

Abstract
Cutaneous hypersensitivity reactions in infants present in a variety of patterns. These skin eruptions can be dramatic, causing alarm in parents and medical personnel. Many of these syndromes have overlapping features, which adds to the confusion and uncertainty regarding diagnosis and management. This review discusses the spectrum of hypersensitivity responses with a focus on their presentation in infants. The clinical findings, pathophysiology, histopathology, management, and complications of these conditions will be reviewed.
Keywords: acute annular urticaria, acute generalized exanthematous pustulosis, acute hemorrhagic edema of infancy, annular erythema of infancy, childhood hypersensitivity reaction, drug reaction with eosinophilia and systemic symptoms, drug‐induced hypersensitivity syndrome, eosinophilic cellulitis, erythema multiforme, Henoch‐Schönlein purpura, Kawasaki disease, serum sickness‐like reaction, Stevens‐Johnson syndrome, Sweet syndrome, toxic epidermal necrolysis
1. INTRODUCTION
Hypersensitivity syndromes are a heterogeneous family of disorders mediated by immunologic mechanisms in response to foreign proteins, most commonly drugs and infectious agents. Four types of hypersensitivity reactions have been described: immunoglobulin E‐mediated (type I), cytotoxic or antibody‐mediated (type II), immune complex‐mediated (type III), and delayed‐type (type IV). Following sensitization during an initial exposure, re‐exposure to the antigen elicits an excessive immune response, resulting in the cutaneous and systemic signs and symptoms that characterize the hypersensitivity syndromes.1 An underlying genetic predisposition is theorized to increase an individual's risk to such processes. The presentation of these conditions can be distinct in the infant population. While many of these diseases are benign and self‐limited, they must be distinguished from disorders with systemic implications. Clinical presentations can often be characteristic; however, overlapping features of many of these conditions may pose a diagnostic dilemma. Dermatologists are critical members of the health care team with the expertise to recognize and differentiate these unusual syndromes. This case presentation and review provides an overview and update of the clinical findings, etiologies, histopathology, management, and complications of cutaneous hypersensitivity reactions in infants (Tables 1 and 2). A brief summary of severe cutaneous adverse drug reactions is included, but an exhaustive review of these conditions is outside of the scope of this review.
Table 1.
Summary of acute hemorrhagic edema of infancy, Henoch‐Schönlein purpura, Kawasaki disease, and Sweet syndrome
| Acute hemorrhagic edema of infancy | Henoch‐Schönlein purpura | Kawasaki disease | Sweet syndrome | |
|---|---|---|---|---|
| Typical age of onset | 6 mo to 2 y | 2‐6 y | 13‐24 mo | 30‐50 y |
| Appearance of lesions | Annular and targetoid, erythematous and purpuric plaques | Urticarial papules progressing to palpable purpura | Nonspecific; morbilliform, urticarial, or scarlatiniform morphologies | Tender, erythematous papules and plaques |
| Typical location | Face, ears, distal extremities | Lower extremities, buttocks | Diffuse | Limbs, face, neck |
| Duration of individual lesions | 1‐3 wk | 5 d | Several days | 3‐9 d (with treatment) |
| Duration of rash | 1‐3 wk | 4‐6 wk | Days to weeks | 3‐9 d (with treatment); recurrence common |
| Mucous membrane involvement | No | No | Yes—lip and tongue erythema | Rarely |
| Facial or acral edema | Yes | Yes | Yes | No |
| Fever | Variable | Variable | Yes | Variable |
| Associated signs/symptoms | Malaise, irritability | Malaise, arthralgias, abdominal pain | Nonexudative conjunctivitis, cervical lymphadenopathy | Malaise, myalgias, arthralgias |
| Inciting factors | Infection, medications, immunizations | Infection, medications, immunizations | Infection | Infection, immunodeficiency, medications, malignancy |
| Pathology | Leukocytoclastic vasculitis; direct immunofluorescence typically negative | Leukocytoclastic vasculitis; direct immunofluorescence may demonstrate IgA deposition | Vasculitis with inflammatory infiltrate of neutrophils and macrophages | Dermal edema, neutrophilic infiltrate, leukocytoclasis without vasculitis |
| Type of hypersensitivity reaction | Type III | Type III | Unknown | Type IV |
| Treatment | Supportive care | Organ‐directed support of renal, GI, joint manifestations | Intravenous immunoglobulin and aspirin | Systemic corticosteroids or potassium iodide |
| Risk for severe complications | Low | Moderate | High | High |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 2.
Summary of acute annular urticaria, serum sickness‐like reaction, annular erythema of infancy, and eosinophilic cellulitis
| Acute annular urticaria | Serum sickness‐like reaction | Annular erythema of infancy | Eosinophilic cellulitis | |
|---|---|---|---|---|
| Typical age of onset | 4 mo to 4 y | 18 mo to 16 y | Birth to 12 mo | Adulthood |
| Appearance of lesions | Polycyclic and annular edematous pink plaques, often with violaceous centers | Erythematous, annular, edematous, urticaria‐like plaques evolving to ecchymotic patches | Annular and arcuate erythematous patches | Erythematous, urticarial or cellulitis‐like plaques; morphology may vary |
| Typical location | Trunk, face, extremities | Trunk, face, extremities | Trunk, face, extremities | Trunk, extremities, rarely face |
| Duration of individual lesions | <24 h | 2‐3 wk | 36‐48 h | 2‐8 wk |
| Duration of rash | Days to weeks | Days to weeks | Episodic over months to years | 2‐8 wk; may recur |
| Mucous membrane involvement | No | No | No | No |
| Facial or acral edema | Yes | Yes | No | No |
| Fever | Variable | Variable | Variable | No |
| Associated signs/symptoms | Pruritus, dermatographism | Malaise, irritability lymphadenopathy, arthralgias, splenomegaly, refusal to walk | None | Prodrome of itching and burning |
| Inciting factors | Infection, medications, immunizations | Infection, medications, immunizations | Unknown | Infection, medications, immunizations, arthropod bite |
| Pathology | Dermal edema with sparse variable inflammatory infiltrate | Dermal edema with mixed inflammatory infiltrate without vasculitis | Perivascular lymphocytic and eosinophilic infiltrate | Diffuse dermal eosinophilic infiltrate with “flame figures” |
| Type of hypersensitivity reaction | Unknown | Type III | Unknown | Unknown |
| Treatment | Antihistamines; systemic corticosteroids if severe | NSAIDs and antihistamines; systemic corticosteroids if severe | None | Topical or systemic corticosteroids if severe |
| Risk for severe complications | Low | Low | Low | Low |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2. CASE REPORT
A 12‐month‐old previously healthy boy presented to the emergency department with a 1‐day history of periorbital edema, swelling and pain of the extremities, fever, and rash. The child had upper respiratory symptoms for several days preceding his presentation and had received his 12‐month vaccines the day prior.
