

Abstract
Definition of functional genomic elements is one of the greater challenges of the genomic era. Traditionally, putative short open reading frames (sORFs) coding for less than 100 amino acids were disregarded due to computational and experimental limitations; however, it has become clear over the past several years that translation of sORFs is pervasive and serves diverse functions. The development of ribosome profiling, allowing identification of translated sequences genome wide, revealed wide spread, previously unidentified translation events. New computational methodologies as well as improved mass spectrometry approaches also contributed to the task of annotating translated sORFs in different organisms. Viruses are of special interest due to the selective pressure on their genome size, their rapid and confining evolution, and the potential contribution of novel peptides to the host immune response. Indeed, many functional viral sORFs were characterized to date, and ribosome profiling analyses suggest that this may be the tip of the iceberg. Our computational analyses of sORFs identified by ribosome profiling in DNA viruses demonstrate that they may be enriched in specific features implying that at least some of them are functional. Combination of systematic genome editing strategies with synthetic tagging will take us into the next step—elucidation of the biological relevance and function of this intriguing class of molecules.
Keywords: micropeptides, ribosome profiling, translation, uORF, virus
1. Introduction
The big challenges of the post‐genomic era, after completing the sequencing of genomes of a wide range of organisms and viruses, are the annotations of functional units in these genomes, including identification of protein coding sequences. Open reading frames (ORFs) are defined as DNA sequences with translation potential, consisting of a string of in‐frame codons flanked by a start codon and a stop codon. Traditionally, the definition of ORFs was sequences that potentially code for peptides of 100 amino acids (aa) or more, this length limit originates from bioinformatic approaches that were used for annotations. Since it was assumed that the majority of coding genes would code for larger proteins and a stretch of 100 codons without a stop codon provides a statistically significant signal, using 100 aa as a cut‐off provided a straightforward strategy to annotate genomes. However, in recent years there have been accumulating evidence for the prevalence of functional translation events from short ORFs (sORFs) encoding proteins with a length of 100 aa or less.1, 2
sORFs were shown to carry out diverse functions and were involved in many biological processes. An important functional subclass of translated sORFs are upstream ORFs (uORFs) that are located at the 5′ leader sequence of mRNAs, upstream of the initiation codon of the main coding ORF. These have been shown to regulate translation efficiency via different mechanisms.3 In most of the cases studied to date, uORFs are cis acting and regulate their downstream ORF. Some examples of uORFs were described, termed peptoswitches, that respond to environmental cues.1 In these cases, small molecules bind to the nascent peptide leading to inhibition of translation of the main ORF by different mechanisms including non‐sense mediated decay4 and ribosome stalling.5 In addition, molecular functions were also assigned to sORF‐encoded peptides (SEPs), these include regulation of post‐translational modifications,6 regulation of metabolite transport,7 and inhibition of kinase activity.8 As expected by their different molecular functions, SEPs are involved in various biological processes including cell communication, signal transduction, transcriptional regulation, and metabolism.
A major advancement in the field was the introduction of ribosome profiling, a powerful experimental strategy to map translation events in vivo.9 This technique is based on early studies showing that each ribosome physically protects a portion of its mRNA template from nuclease digestion.10, 11 The advent of high‐throughput sequencing offered the opportunity to analyze all ribosome‐protected fragments in living cells, thereby providing a snapshot of translation in vivo (Figure 1). Ribosome footprints can indicate the positions of ribosomes with sub‐codon precision. Such high‐resolution information identifies the precise boundaries of translated regions as well as the specific reading frame in which translation occurs. While the ribosome decodes only a single codon at a time, it protects a much larger footprint, typically 28–32 nucleotides.9 The signature of the triplet genetic code is present in these larger footprints, as the footprint positions show a three‐nucleotide periodicity reflecting the translocation steps of the ribosome. This bias in footprint position provides a robust statistical signal that indicates which specific reading frame is being translated on an mRNA.9, 12, 13, 14, 15, 16
Figure 1.

Ribosome profiling allows identification of translated regions. Cells are lysed and subjected to nuclease digestion. Prior to lysis, cells may be treated with cycloheximide to freeze ribosomes on their mRNA targets. Ribosome footprints are isolated and converted to deep sequencing libraries. Reads are then mapped to the genome, this facilitates mapping of translated ORFs in an unbiased quantitative manner.
Translational start sites can be mapped more directly by performing ribosome profiling under conditions that preferentially arrest initiating ribosomes. Harringtonine and lactimidomycin are two drugs that preferentially target eukaryotic initiating ribosomes.12, 13, 17 Both of these drugs lead to strong accumulation of ribosomes precisely at translation initiation sites and depletion of ribosomes over the body of the message. Though their effects are similar, they act through very different mechanisms, and their combination helps to preclude drugs‐related artifacts and results in robust start site detection.13 Harringtonine binds to the peptidyltransferase center in disassembled large ribosomal subunits.18, 19 Lactimidomycin, in contrast, is related to cycloheximide and both bind to the same site on the large ribosomal subunit, but lactimidomycin displays a marked preference for an empty E site, which occurs only in the initiating ribosomes.20, 21 A similar effect can also be achieved by treatment with puromycin, which drives premature termination, thereby removing most elongating ribosomes and leaving footprints predominantly at sites of initiation.22 Furthermore, puromycin can be combined with lactimidomycin in order to stabilize initiating ribosomes while depleting elongating ribosomes in cell lysates.23
Precise mapping of translation initiation and reading frames on RNA provides substantial information that allows accurate analysis of translation events in an unbiased manner. These approaches facilitated new studies that re‐annotated the coding capacity of numerous organisms, revealing widespread translation outside of annotated protein coding sequences.16, 24, 25, 26, 27 Ribosome profiling proved a sensitive methodology with high discovery rate2 providing a robust platform for systematic analysis of translated ORFs. The outstanding challenge is to define the functionality of newly identified translation events and to discriminate between ORFs that provide regulatory or protein‐based functions to products of random translational events.
