

Abstract
Murray Valley encephalitis virus is a member of the flavivirus group, a large family of single‐stranded RNA viruses, which cause serious disease in all regions of the world. Unfortunately, no suitable antivirals are available, and there are commercial vaccines for only three flaviviruses. The solid‐phase synthesis of a library of 400 C‐terminal arginine peptide aldehydes and their screening against Murray Valley encephalitis virus protease are demonstrated. The library was utilised to elucidate several tripeptide sequences that can be used as inhibitors in further SAR studies. Copyright © 2012 European Peptide Society and John Wiley & Sons, Ltd.
Keywords: Murray Valley encephalitis virus (MVEV), peptide aldehyde, protease inhibitors, solid‐phase peptide synthesis, SynPhase lanterns
The solid‐phase synthesis of a library of 400 C‐terminal arginine peptide aldehydes and their screening against Murray Valley encephalitis virus protease are demonstrated. The library was utilised to elucidate several tripeptide sequences that can be used as inhibitors in further SAR studies.

Introduction
C‐terminal peptide aldehydes are important tools for studying serine and cysteine proteases 1, 2, 3. Proteases are involved in the regulation of a wide variety of essential physiological processes, and their dysregulation has been implicated in a number of disorders including cardiovascular disease, rheumatoid arthritis, Alzheimer's disease and cancer, to name only a few. Hence, proteases and their substrates are increasingly viewed as valuable drug targets in disease treatment. We have previously developed a robust and reliable synthetic method for the solid‐phase synthesis of peptide aldehyde protease inhibitors 4. This method was used to synthesise a small library of aldehydes and used to probe the proteolytic sites of hepatitis C virus polyprotein.
Murray Valley encephalitis virus (MVEV) is a member of the flavivirus group, a large family of single‐stranded RNA viruses, which cause serious disease in all regions of the world. Unfortunately, no suitable antivirals are available, and there are commercial vaccines for only three flaviviruses 5. MVEV has been isolated from mosquitoes in Australia and Papua New Guinea and, like West Nile virus, is grouped in the Japanese encephalitis serocomplex 6. Its genome encodes a large polyprotein that is processed by both host proteinases and a virally encoded serine proteinase, non‐structural protein 3 (NS3). NS3, an essential viral enzyme, requires another virally encoded protein cofactor, NS2B, for proteolytic activity 7.
Because the protease is likely to be essential for viral replication, it is an attractive target for inhibitor development. This paper outlines our synthetic method and strategy for the solid‐phase synthesis of a 400 member tripeptide library of C‐terminal arginine peptide aldehydes resulting in a (20 × 20 × 1) combinatorial library (P3, P2 = 20 natural AAs, P1 is fixed as arginine). Arginine is a preferred P1 cleavage site amino acid for many serine and cysteine proteases such as trypsin and thrombin 1, dengue virus NS3 protease 8 and MVEV.
Materials and Methods
Peptides were synthesised on SynPhaseTM‐PA D Series lanterns (loading 15 µmol/lantern), available from Mimotopes Pty Ltd (Melbourne, Australia). These supports had been radiation grafted with methacrylic acid/dimethylacrylamide (polyamide) and functionalized with Fmoc‐Gly. Fmoc‐ l‐α‐amino acids were peptide synthesis grade and purchased from Genzyme Pharmaceuticals (Leistal, Switzerland). HOBt was purchased from GL Biochem (Shanghai, China). THF was Sure‐Seal grade and was obtained from Sigma‐Aldrich (Sydney, Australia). DMF was Guaranteed Reagent (GR) grade and was obtained from Merck (Melbourne, Australia). It was distilled over ninhydrin before further use. DCM, MeOH, triethylamine and piperidine were GR grade and were obtained from Merck. Acetonitrile was HPLC grade and was obtained from Merck. Water was Milli‐Q grade (Merck Millipore, Melbourne, Australia). TFA was BiogradeTM and was obtained from Halocarbon (River Edge, NJ, USA). DIPEA was obtained from Fluka (Sydney, Australia). DIC was obtained from Sigma‐Aldrich.
Mass spectral analysis was performed on a Perkin Elmer Sciex API III (Waltham, MA, USA) ion spray mass spectrometer. The data were processed by software developed at Mimotopes Pty. Ltd. 9.