On examination, the patient was irritable, tachycardic, and febrile to 39.4°C. Skin findings were notable for erythematous and purpuric round to oval annular and targetoid plaques involving the helix of the ears, cheeks, eyelids, back, distal extremities, and buttocks (Figure 1). Swelling of the penis, periorbital regions, and bilateral knees was also present. The left knee was erythematous and tender with limited range of motion. Laboratory evaluation was significant for elevated fibrinogen (18.4 μmol/L), C‐reactive protein (876.2 nmol/L), and D dimer (75.6 nmol/L); prolonged partial thromboplastin time (35.1 seconds) and international normalized ratio (1.2); and anemia (hemoglobin 9.5 g/dL). Respiratory virus panel was positive for coronavirus. Creatinine, hepatic transaminases, white blood cell count, platelets, and immunoglobulin A levels were within normal limits. Peripheral blood cultures were negative, and culture of left knee aspirate did not reveal bacterial growth. Imaging of the left knee showed soft tissue swelling and subcutaneous edema without underlying effusion or osseous abnormality.
Figure 1.
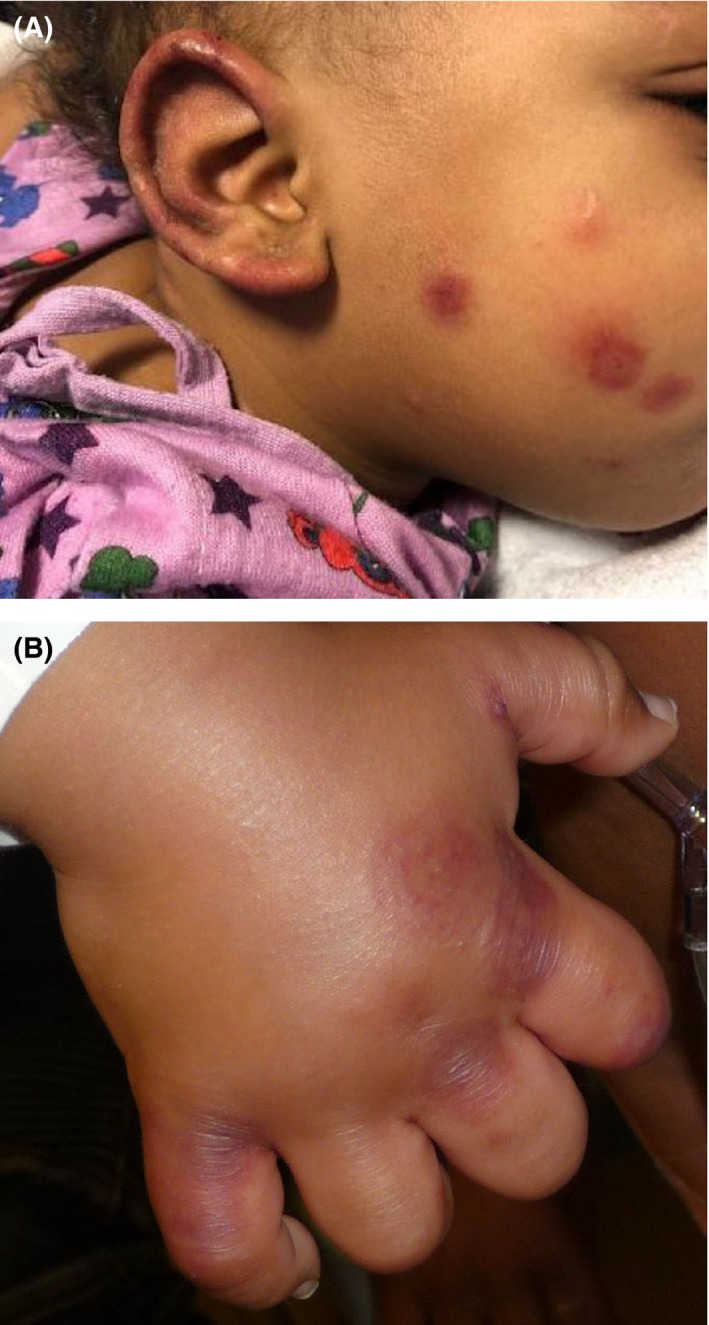
Acute hemorrhagic edema of infancy. A, Erythematous and purpuric annular plaques on the face and ear with associated edema. B, Erythematous and purpuric annular plaques on the hand with associated edema
The patient was diagnosed with acute hemorrhagic edema of infancy (AHEI) given his clinical presentation and history of associated upper respiratory infection and vaccinations. He was treated with prednisolone 2 mg/kg for 6 days due to his severe presentation. Symptoms had resolved by 2 weeks after initial presentation. Physical examination and laboratory abnormalities normalized at the 1‐month follow‐up evaluation.
3. ACUTE HEMORRHAGIC EDEMA OF INFANCY
Acute hemorrhagic edema of infancy is a rare cutaneous leukocytoclastic vasculitis that presents with purpura, edema, and frequently fever in children typically between 6 months and 2 years of age. Though the pathogenesis is not fully understood, AHEI is thought to be caused by an immune complex‐mediated (type III) hypersensitivity reaction in response to infection, vaccination, or medication intake.2 The cutaneous presentation is characterized by tender edema of the face and distal extremities, and annular‐, rosette‐, or targetoid‐shaped, erythematous and purpuric plaques favoring the cheeks, ears, and extremities. Despite the dramatic appearance and rapid onset of the rash, fever tends to be low grade and patients are often well appearing, though some are irritable. Extracutaneous manifestations are rare, but renal, gastrointestinal, and joint involvement has been reported.3 Laboratory findings in AHEI are generally nonspecific, though elevated inflammatory markers, leukocytosis, and eosinophilia may be present. Histopathology shows features of leukocytoclastic vasculitis with a mixed infiltrate of neutrophils and eosinophils; direct immunofluorescence studies are typically negative. AHEI resolves within several weeks with supportive care alone, and rarely recurs.4
4. HENOCH‐SCHÖNLEIN PURPURA
Henoch‐Schönlein purpura (HSP), the most common vasculitis of childhood, appears to be closely related to AHEI.5 HSP is an immune complex‐mediated (type III) hypersensitivity reaction associated with a variety of presumed but unconfirmed precipitants, including group A streptococcus infection.6 It classically presents with the tetrad of palpable purpura, joint pain, abdominal pain, and glomerulonephritis; the presence of fever is variable. The skin rash may be initially urticarial but rapidly becomes purpuric, preferentially affecting the buttocks and lower extremities (Figure 2). HSP primarily affects children between the ages of 2 and 6 years but has been reported as early as 8 months of age.7
Figure 2.