On the computational side, approaches that rely on cross‐species comparisons and conservation, that are adjusted to sORFs, are a valuable tool that could identify sORFs that are likely to produce functional peptides.28, 29
An additional relevant technique, mass spectrometry, is a powerful tool to directly detect proteins and peptides. Detection of SEPs could provide insight into their stability and abundance and therefore can point to functionality. Improvements on the experimental and computational aspects of proteomic approaches30 assisted in discovery of tens of novel SEPs.31 For this, Slavoff et al. used peptidomics, a mass spectrometry–based approach that is augmented for preservation and enrichment of small peptides, mainly by reducing proteolysis during sample processing. Generation of a dataset based on the human transcriptome taking into account out‐of‐frame alternative translation products has led to the discovery of 1259 alternative proteins, many of which derived from sORFs in different tissues and cell lines.32 Interestingly, many of these alternative proteins were found to be secreted. Thus, computational and proteomic approaches still have limitations when applied to search for SEPs, however they are constantly upgraded and improve our discovery capacity of the variety of these peptides. In this review, we will concentrate on sORFs in viruses focusing on known functions of virally encoded short proteins and how viral systems—due to their high transcriptional levels, manipulatable genomes, and measurable phenotypes—could provide opportunities to better decipher sORFs functions.
2. Virally Encoded Short Proteins
Viruses are essentially infectious units that replicate inside a living cell and their life cycle is tailored to support genome amplification and transmission to new hosts. An inevitable requirement of this life cycle is small genome size. Effective strategies to produce diverse functions and to maintain small genome is to produce small proteins and to evolve sophisticated gene expression regulation mechanisms. Studying sORFs in viruses also has the advantage of their rapid and restricting evolution which would imply that sequence conservation is highly indicative of conserved function. Indeed numerous viral functional sORFs were identified and characterized.33 In addition, application of ribosome profiling on infected cells revealed unanticipated complexity in viruses coding capacity, with many novel putative sORFs.
2.1. Short Transmembrane Proteins
A major group of viral SEPs encode short transmembrane proteins.34 These may be advantageous for the virus as they form stable structures to support membrane‐related functions at a minimal burden on genome size. Indeed, short transmembrane proteins were identified in a range of both RNA and DNA animal viruses and were shown to be involved in various viral processes including entry, genome replication, particle assembly and release as well as interfere with host processes. Some of these transmembrane proteins belong to a group of proteins named viroporins35, 36 which contain hydrophobic regions that upon oligomerization form aqueous pores in the host cellular membranes. Viroporins were mainly implicated in viral assembly and release.35 For example, a role in viral release was demonstrated for the human immunodeficiency virus (HIV) vpu protein. Vpu is a ∼80 aa viroporins that oligomerizes to form a selective ion channel37, 38, 39 that may enhance viral release through its ion channel activity and/or by inhibiting tetherin, a cellular factor which inhibits viral release.40, 41 Another extensively characterized viroporin is the 97 aa M2 influenza A virus (IAV) protein which oligomerizes to form an ion channel.42 During infection M2 localizes to the Golgi where it plays an important role in viral replication and assembly by perturbing protein trafficking due to its effect on the ion gradient in the secretory pathway.43 Furthermore, M2 is essential for viral release, mediating membrane curvature and scission.44, 45 M2 may also have a role in viral entry by mediating virion acidification46 which is required for the uncoating of the virus.47 Other short viral transmembrane proteins, which are not part of the viroporin family as they do not oligomerize, are vital for viral entry. The vaccinia 35 aa O3L protein has a hydrophobic domain and is incorporated into the membrane of the mature virion. Importantly, O3L associates with the virus entry/fusion complex and is essential for viral entry,48 a function which is conserved across many other poxviruses.49 Interestingly, vaccinia encodes many more short transmembrane proteins that were shown to have a role in viral entry and biogenesis.34
Short transmembrane proteins were also shown to have effects on host processes. By forming aqueous pores in cellular membranes viroporins perturb membrane permeability leading to alterations in cellular ionic homeostasis and thus to cytopathic effects.35 In addition, some of the short transmembrane viral proteins interfere with various cellular processes. A key role for the HIV vpu proteins is the downregulation of CD4 by targeting newly synthesized CD4 molecules in the ER and mediating its proteosomal degradation. This process is critical for the virus as CD4 expression on the cell surface interferes with HIV viral propagation.50 Short viral transmembrane proteins can also induce cell death. M2, the IAV viroporin, inhibits autophagosome degradation which compromises the survival of infected cells.51 Viruses of the paramyxovirus family encode short hydrophobic (SH) integral membrane proteins 44–60 aa long, which were shown to inhibit apoptosis.52, 53 Papillomaviruses encode e5 proteins, ranging in size from 40 to 85 aa long transmembrane oncoproteins.34 The well‐studied Bovine papillomavirus E5 protein induces stable transformation of cultured fibroblasts by strongly and specifically activating the platelet‐derived growth factor β receptor (PDGFβ‐R). Human papillomaviruses E5 proteins were also shown to have some transforming capabilities as well as immune evasion function via downregulation of MHC class I.54
