

Abstract
Lectins are proteins of nonimmune origin, which are capable of recognizing and binding to glycoconjugate moieties. Some of them can block the interaction of viral glycoproteins to the host cell receptors acting as antiviral agents. Although cyanobacterial lectins have presented broad biotechnological potential, little research has been directed to Amazonian Cyanobacterial diversity. In order to identify new antiviral lectins, we performed genomic analysis in seven cyanobacterial strains from Coleção Amazônica de Cianobactérias e Microalgas (CACIAM). We found 75 unique CDS presenting one or more lectin domains. Since almost all were annotated as hypothetical proteins, we used homology modeling and molecular dynamics simulations to evaluate the structural and functional properties of three CDS that were more similar to known antiviral lectins. Nostoc sp. CACIAM 19 as well as Tolypothrix sp. CACIAM 22 strains presented cyanovirin‐N homologues whose function was confirmed by binding free energy calculations. Asn, Glu, Thr, Lys, Leu, and Gly, which were described as binding residues for cyanovirin, were also observed on those structures. As for other known cyanovirins, those residues in both our models also made favorable interactions with dimannose. Finally, Alkalinema sp. CACIAM 70d presented one CDS, which was identified as a seven‐bladed beta‐propeller structure with binding sites predicted for sialic acid and N‐acetylglucosamine. Despite its singular structure, our analysis suggested this molecule as a new putative antiviral lectin. Overall, the identification and the characterization of new lectins and their homologues are a promising area in antiviral research, and Amazonian cyanobacteria present biotechnological potential to be explored in this regard.
Keywords: Amazonia, antiviral lectins, cyanobacteria, cyanovirin, molecular dynamics
1. INTRODUCTION
Lectins are proteins of nonimmune origin, which are capable of recognizing and binding to glycoconjugate moieties without altering the covalent structure of any of the recognized glycosyl ligands.1 They are present in most organisms, including viruses, bacteria, fungi, plants, and animals and act in many biological processes, such as host‐pathogen interactions, cell‐cell communication, induction of apoptosis, cancer metastasis and differentiation and targeting of cells, as well as recognizing and binding carbohydrates.2 Proteins classified as lectins show major structural differences, and because of that their relation is more functional than structural.
Some lectins have been evaluated as antiviral agents due to their ability to bind to specific carbohydrates in the infection context. Those lectins can block the interaction of viral glycoproteins to host cell receptors, a mechanism which has already been demonstrated for human immunodeficiency virus (HIV), Zaire Ebola virus, Hepatitis C virus, influenza virus, and others.2, 3, 4, 5, 6, 7
The Phylum Cyanobacteria has been shown to be a promising source of antiviral lectins, mainly because of their anti‐HIV activity. Currently, three lectins have gained prominence: (i) Cyanovirin‐N, which was isolated from Nostoc ellipsosporum, may inactivate HIV strains even at low nanomolar concentrations8, 9; (ii) microvirin, isolated from Microcystis aeruginosa, shares 33% of identity with cyanovirin, but it is 50 times less cytotoxic10; and, (iii) scytovirin, which was isolated from Scytonema varium, acts against Zaire Ebola virus, coronavirus, and Cryptococcus fungi besides having anti‐HIV activity.4, 11, 12 In this sense, screening of new cyanobacterial lectins as well their structural improvement is a reasonable strategy in the search for new microbicide candidates.13, 14, 15
Despite the great biological diversity and ecological importance of the region, the first Amazonian cyanobacteria genome was published only in 2014.16 Since then, other genomic studies have been performed with the aim of investigating the genetic potential and diversity of cyanobacteria from this region.17, 18, 19 However, there are few studies based on biotechnological applications for these cyanobacteria. In this sense, genomic screening has been used successfully to access the genetic potential of an individual or an organism group.20, 21, 22 Thus, the aim of this study was to investigate, by genomic analysis, homology modeling, and molecular dynamics simulations, the presence of potential antiviral lectins in cyanobacterial genomes isolated from Amazonian environments.
2. MATERIAL AND METHODS
2.1. Strains
All of the strains analyzed in this study belong to the CACIAM collection (Coleção Amazônica de Cianobactérias e Microalgas) maintained by Laboratório de Tecnologia Biomolecular at Universidade Federal do Pará, Brazil. The genomes of 7 strains were employed for genomic search: Alkalinema sp. CACIAM 70d17, Cyanobium sp. CACIAM 14,16 Limnothrix sp. CACIAM 69d, Microcystis aeruginosa CACIAM 03,18 Nostoc sp. CACIAM 19, Synechococcus sp. CACIAM 66, and Tolypothrix sp. CACIAM 22. These cyanobacteria were isolated from two lakes in the Amazon region: Tucuruí hydroelectric power station reservoir (3°50′04.9″S, 49°42′32.2″W) and Bolonha Lake (1°25′00.7″S, 48°25′52.6″W) both in Pará, Brazil.