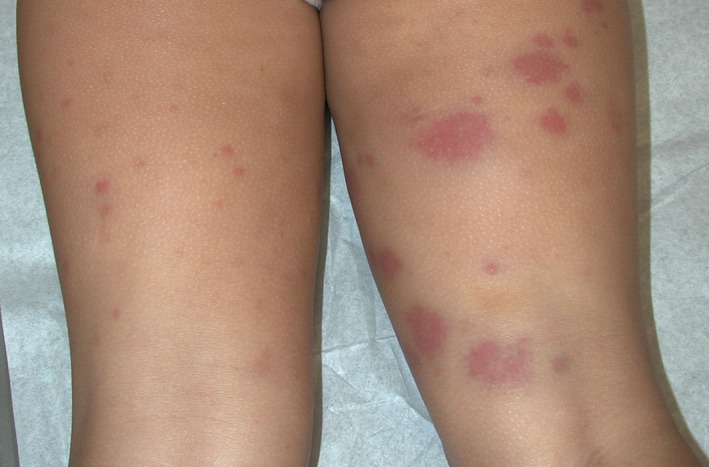
Henoch‐Schonlein purpura. Four‐year‐old girl with erythematous urticarial papules and purpuric plaques on posterior thighs
Systemic manifestations are common and include gastrointestinal symptoms, tender swollen joints, and glomerulonephritis. Less commonly, intussusception may occur, as bowel wall vasculitis can result in a pathologic lead point.8 Routine histology of HSP lesions demonstrates a leukocytoclastic vasculitis. Direct immunofluorescence frequently highlights IgA deposition in vessel walls and/or glomeruli, but this is not a necessary criterion for diagnosis. Serum IgA levels may be elevated in up to one half of patients.9 The typical course of HSP is self‐limited, resolving within 8 weeks.6 HSP appears to be milder in infants, with less systemic involvement and excellent prognosis; this group more commonly presents with edema and facial purpura.7 The goals of management are to minimize symptoms, decrease short‐term complications, and prevent chronic renal insufficiency. Multidisciplinary care in conjunction with nephrology and rheumatology is indicated to optimally manage end‐organ disease.6
5. KAWASAKI DISEASE
Like HSP, Kawasaki disease (KD) is a pediatric vasculitis with frequent cutaneous and systemic manifestations. KD likely represents an immune reaction to an infectious agent in a genetically susceptible individual. Multiple infectious etiologies have been implicated, including Epstein‐Barr virus, Mycoplasma pneumoniae, and varicella zoster virus. The incidence of KD is higher in some ethnic groups, such as Japanese, Chinese, and Koreans, supporting a genetic susceptibility. KD peaks in incidence between 13 and 24 months of age. Diagnosis requires fever for at least 5 days accompanied by four of the following signs: bilateral conjunctivitis, oral mucous membrane changes, peripheral extremity changes, polymorphous rash, and cervical lymphadenopathy. Acral findings may include erythema, edema, and desquamation (typically delayed), and oral involvement is characterized by mucosal erythema, swollen or fissured lips, and a prominent red tongue. Features of the generalized rash are highly variable, including urticarial, scarlatiniform, morbilliform, and dermatitic morphologies (Figure 3).10 Atypical KD and incomplete KD, forms that are more common in infants, may deviate from the classic presentation, thus requiring a high index of suspicion in this population.11
Figure 3.
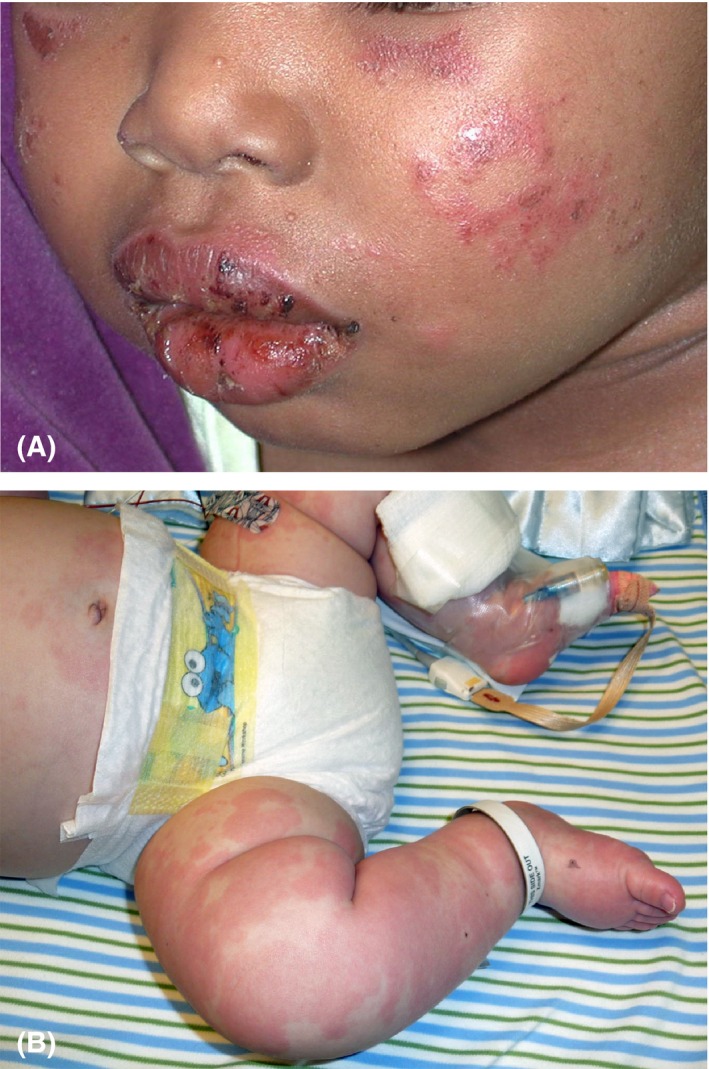
Kawasaki disease. A, Two‐year‐old girl with erythema, edema, erosions, and crusting of the lips and arcuate erythematous scaly plaques on the cheeks. B, Three‐month‐old infant with erythematous geographic urticarial plaques over trunk and extremities with acral edema
Laboratory abnormalities, including leukocytosis, anemia, thrombocytosis, and elevated acute phase reactants, may herald systemic involvement. The most feared complication of KD is coronary artery aneurysm; this may occur in up to 20% of patients in the absence of therapy, and significant risk remains even with treatment.12 Aspirin and intravenous immunoglobulin (IVIG) are the mainstays of therapy and should be promptly initiated.13 Compared to older patients, infants are more likely to develop coronary artery abnormalities and less likely to respond to IVIG, warranting a low threshold for aggressive treatment.11
6. SWEET SYNDROME
Sweet syndrome, a neutrophilic dermatosis more commonly described in the adult population, rarely can present in infants with the acute onset of tender, erythematous papules, and plaques often associated with fever. The earliest reported cases are in the first month of life. Sweet syndrome is thought to be a delayed‐type (type IV) hypersensitivity reaction to infection, medications, or malignancy; in the infant population, bacterial or viral upper respiratory infection has most commonly been associated. Rarely, Sweet syndrome may be a sign of immunodeficiency or manifestation of autoinflammatory disease.14, 15