2.2. Upstream Open Reading Frames
uORFs are sORFs that due to their location upstream of a primary ORF could serve as means of regulating its translation and several examples were found in viruses. In human cytomegalovirus (HCMV), the viral UL4 protein translation is regulated by translation of a uORF.55 This regulation is robust despite the inefficient usage of its AUG, probably through ribosome stalling during translation termination.56 Remarkably, the coding information of the uORF is essential for translation inhibition of UL4 implicating that this regulation is mediated by nascent peptide translated from the uORF.55 In another virus from the herpesviridae, Kaposi's sarcoma‐associated herpesvirus (KSHV), ORF35 and ORF36 protein products are translated from a polycistronic transcript, ORF35–37. The translation of these two proteins is regulated by translation of two uORFs, which inhibit translation from the adjacent ORF35. Interestingly, the second uORF which overlaps ORF35 start codon allows translation of the downstream ORF36 via a reinitiation mechanism and is essential for viral propagation.57, 58
A similar case was demonstrated for hepatitis B virus (HBV), where the polymerase gene is preceded by and partially overlaps the core gene, which is preceded by an upstream AUG, on the pregenomic RNA. Translation from the first AUG was shown to inhibit translation from the core initiation site while allowing reinitiation of translation from the polymerase initiation site.59, 60 Also in mouse hepatitis virus (MHV) a uORF that is present in many coronaviruses was shown to repress translation of the downstream ORF1.61 mRNAs of the Ebola virus (EBOV) have long 5’ UTRs, some of which contain upstream AUGs. Translation from the AUG preceding the l protein coding region, suppresses translation of the primary ORF encoding the L protein and is interestingly responsive to cellular stress. Mutations in this uORF drastically attenuate viral growth, indicating the importance of its regulatory function.62 More complex regulation mechanism was reported in the simian immunodeficiency virus (SIV), where translation from a number of different uORFs present in different splice variants of mRNAs encoding the rev and env genes regulate these genes to different extent.63, 64
Another translation regulation mechanism that is active in different viruses is ribosome shunting. Ribosome shunting is a mechanism in which cap‐dependent translation starts from a short uORF located upstream of a long 5′ UTR stretch that forms a large stem and loop structure. Upon termination, the ribosome is able to bypass the stem and loop structure to resume scanning just 3′ of it, allowing translation from a downstream AUG.33 This mechanism was extensively studied in the cauliflower mosaic virus (CaMV) 35 S RNA,65, 66 but was also demonstrated in additional viruses.33
Functional regulatory uORFs were discovered in different viruses from different families, both DNA and RNA and of different sizes, implying that this regulatory mechanism is probably wide spread. Importantly, these uORFs are found in other strains and sometimes other viruses from the same family, and furthermore, some were shown to be essential for viral propagation, demonstrating their functional importance.
2.3. Additional Functional Viral sORFs
Besides the two groups of functional sORFs described above, that have several examples in several viruses, there are additional examples of functional viral SEPs with various molecular and cellular roles. The HIV Vpr is 96 aa long, conserved between all HIV and SIV and important for viral replication. This small protein has been implicated in different processes during the viral life cycle, including reverse transcription, nuclear import of viral DNA in non‐dividing cells, induction of cell cycle arrest and apoptosis.67 The PB1‐F2 ∼90 aa long protein encoded by IAV localizes to mitochondria and also induces apoptosis.68 Intriguingly, this conserved protein is translated from a +1 reading frame of the PB1 transcript which encodes one of the polymerase subunits.68 In KSHV, a 3 kb polyadenylated RNA is transcribed from the opposite strand of the replication and transcription activator (RTA) encoding ORF50. It has been annotated as a non‐coding RNA following identification because no large ORF was found in the transcript.69 However, later this transcript was found to encode a 48 aa long peptide, designated viral small peptide 1 (vSP‐1) which interacts with RTA and prevents its degradation through the ubiquitin–proteasome pathway, facilitating the virus gene expression and lytic replication.70 A different example is the recent exciting discovery of a communication system in phages that relies on a small peptide secreted to the medium. A 43 aa long peptide is translated from the phage aimP locus and this sORF is then processed to form the mature short communication peptide.71 Significantly, homologs of the aimP gene as well as other components of this communication pathway were found in many other Bacillus phages demonstrating the prevalence of this system.