2.2. Genomic search
In order to identify putative lectin sequences in the 7 CACIAM strain genomes, a conserved domain approach search was applied. First, amino acid sequences from NCBI annotated as lectins that have solved structures were downloaded. Next, the CD‐HIT standalone version23 was used to cluster sequences with >90% identity, making the data nonredundant. The final dataset was submitted to a conserved domain database (CDD) webserver24 for a domain identification procedure. The results obtained were stored to be used as a positive indication of a lectin in the next step.
All coding sequences (CDS) annotated in the 7 CACIAM strains genomes were extracted and translated with Geneious R9,25 using bacterial genetic code. For each genome, the translated CDS were submitted to CDD. A custom Perl script was applied to parse CDD results, retrieving only sequences that had the same domains identified in lectin sequences.
2.3. Homology modeling
To perform the homology modeling, two strategies were used. Sequences having enough identity with templates of Protein Data Bank26 (PDB) were modeled with Modeller9.1627 software. Promals3D28 performed the sequence alignment of the template and the target. A total of 100 models were generated based on the target‐template alignment, considering different conformations, and ranked by molecular probability density function (Molpdf) and DOPE score. Automatic loop refinement was used after model building and the models were generated, satisfying spatial restrictions such as bond lengths, bond angles, dihedral angles, and interactions between nonbonded atoms, and then subjected to validation. Sequences that had no identity with PDB structures were modeled on a I‐TASSER server29 and the best model was selected according to C‐score and alignment quality with the templates.
After the model construction, the stereochemical quality was evaluated using a Ramachandran plot generated in the MolProbity30 server. Verify3D31 determined the quality of folding and finally, Qmean32 was computed to measure the local quality.
2.4. Molecular dynamics
All homology models generated here were submitted to a Molecular Dynamics (MD) refinement simulation at 100 ns. After that the proteins with known ligands were complexed to them for a new MD simulation of 210 ns and binding free energy calculations.
To perform these MD, a PDB2PQR server (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0) was used to determine the protonation state of the protein considering a pH level of 7.0. All steps of preparation and production of MD were produced using the AMBER 16 software package33. The force fields applied were GLYCAM_06j34 and FF14SB35 for the ligand and the protein, respectively. Counter ions Na+ or Cl− were added to neutralize the charges and TIP3P36 water molecules in an octagonal box with 10 Å in each direction of the protein. Energy minimization was performed in five steps, four of these using 3000 cycles of steepest descent and 5000 cycles of conjugate gradients for each one; the heavy atoms were restrained by a harmonic potential of 1000 Kcal/mol*Å2. In the last step, we used 5000 cycles of steepest descent and 30 000 cycles of conjugate gradients and no restraints. The heating and equilibration stage was divided into 14 steps. The temperature was gradually increased, until it reached 300 K. Langevin Dynamics (thermostat) were employed with a collision frequency of 3.0 ps − 1. A harmonic potential of 25 Kcal/mol*Å2 was employed in the initial steps and was turned off during step 13. The heating procedure lasted 650 ps until step 13 and was performed using an NVT ensemble. Afterward, a 2‐ns equilibration phase was employed in an NPT ensemble. The SHAKE algorithm was employed to restrict vibration of the ligations of all hydrogen atoms. The Particle Mesh Ewald method was used for calculating electrostatic interactions using a cutoff value of 10.0 Å.
2.5. Binding free energy calculations
For the systems complexed with a carbohydrate ligand, Molecular Mechanics Generalized Born Surface Area (MM‐GBSA), Molecular Mechanics Poisson‐Boltzmann Surface Area (MM‐PBSA)37 and Solvated Interaction Energy (SIE)38 methods were employed to calculate the binding free energy of the protein‐ligand complexation (ΔGbind). It used the HIV envelope dimannose (MAN‐MAN) to evaluate the lectin affinity for this virus. These calculations were based on five thousand snapshots from the last 10 ns of the molecular dynamics simulations.
3. RESULTS AND DISCUSSION
3.1. Genomic search
A total of 75 unique CDS were returned from the genomic search in the 7 CACIAM strains presenting one or more lectin domains, according to the comparison with the NCBI solved lectin structures dataset (Table 1). More than 50% of these sequences are annotated as hypothetical proteins, which reinforces the importance of structural analysis in discovering new proteins and their functions.
Table 1.
Results of genomic screening showing the number of CDSs predicted with lectins domains
| Strain | Sequences | Smaller | Largest |
|---|---|---|---|
| Alkalinema sp. CACIAM70d | 2 | 177 aa | 264 aa |
| Cyanobium sp. CACIAM14 | 4 | 219 aa | 1257 aa |
| Limnothrix sp. CACIAM69d | 9 | 289 aa | 1652 aa |
| Microcystis sp. CACIAM03 | 20 | 61 aa | 1267 aa |
| Nostoc sp. CACIAM19 | 24 | 128 aa | 1480 aa |
| Synechococcus sp. CACIAM66 | 4 | 358 aa | 784 aa |
| Tolypothrix sp. CACIAM22 | 22 | 99 aa | 1507 aa |
Known antiviral lectins produced by cyanobacteria present less than 200 aa and 2 or more disulfide bonds in their structures.14 Therefore, the best candidates for homology modeling and molecular dynamics analysis were chosen following these criteria. Additionally, two sequences were chosen due to the presence of the CVNH cyanovirin domain; they are from Nostoc sp. CACIAM 19 and Tolypothrix sp. CACIAM 22 strains. Another sequence of 177 aa identified in Alkalinema sp. CACIAM 70d was selected to be modeled because of its length, the presence of 6 cysteine residues, and the CDD results, which identified the VCBS domain (pfam13517), a well characterized lectin domain. The BlastP tool39 classified this sequence as an integrin‐like fungal protein that adopts a seven‐bladed beta‐propeller structure and interacts with monosaccharides and calcium.