Histopathology in Sweet syndrome demonstrates dermal edema, a predominantly neutrophilic infiltrate, and leukocytoclasis without true vasculitis.14 Diagnosis is based upon satisfying the major criteria of clinical presentation and histopathology, with at least two supporting minor criteria, which include the presence of respiratory or gastrointestinal infection, history of preceding vaccination, or concomitant inflammatory disease, malignancy or pregnancy; malaise and fever; elevated acute phase reactants such as erythrocyte sedimentation rate and C‐reactive protein, peripheral blood neutrophilia and leukocytosis; and response to systemic corticosteroid or potassium iodide treatment. The rapid improvement of symptoms upon initiation of systemic corticosteroids is strongly supportive of the diagnosis.16 Cardiac manifestations are a significant cause of morbidity, including pericarditis, dilation of the great vessels, valvular disease, and myocardial infarction. The mortality rate is 9% in the general pediatric population, though even greater in infants and those with cardiac involvement.14
7. ERYTHEMA MULTIFORME
Erythema multiforme (EM) is characterized by acute onset of symmetrically distributed targetoid skin lesions with variable mucosal involvement. EM is rare in infants, but has been reported as early as the first few months of life.17 The pathogenesis involves a virally induced cell‐mediated immune reaction in genetically susceptible individuals; herpes simplex virus (HSV) being the most frequent precipitant. Skin lesions initially appear as erythematous to violaceous macules and evolve to the classic targetoid plaque composed of a peripheral ring of erythema, a zone of edema, and central duskiness heralding epidermal necrosis, which may progress to vesiculation or blistering (Figure 4). Mucosal involvement when present predominantly affects the lips, tongue, and buccal mucosa, with fragile bullae quickly unroofing to reveal tender erosions and crusting of the lips.18
Figure 4.
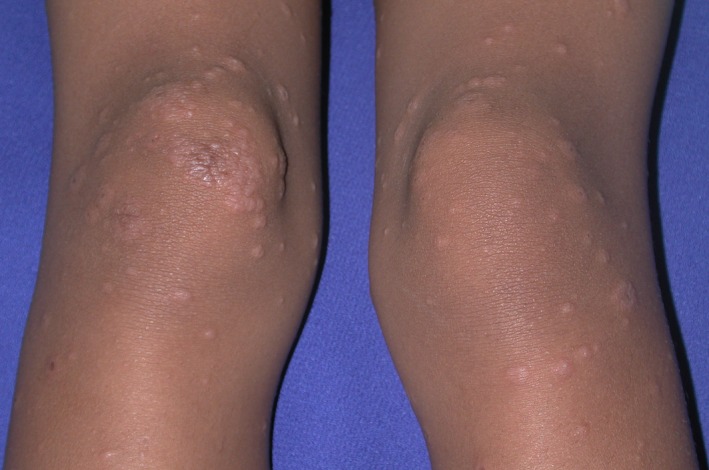
Erythema multiforme. Four‐year‐old girl with symmetric acral edematous papules with central vesiculation and peripheral rim of erythema
Constitutional symptoms are typically limited to generalized malaise, low‐grade fever, and discomfort associated with the eruption. Histopathologic features include an interface dermatitis with degeneration of basal epidermal cells, keratinocyte necrosis, and a superficial dermal perivascular lymphohistiocytic infiltrate.18 Most episodes of EM heal over 2‐3 weeks, but recurrences can become cyclical in a small subset of patients; rare instances of this phenomenon have been described in infants.19 Management includes a careful assessment for systemic infection and supportive measures for pain, poor oral intake, and wound care. Prophylactic therapy with systemic antiviral medication should be considered for patients with HSV‐associated recurrent EM.18
8. URTICARIAL AND URTICARIA‐LIKE HYPERSENSITIVITY SYNDROMES
A subset of the cutaneous hypersensitivity syndromes can be grouped by their shared presentation with urticarial or urticaria‐like lesions. Urticaria, commonly known as hives, is an IgE‐dependent cutaneous vascular reaction characterized by dermal edema (wheal) surrounded by erythema (flare). The lesions are typically well‐defined round, annular or geographical shapes and are often pruritic. Individual papules and plaques characteristically resolve within 24 hours, but new lesions may continue to develop. Urticaria‐like lesions manifest similarly to urticaria but persist longer than 24 hours. Acute annular urticaria, serum sickness‐like reaction, annular erythema of infancy, and eosinophilic cellulitis are reviewed below.