2.4. Novel translated sORFs Discovered by Ribosome Profiling
As discussed above ribosome profiling has great potential for depicting the full variety of translated ORFs and has been successfully applied to a number of viruses in the past few years. HCMV was the first virus to be analyzed by ribosome profiling and revealed numerous previously unidentified ORFs, a large number of which are sORFs.13 A class of uORFs located upstream of canonical ORFs were found, two of them were shown to downregulate translation efficiency of the downstream ORF using a reporter. Interestingly, changes in the 5′ ends of these transcripts along infection led to inclusion of the uORFs in the transcripts, therefore reducing translation from these ORFs at late stages of infection. Translation of many other sORFs was demonstrated, initiating within known ORFs or encoded by distinct transcripts. Multiple sORFs were found to be translated from transcripts previously annotated as non‐coding, more than ten were translated from the long non‐coding RNA b2.7, four of which are highly conserved across different HCMV strains. Translation of two short proteins encoded by RNA1.2 and additional sORFs were confirmed by mass spectrometry. Intriguingly, initiation of translation from a near‐cognate codon was observed for both long and short ORFs. Similarly, in another DNA double stranded herpes virus, KSHV, ribosome profiling identified many uORFs which are widely spread in the KSHV genome.72 Significantly, 24 out of the 85 annotated genes contained 1–6 uORFs, encoding peptides consisting of <100 aa, translating either in or out of frame and in many cases from a non‐canonical start codon. The KSHV data also supported the existence and regulatory role of two uORFs previously described.57, 58 Ribosome profiling analysis was also done on the well characterized bacteriophage lambda during lysogeny and at different time points along the lytic process, revealing translation of tens of previously uncharacterized sORFs.73 In vaccinia virus (VACV), a prototype poxvirus, ribosome profiling revealed translation from 596 unannotated ORFs that add to the 162 annotated ORFs.74 Many of these novel ORFs are sORFs and include uORFs, truncated ORFs that resulted from translation initiation from non‐annotated initiation site, ORFs that result from frameshifting, and ORFs from regions annotated as non‐coding. In MHV, an RNA virus of the Coronavirus family, triplet phasing of the ribosome profiling data allowed precise determination of translated reading frames, revealing several translated sORFs upstream of, or within, annotated virus protein coding regions, some of which are conserved in different MHV strains.14 One of them, a previously reported uORF upstream of ORF161 was confirmed and furthermore, a potential role for it in temporal regulation of replication protein synthesis was suggested.
Overall, all ribosome profiling analyses performed in viruses revealed a wealth of unknown translated sORFs, indicating that this is a prevalent phenomenon and demonstrating the discovery power of this technique. The fact that an ORF is being translated does not necessarily prove that it is functional, however there is a growing list of functional sORFs, thus it is likely that several of these novel translation units indeed have a role. Many of the peptides produced may be non‐functional and may be rapidly degraded, nevertheless, ribosome association or the act of translation may have a role as has been shown for many uORFs. Significantly, dissecting the variety of viral peptides produced during infection is important, as even the non‐functional peptides could be an important part of the immunological repertoire of the virus as major histocompatibility complex (MHC) class I bound peptides. This is evident from the robust cellular immune response that was reported for T cells from human HCMV‐positive donors to peptides translated from several novel sORFs identified by ribosome profiling, including some that are translated from the beta 2.7 transcript, a designated long non‐coding RNA.75
2.5. Analysis of Putative sORFs That Were Discovered by Ribosome Profiling
Since many putative sORFs were identified by ribosome profiling in double stranded DNA viruses—HCMV,13 KSHV,72 and VACV74—we set out to examine whether their sequence can indicate anything about their potential functions. To this end, we applied several sequence‐based analyses that gave us a broad view on the properties of these sORFs.
First, we compared the amino acid composition of sORFs to the composition of long ORFs in the same virus using Composition Profiler,76 showing that there is no significant difference between the groups (Figure 2). This similarity is probably driven by the GC content and the codon usage of each virus, and further suggests similar amino acid selection rates for long and sORFs and no enrichment for specific amino acids in sORFs.
Figure 2.

Distribution of amino acids in short and long ORFs of several DNA viruses. The frequency of amino acids in all short (20 aa> and <100 aa, light color) and long (>100 aa, dark color) viral ORFs is shown for HCMV, KSHV, and VACV. The statistical significance of the differences in amino acid distribution was calculated using Composition Profiler's relative entropy function, and the p‐values are presented.
As discussed above, there are many cases of secreted short proteins. In order to test if these newly discovered sORFs are enriched for signal peptides we used signalP 4.1, a neural network–based method that predicts the probability of the presence of a signal peptide in a protein sequence.77 The ratio of sORFs that contained a signal peptide was compared to the distribution of these ratios in a set of random sequences of the same length and aa composition. The long canonical ORFs showed significant enrichment in signal peptides prediction compared to shuffled sequences (p‐value < 1 × 10–5). In the sORFs, KSHV had no predicted signal peptides, while in the shuffled sequences some signal peptides were generated. In sORFs from HCMV and VACV, we found that signal peptides are weakly enriched compared to what is observed in the set of random sequences (p‐value: VACV 0.016, HCMV 0.2, Figure 3A), suggestive of selection for sORFs which are destined for the secretory pathway.
Figure 3.

Functional predictions of non‐canonical viral short ORFs that were discovered by ribosome profiling. Bioinformatic tools were used to predict protein characteristics for short and long viral ORFs, as annotated by ribosome profiling in HCMV, KSHV, and VACV. The results were compared to a distribution created by performing the same predictions of a set of 100 random sequences of the same size and aa composition. A) The presence or absence of a signal peptide in each ORF was predicted using SignalP. The fraction of signal peptide containing ORFs in each group is marked by an orange square, and the distribution of the sums in the random set is shown is shown in boxplots. B) The number of transmembrane domains in each ORF group was predicted using TMHMM. The sum of domains for each ORF group is shown as an orange square, and the distribution of the sums in the random set is shown in boxplots.
