cells, while thick lines represent mock‐treated control cells. The shaded areas represent background staining. Modified from de Lang et al 94, with permission
Conclusions and future perspectives
ACE2 is now implicated in a variety of (patho) physiological processes. Further understanding of its role in disease will hopefully lead to the exploration of novel therapeutic options. Functional studies using ACE2 inhibitors will be essential to elucidate the regulatory mechanisms of ACE2. Moreover, relatively little progress has been made on the development of such specific ACE2 inhibitors, largely because of lack of a clear therapeutic target. MLN4760 is the most potent and selective ACE2 inhibitor currently available 98. Structure‐based screening programmes have been applied to the identification of novel and selective ACE2 inhibitors with some success 99, 100. Since ACE2 opposes the vasoactive and proliferative actions of Ang II, up‐regulation of ACE2 expression or activity is a much more desirable characteristic. Modification of ACE2 levels by stimulating its expression or exogenous administration of recombinant ACE2 may have beneficial effects in several disease conditions and should be the scope of future studies.
Acknowledgements
We thank Mirjan van Timmeren for critically reading the manuscript.
No conflicts of interest were declared.
References
- 1. Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin‐converting enzyme 2 and angiotensin‐(1–7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol 2005; 289: H2281–H2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res 2000; 87: E1–E9. [DOI] [PubMed] [Google Scholar]
- 3. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem 2000; 275: 33238–33243. [DOI] [PubMed] [Google Scholar]
- 4. Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin–angiotensin system. Am J Physiol Regul Integr Comp Physiol 2007; 292: R373–R381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gembardt F, Sterner‐Kock A, Imboden H, Spalteholz M, Reibitz F, Schultheiss HP, et al. Organ‐specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 2005; 26: 1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamming I, Timens W, Bulthuis M, Lely A, Navis G, Van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004; 203: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harmer D, Gilbert M, Borman R, Clark KL. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 2002; 532: 107–110. [DOI] [PubMed] [Google Scholar]
- 8. Komatsu T, Suzuki Y, Imai J, Sugano S, Hida M, Tanigami A, et al. Molecular cloning, mRNA expression and chromosomal localization of mouse angiotensin‐converting enzyme‐related carboxypeptidase (mACE2). DNA Seq 2002; 13: 217–220. [DOI] [PubMed] [Google Scholar]
- 9. Itoyama S, Keicho N, Hijikata M, Quy T, Phi NC, Long HT, et al. Identification of an alternative 5′‐untranslated exon and new polymorphisms of angiotensin‐converting enzyme 2 gene: lack of association with SARS in the Vietnamese population. Am J Med Genet A 2005; 136: 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang H, Wada J, Hida K, Tsuchiyama Y, Hiragushi K, Shikata K, et al. Collectrin, a collecting duct‐specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem 2001; 276: 17132–17139. [DOI] [PubMed] [Google Scholar]
- 11. Akpinar P, Kuwajima S, Krutzfeldt J, Stoffel M. Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab 2005; 2: 385–397. [DOI] [PubMed] [Google Scholar]
- 12. Fukui K, Yang Q, Cao Y, Takahashi N, Hatakeyama H, Wang H, et al. The HNF‐1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab 2005; 2: 373–384. [DOI] [PubMed] [Google Scholar]
- 13. Malakauskas SM, Quan H, Fields TA, McCall SJ, Yu MJ, Kourany WM, et al. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am J Physiol Renal Physiol 2006; 292: F533–F544. [DOI] [PubMed] [Google Scholar]
- 14. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J 2004; 383: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brosnihan KB, Neves LA, Joyner J, Averill DB, Chappell MC, Sarao R, et al. Enhanced renal immunocytochemical expression of ANG‐(1–7) and ACE2 during pregnancy. Hypertension 2003; 42: 749–753. [DOI] [PubMed] [Google Scholar]
- 16. Campbell DJ, Zeitz CJ, Esler MD, Horowitz JD. Evidence against a major role for angiotensin converting enzyme‐related carboxypeptidase (ACE2) in angiotensin peptide metabolism in the human coronary circulation. J Hypertens 2004; 22: 1971–1976. [DOI] [PubMed] [Google Scholar]