Therefore, these three sequences (the two cyanovirin homologues and the Alkalinema sp. CACIAM 70d sequence) were modeled with the aim of investigating their structural properties.
3.2. Homology modeling
Cyanovirin tridimensional structures of Nostoc sp. CACIAM 19 and Tolypothrix sp. CACIAM 22 were modeled with Modeller9.16. Nostoc sp. CACIAM 19 cyanovirin homologue presented 103 amino acid residues after the cleavage of a signal peptide predicted by the server SignalP.40 The best alignment with PDB structures showed 60% of identity with the cyanovirin of Cyanothece sp. PCC 7424 (PDB ID: 5 K79),41 which was chosen as template. The root‐mean‐square deviation (RMSD) of these structures was 0.1 Å (Figure 1(A)). The model presented two disulfide bonds between Cys8–Cys22 and Cys59–Cys74.
Figure 1.
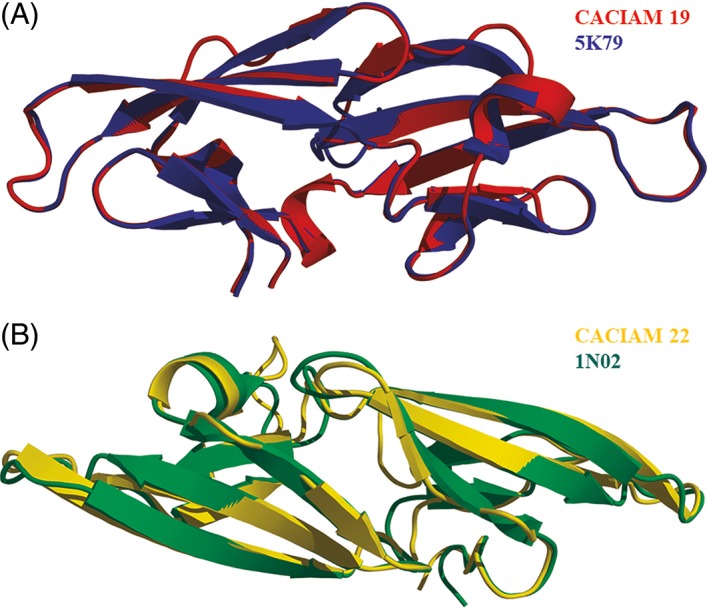
– Structural alignment of Nostoc sp. CACIAM 19 cyanovirin and its template (a) and Tolypothrix sp. CACIAM 22 cyanovirin and its template (B) [Color figure can be viewed at http://wileyonlinelibrary.com]
Tolypothrix sp. CACIAM 22 cyanovirin homologue presented 105 amino acid residues with no signal peptide. Its alignment with PDB structures showed 39% of identity with a potent variant of cyanovirin from Nostoc ellipsosporum (PDB ID: 1 N02).42 RMSD of target and template structures was 0.4 Å (Figure 1(B)). This model showed only one disulfide bond between Cys9 and Cys23.
I‐TASSER server generated the Alkalinema sp. CACIAM 70d lectin model. The sequence submitted presented 147 amino acid residues after the identification of a signal peptide by the server SignalP.40 This model has 3 disulfide bonds between the cysteine pairs Cys4–Cys26, Cys51–Cys73, and Cys105–Cys127. COACH meta‐server43 present in the I‐TASSER server predicted two relevant putative bind sites for this model around residue Lys140 for sialic acid in C‐terminal portion and another one around residue Cys105 for N‐acetylglucosamine (Figure 2). Sialic acid is derived from neuraminic acid that occurs in polysaccharides, glycoproteins, and glycolipids in bacteria and animals. N‐acetylglusamine is a common carbohydrate in mammal glycoproteins added by cotranslational or posttranslational modifications acting in different biological process.44 Both ligands are related to viral infections in humans; for example, influenza virus hemagglutinin uses sialic acid receptors to attach to human cells,45 and dengue virus uses N‐acetylglucosamine receptors during the infection process.46
Figure 2.