8.1. Acute annular urticaria
Acute annular urticaria (AAU), also referred to as urticaria multiforme, describes a distinct subtype of acute urticaria noted most commonly in infants and preschool‐aged children. This entity can be mistaken for EM based on the dramatically annular configuration of the lesions. AAU is thought to result from an allergic hypersensitivity reaction that may be IgE dependent or independent, in association with infection, medication, or immunization.20 Patients present with polycyclic and annular edematous pink plaques often with violaceous centers, affecting the trunk, face, and extremities (Figure 5). Edema of the face, hands, and feet is particularly prominent in infants. Patients usually complain of itch, and the condition may be accompanied by fever or dermatographism. As with typical urticaria, individual skin lesions tend to resolve within 24 hours, while there may be ongoing eruption of new lesions for 1‐2 weeks. Histology is similar to that of acute urticaria, demonstrating dermal edema and sparse perivascular and interstitial infiltrate of neutrophils and eosinophils. AAU can be differentiated from EM by the presence of pruritus rather than pain, the absence of skin necrosis and blistering, and transient nature of individual lesions.20 Management of the eruption is achieved with systemic antihistamines; evaluation for a precipitating infectious source is warranted and any implicated medications should be discontinued. Corticosteroids are generally reserved for severe or refractory cases.21
Figure 5.
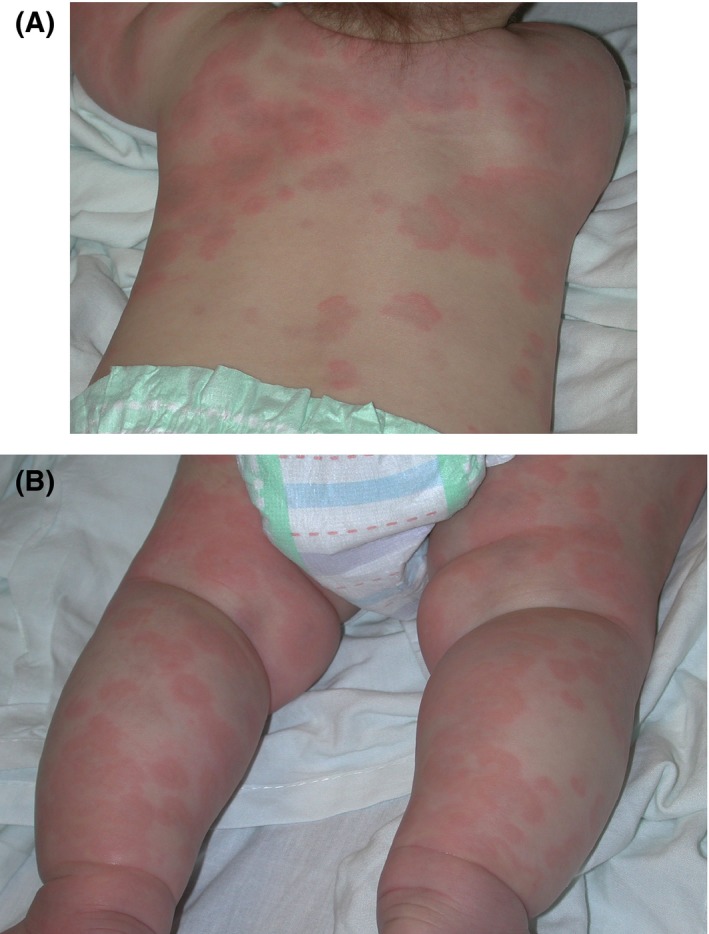
Acute annular urticaria. A, Five‐month‐old boy with polycyclic and annular erythematous plaques with violaceous centers. B, Five‐month‐old boy with polycyclic and annular erythematous plaques with violaceous centers and acral edema
8.2. Serum sickness‐like reaction
Serum sickness‐like reaction (SSLR) falls within the spectrum of urticaria‐like eruptions, and unlike classic serum sickness is not a true vasculitis.22 An immune complex‐mediated (type III) hypersensitivity reaction originally described in the setting of exposure to the cephalosporin cefaclor, SSLR has also been linked to other medications, viral infections, and vaccinations.23, 24 The eruption is composed of erythematous, annular, edematous plaques similar to that of urticaria, though the plaques generally evolve to a lilac or ecchymotic hue which may persist for days to weeks, giving rise to the term “purple urticaria” (Figure 6). These patients also frequently present with acral edema, lymphadenopathy, and arthralgias, often refusing to walk; the presence of fever is also variable.22 The notable irritability of these patients, the absence of pruritus, and the fixed nature of individual lesions distinguishes this entity from AAU. SSLR can present at any age, with multiple cases reported in patients younger than 2 years.23
Figure 6.
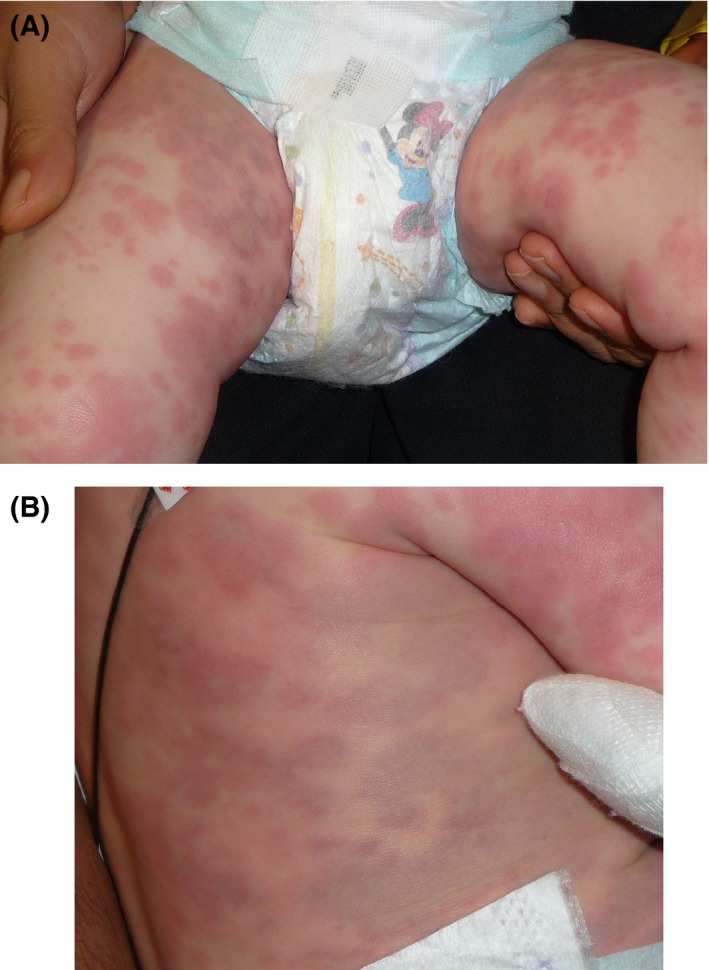
Serum sickness‐like reaction. A, One‐year‐old boy with erythematous edematous papules and plaques evolving to ecchymotic “purple urticaria” on the extremities. B, One‐year‐old boy with erythematous edematous papules and plaques evolving to ecchymotic “purple urticaria” on the trunk
Laboratory findings may include leukocytosis, elevated erythrocyte sedimentation rate, and mild proteinuria or hematuria; unlike true serum sickness, complement levels are normal.25 Histopathology is marked by a mixed dermal inflammatory infiltrate and edema without vasculitis.22 SSLR is managed with discontinuation of any predisposing medications with gradual resolution of symptoms over weeks to months. Supportive care with antihistamines and pain management is generally sufficient; however, systemic corticosteroids may be considered if symptoms are severe.25
8.3. Annular erythema of infancy
Annular erythema of infancy (AEI) is a condition exclusive to the infant population. It is characterized by cyclic, asymptomatic, self‐limited eruptions of annular, and arcuate erythematous patches. Individual lesions resolve after 36‐48 hours without scarring.26 Histopathology reveals a moderate perivascular lymphocytic and eosinophilic infiltrate.27 Etiology is poorly understood, though one case of AEI associated with intestinal Candida colonization has been reported.28 Spontaneous resolution of the episodic flares has been observed over 1 to 2 years.27 AEI can be distinguished from AAU by the longer duration of individual lesions, variable fever, and lack of edema or pruritus.