- 17. Elased KM, Cunha TS, Gurley SB, Coffman TM, Morris M. New mass spectrometric assay for angiotensin‐converting enzyme 2 activity. Hypertension 2006; 47: 1010–1017. [DOI] [PubMed] [Google Scholar]
- 18. Ferrario CM, Averill DB, Brosnihan KB, Chappell MC, Iskandar SS, Dean RH, et al. Vasopeptidase inhibition and Ang‐(1–7) in the spontaneously hypertensive rat. Kidney Int 2002; 62: 1349–1357. [DOI] [PubMed] [Google Scholar]
- 19. Li N, Zimpelmann J, Cheng K, Wilkins JA, Burns KD. The role of angiotensin converting enzyme 2 in the generation of angiotensin 1–7 by rat proximal tubules. Am J Physiol Renal Physiol 2005; 288: F353–F362. [DOI] [PubMed] [Google Scholar]
- 20. Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, et al. Increased angiotensin‐(1–7)‐forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin‐converting enzyme homologue ACE2. Circulation 2003; 108: 1707–1712. [DOI] [PubMed] [Google Scholar]
- 21. Ocaranza MP, Godoy I, Jalil JE, Varas M, Collantes P, Pinto M, et al. Enalapril attenuates downregulation of angiotensin‐converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 2006; 48: 572–578. [DOI] [PubMed] [Google Scholar]
- 22. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin‐converting enzyme‐related carboxypeptidase. J Biol Chem 2002; 277: 14838–14843. [DOI] [PubMed] [Google Scholar]
- 23. Warner FJ, Lew RA, Smith AI, Lambert DW, Hooper NM, Turner AJ. Angiotensin‐converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J Biol Chem 2005; 280: 39353–39362. [DOI] [PubMed] [Google Scholar]
- 24. Ren X, Glende J, Al Falah M, de Vries V, Schwegmann‐Wessels C, Qu X, et al. Analysis of ACE2 in polarized epithelial cells: surface expression and function as receptor for severe acute respiratory syndrome‐associated coronavirus. J Gen Virol 2006; 87: 1691–1695. [DOI] [PubMed] [Google Scholar]
- 25. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor‐alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe‐acute respiratory syndrome‐coronavirus (SARS‐CoV) receptor, angiotensin‐converting enzyme‐2 (ACE2). J Biol Chem 2005; 280: 30113–30119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaltout HA, Westwood B, Averill DB, Ferrario CM, Figueroa J, Diz DI, et al. Angiotensin metabolism in renal proximal tubules, urine and serum of sheep: evidence for ACE2‐dependent processing of angiotensin II. Am J Physiol Renal Physiol 2006; 292: F82–F91. [DOI] [PubMed] [Google Scholar]
- 27. Burrell LM, Risvanis J, Kubota E, Dean RG, MacDonald PS, Lu S, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J 2005; 26: 369–375. [DOI] [PubMed] [Google Scholar]
- 28. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature 2002; 417: 822–828. [DOI] [PubMed] [Google Scholar]
- 29. Gurley SB, Allred A, Le TH, Griffiths R, Mao L, Philip N, et al. Altered blood pressure responses and normal cardiac phenotype in ACE2‐null mice. J Clin Invest 2006; 116: 2218–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamamoto K, Ohishi M, Katsuya T, Ito N, Ikushima M, Kaibe M, et al. Deletion of angiotensin‐converting enzyme 2 accelerates pressure overload‐induced cardiac dysfunction by increasing local angiotensin II. Hypertension 2006; 47: 718–726. [DOI] [PubMed] [Google Scholar]
- 31. Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N, et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol 2003; 35: 1043–1053. [DOI] [PubMed] [Google Scholar]
- 32. Goulter AB, Goddard MJ, Allen JC, Clark KL. ACE2 gene expression is up‐regulated in the human failing heart. BMC Med 2004; 2: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin‐converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004; 43: 970–976. [DOI] [PubMed] [Google Scholar]
- 34. Karram T, Abbasi A, Keidar S, Golomb E, Hochberg I, Winaver J, et al. Effects of spironolactone and eprosartan on cardiac remodeling and angiotensin‐converting enzyme isoforms in rats with experimental heart failure. Am J Physiol Heart Circ Physiol 2005; 289: H1351–H1358. [DOI] [PubMed] [Google Scholar]
- 35. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin‐converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin‐converting enzyme 2. Circulation 2005; 111: 2605–2610. [DOI] [PubMed] [Google Scholar]
