

Abstract
The development of clinically useful peptide‐based vaccines remains a long‐standing goal. This review highlights that intrinsically disordered protein antigens, which lack an ordered three‐dimensional structure, represent excellent starting points for the development of such vaccines. Disordered proteins represent an important class of antigen in a wide range of human pathogens, and, contrary to widespread belief, they are frequently targets of protective antibody responses. Importantly, disordered epitopes appear invariably to be linear epitopes, rendering them ideally suited to incorporation into a peptide vaccine. Nonetheless, the conformational properties of disordered antigens, and hence their recognition by antibodies, frequently depend on the interactions they make and the context in which they are presented to the immune system. These effects must be considered in the design of an effective vaccine. Here we discuss these issues and propose design principles that may facilitate the development of peptide vaccines targeting disordered antigens.
Keywords: design, intrinsically disordered antigen, malaria, membrane interactions, peptide epitope, structure
1. INTRODUCTION
Vaccines are an indispensable tool in the fight against disease and have had a significant impact on public health over several decades.1 Traditionally, vaccines have relied on the use of live‐attenuated or inactivated forms of the pathogen to induce a protective immune response. However, in some cases, such as malaria, the pathogen is difficult to culture in vitro at large scale. Moreover, the use of whole organisms as vaccines introduces a high antigenic load when often only a small subset of antigens is driving protection.2 There is also the possibility of adverse allergenic reactions to certain antigens in these preparations. These obstacles have led to an increased interest in subunit vaccines, in which single, or a select few, proteins are used in a vaccine formulation to induce protective immunity.3
The use of peptides as vaccines takes this rationale further, as even a single protein antigen may have many epitopes, not all of which contribute to protective immunity. Peptide vaccines offer a means to formulate vaccines containing only epitopes that are capable of inducing a positive and efficient immune response.2, 4 The ease of peptide synthesis makes large scale production feasible whilst also offering a cleaner vaccine preparation lacking biological contaminations commonly associated with recombinant expression or whole organism vaccines. The specificity of peptides also allows for improved customisability, facilitating, for example, a multiepitope approach to target different strains or stages in the life cycle of the pathogen.
For these reasons, there has been long‐standing interest in this area of vaccine design, with over 500 peptide vaccines reaching clinical trials, targeting a wide range of indications (Table 1). Seven of these vaccines have reached Phase III trial (Table 2). Much of this recent interest has focussed on T‐cell epitopes, with all of the vaccines to reach Phase III being T‐cell based. For many diseases, however, B‐cell responses also play an important role in immune protection, but peptide‐based vaccines based on B‐cell epitopes have proved more challenging for a number of reasons. Owing to the small size of peptides, they are generally poorly immunogenic. In addition, T‐cell epitopes required for the establishment of a robust immune response may also be absent in smaller peptides. To address these issues, carrier proteins or adjuvants are typically included in the vaccine formulation. Peptides are also prone to enzymatic degradation; to combat this, the epitopes can be modified by conformational stabilization or cyclisation, or peptide mimetics that are resistant to proteolysis can be used.5 Finally, and perhaps most importantly, peptide‐based approaches are limited because the antibody response to many protein antigens is dominated by conformational epitopes, which are difficult or impossible to capture effectively in a peptide‐based design.
Table 1.
Peptide vaccines in clinical trials
| Number of active or completed clinical trialsa | Conditions being treated with peptide vaccines | |
|---|---|---|
| Phase III | 7 | Cancer immunotherapies, Multiple sclerosis, Type 1 diabetes |
| Phase II | 203 | Cancer immunotherapies, Myelodysplastic syndrome, HIV, HBV, HCV, Cytomegalovirus, Myasthenia gravis, Influenza, Malaria (falciparum) |
| Phase I and Early Phase 1 | 307 | Cancer immunotherapies, HIV, HPV, HBV, HCV, Age‐related macular degeneration, Respiratory syncytial virus, Malaria (vivax), Malaria (falciparum), Hand foot and mouth disease, Influenza, Multiple sclerosis, Alzheimer's disease, Listeria, Cat allergy, Ragweed allergy, House dust mite allergy, Grass allergy |
Clinical studies were found using “peptide vaccine” as a search term on ClinicalTrials.gov, withdrawn studies were excluded.
Table 2.
Phase III clinical trials currently active or completed
| Candidate | Construct | Condition | Clinical Trials Identifier |
|---|---|---|---|
| MDX‐1379 | Two peptides from gp100 melanocyte protein | Metastatic melanoma | NCT00094653 |
| PR1 leukaemia peptide vaccine | Derived from proteinase 3 and neutrophil elastase | Acute myeloid leukemia | NCT00454168 |
| Telomerase peptide vaccine GV1001 | Derived from reverse transcriptase subunit of telomerase (hTERT) | Pancreatic cancer | NCT00425360 |
| NeuVax | Derived from human leukocyte antigen HER2 | Breast cancer | NCT01479244 |
| NeuroVax | Two peptides from T‐cell receptor | Multiple sclerosis | NCT02057159 |
| MAGE‐A3 and NY‐ESO‐1 Immunotherapy | Peptides from MAGE‐A3 and NY‐ESO‐1 proteins | Multiple myeloma | NCT00090493 |
| Diapep277 | T‐cell epitope of heat shock protein 60 | Type 1 diabetes | NCT01281072 |
In this review, we argue that intrinsically disordered protein antigens, which lack an ordered three‐dimensional structure, represent excellent starting points for the development of peptide vaccines. Our argument is structured as follows: first, we establish that disordered proteins represent an important class of antigen in a wide range of human pathogens. Second, we describe the unique features of these antigens and their interactions with antibodies, highlighting those properties that render them particularly suited to the development of peptide vaccines. In particular, we note that disordered epitopes appear invariably to be linear epitopes, so the challenge of capturing conformational epitopes in a peptide vaccine may not apply. We then outline some of the opportunities and challenges presented by the peptide‐based approach to vaccine development, as exemplified by efforts to develop a vaccine based on the disordered malaria antigen MSP2. Finally, we speculate on future developments, within peptide science and beyond, that will more fully enable the promise of this approach to be realized.