We next used the TMHMM 2.0 to test viral sORFs for enrichment in transmembrane domains.78 This analysis revealed that similarly to the signal peptide prediction, the long canonical ORFs had a higher number of predicted transmembrane domains compared to random sequences (p‐value: VACV 0.02, HCMV and KSHV < 1 × 10–5) while for sORFs significantly high numbers of transmembrane domains were found only for HCMV (p‐value = 1.201 × 10–5, Figure 3B).
Whether sORFs are translated as a regulatory mechanism or into a functional protein, they can generate peptides that will be presented on MHC molecules and be recognized by the immune system. To explore if there is any selection for the immunogenicity of viral sORFs we used netMHC 4.0, which is an artificial neural network trained platform for predicting MHC class I binding affinities of peptides, based on their sequence.79, 80 The number of fragments that strongly bind MHC‐I (high binders) was counted for each ORF. To take into account the effect of the ORF length, we calculated the ratio between the number of high binders in each ORF and its random set as a function of its length. We calculated the spearmen correlation and although some specific sORFs showed deviation from the median of random sequences, we found no significant correlation (Figure 4), suggesting that MHC‐I presentation is not a dominating negative selection force for sORFs translation in these viruses.
Figure 4.
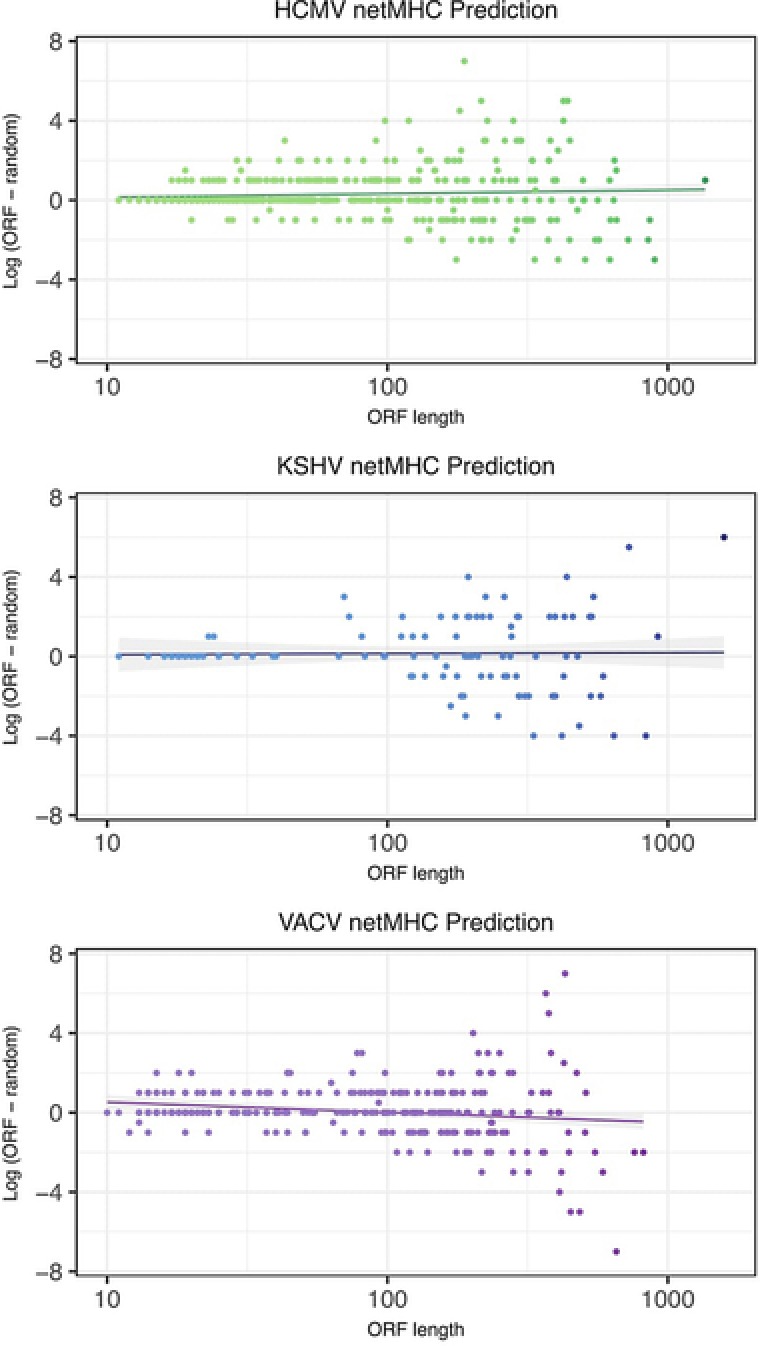
Prediction of MHC‐I peptide presentation of viral ORFs. For each ORF, the number of nine aa long fragments that have the ability to strongly bind MHC‐I molecules was predicted using netMHC. The difference between the number of predicted peptides in each ORF and the median of predicted peptides in a set of random sequences of the same size and aa composition is presented for HCMV, KSHV, and VACV.