- 36. Keidar S, Gamliel‐Lazarovich A, Kaplan M, Pavlotzky E, Hamoud S, Hayek T, et al. Mineralocorticoid receptor blocker increases angiotensin‐converting enzyme 2 activity in congestive heart failure patients. Circ Res 2005; 97: 946–953. [DOI] [PubMed] [Google Scholar]
- 37. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309–1321. [DOI] [PubMed] [Google Scholar]
- 38. Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin‐(1–7) is an endogenous ligand for the G protein‐coupled receptor Mas. Proc Natl Acad Sci U S A 2003; 100: 8258–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, et al. Protection from angiotensin II‐induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol 2005; 90: 783–790. [DOI] [PubMed] [Google Scholar]
- 40. Diez‐Freire C, Vazquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, et al. ACE2 gene transfer attenuates hypertension‐linked pathophysiological changes in the SHR. Physiol Genomics 2006; 27: 12–19. [DOI] [PubMed] [Google Scholar]
- 41. Yagil Y, Yagil C. Hypothesis: ACE2 modulates blood pressure in the mammalian organism. Hypertension 2003; 41: 871–873. [DOI] [PubMed] [Google Scholar]
- 42. Hilbert P, Lindpaintner K, Beckmann JS, Serikawa T, Soubrier F, Dubay C, et al. Chromosomal mapping of two genetic loci associated with blood‐pressure regulation in hereditary hypertensive rats. Nature 1991; 353: 521–529. [DOI] [PubMed] [Google Scholar]
- 43. Yagil C, Sapojnikov M, Kreutz R, Zurcher H, Ganten D, Yagil Y. Role of chromosome X in the Sabra rat model of salt‐sensitive hypertension. Hypertension 1999; 33: 261–265. [DOI] [PubMed] [Google Scholar]
- 44. Zhong JC, Huang DY, Yang YM, Li YF, Liu GF, Song XH, et al. Upregulation of angiotensin‐converting enzyme 2 by all‐trans retinoic acid in spontaneously hypertensive rats. Hypertension 2004; 44: 907–912. [DOI] [PubMed] [Google Scholar]
- 45. Tikellis C, Cooper ME, Bialkowski K, Johnston CI, Burns WC, Lew RA, et al. Developmental expression of ACE2 in the SHR kidney: a role in hypertension? Kidney Int 2006; 70: 34–41. [DOI] [PubMed] [Google Scholar]
- 46. Hamming I, Kreutz R, Sluimer J, Bolbrinker J, Bernardy C, Walther T, et al. Renal angiotensin‐converting enzyme 2 is unaltered in experimental hypertension. (Abstract.) J Am Soc Nephrol 2005; 16: TH–PO294. [Google Scholar]
- 47. Benjafield AV, Wang WY, Morris BJ. No association of angiotensin‐converting enzyme 2 gene (ACE2) polymorphisms with essential hypertension. Am J Hypertens 2004; 17: 624–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yi L, Gu YH, Wang XL, An LZ, Xie XD, Shao W, et al. Association of ACE, ACE2 and UTS2 polymorphisms with essential hypertension in Han and Dongxiang populations from north‐western China. J Int Med Res 2006; 34: 272–283. [DOI] [PubMed] [Google Scholar]
- 49. Zhong J, Yan Z, Liu D, Ni Y, Zhao Z, Zhu S, et al. Association of angiotensin‐converting enzyme 2 gene A/G polymorphism and elevated blood pressure in Chinese patients with metabolic syndrome. J Lab Clin Med 2006; 147: 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, Van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004; 203: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lely AT, Hamming I, Van Goor H, Navis GJ. Renal ACE2 expression in human kidney disease. J Pathol 2004; 204: 587–593. [DOI] [PubMed] [Google Scholar]
- 52. Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of angiotensin‐converting enzyme 2 and angiotensin‐converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 2006; 17: 3067–3075. [DOI] [PubMed] [Google Scholar]
- 53. Ye M, Wysocki J, Naaz P, Salabat MR, LaPointe MS, Batlle D. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice. A renoprotective combination? Hypertension 2004; 43: 1120–1125. [DOI] [PubMed] [Google Scholar]
- 54. Dzau VJ, Bernstein K, Celermajer D, Cohen J, Dahlof B, Deanfield J, et al. The relevance of tissue angiotensin‐converting enzyme: manifestations in mechanistic and endpoint data. Am J Cardiol 2001; 88: 1L–20L. [DOI] [PubMed] [Google Scholar]
- 55. Huang W, Gallois Y, Bouby N, Bruneval P, Heudes D, Belair MF, et al. Genetically increased angiotensin I‐converting enzyme level and renal complications in the diabetic mouse. Proc Natl Acad Sci U S A 2001; 98: 13330–13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rook M, Lely AT, Kramer AB, Van Goor H, Navis G. Individual differences in renal ACE activity in healthy rats predict susceptibility to adriamycin‐induced renal damage. Nephrol Dial Transplant 2005; 20: 59–64. [DOI] [PubMed] [Google Scholar]