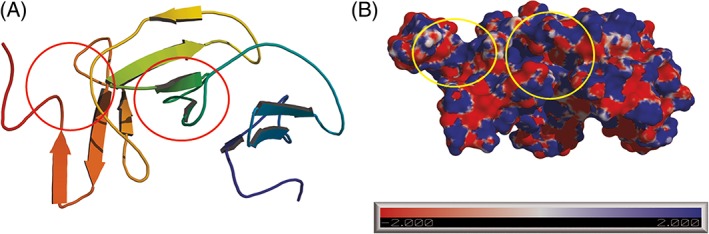
– (A) Alkalinema sp. CACIAM 70d lectin model showing two regions predicted as putative binding sites. (B) Electrostatic surface of the model showing the cavities predicted by COACH. Sialic acid binding sites are on the left and N‐acetylglucosamine binding sites are at the center [Color figure can be viewed at http://wileyonlinelibrary.com]
3.3. Molecular dynamics
A 100‐ns MD simulation was performed for refining the models. After that, they were validated and the results are presented in Table 2. Conformational changes observed in these simulations were fundamental for improving the validation tests of the models, which were constructed based on crystallographic data. Alkalinema sp. CACIAM 70d lectin showed the highest RMSD values but its Ramachandran evaluation went up from 74% to 96.5% after the 100 ns simulation (Figure 3(A)). The CDD identification, the validated model, and the binding site prediction suggest that this ORF (OUC12179.1) annotated as a hypothetical protein in the Alkalinema sp. CACIAM 70d genome is probably a new putative antiviral lectin, the first described for this new genus.17
Table 2.
– Models identification and validation. Ramachandran values show the residues in favorable regions. SignalP server was used to predict the presence of signal peptide (http://www.cbs.dtu.dk/services/SignalP)
| Model | Ramachandran | Verify3D | Qmean | Signal P | Domain | Annotation |
|---|---|---|---|---|---|---|
| Alkalinema sp. CACIAM70d | 96.5% | 100.0% | −2.87 | Yes | VCBS | Hypothetical protein |
| Nostoc sp. CACIAM19 | 96.0% | 100.0% | −1.12 | Yes | CVNH | Cyanovirin‐N (CV‐N) |
| Tolypothrix sp. CACIAM22 | 94.9% | 89.4% | 0.39 | No | CVNH | Hypothetical protein |
Figure 3.
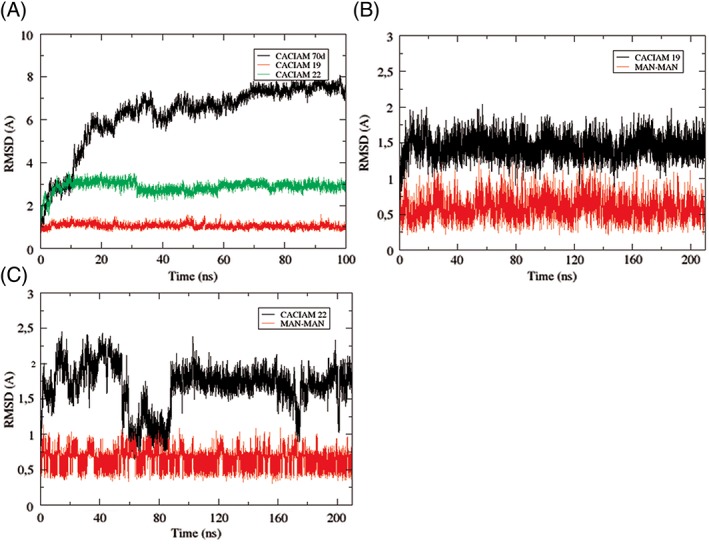
– RMSD graphs of MD simulations. (A) 100 ns refinement simulation of the models. (B) 210 ns simulation of Nostoc sp. CACIAM 19 cyanovirin complexed with MAN–MAN. (C) 210 ns simulation of Tolypothrix sp. CACIAM 22 cyanovirin complexed with MAN–MAN
The structural coordinates of crystallographic dimannose (MAN‐MAN) were obtained from cyanovirin 2RDK deposited in PDB47 and were employed as a template to complex this ligand to the cyanovirin models and their respective templates used in the homology modeling step. MD simulations of 210 ns were produced for these four systems and the structural stability of the models is presented in Figure 3(B) and (C). All complexes remained stable during the simulation and the ligand MAN‐MAN showed RMSD values less than 1 Å in the four cases. The final state of the MD simulation for each system is presented in Figure 4. The binding site affinity and the individual contribution of residues were estimated through binding free energy calculations.
Figure 4.