It is noteworthy that in ribosome profiling as with other high‐throughput methodologies, different approaches may introduce background noise originating from technical issues and thus might lead to biases in the detection of ORFs. False detection of spurious ORFs as well as falsely missing real ORFs are bound to occur. The analyses presented above were performed on data from three ribosome profiling studies. These studies used similar experimental approaches to generate the libraries but different computational approaches to predict the translated ORFs. Stern‐Ginossar et al. and Arias et al. used a machine learning approach and the false negative rate was assessed as 13 and 36% for the HCMV and KSHV data, respectively and 1% false positive rate for the HCMV data.13, 72 Yang et al. used a rule‐based approach hence it is harder to assess the false discovery rate.74 We estimate that the use of different computational approaches in these studies has no major impact on the analyses we performed, however, further studies defining new viral sORFs will help to shed more light on the characteristics of these fascinating group of molecules.
3. Conclusions and Future Perspectives
Recent advances in computational and experimental techniques have revealed wide spread translation outside of canonical ORFs. It has been demonstrated that translated sORFs have essential roles during viral infection, however, the overwhelming majority of them remain to be characterized. To date, biological roles have been assigned to a small fraction of the translation products that have been mapped and a huge amount of work remains to be done to prove their existence and elucidate their functions. The outstanding challenge is to discriminate between ORFs that provide regulatory or protein‐based functions to random translational events, and to identify their roles. Using sequence predictions, we show that viral sORFs are enriched for specific functional features, suggesting that some of these translation products may act at the protein level. Notably, differences between the functional enrichments we discovered in the different viruses can stem from genuine biological differences or may be due to variations in experimental approaches that were used to generate these datasets. Designing mutations that will distinguish between the function of the translated product and the act of translation itself and studying their phenotypic consequences could provide an important platform for future studies. Advancement in gene editing strategies and use of reporters provide powerful strategies to study the effects of mutating these translated regions and validating their expression.
Importantly, widespread translation outside of annotated protein coding genes was also found in many organisms including mammalians cells. As for many molecular biology principles, viral infection could provide a powerful model for studying functions of translated sORFs, due to robust expression levels and quantifiable phenotypes.
4. Methods
4.1. Data
Sequences of viral sORFs (encoding for peptides shorter than 100 aa) were obtained from three published papers presenting ribosome profiling data of DNA viruses13, 72, 74 515 HCMV, 50 KSHV, and 506 VACV sORFs were analyzed. Sequences of all previously annotated KSHV and VACV proteins longer than 100 aa were obtained from Uniprot manually annotated protein lists.81 The full list of HCMV proteins longer than 100 aa of HCMV was taken from Stern‐Ginossar et al.13
4.2. Analysis
We calculated the frequency of amino acids in short and long ORFs for each virus. This was done by dividing the number of appearances of each amino acid in the sequences by the total combined length of the sequences. To test the significance of the difference between the frequencies in the short and long ORFs the relative entropy function in the Composition Profiler web‐based tool was used, designed to compare aa distributions.76
Predictions of signal peptides and transmembrane domains were performed using SignalP77 and TMHMM78 web‐based tools, respectively, using default parameters. Additionally, we used netMHC80 to predict the number of all possible putative peptides that efficiently bind MHC‐I molecules, using the recommended peptide length—9 aa. The predictions were done on the sets of short and long ORFs for each virus. For statistical comparison, the same tests were performed on random sets containing 100 versions for each sORF, created by shuffling the order of amino acids.
Conflict of Interest
The authors have declared no conflict of interest.
Acknowledgements
N.S.G. acknowledges funding from the Israeli Science Foundation (1073/14), the European Research Council starting grant (StG‐2014‐638142), and Marie Curie career integration grant (2013‐631003).
Finkel Y., Stern‐Ginossar N., Schwartz M., Proteomics 2018, 18, 1700255 10.1002/pmic.201700255
REFERENCES
- 1. Andrews S. J., Rothnagel J. A., Nat. Rev. Genet. 2014, 15, 193. [DOI] [PubMed] [Google Scholar]
- 2. Pueyo J. I., Magny E. G., Couso J. P., Trends Biochem. Sci. 2016, 41, 665. [DOI] [PubMed] [Google Scholar]
- 3. Barbosa C., Peixeiro I., Romão L., PLoS Genet. 2013, 9, e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaba A., Jacobson A., Sachs M. S., Mol. Cell 2005, 20, 449. [DOI] [PubMed] [Google Scholar]