2. DISORDERED PROTEINS ARE IMPORTANT ANTIGENS
2.1. Disordered proteins are abundant in a range of pathogens
Intrinsically disordered proteins are found throughout biology,6 and are particularly enriched in the context of molecular signaling and regulatory functions.7 Consistent with this regulatory role, the extent of disorder within the proteome of individual species tends to increase with the environmental complexity of the organisms life‐cycle.8 Thus, organisms that live in diverse or highly variable environments tend to have highly disordered proteomes, reflecting the need for sophisticated regulatory mechanisms to survive such environmental diversity. As a result, disordered proteins are particularly abundant in a range of pathogenic organisms, and in some viruses,9, 10, 11, 12, 13 which must survive diverse and often hostile host environments. In these contexts, disordered proteins play important roles in host‐pathogen interactions,11 and accordingly, disordered proteins are often exposed to the host immune system.12 Even amongst bacteria, which have significantly more ordered proteomes than eukaryotic species, many proteins involved in pathogenic effector mechanisms appear to be disordered.11
It is often assumed that disordered antigens impede the development of an effective immune response, and hence that their abundance represents an adaptive mechanism enabling the pathogen to evade the immune response.14, 15, 16, 17, 18 Mechanisms proposed to account for this immune evasion are diverse and contested, and we critically assess several of these mechanisms in section 3 below. Despite the fact that these hypotheses have received limited direct experimental support, the underlying assumption that repetitive or disordered antigens will fail to elicit an effective antibody response continues to enjoy significant currency.19, 20, 21 However, recent sequencing of the genomes of several species of parasite has demonstrated that low‐complexity and disordered sequences are unusually abundant in a wide range of genes, including many that do not appear to be antigenically important.22 This suggests that the presence of such sequences in these parasites is not primarily a strategy to evade the host immune response. This has prompted significant debate surrounding the mechanisms responsible for generating and maintaining this abundance of low‐complexity sequence.23, 24, 25 In fact, it seems likely that distinct classes of low‐complexity sequences exist within parasite genomes, suggesting a diversity of contributory mechanisms.25
2.2. Disordered antigens are important targets in vaccine development
As noted above, the abundance of disordered and low‐complexity sequences in the Plasmodium species that are responsible for malaria and in related parasites was first identified through analysis of antigenically important proteins. It is not surprising, then, that a substantial fraction of malaria vaccine candidates contain regions of disorder, and that these regions are often important targets of antibodies. The most prominent example is the circumsporozoite protein (CSP), the dominant antigen on the surface of the sporozoite, the parasite form that is injected into the host by the mosquito and which must replicate within host hepatocytes prior to the establishment of the symptomatic blood‐stage infection.26 CSP has been extensively studied as an antigen for inclusion in a malaria vaccine, and the recently‐approved vaccine RTS,S includes the C‐terminal half of CSP, fused to the hepatitis B S‐antigen and assembled as a virus‐like particle.27 The RTS,S antigen includes the thrombospondin repeat domain at the C‐terminus of CSP, although it is not clear that this domain is correctly folded in the vaccine formulation.28 The rest of CSP is predicted to be disordered, and these predictions have been confirmed experimentally for the tetra‐peptide repeats of CSP that appear to be immunodominant and are targets of protective antibodies induced by RTS,S.29, 30, 31, 32
A second important malaria vaccine candidate that is entirely disordered is merozoite surface protein 2 (MSP2). Antibodies targeting MSP2 arise in the course of malaria infection, and the presence of these antibodies correlates with protection from subsequent infection.33, 34, 35 MSP2 also induces robust antibody responses when used as a vaccine, and these responses were shown to mediate protection in a Phase IIb trial in Papua New Guinea.36, 37 The conformational and antigenic properties of MSP2 have been the subject of extensive study,38, 39, 40, 41 providing important insights into the interplay between antigenicity and disorder, to which we will return in section 4 below.
Many additional antigens of significant interest as vaccine candidates against malaria and other diseases are known or predicted to be disordered, and we highlight a number of examples in Table 3. It is apparent, however, that in many cases the disordered nature of these antigens is not widely appreciated, raising the possibility that many important antigens are yet to be recognized as disordered.
Table 3.
Selected disordered antigens under development as vaccines
| Disordered antigen | Pathogen | Vaccine type | Stage of development | Refs |
|---|---|---|---|---|
| Neisserial heparin binding antigen | Neisseria meningitidis Serogroup B | Subunit combination | Licenced (Bexsero) | 85,86 |
| CSP | Plasmodium falciparum | Virus‐like particle | Phase IV (RTS,S) | 29,87 |
| MSP2 | Plasmodium falciparum | Subunit combination | Phase IIb | 36, 37 |
| KMP‐11 | Leishmania amazonensis | Subunit combination | Phase I | 88 |
| P27A | Plasmodium falciparum | Peptide | Phase I | 83 |
| preS Antigen | Hepatitis B | Virus‐like particle | Preclinical | 89 |
| HSP90 | Candida albicans | Peptide | Preclinical | 90 |
| SAPA | Trypanosoma cruzi | Peptide/fusion‐protein | Preclinical | 91 |
| Nucleocapsid protein | SARS coronavirus Urbani | Protein | Preclinical | 92 |
| Protease precursor | Porphyromonas gingivalis | Peptide | Preclinical | 93 |
| Protective recombinant antigen | Taenia crassiceps | Peptide/protein | Preclinical | 94,95 |
| Glycoprotein G | Herpes simplex virus | Peptide | Preclinical | 96 |
| Glycoprotein D | Herpes simplex virus | Peptide/Fusion protein | Preclinical | 97,98 |
| Phosphoprotein 150 | Cytomegalovirus | Peptide | Preclinical | 99 |
| VP1 | Human parvovirus B19 | Virus‐like particle | Preclinical | 100,101 |
| Glycoprotein G | RSV | Virus‐like particle | Preclinical | 102,103 |
| Protein P | RSV | Peptide | Preclinical | 104,105 |
| Gag region encoded protein | Human T‐lymphotropic Virus Type‐1 | Protein subunit/peptide | Preclinical | 106,107 |
3. ANTIBODY INTERACTIONS OF DISORDERED ANTIGENS
3.1. Disordered antigens are targets of antibody recognition
The preceding discussion has established that disordered proteins are important antigens in a broad range of immunological contexts. It would be natural to suppose, then, that antibodies are able to effectively recognize disordered antigens. On the other hand, we have also alluded to a number of arguments which suggest that the antibody response to disordered antigens will be dysfunctional or even absent.14, 15, 19, 20, 21, 42 To address this apparent contradiction, we recently assembled a large dataset43 of well‐characterized protein antigens from the Immune Epitope Database (IEDB).44 Epitopes arising from protein regions predicted to be disordered are well represented in this dataset. In fact, we found a positive correlation between predicted disorder and the likelihood that any putative epitope will test positive in an antibody binding assay reported in the IEDB, establishing that disorder is no impediment to effective antibody recognition (Figure 1A). Others have recently made the same observation, and extended it to show that predictors of protein disorder out‐perform dedicated sequence‐based predictors for the identification of B‐cell epitopes.45 This may reflect the fact that residues that score highly in disorder predictors are more likely to be solvent‐exposed,43 as solvent exposure is both a prerequisite for, and established predictor of, antigenicity.46
Figure 1.