8.4. Eosinophilic cellulitis
Eosinophilic cellulitis, or Wells syndrome, is characterized by a prodrome of itching or burning followed by an eruption of erythematous, urticarial or cellulitis‐like plaques on the trunk and extremities, and rarely face (Figure 7).29 The etiology is unconfirmed but may involve a hypersensitivity reaction to infection, medication exposure, vaccination, or arthropod bite, resulting in eosinophil degranulation.30 The preponderance of cases occurs in adults, but rare cases have been reported in infants.31 Congenital Wells syndrome has also been described, presenting as subcutaneous nodules shortly after birth with the more classic lesions appearing after several months.32 Histopathology is characteristic, demonstrating a diffuse dermal eosinophilic infiltrate with the presence of degranulating eosinophils (flame figures). Peripheral eosinophilia may also be present.31 The acute lesions of Wells syndrome fade over the course of several weeks, often replaced by atrophy and hyperpigmentation. Recurrence is common, but overall prognosis is favorable. Treatment of the precipitating factor may be sufficient, but oral and/or topical corticosteroids may be additionally necessary.31
Figure 7.
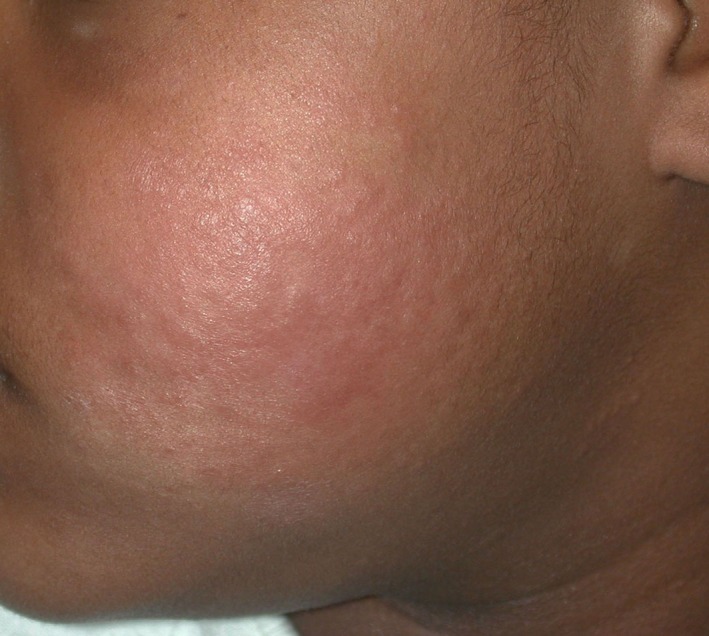
Eosinophilic cellulitis. Ill‐defined erythematous edematous firm plaque on the cheek
9. SEVERE CUTANEOUS ADVERSE DRUG REACTIONS
9.1. Stevens‐Johnson syndrome/toxic epidermal necrolysis
Stevens‐Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are the hypersensitivity syndromes with the most severe cutaneous manifestations, existing on a spectrum characterized by extent of epidermal necrosis.33 These conditions are extremely rare in infants.34 In this population, SJS is the more common presentation and most often occurs in the setting of antiepileptic medications.35 Skin findings may include targetoid lesions, dusky erythematous macules, and bullae; epidermal detachment may be spontaneous or elicited by lateral pressure.33 Involvement of two or more mucosal surfaces is a defining feature. Necrotic keratinocytes are the primary finding on histopathology.36 Beyond the cutaneous and mucosal manifestations, the risk of systemic dysfunction, infection, malnutrition, and pain are prominent. Long‐term complications include ocular sequelae, dental effects, genitourinary scarring, and pulmonary disease.37 The first step in management consists of discontinuing the offending drug and providing intensive multisystem supportive care, typically best accomplished in an intensive care or burn unit. Several small retrospective studies have failed to show benefit from systemic corticosteroids or intravenous immunoglobulin in children with SJS/TEN.38, 39 Cyclosporine and tumor necrosis factor inhibitors have shown promise in case reports and preliminary studies in adults.40
Diffuse exfoliating rash, extensive mucosal involvement, and internal organ injury differentiate SJS/TEN from the other cutaneous hypersensitivity reactions discussed above.