- 5. Fang P., Spevak C. C., Wu C., Sachs M. S., Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kondo T., Plaza S., Zanet J., Benrabah E., Valenti P., Hashimoto Y., Kobayashi S., Payre F., Kageyama Y., Science 2010, 329, 336. [DOI] [PubMed] [Google Scholar]
- 7. Magny E. G., Pueyo J. I., Pearl F. M. G., Cespedes M. A., Niven J. E., Bishop S. A., Couso J. P., Science 2013, 341, 1116. [DOI] [PubMed] [Google Scholar]
- 8. Pueyo J. I., Magny E. G., Sampson C. J., Amin U., Evans I. R., Bishop S. A., Couso J. P., PLoS Biol. 2016, 14, e1002395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ingolia N. T., Ghaemmaghami S., Newman J. R. S., Weissman J. S., Science 2009, 324, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolin S. L., Walter P., EMBO J. 1988, 7, 3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steitz J. A., Nature 1969, 224, 957. [DOI] [PubMed] [Google Scholar]
- 12. Ingolia N. T., Lareau L. F., Weissman J. S., Cell 2011, 147, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stern‐Ginossar N., Weisburd B., Michalski A., Le V. T. K., Hein M. Y., Huang S.‐X., Ma M., Shen B., Qian S.‐B., Hengel H., Mann M., Ingolia N. T., Weissman J. S., Science. 2012, 338, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Irigoyen N., Firth A. E., Jones J. D., Chung B. Y. W., Siddell S. G., Brierley I., PLoS Pathog. 2016, 12, e1005473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chew G.‐L., Pauli A., Rinn J. L., Regev, A. F. Schier A., Valen E., Development 2013, 140, 2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bazzini A. A., Johnstone T. G., Christiano R., MacKowiak S. D., Obermayer B., Fleming E. S., Vejnar C. E., Lee M. T., Rajewsky N., Walther T. C., Giraldez A. J., EMBO J. 2014, 33, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S., Liu B., Lee S., Huang S.‐X., Shen B., Qian S.‐B., Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fresno M., Jiménez A., Vázquez D., Eur. J. Biochem. 1977, 72, 323. [DOI] [PubMed] [Google Scholar]
- 19. Robert F., Carrier M., Rawe S., Chen S., Lowe S., Pelletier J., PLoS One 2009, 4, e5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schneider‐Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., Liu J. O., Nat. Chem. Biol. 2010, 6, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garreau de Loubresse N., Prokhorova I., Holtkamp W., Rodnina M. V., Yusupova G., Yusupov M., Nature 2014, 513, 517. [DOI] [PubMed] [Google Scholar]
- 22. Fritsch C., Herrmann A., Nothnagel M., Szafranski K., Huse K., Schumann F., Schreiber S., Platzer M., Krawczak M., Hampe J., Brosch M., Genome Res. 2012, 22, 2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao X., Wan J., Liu B., Ma M., Shen B., Qian S.‐B., Nat. Methods 2014, 12, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fields A. P., Rodriguez E. H., Jovanovic M., Stern‐Ginossar N., Haas B. J., Mertins P., Raychowdhury R., Hacohen N., Carr S. A., Ingolia N. T., Regev A., Weissman J. S., Mol. Cell 2015, 60, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chew G.‐L., Pauli A., Schier A. F., Nat. Commun. 2016, 7, 11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raj A., Wang S. H., Shim H., Harpak A., Li Y. I., Engelmann B., Stephens M., Gilad Y., Pritchard J. K., Elife 2016, 5, e13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Calviello L., Mukherjee N., Wyler E., Zauber H., Hirsekorn A., Selbach M., Landthaler M., Obermayer B., Ohler U., Nat. Methods 2015, 13, 165. [DOI] [PubMed] [Google Scholar]
- 28. Frith M. C., Forrest A. R., Nourbakhsh E., Pang K. C., Kai C., Kawai J., Carninci P., Hayashizaki Y., Bailey T. L., Grimmond S. M., PLoS Genet. 2006, 2, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackowiak S. D., Zauber H., Bielow C., Thiel D., Kutz K., Calviello L., Mastrobuoni G., Rajewsky N., Kempa S., Selbach M., Obermayer B., Genome Biol. 2015, 16, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krug K., Nahnsen S., Macek B., Mol. BioSyst. 2011, 7, 284. [DOI] [PubMed] [Google Scholar]
- 31. Slavoff S. A., Mitchell A. J., Schwaid A. G., Cabili M. N., Ma J., Levin J. Z., Karger A. D., Budnik B. A., Rinn J. L., Saghatelian A., Nat. Chem. Biol. 2012, 9, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vanderperre B., Lucier J. F., Bissonnette C., Motard J., Tremblay G., Vanderperre S., Wisztorski M., Salzet M., Boisvert F. M., Roucou X., PLoS One 2013, 8, e70698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Firth A. E., Brierley I., J. Gen. Virol. 2012, 93, 1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DiMaio D., Annu. Rev. Microbiol. 2014, 68, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nieva J. L., Madan V., Carrasco L., Nat Rev Microbiol. 2012, 10, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sze C. W., Tan Y. J., Viruses 2015, 7, 3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strebel K., Klimbait T., Martin M. A., Science 1988, 241, 1221. [DOI] [PubMed] [Google Scholar]
- 38. Wang S., Huang B., Wang Z., Liu Y., Wei W., Qin X., Zhang X., Dai Y., Dalton Trans. 2011, 40, 12670. [DOI] [PubMed] [Google Scholar]
- 39. Schubert U., Ferrer‐Montiel A. V., Oblatt‐Montal M., Henklein P., Strebel K., Montal M., FEBS Lett. 1996, 398, 12. [DOI] [PubMed] [Google Scholar]
- 40. González M. E., Viruses 2015, 7, 4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roy N., Pacini G., Berlioz‐Torrent C., Janvier K., Front. Microbiol. 2014, 5, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cardé R. T., Cardé A. M., Hill A. S., Roelofs W. L., J. Chem. Ecol. 1977, 3, 71. [Google Scholar]