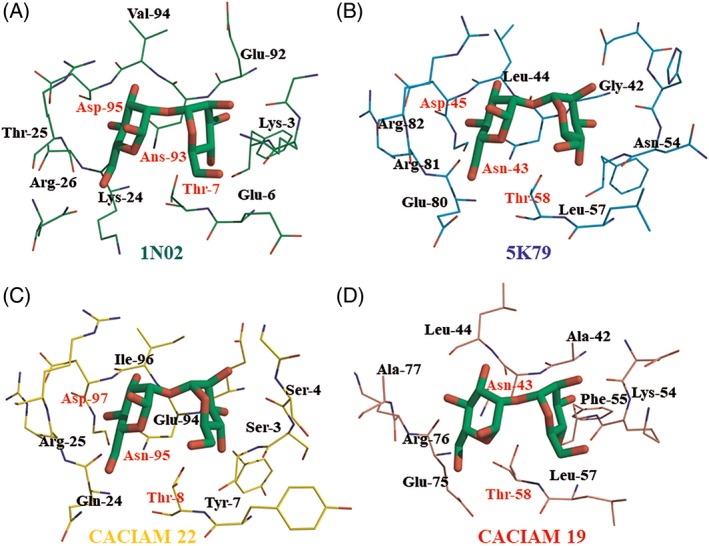
– Final coordinates of MD simulation of (A) Nostoc ellipsosporum (1 N02) template cyanovirin, (B) Cyanothece sp. PCC 7424 (5 K79) template cyanovirin, (C) Tolypothrix sp. CACIAM 22 model cyanovirin, and (D) Nostoc sp. CACIAM 19 model cyanovirin. The residues labeled were the ones most important to the binding process according to the binding free energy calculations
3.4. Binding free energy calculations
The last MD 5000 frames were used for calculating the binding free energy by MM‐GBSA, MM‐PBSA, and SIE methods. The results are presented in Table 3. According to these results, Tolypothrix sp. CACIAM 22 cyanovirin and its template Cyanothece sp. PCC 7424 (5 K79) cyanovirin presented higher affinity to the MAN‐MAN ligand. The individual contribution of residues was evaluated by decomposition of the MM‐GBSA method and the results are presented in Figure 5. Asn, Glu, Thr, Lys, Leu, and Gly, which were described as binding residues for cyanovirin, were also observed on those structures. As to other known cyanovirins, those residues in both of our models also made favorable interactions with dimannose.
Table 3.
– Bind free energy calculations results based on last 10 ns of MD simulation. All energy values are in kcal Mol−1
| MM‐GBSA | Std. dev. | Std. error | MM‐PBSA | Std. dev. | Std. error | SIE | Std. dev. | Std. error | |
|---|---|---|---|---|---|---|---|---|---|
| Nostoc ellipsosporum (1 N02) | −35.02 | 4.41 | 0.14 | −31.88 | 5.74 | 0.18 | −7.97 | 0.41 | 0.03 |
| Cyanothece sp. PCC 7424 (5 K79) | −26.74 | 3.61 | 0.11 | −30.50 | 3.60 | 0.11 | −7.59 | 0.33 | 0.02 |
| Tolypothrix sp. CACIAM 22 | −31.31 | 3.39 | 0.11 | −31.97 | 3.58 | 0.11 | −7.93 | 0.30 | 0.02 |
| Nostoc sp. CACIAM 19 | −18.01 | 2.91 | 0.09 | −19.75 | 3.20 | 0.10 | −6.85 | 0.25 | 0.02 |
Figure 5.
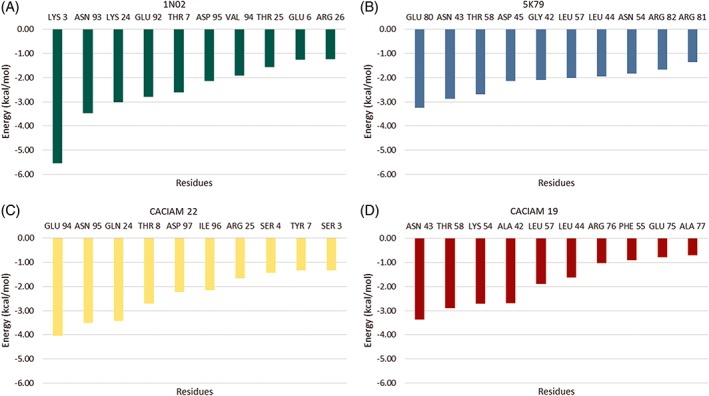
– Individual energy contribution by residues according to the MM‐GBSA method in cyanovirin‐ligand complexation [Color figure can be viewed at http://wileyonlinelibrary.com]
Despite the structural differences among four cyanovirins evaluated here, it was possible to observe the structural conservation of a group of residues. The aspartate, asparagine, and threonine triad appeared in three of the four systems and the Nostoc sp. CACIAM 19 cyanovirin system, which had not conserved the aspartate residue, showed the worst results in binding free energy calculations (Figure 4 and Table 3). Besides that, this triad seems to be fundamental to complexation with dimannose of microvirin, another cyanobacterial lectin.48 Arginine residues were also conserved at the binding site and they presented relevant energy values in decomposition analysis (Figures 4 and 5). In general, polar residues (Glu, Asp, Arg, Lys), capable of making favorable electrostatic interactions with dimannose, collaborated together with the aspartate, asparagine, and threonine triad in the cyanovirin‐ligand complexation. Lys3, Glu80, and Glu94 were the most contributing residues in Nostoc ellipsosporum (1 N02), Cyanothece sp. PCC 7424 (5 K79), and Tolypothrix sp. CACIAM 22 cyanovirin, respectively (Figure 5).