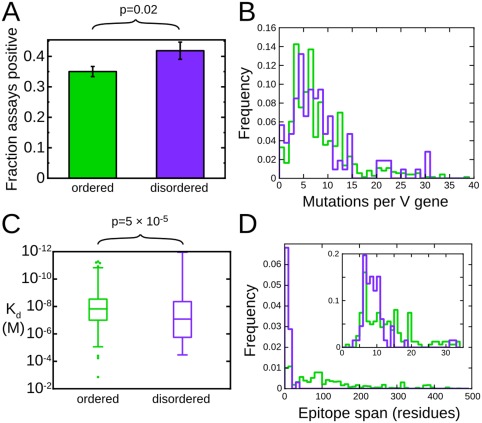
Disordered antigens are bona fide targets of antibody recognition. A, Epitopes within disordered protein regions are more likely to be targets of positive antibody binding assays. B, Antibodies to disordered epitopes (purple) and ordered epitopes (green) are subject to similar levels of somatic hypermutation. C, Disorder accounts for only a small fraction of the variability in antibody affinity. D, Disordered epitopes (purple) are exclusively short linear epitopes, while ordered epitopes (green) are predominantly conformational epitopes spanning many residues in the primary sequence. Modified with permission from Ref. 43
3.2. Affinity maturation of antibodies to disordered antigens
It has been argued that disordered antigens may impede the development of an effective immune response by interfering with the process by which B‐cells mature and differentiate into memory or antibody producing cells.14, 15, 40, 42 In the process of this maturation, antibodies undergo somatic hypermutation and selection for high‐affinity antigen binding. This affinity maturation is essential to the development of a high‐affinity and long‐lived antibody response. If the affinity maturation of antibodies to disordered antigens were significantly impeded, we would expect such antibodies to have fewer mutations with respect to germline antibody sequences than do antibodies to ordered antigens. In fact, when we compared the available antibody sequences to the corresponding germline sequences, we detected no difference in the number of V‐gene mutations between the two classes of antibodies (Figure 1B), implying that affinity maturation proceeds with equivalent efficacy for antibodies to ordered and disordered antigens.43
Highly repetitive antigens are capable of eliciting B‐cell responses that are independent of the T‐cell help normally required for B‐cell maturation. Such responses are characterized by defective affinity maturation and a failure to establish a conventional memory response.47 As such, this has been proposed as one reason that B‐cells to repetitive and disordered antigens may fail to mature.15 Indeed, the repetitive malaria antigen CSP can induce a B‐cell response in the absence of T‐cell help, but this response is much weaker and more transient than that in the presence of T‐cells. Thus, the normal response to this prototypical repetitive antigen is T‐cell dependent.48
Additional specific evidence for affinity maturation in antibodies targeting this epitope comes from recent studies of the antibody responses induced in clinical trials of the RTS,S vaccine,49 and in the context of natural infection.50 These studies have established the presence of extensive somatic hyper‐mutation in both infection‐ and vaccine‐derived antibody responses. Hyper‐mutation in the vaccine‐induced response was influenced strongly by the vaccination schedule and correlated with antibody avidity, confirming that affinity maturation is effective in this context. As such, it is clear that the presence of a transient T‐cell independent response to repetitive and disordered antigens such as CSP does not preclude the development of a classical T‐cell dependent response and accompanying affinity maturation.
3.3. Effect of disorder on the affinity of antigen–antibody interactions
A further basis for the misconception that disordered proteins will be poorly recognized by antibodies arises from the fact that disordered epitopes, by definition, fail to adopt a single well‐defined conformation in the context of the native protein that might be recognized by an antibody. It is clear, however, that disordered proteins are capable of being recognized by protein binding partners, antibodies or otherwise. To achieve this, disordered proteins often undergo a process of coupled folding and binding in which the binding partner acts effectively as a template into which the disordered protein can fold into a relatively well‐defined bound conformation.51 Nonetheless, coupled folding and binding places both kinetic and thermodynamic constraints on the interaction.52 In particular, the significant entropic cost of folding reduces the overall binding affinity of these interactions, unless this cost can be offset by other means.53 Thus, the interactions of disordered proteins are often of relatively low affinity, despite maintaining high specificity. These properties are ideally suited to mediators of signals that must be switched on or off rapidly, accounting for the enrichment of disorder in proteins involved in signal transduction and regulation.7 These interactions are quite distinct, however, from typical antibody‐antigen interactions, which are selected for high affinity and slow dissociation.
These considerations raise the possibility that antibodies that recognize disordered antigens may do so with reduced affinity, reflecting the substantial entropic cost that must accompany the disordered antigen adopting a single antibody‐bound conformation.54, 55, 56 In fact, using our dataset of ordered and disordered epitopes from the IEDB, we have shown that disorder accounts for very little of the wide variation observed in the affinities of antibodies for protein antigens.43 Specifically, we observed an approximately seven‐fold difference in median affinity between antibodies that recognize ordered epitopes and those that recognize disordered epitopes, a loss of affinity that is very much smaller than might be expected on the basis of the expected entropic cost of coupled folding and binding (Figure 1C).54, 55
To investigate the structural basis for the relatively high affinity with which antibodies recognize disordered antigens, we cross‐referenced our IEDB‐derived dataset of ordered and disordered antigens with the Protein Databank (PDB),57 resulting in a set of 872 structures of antibody–protein complexes, of which 69 represent disordered epitopes.43 Comparison of these structures revealed that disordered epitopes involve substantially fewer residues than ordered epitopes, perhaps as a means to minimize the entropic cost of binding (Figure 1D). Thus, disordered epitopes rarely span more than 12 residues of the antigen primary sequence, and the majority of these residues make direct interactions with antibody. These structural observations are consistent with epitope mapping experiments with a large number of antibodies against several disordered antigens,38 which imply that disordered epitopes are overwhelmingly linear epitopes, and that antibody recognition is determined largely by the local antigen sequence. This stands in contrast to more conventional ordered antigens, in which a large fraction of the antibody response is typically directed to conformational epitopes.
A further implication of the reduction in size of disordered epitopes is that disordered epitopes are much more efficient than ordered epitopes in their interactions with antibody, in the sense that they achieve more binding free energy per unit of interaction surface area, or per epitope residue.43 Comparison of the structures of antibody‐bound ordered and disordered antigens reveals a number of features of the antibody‐antigen interaction that may contribute to this efficiency, including a more concave antigen binding site with better complementarity to the shape of the antigen, as well as a two‐fold increase in the number of inter‐molecular H‐bonds per epitope residue. These structural features imply that antibodies to disordered epitopes are better matched to the bound conformation of that epitope than are antibodies to ordered epitopes. It is likely that this reflects to some extent the ability of the disordered epitope to adapt to its binding site on the antibody. Indeed, this flexibility permits some disordered epitopes to adopt distinct conformations bound to different antibodies.50, 58, 59 Nonetheless, this exquisite complementarity is also likely to require adaptation on the part of the antibody.50 As such, it may also be seen as evidence to support the conclusion that affinity maturation of antibodies to disordered epitopes is at least as effective as is affinity maturation targeting ordered epitopes.
4. The IMPLICATIONS OF DISORDER FOR VACCINE DEVELOPMENT: EXAMPLES FROM THE MALARIA ANTIGEN MSP2
We now place the preceding general considerations on a more specific footing, by outlining their implications in the context of vaccine development efforts against the disordered malaria antigen MSP2.39, 60 MSP2 is a highly abundant, C‐terminally glycosylphosphatidylinositol (GPI)‐anchored blood‐stage surface antigen of the malaria parasite P. falciparum and a potential component of a malaria vaccine.36, 61, 62, 63 MSP2 has conserved N‐ and C‐terminal regions, flanking a central region comprising polymorphic repetitive sequences and non‐repetitive sequences that are predominantly dimorphic (Figure 2). These dimorphic sequences within the central variable region differentiate MSP2 variants into two allelic families, 3D7 and FC27 MSP2.64, 65 The Combination B vaccine, which included 3D7 MSP2 as one of three protein antigens, yielded evidence for protection in a clinical trial in Papua New Guinea, but this protection was strain‐specific, with vaccines not being protected from malarial strains expressing FC27‐family MSP2 sequences.37 Thus, a primary concern for vaccine development against MSP2, like many other malaria antigens, has been to address the polymorphism in the antigen.66
Figure 2.