- 43. Pielak R. M., Chou J. J., Biochim. Biophys. Acta 2011, 1808, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rossman J. S., Jing X., Leser G. P., Lamb R. A., Cell 2010, 142, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roberts K. L., Leser G. P., Ma C., Lamb R. A., J. Virol. 2013, 87, 9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Qiu J. X., Soto C., Degrado W. F., Curr. Opin. Struct. Biol. 2011, 21, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Helenius A., Cell 1992, 69, 577. [DOI] [PubMed] [Google Scholar]
- 48. Satheshkumar P. S., Moss B., J. Virol. 2009, 83, 12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Satheshkumar P. S., Moss B., J. Virol. 2012, 86, 1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dubé M., Bego M. G., Paquay C., Cohen É. A., Retrovirology 2010, 7, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gannagé M., Dormann D., Albrecht R., Dengjel J., Torossi T., Rämer P. C., Lee M., Strowig T., Arrey F., Conenello G., Pypaert M., Andersen J., García‐Sastre A., Münz C., Cell Host Microbe 2009, 6, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin Y., Bright A. C., Rothermel T. A., He B., J. Virol. 2003, 77, 3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilson R. L., Fuentes S. M., Wang P., Taddeo E. C., Klatt A., Henderson A. J., He B., J. Virol. 2006, 80, 1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Venuti A., Paolini F., Nasir L., Corteggio A., Roperto S., Campo M. S., Borzacchiello G., Mol. Cancer 2011, 10, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Degnin C. R., Schleiss M. R., Cao J., Geballe A. P., J. Virol. 1993, 67, 5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cao J., Geballe A. P., J. Virol. 1995, 69, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kronstad L. M., Brulois K. F., Jung J. U., Glaunsinger B. A., PLoS Pathog. 2013, 9, e1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kronstad L. M., Brulois K. F., Jung J. U., Glaunsinger B. A., J. Virol. 2014, 88, 6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen A., Kao Y. F., Brown C. M., Nucleic Acids Res. 2005, 33, 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zong L., Qin Y., Jia H., Ye L., Wang Y., Zhang J., Wands J. R., Tong S., Li J., Virology 2017, 505, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu H.‐Y., Guan B.‐J., Su Y.‐P., Fan Y.‐H., Brian D. A., J. Virol. 2014, 88, 846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shabman R. S., Hoenen T., Groseth A., Jabado O., Binning J. M., Amarasinghe G. K., Feldmann H., Basler C. F., PLoS Pathog. 2013, 9, e1003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van der Velden G. J., Klaver B., Das A. T., Berkhout B., J. Virol. 2012, 86, 12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. van der Velden G. J., Vink M. A., Klaver B., Das A. T., Berkhout B., Virology 2013, 436, 191. [DOI] [PubMed] [Google Scholar]
- 65. Ryabova L. A., Pooggin M. M., Dominguez D. I., Hohn T., J. Biol. Chem. 2000, 275, 37278. [DOI] [PubMed] [Google Scholar]
- 66. Fütterer J., Gordon K., Sanfaçon H., Bonneville J. M., Hohn T., EMBO J. 1990, 9, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guenzel C. A., Hérate C., Benichou S., Front. Microbiol. 2014, 5, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen W., Calvo P. A. A., Malide D., Gibbs J., Schubert U., Bacik I., Basta S., O'Neill R., Schickli J., Palese P., Henklein P., Bennink J. R. R., Yewdell J. W. W., Nat. Med. 2001, 7, 1306. [DOI] [PubMed] [Google Scholar]
- 69. Saveliev A., Zhu F., Yuan Y., Virology 2002, 299, 301. [DOI] [PubMed] [Google Scholar]
- 70. Jaber T., Yuan Y., J. Virol. 2013, 87, 3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Erez Z., Steinberger‐Levy I., Shamir M., Doron S., Stokar‐Avihail A., Pekeg Y., Melamed S., Leavitt A., Savidor A., Albeck S., Amitai G., Sorek R., Nature 2017, 541, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arias C., Weisburd B., Stern‐Ginossar N., Mercier A., Madrid A. S., Bellare P., Holdorf M., Weissman J. S., Ganem D., PLoS Pathog. 2014, 10, e1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu X., Jiang H., Gu Z., Roberts J. W., Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang Z., Cao S., Martens C. A., Porcella S. F., Xie Z., Ma M., Shen B., Moss B., J. Virol. 2015, 89, 6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ingolia N. T., Brar G. A., Stern‐Ginossar N., Harris M. S., Talhouarne G. J. S., Jackson S. E., Wills M. R., Weissman J. S., Cell Rep. 2014, 8, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vacic V., Uversky V. N., Dunker A. K., Lonardi S., BMC Bioinformatics 2007, 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Petersen T. N., Brunak S., von Heijne G., Nielsen H., Nat. Methods 2011, 8, 785. [DOI] [PubMed] [Google Scholar]
- 78. Sonnhammer E. L., von Heijne G., Krogh A., Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175. [PubMed] [Google Scholar]
- 79. Andreatta M., Nielsen M., Bioinformatics 2015, 32, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nielsen M., Lundegaard C., Worning P., Lauemøller S. L., Lamberth K., Buus S., Brunak S., Lund O., Protein Sci. 2003, 12, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. The UniProt Consortium , Nucleic Acids Res. 2017, 45, D158. [DOI] [PMC free article] [PubMed] [Google Scholar]