- 57. Ruiz‐Ortega M, Ruperez M, Esteban V, Rodriguez‐Vita J, Sanchez‐Lopez E, Carvajal G, et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant 2006; 21: 16–20. [DOI] [PubMed] [Google Scholar]
- 58. Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int 2005; 67: 799–812. [DOI] [PubMed] [Google Scholar]
- 59. Tikellis C, Johnston CI, Forbes JM, Burns WC, Burrell LM, Risvanis J, et al. Characterization of renal angiotensin‐converting enzyme 2 in diabetic nephropathy. Hypertension 2003; 41: 392–397. [DOI] [PubMed] [Google Scholar]
- 60. Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell M, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren‐2 rats. Am J Physiol Heart Circ Physiol 2006; 291: H2166–H2172. [DOI] [PubMed] [Google Scholar]
- 61. Hamming I, Navis G, Akkermans KE, Van Goor H. ACE inhibition during low salt intake decreases renal ACE2 expression in healthy and nephrotic rats. (Abstract.) J Hypertension 2006; 24: S295. [Google Scholar]
- 62. Hamming I, Navis G, Kocks M, Van Goor H. ACE inhibition has adverse renal effects during dietary sodium restriction in proteinuric and healthy rats. J Pathol 2006; 209: 129–139. [DOI] [PubMed] [Google Scholar]
- 63. Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R, et al. Loss of angiotensin‐converting enzyme‐2 leads to the late development of angiotensin II‐dependent glomerulosclerosis. Am J Pathol 2006; 168: 1808–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wysocki J, Ye M, Soler MJ, Gurley SB, Xiao HD, Bernstein KE, et al. ACE and ACE2 activity in diabetic mice. Diabetes 2006; 55: 2132–2139. [DOI] [PubMed] [Google Scholar]
- 65. Konoshita T, Wakahara S, Mizuno S, Motomura M, Aoyama C, Makino Y, et al. Tissue gene expression of renin–angiotensin system in human type 2 diabetic nephropathy. Diabetes Care 2006; 29: 848–852. [DOI] [PubMed] [Google Scholar]
- 66. Frojdo S, Sjolind L, Parkkonen M, Makinen VP, Kilpikari R, Pettersson‐Fernholm K, et al. Polymorphisms in the gene encoding angiotensin I converting enzyme 2 and diabetic nephropathy. Diabetologia 2005; 48: 2278–2281. [DOI] [PubMed] [Google Scholar]
- 67. Valdes G, Neves LA, Anton L, Corthorn J, Chacon C, Germain AM, et al. Distribution of angiotensin‐(1–7) and ACE2 in human placentas of normal and pathological pregnancies. Placenta 2006; 27: 200–207. [DOI] [PubMed] [Google Scholar]
- 68. Marshall RP. The pulmonary renin–angiotensin system. Curr Pharm Des 2003; 9: 715–722. [DOI] [PubMed] [Google Scholar]
- 69. Orte C, Polak JM, Haworth SG, Yacoub MH, Morrell NW. Expression of pulmonary vascular angiotensin‐converting enzyme in primary and secondary plexiform pulmonary hypertension. J Pathol 2000; 192: 379–384. [DOI] [PubMed] [Google Scholar]
- 70. Morrell NW, Morris KG, Stenmark KR. Role of angiotensin‐converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol 1995; 269: H1186–H1194. [DOI] [PubMed] [Google Scholar]
- 71. Loddenkemper R, Kloppenborg A, Schoenfeld N, Grosser H, Costabel U. Clinical findings in 715 patients with newly detected pulmonary sarcoidosis—results of a cooperative study in former West Germany and Switzerland. WATL Study Group. Wissenschaftliche Arbeitsgemeinschaft fur die Therapie von Lungenkrankheitan. Sarcoidosis Vasc Diffuse Lung Dis 1998; 15: 178–182. [PubMed] [Google Scholar]
- 72. Csaszar A, Halmos B, Palicz T, Szalai C, Romics L. Interpopulation effect of ACE I/D polymorphism on serum concentration of ACE in diagnosis of sarcoidosis. Lancet 1997; 350: 518. [DOI] [PubMed] [Google Scholar]
- 73. Specks U, Martin WJ, Rohrbach MS. Bronchoalveolar lavage fluid angiotensin‐converting enzyme in interstitial lung diseases. Am Rev Respir Dis 1990; 141: 117–123. [DOI] [PubMed] [Google Scholar]
- 74. Fourrier F, Chopin C, Wallaert B, Mazurier C, Mangalaboyi J, Durocher A. Compared evolution of plasma fibronectin and angiotensin‐converting enzyme levels in septic ARDS. Chest 1985; 87: 191–195. [DOI] [PubMed] [Google Scholar]
- 75. Idell S, Kueppers F, Lippmann M, Rosen H, Niederman M, Fein A. Angiotensin converting enzyme in bronchoalveolar lavage in ARDS. Chest 1987; 91: 52–56. [DOI] [PubMed] [Google Scholar]