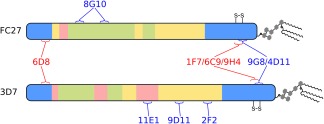
Schematic of the primary structure of the two allelic families of MSP2. Regions of conserved (blue), repetitive (green), dimorphic (yellow), and polymorphic (pink) sequence are shown. The epitopes of a panel of monoclonal antibodies are also shown, with antibodies that strongly recognize the parasite antigen in blue text, and those that do so only weakly or not at all in red
4.1. Disorder and antigenicity in MSP2
Although the studies described above have established that disorder is no impediment to the development of a robust antibody response, the interplay between disorder and antigenicity may be more complicated at the level of a single antigen. In our studies of MSP2, we have compared experimental measures of conformational disorder derived from NMR relaxation rates with observed patterns of antigenicity inferred from experimental immunizations of animals and humans.40 The result of that study, in contrast to the bioinformatic analysis,43, 45 was a negative correlation between antigenicity and disorder, with the most flexible regions of MSP2 being the least likely to be recognized by antibodies in MSP2‐immune sera, and with regions that show slight conformational restriction apparently dominating the antibody response in these immunizations. It is important to note that for MSP2 the experimental measures of disorder used in this study correlate poorly with the disorder prediction scores used in the bioinformatics analysis, reflecting the uniformly high levels of disorder across MSP2 (>85% of MSP2 residues have IUPred scores >0.8). Indeed, the most flexible regions of MSP2 are extremely disordered, being composed exclusively of the small amino acids Gly, Ser, and Ala. Thus, it may be that this extreme level of disorder and attendant low sequence complexity is sufficient to significantly impede the development of a strong antibody response, whereas the levels of disorder more typically encountered in disordered antigens are compatible with a robust response, as outlined above.
Importantly, these most flexible and dynamic regions in MSP2 are also the most polymorphic regions in the antigen, and this, together with their low sequence complexity, renders them unattractive targets for vaccine development. As such, the greater immunogenicity of other disordered but less flexible regions of the protein is likely to be advantageous in the context of vaccine development. Moreover, a key advantage afforded by peptide‐based approaches to vaccine development is the ability to omit ineffective regions such as these polymorphic regions entirely, ensuring that the response is focused on conserved epitopes that are more likely to elicit broadly protective responses. This is exemplified by the long synthetic peptides derived from the C‐terminal half of MSP2, which have been considered potential components of a malaria vaccine.41, 67 These peptides include both conserved and dimorphic family‐specific sequences, but exclude the highly polymorphic regions of MSP2.
4.2. Lipid interactions modulate the conformation and antigenicity of MSP2
Beyond the specificity of the immediate epitope‐antibody interaction, the broader context in which the antibody encounters the antigen is likely to influence the way in which antibodies are able to recognize disordered antigens. Such effects are well‐known in the case of ordered antigens, and can arise when, for example, molecular interactions lead to conformational changes that mask or disrupt epitopes or otherwise render them cryptic.68, 69 Similar effects are likely in disordered antigens; indeed, the conformational properties of disordered proteins are uniquely sensitive to modulation by interactions, post‐translational modifications and other mechanisms. As such, antibody recognition of disordered epitopes may be expected to be particularly sensitive to the context in which the antibody is recognized.
An example of this is seen in the masking of conserved epitopes in MSP2. Amongst a panel of monoclonal mouse antibodies raised against recombinant MSP2, all of those targeting variable epitopes also recognized MSP2 on the parasite surface, whereas many antibodies targeting conserved epitopes recognized the parasites weakly or not at all (Figure 2).38 Together, these data imply that at least some conserved epitopes within MSP2 may be masked by the conformations or interactions adopted by the antigen on the parasite surface.
The structural basis of this masking has recently been characterized in the case of one such epitope. 6D8, a murine monoclonal antibody (mAb) recognizing an epitope in the conserved N‐terminal region binds recombinant MSP2 and epitope‐derived peptides with high affinity, yet it fails to recognize the parasite surface.38 The N‐terminal region of MSP2, including the 6D8 epitope, binds lipids, adopting an amphipathic helical conformation that is expected to be heavily populated in parasite MSP2 given that it is GPI‐anchored to the parasite membrane.70 This helical conformation is incompatible with the conformation that is recognized by 6D8, as revealed by X‐ray crystallography (Figure 3A,B).71 Moreover, the 6D8 epitope is lost in recombinant MSP2 when the protein is C‐terminally tethered to lipid vesicles,71 indicating that the N‐terminal interaction with lipid and the accompanying conformational change are sufficient to account for the observed masking of the 6D8 epitope on the parasite surface.
Figure 3.
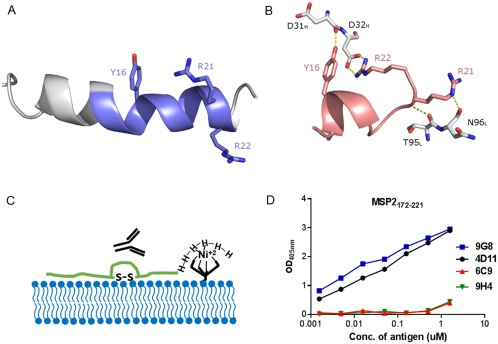
Interaction of different MSP2‐based conserved regions specific peptides with antibody and lipid. Comparison between A, lipid‐bound N‐terminal MSP21–25 70 and B, 6D8 mAb‐bound MSP214–22 (PDB ID 4QYO);71 key 6D8 paratope residues involved in binding are shown in white. The α‐helical configuration of the lipid‐bound peptide removes the backbone flexibility required for Arg22 to access Tyr16, which provides a structural rationale for 6D8 epitope masking at the parasite membrane. C, Schematic of lipid tethering of C‐terminal region of MSP2 (MSP2172–221) where MSP2172–221 was synthesized with a C‐terminal His6‐tag to immobilize on nickel bound to nickel‐chelating lipid. D, ELISA showing the effects of lipid tethering on the binding of four C‐terminal region‐specific mouse mAbs for MSP2172–221.72
To explore the impact of lipid conjugation on the C‐terminal domain of MSP2, a shorter construct of 50 residues, MSP2172–221, consisting of only the conserved C‐terminal region, was generated.72 Our NMR data suggest that MSP2172–221 is unstructured in solution and that the conformational properties of the C‐terminal region are identical in MSP2172–221, 3D7 and FC27 MSP2. NMR studies also indicate that many residues in MSP2172–221 interact with lipid, including some in epitopes recognized by C‐terminal region specific monoclonal antibodies (mAbs). In contrast to the N‐terminal region however, there is no indication of stable helical conformation within lipid‐bound MSP2177–221.71, 72 Nonetheless, the antigenicity of liposome‐conjugated MSP2172–221 and full‐length MSP2 are altered in comparison to the lipid‐free recombinant peptides (Figure 3C). Importantly, it also appears that the binding pattern of C‐terminal specific mAbs for liposome‐conjugated antigens (Figure 3D) is consistent with that of the mAbs for parasite lysate. Thus, lipid tethering appears to alter the antigenic state of these MSP2 constructs, rendering it more similar to that of the antigen on the parasite surface. Lipid‐conjugation of MSP2 based vaccines may therefore provide a more effective antibody response.
Similar masking of potentially protective epitopes is seen in the pre‐erythrocytic malaria antigen CSP. Although the repetitive sequences in CSP have long been the focus of vaccine development efforts, recent studies have identified epitopes within the conserved N‐terminal region that appear to contribute to the protective response to CSP in the context of natural immunity and vaccination.73, 74, 75 These antibodies act synergistically with antibodies targeting the CSP repeats.75 In the course of parasite development within the mosquito and subsequent migration through the human host to the liver, CSP undergoes proteolytic processing, which is essential for the establishment of a liver‐stage infection. This proteolysis exposes otherwise cryptic epitopes in both the N‐ and C‐terminal regions of CSP, indicating conformational changes that appear to involve long‐range interactions within CSP, which in turn determine the accessibility of some of these N‐terminal epitopes.74, 76, 77 The nature of these interactions and the associated conformational changes remain largely obscure. In light of the efficacy of antibodies targeting N‐terminal and possibly also C‐terminal epitopes, it is likely that a better understanding of these interactions will facilitate the design of improved CSP‐based vaccine constructs.