Cyanovirin has been tested against different viral targets and promising results have already been found for human and simian immunodeficiency virus, parainfluenza virus, herpes simplex virus, Epstein‐Barr virus, human herpes virus, bovine viral diarrhea virus, influenza A virus, and others.5, 6, 7, 9, 14 Due to this fact, several studies have been developed with the aim of creating an efficient heterologous expression system for obtaining and purifying cyanovirin. Soy seeds, Pichia pastoris, Nicotiana tabacum, and Althaea officinalis are examples of eukaryotic hosts used to express cyanovirin and its homologues.49, 50, 51, 52
In this sense, the search for new forms of cyanovirin obtained from other cyanobacteria could reveal new applications for this protein, including the reduction of adverse reactions caused by some protein variants. In fact, an identity of approximately 33% is capable of reducing the cytotoxic 50‐fold and maintaining the antiviral activity in microvirin10. Cyanovirin is more active than microvirin due its bivalent iterations with the viral envelopes53, so new cyanovirin forms may present combined properties to be potent inhibitors and less cytotoxic than current variants at the same time. Additionally, the detailed study of residue conservation and binding interactions helps to select the best candidates for antiviral applications.
4. CONCLUSION
This work was the first attempt to identify antiviral lectins in Amazonian cyanobacterial diversity. A genomic search approach returned a reasonable number of sequences with lectin domains and this pipeline may be replicated in different organisms to help in lectin identification, given that the number of sequences annotated as hypothetical protein was superior to 50%. It was also possible to identify two cyanovirin homologues with average sequence identity with PDB proteins, one annotated as cyanovirin and the other as hypothetical protein, both with the binding property demonstrated by molecular dynamics analysis. Additionally, the genomic search allied with homology modeling suggests that the Alkalinema sp. CACIAM 70d lectin model validated here is a binding sugar lectin, the first one reported for this cyanobacteria genus. Thus, the identification and the characterization of new lectins and their homologues are a promising area in antiviral research, and Amazonian cyanobacteria present biotechnological potential to be explored in this regard.
CONFLICT OF INTERESTS
The authors declare that there is no conflict of interest.
ACKNOWLEDGEMENTS
We acknowledge Fundação Amazônia de Amparo a Estudos e Pesquisas do Pará (FAPESPA) for financially supporting (ICAAF 099/2014) our project. Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) also supported an individual author through grant 311686/2015‐0 (ECG).
Siqueira AS, Lima ARJ, Aguiar DCF, Santos AS, Vianez Júnior João Lídio da Silva Gonçalves, Gonçalves EC. Genomic screening of new putative antiviral lectins from Amazonian cyanobacteria based on a bioinformatics approach. Proteins. 2018;86:1047–1054. 10.1002/prot.25577
Funding information Fundação Amazônia Paraense de Amparo à Pesquisa, Grant/Award Number: ICAAF 099/2014; Conselho Nacional de Desenvolvimento Científico e Tecnológico, Grant/Award Number: 311686/2015‐0
REFERENCES
- 1. Sharon N, Lis H. Lectins as cell recognition molecules. Science (New York, N.Y.). 1989;246(4927):227‐234. [DOI] [PubMed] [Google Scholar]
- 2. Ziółkowska NE, Wlodawer A. Structural studies of algal lectins with anti‐HIV activity. Acta Biochim Pol. 2006;53(4):617‐626. [PubMed] [Google Scholar]
- 3. Huskens D, Schols D. Algal lectins as potential HIV microbicide candidates. Mar Drugs. 2012;10(7):1476‐1497. 10.3390/md10071476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garrison AR, Giomarelli BG, Lear‐Rooney CM, et al. The cyanobacterial lectin scytovirin displays potent in vitro and in vivo activity against Zaire Ebola virus. Antiviral Res. 2014;112:1‐7. 10.1016/j.antiviral.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Helle F, Wychowski C, Vu‐Dac N, Gustafson KR, Voisset C, Dubuisson J. Cyanovirin‐N inhibits hepatitis C virus entry by binding to envelope protein glycans. J Biol Chem. 2006;281(35):25177‐25183. 10.1074/jbc.M602431200. [DOI] [PubMed] [Google Scholar]
- 6. O'Keefe BR, Smee DF, Turpin JA, et al. Potent anti‐influenza activity of Cyanovirin‐N and interactions with viral hemagglutinin. Antimicrob Agents Chemother. 2003;47(8):2518‐2525. 10.1128/AAC.47.8.2518-2525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dey B, Lerner DL, Lusso P, Boyd MR, Elder JH, Berger EA. Multiple antiviral activities of cyanovirin‐N: blocking of human immunodeficiency virus type 1 gp120 interaction with CD4 and coreceptor and inhibition of diverse enveloped viruses. J Virol. 2000;74(10):4562‐4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyd MR, Gustafson KR, McMahon JB, et al. Discovery of cyanovirin‐N, a novel human immunodeficiency virus‐inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob Agents Chemother. 