9.2. Drug‐induced hypersensitivity syndrome
Drug‐induced hypersensitivity syndrome (DIHS), also known as drug reaction with eosinophilia and systemic symptoms (DRESS), presents as a rash in the setting of fever, eosinophilia, and internal organ involvement. In the infant population, DIHS/DRESS occurs almost exclusively in the context of antiepileptic therapy.41 Skin findings are characterized by facial edema and a pruritic, diffuse rash of variable morphology including morbilliform, pustular, scarlatiniform, urticarial, targetoid, or full erythroderma; mucosal involvement is typically mild, in contrast to SJS/TEN.42 The histopathology of DIHS/DRESS is nonspecific.43 Visceral manifestations appear 2‐6 weeks after onset, with hepatitis being the most common, and thyroid dysfunction presenting as a later sequela.44 In addition to discontinuation of the inciting medication, systemic corticosteroids historically have comprised the mainstay of treatment, but recent work suggests topical steroids may be effective and sufficient in patients with limited disease.45
9.3. Acute generalized exanthematous pustulosis
Acute generalized exanthematous pustulosis (AGEP) is the eruption of numerous small sterile pustules on a background of diffuse erythema, typically in the setting of fever, and rarely accompanied by systemic symptoms. The pustules resolve with prominent desquamation.46 Very few cases have been reported in infants and pustular psoriasis should be strongly considered as an alternative diagnosis. AGEP may be precipitated by medications (most commonly antibiotics), infection, or vaccination.47 Histopathology is characterized by neutrophil exocytosis, pustules, papillary dermal edema, and a neutrophilic and eosinophilic dermal infiltrate.46 Management consists of removal of the offending drug and supportive care, with resolution in 4‐10 days. Topical steroid therapy has also been reported as beneficial in affected infants.47
10. SUMMARY
This review of cutaneous hypersensitivity reactions as they affect infants illustrates the breadth of conditions and diversity of presentations considered within this category. As there is much overlap in clinical presentation, accurate diagnosis is facilitated by recognizing the similarities and differences between these entities. While many of these conditions are benign and self‐limited, others may have serious consequences, and early diagnosis is essential to improving outcomes. Evaluation for an inciting infection should be undertaken when appropriate, given the relatively immunocompromised state of young infants and the likelihood that many of these entities may be triggered by infectious agents. However, the adverse effects of overly aggressive systemic therapy may outweigh more limited potential benefits, and accurate diagnosis will spare unnecessary investigations and treatment. The dermatologist will play a critical role in providing the expertise necessary to recognize these rare syndromes and confirming uncertain diagnoses via skin biopsy when indicated. Further work remains to be done, particularly in the infant population, to better understand pathophysiology and optimize management of these rare conditions.
Arnold KA, Gao J, Stein SL. A review of cutaneous hypersensitivity reactions in infants: From common to concerning. Pediatr Dermatol. 2019;36:274–282. 10.1111/pde.13827
This study conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
REFERENCES
- 1. Noguera‐Morel L, Hernández‐Martín Á, Torrelo A. Cutaneous drug reactions in the pediatric population. Pediatr Clin North Am. 2014;61(2):403‐426. [DOI] [PubMed] [Google Scholar]
- 2. Savino F, Lupica MM, Tarasco V, et al. Acute hemorrhagic edema of infancy: a troubling cutaneous presentation with a self‐limiting course. Pediatr Dermatol. 2013;30(6):e149‐e152. [DOI] [PubMed] [Google Scholar]
- 3. Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59(4):684‐695. [DOI] [PubMed] [Google Scholar]
- 4. Krause I, Lazarov A, Rachmel A, et al. Acute haemorrhagic oedema of infancy, a benign variant of leucocytoclastic vasculitis. Acta Paediatr. 1996;85(1):114‐117. [DOI] [PubMed] [Google Scholar]
- 5. Saraclar Y, Tinaztepe K, Adalioğlu G, Tuncer A. Acute hemorrhagic edema of infancy (AHEI)–a variant of Henoch‐Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86(4 Pt 1):473‐483. [DOI] [PubMed] [Google Scholar]
- 6. McCarthy HJ, Tizard EJ. Clinical practice: diagnosis and management of Henoch‐Schönlein purpura. Eur J Pediatr. 2010;169(6):643‐650. [DOI] [PubMed] [Google Scholar]
- 7. Al‐Sheyyab M, El‐Shanti H, Ajlouni S, Sawalha D, Daoud A. The clinical spectrum of Henoch‐Schönlein purpura in infants and young children. Eur J Pediatr. 1995;154(12):969‐972. [DOI] [PubMed] [Google Scholar]
- 8. Yang Y‐H, Yu H‐H, Chiang B‐L. The diagnosis and classification of Henoch‐Schönlein purpura: an updated review. Autoimmun Rev. 2014;13(4–5):355‐358. [DOI] [PubMed] [Google Scholar]
- 9. Reid‐Adam J. Henoch‐Schonlein purpura. Pediatr Rev. 2014;35(10):447‐449; discussion 449. [DOI] [PubMed] [Google Scholar]
- 10. Bayers S, Shulman ST, Paller AS. Kawasaki disease: part I. Diagnosis, clinical features, and pathogenesis. J Am Acad Dermatol. 2013;69(4):501.e1‐501.e11; quiz 511‐512. [DOI] [PubMed] [Google Scholar]
- 11. Manlhiot C, Yeung RSM, Clarizia NA, Chahal N, McCrindle BW. Kawasaki disease at the extremes of the age spectrum. Pediatrics. 2009;124(3):e410‐e415. [DOI] [PubMed] [Google Scholar]
- 12. Scuccimarri R. Kawasaki disease. Pediatr Clin North Am. 2012;59(2):425‐445. [DOI] [PubMed] [Google Scholar]
- 13. Bayers S, Shulman ST, Paller AS. Kawasaki disease: part II. Complications and treatment. J Am Acad Dermatol. 2013;69(4):513.e1‐513.e8; quiz 521‐522. [DOI] [PubMed] [Google Scholar]
- 14. Halpern J, Salim A. Pediatric sweet syndrome: case report and literature review. Pediatr Dermatol. 2009;26(4):452‐457. [DOI] [PubMed] [Google Scholar]
- 15. Satoh TK, Mellett M, Contassot E, French LE. Are neutrophilic dermatoses autoinflammatory disorders? Br J Dermatol. 2018;178(3):603‐613. [DOI] [PubMed] [Google Scholar]
- 16. von den Driesch P. Sweet's syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31(4):535‐556; quiz 557‐560. [DOI] [PubMed] [Google Scholar]
- 17. Siedner‐Weintraub Y, Gross I, David A, Reif S, Molho‐Pessach V. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2017;97(4):489‐492. [DOI] [PubMed] [Google Scholar]
- 18. Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51(8):889‐902. [DOI] [PubMed] [Google Scholar]
- 19. Heinze A, Tollefson M, Holland KE, Chiu YE. Characteristics of pediatric recurrent erythema multiforme. Pediatr Dermatol. 2018;35(1):97‐103. [DOI] [PubMed] [Google Scholar]
- 20. Shah KN, Honig PJ, Yan AC. “Urticaria multiforme”: a case series and review of acute annular urticarial hypersensitivity syndromes in children. Pediatrics. 2007;119(5):e1177‐e1183. [DOI] [PubMed] [Google Scholar]
- 21. Starnes L, Patel T, Skinner RB. Urticaria multiforme–a case report. Pediatr Dermatol. 2011;28(4):436‐438. [DOI] [PubMed] [Google Scholar]
- 22. Tolpinrud WL, Bunick CG, King BA. Serum sickness‐like reaction: histopathology and case report. J Am Acad Dermatol. 2011;65(3):e83‐e85. [DOI] [PubMed] [Google Scholar]
- 23. Serum sickness–like reaction in children: a retrospective review ‐ Journal of the American Academy of Dermatology. http://www.jaad.org/article/S0190-9622(08)01478-3/abstract. Accessed February 19, 2018.