4.3. Interactions outside the defined epitope may modulate the specificity of antibodies targeting disordered antigens
Disordered proteins typically adopt a well‐defined structure when interacting with binding partners, as described above. Nonetheless, in some cases significant disorder persists in these interactions, giving rise to what are known as “fuzzy interactions.”78 It is becoming increasingly clear that these fuzzy interactions modulate the function of disordered proteins in several different ways. In particular, transient interactions involving the disordered regions flanking the structurally defined binding site can modulate the affinity with which disordered proteins are recognized by their binding partners.79 The extent to which such interactions modulate the antibody recognition of disordered antigens remains largely unexplored, but we have recently characterized one example which suggests that such modulation is feasible, and possibly quite general. The mAb 6D8, introduced above, recognizes a conserved epitope near the N‐terminus of MSP2, yet 6D8 binds the two allelic forms of MSP2 with affinities that differ by approximately five fold.71 NMR experiments provide evidence of transient interactions that are not resolved in the crystal structure and which involve variable residues that are distal to the structurally defined epitope. These transient or fuzzy interactions thus appear to modulate the specificity of 6D8, rendering the antibody somewhat strain‐specific in spite of the conserved nature of its epitope.71, 80 Although the generality of this effect remains to be established, it appears to originate from largely non‐specific interactions that occur, almost inevitably, as a result of the high effective concentration of the disordered regions that immediately flank the structurally defined epitope.80 As such, it is expected that similar fuzzy interactions may contribute to antibody recognition of many disordered antigens.
4.4. Toward a structural vaccinology of disordered antigens
The conserved C‐terminal region of MSP2 is particularly immunodominant, with a total of five mouse mAbs able to recognize an overlapping epitope spanning MSP2207–224.38 As discussed above, antibodies targeting the conserved regions have shown differing ability to recognize parasite MSP2 (Figure 2). The mAbs 4D11 and 9G8 are able to bind to parasite MSP2 by western blot and immunofluorescence assay (IFA) whilst the mAbs 9H4, 6C9 and 1F7, show significantly weaker signal by IFA and no binding by western blot, despite the five antibodies recognizing overlapping linear epitopes. The close proximity of the C‐terminal region to the parasite surface via its GPI tether may influence the local conformation, making parts of the epitope inaccessible to antibody binding. Understanding the conformation of epitopes bound to antibodies such as 4D11 and 9G8 affords a good platform for further peptide vaccine design. By presenting the immune system with epitopes that are accessible on the parasite surface whilst removing distracting sequences, a more efficient and specific immune response may be elicited.
Recently, the crystal structure of 4D11 Fv bound to its minimal binding epitope MSP2215–222, which it also shares with 9G8, was solved at 2.2 Å.81 Upon antibody binding, the 8‐residue peptide epitope was found to adopt a β‐bend ribbon conformation, characterized by consecutive overlapping β‐turns (Figure 4). The two β‐turns in the epitope were stabilized by hydrogen bonds involving Asn215‐Asn218 and Glu217‐Gly220, and allowed key interactions of residues Lys216, Glu217, and Asn218 with the 4D11 paratope. This structural information suggests strategies by which stabilized analogues of the epitope could be optimized for use as a peptide vaccine, with a view to guiding the specificity of the antibody response towards 4D11‐like epitopes that are both conserved and accessible on the parasite surface, and thus are more likely to contribute to a broadly protective immune response.
Figure 4.
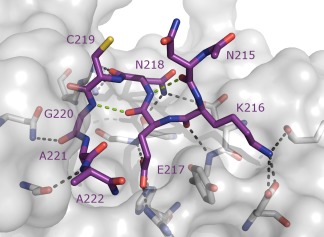
Murine mAb 4D11 Fv in complex with its cognate 8‐residue epitope (PDB ID 5TBD).81 Intramolecular hydrogen bonds are indicated by green dashed lines. Interactions with 4D11 Fv paratope are shown in black dashed lines
5. CONCLUSIONS AND FUTURE DIRECTIONS
Disordered proteins are highly abundant in infectious organisms across all kingdoms of life, and these proteins are bona fide targets of antibody responses in the context of natural infection and vaccination.43 In contrast to widely‐held views that disordered antigens will fail to elicit an effective antibody response,14, 15, 16, 17, 18, 19, 20, 21 it is clear that they play an important role as protective antigens in a range of diseases (Table 3). Epitopes from disordered antigens are typically short linear epitopes, not the more complex conformational epitopes dominant in ordered antigens. As such, disordered antigens are well suited as targets of peptide vaccines.
Despite the potential promise of this approach, careful consideration must be paid to the context in which target epitopes are recognized by the immune system. The conformational and antigenic properties of disordered antigens can be strongly modulated by their interactions, both in their native parasite context and in any vaccine formulation. Mismatch between the parasite and vaccine context has probably contributed to the failure of a number of peptide‐based vaccine strategies.82 The use of long peptides in a vaccine formulation, potentially incorporating several protective epitopes within their native sequence, may offer one strategy to address this issue.67, 83 Potentially a more effective approach will be to define the structures of disordered peptide epitopes bound to their cognate antibodies, and to use this structural information as a basis for design of peptides in which these structures are stabilized. To this end, the range of available strategies for stabilizing the conformation of synthetic peptides represents a clear advantage to the peptide vaccine approach. Future developments in this area, enabling the stabilization of a wider range of peptide conformations while limiting perturbation of residues essential for antibody binding, will serve to broaden the scope of the strategy further.
It is clear that the targeted vaccine development strategy described here requires a detailed understanding of the functional determinants of immune protection, and of the specific antigens and epitopes responsible for mediating these functions. While this is well understood for some pathogens, for other less studied or immunologically complex pathogens, such information may still be emerging. In this context, new strategies for high‐throughput profiling of immunological responses and identification of correlates of immune protection84 represent a welcome advance that promises to allow peptide‐based vaccine development against diseases for which no effective vaccine currently exists.
ACKNOWLEDGMENTS
This work was supported in part by the Australian National Health and Medical Research Council (project grants 1042520 and 1125788 to CAM and RSN). RSN acknowledges fellowship support from the NHMRC. The authors thank Robin Anders and Jack Richards for many helpful discussions in the course of their studies on MSP2.
Biographies
From left to right: Chris MacRaild, Sreedam Das, Jeff Seow, Ray Nortondr chris macraild received his Ph.D. from the University of Melbourne in 2004. His recent research has addressed diverse aspects of the structural biology of the malaria parasite, with the ultimate goal of establishing new therapeutic opportunities against the devastating disease it causes. A particular ongoing interest is the structural and antigenic properties of the disordered proteins which constitute an important class of malaria antigen.

professor ray norton holds a personal chair at the Monash Institute of Pharmaceutical Sciences. He graduated with BSc(Honours) from the University of Melbourne and obtained his Ph.D. in chemistry from the Australian National University. His group at Monash employs a range of biophysical approaches (NMR, SPR, ITC and X‐ray crystallography) in studies of peptide and protein toxins and infectious diseases. He has published over 350 articles, received numerous national awards, and is an inventor on several patents.
MacRaild CA, Seow J, Das SC, Norton RS. Disordered epitopes as peptide vaccines. Peptide Science. 2018;110:e24067. 10.1002/pep2.24067
Funding information Australian National Health and Medical Research Council, Grants Numbers: 1042520, 1125788 NHMRC
Contributor Information
Christopher A. MacRaild, Email: chris.macraild@monash.edu.