1997;41(7):1521‐1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mori T, Boyd MR. Cyanovirin‐N, a potent human immunodeficiency virus‐inactivating protein, blocks both CD4‐dependent and CD4‐independent binding of soluble gp120 (sgp120) to target cells, inhibits sCD4‐induced binding of sgp120 to cell‐associated CXCR4, and dissociates bound sgp120 from target cells. Antimicrob Agents Chemother. 2001;45(3):664‐672. 10.1128/AAC.45.3.664-672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huskens D, Férir G, Vermeire K, et al. Microvirin, a novel alpha(1,2)‐mannose‐specific lectin isolated from Microcystis aeruginosa, has anti‐HIV‐1 activity comparable with that of cyanovirin‐N but a much higher safety profile. J Biol Chem. 2010;285(32):24845‐24854. 10.1074/jbc.M110.128546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bokesch HR, O'Keefe BR, McKee TC, et al. A potent novel anti‐HIV protein from the cultured cyanobacterium Scytonema varium. Biochemistry. 2003;42(9):2578‐2584. 10.1021/bi0205698. [DOI] [PubMed] [Google Scholar]
- 12. Jones TH, McClelland EE, McFeeters H, McFeeters RL. Novel antifungal activity for the lectin Scytovirin: inhibition of Cryptococcus neoformans and Cryptococcus gattii. Front Microbiol. 2017;8(755):1–9. 10.3389/fmicb.2017.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McFeeters H, Gilbert MJ, Wood AM, Haggenmaker CB, Jones J, Kutsch O, RL McFeeters. Scytovirin engineering improves carbohydrate affinity and HIV‐1 entry inhibition. Biochem Physiol. 2013;S2(003):1–5. https://www.omicsonline.org/open-access/scytovirin-engineering-improves-carbohydrate-affinity-and-hiv-1-entry-inhibition-2168-9652.S2-003.php?aid=13502. doi: 10.4172/2168-9652.S2-003, [accessed 2017 Aug 31] [DOI] [Google Scholar]
- 14. Singh RS, Walia AK, Khattar JS, Singh DP, Kennedy JF. Cyanobacterial lectins characteristics and their role as antiviral agents. Int J Biol Macromol. 2017;102:475‐496. 10.1016/j.ijbiomac.2017.04.041. [DOI] [PubMed] [Google Scholar]
- 15. Singh RS, Walia AK, Pratibha, Khattar JS , Singh DP. New cell surface lectins with complex carbohydrate specificity from cyanobacteria. IJEB July 2017;55(07):514‐522. http://nopr.niscair.res.in/handle/123456789/42341, [accessed 2017 Aug 22] [Google Scholar]
- 16. Lima ARJ, Siqueira AS, Santos BGS dos, Silva FDF da, Lima CP, Cardoso JF, da SGV Júnior JL, Dall'Agnol LT, JA McCulloch, MRT Nunes, et al. Draft genome sequence of the Brazilian Cyanobium sp. strain CACIAM 14. Genome Announc 2014;2(4):e00669–14. doi: 10.1128/genomeA.00669-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lima ARJ, Castro W de O, Moraes PHG, Siqueira AS, Aguiar DCF, Lima CPS de, Vianez‐Júnior JLSG, Nunes MRT, Dall'Agnol LT, Gonçalves EC. Draft genome sequence of Alkalinema sp. strain CACIAM 70d, a cyanobacterium isolated from an Amazonian freshwater environment. Genome Announc 2017;5(28):e00635–17. doi: 10.1128/genomeA.00635-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castro WO, Lima ARJ, Moraes PHG, et al. Draft genome sequence of Microcystis aeruginosa CACIAM 03, a cyanobacterium isolated from an Amazonian freshwater environment. Genome Announc. 2016;4(6):e01299‐e01216. 10.1128/genomeA.01299-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fiore M de F, Neilan BA, Copp JN, Rodrigues JLM, Tsai SM, Lee H, Trevors JT. Characterization of nitrogen‐fixing cyanobacteria in the Brazilian Amazon floodplain. Water Res 2005;39(20):5017–5026. doi: 10.1016/j.watres.2005.10.002 [DOI] [PubMed] [Google Scholar]
- 20. Drickamer K, Dodd RB. C‐type lectin‐like domains in Caenorhabditis elegans: predictions from the complete genome sequence. Glycobiology. 1999;9(12):1357‐1369. 10.1093/glycob/9.12.1357. [DOI] [PubMed] [Google Scholar]
- 21. Abhinav KV, Samuel E, Vijayan M. Archeal lectins: an identification through a genomic search. Proteins: Structure, Function, and Bioinformatics. 2016;84(1):21‐30. 10.1002/prot.24949. [DOI] [PubMed] [Google Scholar]
- 22. Abhinav KV, Sharma A, Vijayan M. Identification of mycobacterial lectins from genomic data. Proteins: Structure, Function, and Bioinformatics. 2013;81(4):644‐657. 10.1002/prot.24219. [DOI] [PubMed] [Google Scholar]
- 23. Li W, Godzik A. Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics (Oxford, England). 2006;22(13):1658‐1659. 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 24. Marchler‐Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015;43(Database issue):D222‐D226. 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kearse M, Moir R, Wilson A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics (Oxford, England). 2012;28(12):1647‐1649. 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rose PW, Prlić A, Altunkaya A, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017;45(D1):D271‐D281. 10.1093/nar/gkw1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1‐5.6.37. 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pei J, Tang M, Grishin NV. PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Research. 2008;36(suppl_2):W30‐W34. 10.1093/nar/gkn322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I‐TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7‐8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davis IW, Leaver‐Fay A, Chen VB, et al. MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Research. 2007;35(suppl_2):W375‐W383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three‐dimensional profiles. Nature. 1992;356(6364):83‐85. 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 32. Benkert P, Tosatto SCE, Schomburg D. QMEAN: a comprehensive scoring function for model quality assessment. Proteins. 2008;71(1):261‐277. 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 33. The Amber Molecular Dynamics Package . [accessed 2017 Aug 23]. http://ambermd.org/