- 24. Chiong FJK, Loewenthal M, Boyle M, Attia J. Serum sickness‐like reaction after influenza vaccination. BMJ Case Rep. 2015;2015:bcr2015211917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yorulmaz A, Akın F, Sert A, Ağır MA, Yılmaz R, Arslan Ş. Demographic and clinical characteristics of patients with serum sickness‐like reaction. Clin Rheumatol. 2018;37(5):1389‐1394. [DOI] [PubMed] [Google Scholar]
- 26. Peterson AO, Jarratt M. Annular erythema of infancy. Arch Dermatol. 1981;117(3):145‐148. [PubMed] [Google Scholar]
- 27. Hebert AA, Esterly NB. Annular erythema of infancy. J Am Acad Dermatol. 1986;14(2 Pt 2):339‐343. [DOI] [PubMed] [Google Scholar]
- 28. Stachowitz S, Abeck D, Schmidt T, Ring J. Persistent annular erythema of infancy associated with intestinal Candida colonization. Clin Exp Dermatol. 2000;25(5):404‐405. [DOI] [PubMed] [Google Scholar]
- 29. Kuwahara RT, Randall MB, Eisner MG. Eosinophilic cellulitis in a newborn. Pediatr Dermatol. 2001;18(1):89‐90. [DOI] [PubMed] [Google Scholar]
- 30. Wells GC, Smith NP. Eosinophilic cellulitis. Br J Dermatol. 1979;100(1):101‐109. [DOI] [PubMed] [Google Scholar]
- 31. Gilliam AE, Bruckner AL, Howard RM, Lee BP, Wu S, Frieden IJ. Bullous “cellulitis” with eosinophilia: case report and review of Wells’ syndrome in childhood. Pediatrics. 2005;116(1):e149‐e155. [DOI] [PubMed] [Google Scholar]
- 32. Garty BZ, Feinmesser M, David M, Gayer S, Danon YL. Congenital Wells syndrome. Pediatr Dermatol. 1997;14(4):312‐315. [DOI] [PubMed] [Google Scholar]
- 33. Dodiuk‐Gad RP, Chung W‐H, Valeyrie‐Allanore L, Shear NH. Stevens‐Johnson syndrome and toxic epidermal necrolysis: an update. Am J Clin Dermatol. 2015;16(6):475‐493. [DOI] [PubMed] [Google Scholar]
- 34. Raucci U, Rossi R, Da Cas R, et al. Stevens‐Johnson syndrome associated with drugs and vaccines in children: a case‐control study. PLoS ONE. 2013;8(7):e68231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Finkelstein Y, Macdonald EM, Li P, Hutson JR, Juurlink DN. Recurrence and mortality following severe cutaneous adverse reactions. JAMA. 2014;311(21):2231‐2232. [DOI] [PubMed] [Google Scholar]
- 36. Rzany B, Hering O, Mockenhaupt M, et al. Histopathological and epidemiological characteristics of patients with erythema exudativum multiforme major, Stevens‐Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 1996;135(1):6‐11. [PubMed] [Google Scholar]
- 37. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69(2):187.e1‐187.e16; quiz 203‐204. [DOI] [PubMed] [Google Scholar]
- 38. Koh MJ‐A, Tay Y‐K. Stevens‐Johnson syndrome and toxic epidermal necrolysis in Asian children. J Am Acad Dermatol 2010;62(1):54‐60. [DOI] [PubMed] [Google Scholar]
- 39. Dore J, Salisbury RE. Morbidity and mortality of mucocutaneous diseases in the pediatric population at a tertiary care center. J Burn Care Res. 2007;28(6):865‐870. [DOI] [PubMed] [Google Scholar]
- 40. Schneider JA, Cohen PR. Stevens‐Johnson syndrome and toxic epidermal necrolysis: a concise review with a comprehensive summary of therapeutic interventions emphasizing supportive measures. Adv Ther. 2017;34(6):1235‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carroll MC, Yueng‐Yue KA, Esterly NB, Drolet BA. Drug‐induced hypersensitivity syndrome in pediatric patients. Pediatrics. 2001;108(2):485‐492. [DOI] [PubMed] [Google Scholar]
- 42. Kardaun SH, Sekula P, Valeyrie‐Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169(5):1071‐1080. [DOI] [PubMed] [Google Scholar]
- 43. Gonçalo MM, Cardoso JC, Gouveia MP, et al. Histopathology of the exanthema in DRESS is not specific but may indicate severity of systemic involvement. Am J Dermatopathol. 2016;38(6):423‐433. [DOI] [PubMed] [Google Scholar]
- 44. Avancini J, Maragno L, Santi CG, Criado PR. Drug reaction with eosinophilia and systemic symptoms/drug‐induced hypersensitivity syndrome: clinical features of 27 patients. Clin Exp Dermatol. 2015;40(8):851‐859. [DOI] [PubMed] [Google Scholar]
- 45. Funck‐Brentano E, Duong T‐A, Bouvresse S, et al. Therapeutic management of DRESS: a retrospective study of 38 cases. J Am Acad Dermatol. 2015;72(2):246‐252. [DOI] [PubMed] [Google Scholar]
- 46. Alniemi DT, Wetter DA, Bridges AG, et al. Acute generalized exanthematous pustulosis: clinical characteristics, etiologic associations, treatments, and outcomes in a series of 28 patients at Mayo Clinic, 1996‐2013. Int J Dermatol. 2017;56(4):405‐414. [DOI] [PubMed] [Google Scholar]
- 47. Ersoy S, Paller AS, Mancini AJ. Acute generalized exanthematous pustulosis in children. Arch Dermatol. 2004;140(9):1172‐1173. [DOI] [PubMed] [Google Scholar]