Raymond S. Norton, Email: ray.norton@monash.edu.
REFERENCES
- 1. Stern A. M., Markel H., Health Aff (Millwood) 2005, 24, 611. [DOI] [PubMed] [Google Scholar]
- 2. Skwarczynski M., Toth I., Chem. Sci. 2016, 7, 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anders R. F., Trends Parasitol. 2011, 27, 330. [DOI] [PubMed] [Google Scholar]
- 4. Kumai T., Fan A., Harabuchi Y., Celis E., Curr. Opin. Immunol. 2017, 47, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robinson J. A., J. Pept. Sci. 2013, 19, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dunker A. K., Obradovic Z., Romero P., Garner E. C., Brown C. J., Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 161. [PubMed] [Google Scholar]
- 7. Wright P. E., Dyson H. J., Nat. Rev. Mol. Cell Biol. 2015, 16, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pancsa R., Tompa P., PLoS One 2012, 7, e34687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xue B., Blocquel D., Habchi J., Uversky A. V., Kurgan L., Uversky V. N., Longhi S., Chem. Rev. 2014, 114, 6880. [DOI] [PubMed] [Google Scholar]
- 10. Mohan A., W. J. Sullivan, Jr. , Radivojac P., Dunker A. K., Uversky V. N., Mol. Biosyst. 2008, 4, 328. [DOI] [PubMed] [Google Scholar]
- 11. Marin M., Uversky V. N., Ott T., Plant Cell 2013, 25, 3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guy A. J., Irani V., MacRaild C. A., Anders R. F., Norton R. S., Beeson J. G., Richards J. S., Ramsland P. A., PLoS One 2015, 10, e0141729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng Z. P., Zhang X., Han P., Arora N., Anders R. F., Norton R. S., Mol. Biochem. Parasitol. 2006, 150, 256. [DOI] [PubMed] [Google Scholar]
- 14. Anders R. F., Parasite Immunol. 1986, 8, 529. [DOI] [PubMed] [Google Scholar]
- 15. Schofield L., Parasitol Today 1991, 7, 99. [DOI] [PubMed] [Google Scholar]
- 16. Mendes T. A., Lobo F. P., Rodrigues T. S., Rodrigues‐Luiz G. F., daRocha W. D., Fujiwara R. T., Teixeira S. M., Bartholomeu D. C., Mol. Biol. Evol. 2013, 30, 951. [DOI] [PubMed] [Google Scholar]
- 17. Ferreira M. U., da Silva Nunes M., Wunderlich G., Clin. Diagn. Lab. Immunol. 2004, 11, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hughes A. L., J. Mol. Evol. 2004, 59, 528. [DOI] [PubMed] [Google Scholar]
- 19. Penkett C. J., Redfield C., Jones J. A., Dodd I., Hubbard J., Smith R. A., Smith L. J., Dobson C. M., Biochemistry 1998, 37, 17054. [DOI] [PubMed] [Google Scholar]
- 20. Dunker A. K., Brown C. J., Lawson J. D., Iakoucheva L. M., Obradovic Z., Biochemistry 2002, 41, 6573. [DOI] [PubMed] [Google Scholar]
- 21. Kwong P. D., Doyle M. L., Casper D. J., Cicala C., Leavitt S. A., Majeed S., Steenbeke T. D., Arthos J., Nature 2002, 420, 678. [DOI] [PubMed] [Google Scholar]
- 22. Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., Carlton J. M., Pain A., Nelson K. E., Bowman S., Paulsen I. T., James K., Eisen J. A., Rutherford K., Salzberg S. L., Craig A., Kyes S., Chan M. S., Nene V., Shallom S. J., Suh B., Peterson J., Angiuoli S., Pertea M., Allen J., Selengut J., Haft D., Mather M. W., Vaidya A. B., Martin D. M., Fairlamb A. H., Fraunholz M. J., Roos D. S., Ralph S. A., McFadden G. I., Cummings L. M., Subramanian G. M., Mungall C., Venter J. C., Carucci D. J., Hoffman S. L., Newbold C., Davis R. W., Fraser C. M., Barrell B., Nature 2002, 419, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DePristo M. A., Zilversmit M. M., Hartl D. L., Gene 2006, 378, 19. [DOI] [PubMed] [Google Scholar]
- 24. Xue H. Y., Forsdyke D. R., Mol. Biochem. Parasitol. 2003, 128, 21. [DOI] [PubMed] [Google Scholar]
- 25. Zilversmit M. M., Volkman S. K., DePristo M. A., Wirth D. F., Awadalla P., Hartl D. L., Mol. Biol. Evol. 2010, 27, 2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aly A. S., Vaughan A. M., Kappe S. H., Annu. Rev. Microbiol. 2009, 63, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crompton P. D., Pierce S. K., Miller L. H., J. Clin. Invest. 2010, 120, 4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doud M. B., Koksal A. C., Mi L.‐Z., Song G., Lu C., Springer T. A., Proc. Natl. Acad. Sci. USA 2012. [Google Scholar]
- 29. Foquet L., Hermsen C. C., van Gemert G. J., Van Braeckel E., Weening K. E., Sauerwein R., Meuleman P., Leroux‐Roels G., J Clin Invest 2014, 124, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olotu A., Lusingu J., Leach A., Lievens M., Vekemans J., Msham S., Lang T., Gould J., Dubois M. C., Jongert E., Vansadia P., Carter T., Njuguna P., Awuondo K. O., Malabeja A., Abdul O., Gesase S., Mturi N., Drakeley C. J., Savarese B., Villafana T., Lapierre D., Ballou W. R., Cohen J., Lemnge M. M., Peshu N., Marsh K., Riley E. M., von Seidlein L., Bejon P., Lancet Infect Dis. 2011, 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moorthy V. S., Ballou W. R., Malar. J. 2009, 8, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dyson H. J., Satterthwait A. C., Lerner R. A., Wright P. E., Biochemistry 1990, 29, 7828. [DOI] [PubMed] [Google Scholar]
- 33. Taylor R. R., Allen S. J., Greenwood B. M., Riley E. M., Am. J. Trop. Med. Hyg. 1998, 58, 406. [DOI] [PubMed] [Google Scholar]
- 34. Al‐Yaman F., Genton B., Anders R. F., Falk M., Triglia T., Lewis D., Hii J., Beck H. P., Alpers M. P., Am. J. Trop. Med. Hyg. 1994, 51, 593. [DOI] [PubMed] [Google Scholar]
- 35. Metzger W. G., Okenu D. M., Cavanagh D. R., Robinson J. V., Bojang K. A., Weiss H. A., McBride J. S., Greenwood B. M., Conway D. J., Parasite Immunol. 2003, 25, 307. [DOI] [PubMed] [Google Scholar]
- 36. Genton B., Betuela I., Felger I., Al‐Yaman F., Anders R. F., Saul A., Rare L., Baisor M., Lorry K., Brown G. V., Pye D., Irving D. O., Smith T. A., Beck H. P., Alpers M. P., J. Infect. Dis. 2002, 185, 820. [DOI] [PubMed] [Google Scholar]