- 34. Tessier MB, DeMarco ML, Yongye AB, Woods RJ. Extension of the GLYCAM06 biomolecular force field to lipids. Lipid Bilayers and Glycolipids Molecular simulation. 2008;34(4):349‐363. 10.1080/08927020701710890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of chemical theory and computation. 2015;11(8):3696‐3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Price DJ, Brooks CL. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121(20):10096‐10103. 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 37. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand‐binding affinities. Expert Opin Drug Discovery. 2015;10(5):449‐461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sulea T, Vivcharuk V, Corbeil CR, Deprez C, Purisima EO. Assessment of solvated interaction energy function for ranking antibody‐antigen binding affinities. J Chem Inf Model. 2016;56(7):1292‐1303. 10.1021/acs.jcim.6b00043. [DOI] [PubMed] [Google Scholar]
- 39. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785‐786. 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 41. Matei E, Basu R, Furey W, et al. Structure and glycan binding of a new Cyanovirin‐N homolog. J Biol Chem. 2016;291(36):18967‐18976. 10.1074/jbc.M116.740415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barrientos LG, Louis JM, Ratner DM, Seeberger PH, Gronenborn AM. Solution structure of a circular‐permuted variant of the potent HIV‐inactivating protein cyanovirin‐N: structural basis for protein stability and oligosaccharide interaction. J Mol Biol. 2003;325(1):211‐223. [DOI] [PubMed] [Google Scholar]
- 43. Yang J, Roy A, Zhang Y. Protein–ligand binding site recognition using complementary binding‐specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29(20):2588‐2595. 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki T. Catabolism of N‐glycoproteins in mammalian cells: molecular mechanisms and genetic disorders related to the processes. Molecular Aspects of Medicine. 2016;51:89–103. (Molecular Role of Glycoproteins in Disease). doi: 10.1016/j.mam.2016.05.004 [DOI] [PubMed] [Google Scholar]
- 45. Horiguchi Y, Goda T, Matsumoto A, Takeuchi H, Yamaoka S, Miyahara Y. Direct and label‐free influenza virus detection based on multisite binding to sialic acid receptors. Biosens Bioelectron. 2017;92:234‐240. 10.1016/j.bios.2017.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Diamond MS, Pierson TC. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell. 2015;162(3):488‐492. 10.1016/j.cell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fromme R, Katiliene Z, Fromme P, Ghirlanda G. Conformational gating of dimannose binding to the antiviral protein cyanovirin revealed from the crystal structure at 1.35 Å resolution. Protein Science : A Publication of the Protein Society. 2008;17(5):939‐944. 10.1110/ps.083472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Souza RC de, Muniz G de M, Siqueira AS, Lima A de M, Silva AP da, Gonçalves EC, Júnior JL da SGV. Investigating the effects of point mutations on the affinity between the cyanobacterial lectin microvirin and high mannose‐type glycans present on the HIV envelope glycoprotein. Journal of Molecular Modeling. 2016;22(11):269. doi: 10.1007/s00894-016-3137-3 [DOI] [PubMed] [Google Scholar]
- 49. Mori T, Barrientos LG, Han Z, Gronenborn AM, Turpin JA, Boyd MR. Functional homologs of cyanovirin‐N amenable to mass production in prokaryotic and eukaryotic hosts. Protein Expr Purif. 2002;26(1):42‐49. 10.1016/S1046-5928(02)00513-2. [DOI] [PubMed] [Google Scholar]
- 50. Elghabi Z, Karcher D, Zhou F, Ruf S, Bock R. Optimization of the expression of the HIV fusion inhibitor cyanovirin‐N from the tobacco plastid genome. Plant Biotechnol J. 2011;9(5):599‐608. 10.1111/j.1467-7652.2011.00598.x. [DOI] [PubMed] [Google Scholar]
- 51. O'Keefe BR, Murad AM, Vianna GR, et al. Engineering soya bean seeds as a scalable platform to produce cyanovirin‐N, a non‐ARV microbicide against HIV. Plant Biotechnol J. 2015;13(7):884‐892. 10.1111/pbi.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drake PMW, Madeira L de M, Szeto TH, Ma JK‐C. Transformation of Althaea officinalis L. by agrobacterium rhizogenes for the production of transgenic roots expressing the anti‐HIV microbicide cyanovirin‐N. Transgenic Res 2013;22(6):1225–1229. doi: 10.1007/s11248-013-9730-7 [DOI] [PubMed] [Google Scholar]
- 53. Shahzad‐ul‐Hussan S, Gustchina E, Ghirlando R, Clore GM, Bewley CA. Solution structure of the monovalent lectin Microvirin in complex with Manα(1–2)man provides a basis for anti‐HIV activity with low toxicity. J Biol Chem. 2011;286(23):20788‐20796. 10.1074/jbc.M111.232678. [DOI] [PMC free article] [PubMed] [Google Scholar]