- 76. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature 2005; 436: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kuba K, Imai Y, Penninger JM. Angiotensin‐converting enzyme 2 in lung diseases. Curr Opin Pharmacol 2006; 6: 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pohlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A 2005; 102: 7988–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426: 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Weiss SR, Navas‐Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 2005; 69: 635–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003; 361: 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science 2005; 309: 1864–1868. [DOI] [PubMed] [Google Scholar]
- 83. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193‐amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin‐converting enzyme 2. J Biol Chem 2004; 279: 3197–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Han DP, Penn‐Nicholson A, Cho MW. Identification of critical determinants on ACE2 for SARS‐CoV entry and development of a potent entry inhibitor. Virology 2006; 350: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mossel EC, Huang C, Narayanan K, Makino S, Tesh RB, Peters CJ. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. J Virol 2005; 79: 3846–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nature Med 2005; 11: 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. He L, Ding Y, Zhang Q, Che X, He Y, Shen H, et al. Expression of elevated levels of pro‐inflammatory cytokines in SARS‐CoV‐infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J Pathol 2006; 210: 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2‐expressing cells. J Biol Chem 2006; 281: 3198–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 2005; 102: 11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jeffers SA, Tusell SM, Gillim‐Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L‐SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A 2004; 101: 15748–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li W, Greenough TC, Moore MJ, Vasilieva N, Somasundaran M, Sullivan JL, et al. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin‐converting enzyme 2. J Virol 2004; 78: 11429–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, et al. Receptor and viral determinants of SARS‐coronavirus adaptation to human ACE2. EMBO J 2005; 24: 1634–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liang G, Chen Q, Xu J, Liu Y, Lim W, Peiris JS, et al. Laboratory diagnosis of four recent sporadic cases of community‐acquired SARS, Guangdong Province, China. Emerg Infect Dis 2004; 10: 1774–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. de Lang A, Osterhaus AD, Haagmans BL. Interferon‐gamma and interleukin‐4 downregulate expression of the SARS coronavirus receptor ACE2 in Vero E6 cells. Virology 2006; 353: 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol 2005; 79: 14614–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chou CF, Loh CB, Foo YK, Shen S, Fielding BC, Tan TH, et al. ACE2 orthologues in non‐mammalian vertebrates (Danio, Gallus, Fugu, Tetraodon and Xenopus). Gene 2006; 377: 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chiu RW, Tang NL, Hui DS, Chung GT, Chim SS, Chan KC, et al. ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome. Clin Chem 2004; 50: 1683–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dales NA, Gould AE, Brown JA, Calderwood EF, Guan B, Minor CA, et al. Substrate‐based design of the first class of angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) inhibitors. J Am Chem Soc 2002; 124: 11852–11853. [DOI] [PubMed] [Google Scholar]
- 99. Huentelman MJ, Zubcevic J, Hernandez Prada JA, Xiao X, Dimitrov DS, Raizada MK, et al. Structure‐based discovery of a novel angiotensin‐converting enzyme 2 inhibitor. Hypertension 2004; 44: 903–906. [DOI] [PubMed] [Google Scholar]
- 100. Rella M, Rushworth CA, Guy JL, Turner AJ, Langer T, Jackson RM. Structure‐based pharmacophore design and virtual screening for novel angiotensin converting enzyme 2 inhibitors. J Chem Inf Model 2006; 46: 708–716. [DOI] [PubMed] [Google Scholar]
- 101. Warner FJ, Smith AI, Hooper NM, Turner AJ. Angiotensin‐converting enzyme‐2: a molecular and cellular perspective. Cell Mol Life Sci 2004; 61: 2704–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Turner AJ, Hooper NM. The angiotensin‐converting enzyme gene family: genomics and pharmacology. Trends Pharmacol Sci 2002; 23: 177–183. [DOI] [PubMed] [Google Scholar]