- 37. Flück C., Smith T., Beck H. P., Irion A., Betuela I., Alpers M. P., Anders R., Saul A., Genton B., Felger I., Infect. Immun. 2004, 72, 6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adda C. G., MacRaild C. A., Reiling L., Wycherley K., Boyle M. J., Kienzle V., Masendycz P., Foley M., Beeson J. G., Norton R. S., Anders R. F., Infect. Immun. 2012, 80, 4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X., Perugini M. A., Yao S., Adda C. G., Murphy V. J., Low A., Anders R. F., Norton R. S., J. Mol. Biol. 2008, 379, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacRaild C. A., Zachrdla M., Andrew D., Bankala K., Nováček J., Žídek L., Sklenář V., Richards J. S., Beeson J. G., Anders R. F., Norton R. S., PLoS One 2015, 10, e0119899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Balam S., Olugbile S., Servis C., Diakité M., D'Alessandro A., Frank G., Moret R., Corradin G., Malar. J. 2014, 13, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carl P. L., Temple B. R., Cohen P. L., Arthritis Res. Ther. 2005, 7, R1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. MacRaild C. A., Richards J. S., Anders R. F., Norton R. S., Structure 2016, 24, 148. [DOI] [PubMed] [Google Scholar]
- 44. Vita R., Overton J. A., Greenbaum J. A., Ponomarenko J., Clark J. D., Cantrell J. R., Wheeler D. K., Gabbard J. L., Hix D., Sette A., Peters B., Nucleic Acids Res. 2015, 43, D405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rahman K. S., Chowdhury E. U., Sachse K., Kaltenboeck B., J. Biol. Chem. 2016, 291, 14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Novotny J., Handschumacher M., Haber E., Bruccoleri R. E., Carlson W. B., Fanning D. W., Smith J. A., Rose G. D., Proc. Natl. Acad. Sci. USA 1986, 83, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Defrance T., Taillardet M., Genestier L., Curr. Opin. Immunol. 2011, 23, 330. [DOI] [PubMed] [Google Scholar]
- 48. Fisher C. R., Sutton H. J., Kaczmarski J. A., McNamara H. A., Clifton B., Mitchell J., Cai Y., Dups J. N., D'Arcy N. J., Singh M., Chuah A., Peat T. S., Jackson C. J., Cockburn I. A., PLoS Pathog. 2017, 13, e1006469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Regules J. A., Cicatelli S. B., Bennett J. W., Paolino K. M., Twomey P. S., Moon J. E., Kathcart A. K., Hauns K. D., Komisar J. L., Qabar A. N., Davidson S. A., Dutta S., Griffith M. E., Magee C. D., Wojnarski M., Livezey J. R., Kress A. T., Waterman P. E., Jongert E., Wille‐Reece U., Volkmuth W., Emerling D., Robinson W. H., Lievens M., Morelle D., Lee C. K., Yassin‐Rajkumar B., Weltzin R., Cohen J., Paris R. M., Waters N. C., Birkett A. J., Kaslow D. C., Ballou W. R., Ockenhouse C. F., Vekemans J., J. Infect. Dis. 2016, 214, 762. [DOI] [PubMed] [Google Scholar]
- 50. Triller G., Scally S. W., Costa G., Pissarev M., Kreschel C., Bosch A., Marois E., Sack B. K., Murugan R., Salman A. M., Janse C. J., Khan S. M., Kappe S. H. I., Adegnika A. A., Mordmuller B., Levashina E. A., Julien J. P., Wardemann H., Immunity 2017, 47, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sugase K., Dyson H. J., Wright P. E., Nature 2007, 447, 1021. [DOI] [PubMed] [Google Scholar]
- 52. Uversky V. N., Biochim. Biophys. Acta 2013, 1834, 932. [DOI] [PubMed] [Google Scholar]
- 53. Pancsa R., Fuxreiter M., IUBMB Life 2012, 64, 513. [DOI] [PubMed] [Google Scholar]
- 54. Bracken C., Carr P. A., Cavanagh J., Palmer A. G., III, J. Mol. Biol. 1999, 285, 2133. [DOI] [PubMed] [Google Scholar]
- 55. Baxa M. C., Haddadian E. J., Jumper J. M., Freed K. F., Sosnick T. R., Proc. Natl. Acad. Sci. USA 2014, 111, 15396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Papadakos G., Sharma A., Lancaster L. E., Bowen R., Kaminska R., Leech A. P., Walker D., Redfield C., Kleanthous C., J. Am. Chem. Soc. 2015, 137, 5252. [DOI] [PubMed] [Google Scholar]
- 57. Berman H. M., Battistuz T., Bhat T. N., Bluhm W. F., Bourne P. E., Burkhardt K., Feng Z., Gilliland G. L., Iype L., Jain S., Fagan P., Marvin J., Padilla D., Ravichandran V., Schneider B., Thanki N., Weissig H., Westbrook J. D., Zardecki C., Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899. [DOI] [PubMed] [Google Scholar]
- 58. Deng L., Ma L., Virata‐Theimer M. L., Zhong L., Yan H., Zhao Z., Struble E., Feinstone S., Alter H., Zhang P., Proc. Natl. Acad. Sci. USA 2014, 111, 10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chu H. M., Wright J., Chan Y. H., Lin C. J., Chang T. W., Lim C., Nat. Commun. 2014, 5, 3139. [DOI] [PubMed] [Google Scholar]
- 60. Adda C. G., Murphy V. J., Sunde M., Waddington L. J., Schloegel J., Talbo G. H., Vingas K., Kienzle V., Masciantonio R., Howlett G. J., Hodder A. N., Foley M., Anders R. F., Mol. Biochem. Parasitol. 2009, 166, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smythe J. A., Peterson M. G., Coppel R. L., Saul A. J., Kemp D. J., Anders R. F., Mol. Biochem. Parasitol. 1990, 39, 227. [DOI] [PubMed] [Google Scholar]
- 62. Gilson P. R., Nebl T., Vukcevic D., Moritz R. L., Sargeant T., Speed T. P., Schofield L., Crabb B. S., Mol. Cell Proteom. 2006, 5, 1286. [DOI] [PubMed] [Google Scholar]
- 63. McCarthy J. S., Marjason J., Elliott S., Fahey P., Bang G., Malkin E., Tierney E., Aked‐Hurditch H., Adda C., Cross N., Richards J. S., Fowkes F. J. I., Boyle M. J., Long C., Druilhe P., Beeson J. G., Anders R. F., PLoS One 2011, 6, e24413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Smythe J. A., Coppel R. L., Day K. P., Martin R. K., Oduola A. M., Kemp D. J., Anders R. F., Proc. Natl. Acad. Sci. USA 1991, 88, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fenton B., Clark J. T., Khan C. M., Robinson J. V., Walliker D., Ridley R., Scaife J. G., McBride J. S., Mol. Cell Biol. 1991, 11, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Anders R. F., Adda C. G., Foley M., Norton R. S., Hum. Vaccin. 2010, 6, 39. [DOI] [PubMed] [Google Scholar]
- 67. Flueck C., Frank G., Smith T., Jafarshad A., Nebie I., Sirima S. B., Olugbile S., Corradin G., Vaccine 2009, 27, 2653. [DOI] [PubMed] [Google Scholar]
- 68. Zarnitsyna V. I., Ellebedy A. H., Davis C., Jacob J., Ahmed R., Antia R., Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370, 20140248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mould A. P., Askari J. A., Byron A., Takada Y., Jowitt T. A., Humphries M. J., J. Biol. Chem. 2016, 291, 20993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. MacRaild C. A., Pedersen M. Ø., Anders R. F., Norton R. S., Biochim. Biophys. Acta 2012, 1818, 2572. [DOI] [PubMed] [Google Scholar]
- 71. Morales R. A. V., MacRaild C. A., Seow J., Krishnarjuna B., Drinkwater N., Rouet R., Anders R. F., Christ D., McGowan S., Norton R. S., Sci. Rep. 2015, 5, 10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Das S. C., Morales R. A. V., Seow J., Krishnarjuna B., Dissanayake R., Anders R. F., MacRaild C. A., Norton R. S., FEBS J 2017, 284, 2649. [DOI] [PubMed] [Google Scholar]
- 73. Bongfen S. E., Ntsama P. M., Offner S., Smith T., Felger I., Tanner M., Alonso P., Nebie I., Romero J. F., Silvie O., Torgler R., Corradin G., Vaccine 2009, 27, 328. [DOI] [PubMed] [Google Scholar]
- 74. Rathore D., Nagarkatti R., Jani D., Chattopadhyay R., de la Vega P., Kumar S., McCutchan T. F., J. Biol. Chem. 2005, 280, 20524. [DOI] [PubMed] [Google Scholar]
- 75. Espinosa D. A., Gutierrez G. M., Rojas‐Lopez M., Noe A. R., Shi L., Tse S. W., Sinnis P., Zavala F., J. Infect. Dis. 2015, 212, 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Herrera R., Anderson C., Kumar K., Molina‐Cruz A., Nguyen V., Burkhardt M., Reiter K., R. Shimp, Jr. , Howard R. F., Srinivasan P., Nold M. J., Ragheb D., Shi L., DeCotiis M., Aebig J., Lambert L., Rausch K. M., Muratova O., Jin A., Reed S. G., Sinnis P., Barillas‐Mury C., Duffy P. E., MacDonald N. J., Narum D. L., Infect. Immun. 2015, 83, 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Coppi A., Natarajan R., Pradel G., Bennett B. L., James E. R., Roggero M. A., Corradin G., Persson C., Tewari R., Sinnis P., J. Exp. Med. 2011, 208, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tompa P., Fuxreiter M., Trends Biochem. Sci. 2008, 33, 2. [DOI] [PubMed] [Google Scholar]
- 79. Fuxreiter M., Tompa P., Adv. Exp. Med. Biol. 2012, 725, 1. [DOI] [PubMed] [Google Scholar]
- 80. Krishnarjuna B., Suguki T., Morales R. A. V., Seow J., Fujiwara T., Wilde K. L., Norton R. S., MacRaild C. A., Submitted 2018. [DOI] [PMC free article] [PubMed]
- 81. Seow J., Morales R. A., MacRaild C. A., Krishnarjuna B., McGowan S., Dingjan T., Jaipuria G., Rouet R., Wilde K. L., Atreya H. S., Richards J. S., Anders R. F., Christ D., Drinkwater N., Norton R. S., J. Mol. Biol. 2017, 429, 836. [DOI] [PubMed] [Google Scholar]
- 82. Irimia A., Sarkar A., Stanfield R. L., Wilson I. A., Immunity 2016, 44, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Olugbile S., Kulangara C., Bang G., Bertholet S., Suzarte E., Villard V., Frank G., Audran R., Razaname A., Nebie I., Awobusuyi O., Spertini F., Kajava A. V., Felger I., Druilhe P., Corradin G., Infect. Immun. 2009, 77, 5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sette A., Rappuoli R., Immunity 2010, 33, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Esposito V., Musi V., de Chiara C., Veggi D., Serruto D., Scarselli M., Kelly G., Pizza M., Pastore A., J Biol Chem 2011, 286, 41767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Serruto D., Spadafina T., Ciucchi L., Lewis L. A., Ram S., Tontini M., Santini L., Biolchi A., Seib K. L., Giuliani M. M., Donnelly J. J., Berti F., Savino S., Scarselli M., Costantino P., Kroll J. S., O'Dwyer C., Qiu J., Plaut A. G., Moxon R., Rappuoli R., Pizza M., Arico B., Proc Natl Acad Sci U S A 2010, 107, 3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.RTS, S. C. T. P. Lancet 2015.
- 88. Osman M., Mistry A., Keding A., Gabe R., Cook E., Forrester S., Wiggins R., Di Marco S., Colloca S., Siani L., Cortese R., Smith D. F., Aebischer T., Kaye P. M., Lacey C., J. PLoS Negl Trop Dis 2017, 11, e0005527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cai X., Zheng W., Pan S., Zhang S., Xie Y., Guo H., Wang G., Li Z., Luo M., Antiviral Res 2017, 149, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang G., Sun M., Fang J., Yang Q., Tong H., Wang L., Vaccine 2006, 24, 6065. [DOI] [PubMed] [Google Scholar]
- 91. Alvarez P., Leguizamon M. S., Buscaglia C. A., Pitcovsky T. A., Campetella O., Infect Immun 2001, 69, 7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu S. J., Leng C. H., Lien S. P., Chi H. Y., Huang C. Y., Lin C. L., Lian W. C., Chen C. J., Hsieh S. L., Chong P., Vaccine 2006, 24, 3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kelly C. G., Booth V., Kendal H., Slaney J. M., Curtis M. A., Lehner T., Clin Exp Immunol 1997, 110, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gevorkian G., Manoutcharian K., Larralde C., Hernandez M., Almagro J. C., Viveros M., Sotelo J., Garcia E., Sciutto E., Immunol Lett 1996, 49, 185. [DOI] [PubMed] [Google Scholar]
- 95. Valdez F., Hernandez M., Govezensky T., Fragoso G., Sciutto E., J Parasitol 1994, 80, 931. [PubMed] [Google Scholar]
- 96. Tunback P., Liljeqvist J. A., Lowhagen G. B., Bergstrom, T. J Gen Virol 2000, 81, 1033. [DOI] [PubMed] [Google Scholar]
- 97. Strynadka N. C., Redmond M. J., Parker J. M., Scraba D. G., Hodges R. S., J Virol 1988, 62, 3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zalewska B., Piatek R., Konopa G., Nowicki B., Nowicki S., Kur J., Infect Immun 2003, 71, 5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Landini M. P., Ripalti A., Sra K., Pouletty P., J Clin Microbiol 1991, 29, 1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Franssila R., Hedman K., Vaccine 2004, 22, 3809. [DOI] [PubMed] [Google Scholar]
- 101. Corcoran A., Mahon B. P., Doyle S., J Infect Dis 2004, 189, 1873. [DOI] [PubMed] [Google Scholar]
- 102. Norrby E., Mufson M. A., Alexander H., Houghten R. A., Lerner R. A., Proc Natl Acad Sci U S A 1987, 84, 6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cane P. A., J Med Virol 1997, 51, 297. [DOI] [PubMed] [Google Scholar]
- 104. Leonov S. V., Waris M., Norrby E., J Gen Virol 1995, 76(Pt 2), 357. [DOI] [PubMed] [Google Scholar]
- 105. Leonova I. V., Leonov S. V., Waris M., Russi J. C., Grandien M., Norrby E., J Clin Virol 1998, 11, 137. [DOI] [PubMed] [Google Scholar]
- 106. Lal R. B., Buckner C., Khabbaz R. F., Kaplan J. E., Reyes G., Hadlock K., Lipka J., Foung S. K., Chan L., Coligan J. E., Clin Immunol Immunopathol 1993, 67, 40. [DOI] [PubMed] [Google Scholar]
- 107. Palker T. J., Scearce R. M., Copeland T. D., Oroszlan S., Haynes B. F., J Immunol 1986, 136, 2393. [PubMed] [Google Scholar]