Synthesis of the protected amino acid aldehyde Fmoc‐Arg(Pbf)‐H was achieved via reduction of the corresponding N,O‐dimethylhydroxamate with LiAlH4 as previously described 4.
Preparation of Support‐Bound Threonine Aldehyde Linker 1
To a glass bottle (500 ml), 700 SynPhaseTM lanterns (loading 15 µmol/lantern) derivatised with Fmoc‐Gly‐OH were added. The Fmoc group was removed with 20% piperidine/DMF (400 ml) for 30 min. The lanterns were washed (all washes with lanterns were 5 min) with DMF (2 × 400 ml), MeOH (2 × 400 ml) and air dried. To the bottle containing the lanterns, a solution of Fmoc‐Thr‐OH (13.64 g, 40 mmol), DIC (6.08 ml, 40 mmol) and HOBt (6.12 g, 40 mmol) in DMF (400 ml), 0.1 M or 6–7 equivalents, was added. The reaction was allowed to proceed for 2 h at room temperature after which the reaction mixture was drained of solution, and the lanterns were washed with DMF (2 × 400 ml). To the washed lanterns, a solution of 20% piperidine in DMF (400 ml) was added, and the reaction was allowed to proceed for 30 min at room temperature. The reaction mixture was drained of solution, and the lanterns were washed with DMF (3 × 400 ml), MeOH (2 × 400 ml) and air dried to yield lanterns derivatized with threonine aldehyde linker 1.
Synthesis of Support‐Bound Fmoc‐Arg(Pbf)‐H Oxazolidine Aldehyde Linker 2
Lanterns (500) 1 were placed into sealable glass bottle. The aldehyde Fmoc‐Arg(Pbf)‐H (15.48 g, 24.46 mmol) was dissolved in a 1% DIPEA/MeOH solution (250 ml) to give a 0.1‐ m solution. This solution was added to the lanterns and the mixture heated at 60 °C for 2 h (with occasional swirling). The reaction mixture was drained of solution and the lanterns washed with MeOH (3 × 250 ml). The yield of attachment of Fmoc‐Arg(Pbf)‐H to the threonine linker can be determined spectrophotometrically by cleaving the Fmoc protecting group with 20% piperidine in DMF and measuring absorbance at 301 nm. A single lantern was treated with 20% piperidine/DMF (10 ml) for 30 min at 20 °C, and the absorbance of the solution was determined at 301 nm after diluting the deprotection solution 1 : 10 in 20% piperidine/DMF and using the following equation: loading (nmol/crown) = (Abs/0.0078) × 110. The loading was determined to be 47% (7.0 µmol).
Peptide Synthesis Protocol
The lanterns (400 for library and 10 for controls) were subjected to Fmoc peptide synthesis using the following conditions. All reactions utilising lanterns require 0.5 ml of solution per lantern, either when reacted in bulk or in the microtitre plate format. (i) Fmoc deprotection: 20% piperidine in DMF for 30 min, followed by washing with DMF (3 × 3 min) and DCM (2× 3 min). (ii) Coupling conditions: Fmoc‐protected amino acid, DIC, HOBt in DMF (0.1 m), 2 h at 25 °C. Following all couplings, the lanterns were washed with DMF (2 × 3 min), DCM (2 × 3 min) and air dried. Acetylation of the N‐terminus was achieved with acetic anhydride : DIPEA : DMF (ratio 3 : 1 : 96 v:v:v) at 4 °C for 5 min only. The lanterns were washed with DMF (2 × 30 s), DMF (2× 3 min), DCM (2 × 3 min) and air dried. (iii) Side‐chain deprotection and cleavage: each lantern was treated with TFA (0.5 ml per lantern) for 30 min after which the lanterns were washed with DCM (2 × 2 min) and air dried. The peptide aldehydes were cleaved with TFA : MeCN : H2O (0.1 : 60 : 40, v:v:v) (2.5 ml per lantern) at 60 °C for 30 min. Each lantern was removed and the cleavage solution lyophilized to give peptide aldehydes as white solids.
Protease Assay
The inhibition of MVEV protease by different acetyl‐tripeptide‐aldehyde inhibitors was performed in a reaction buffer with 10‐m m Tris–HCl, pH 8.0, 1‐mM CHAPS (3‐[(3‐Cholamidopropyl) dimethylammonio]‐1‐propane sulfonate) and 30% glycerol. 40 n m of the enzyme and varying concentrations of the inhibitor were pre‐incubated in a reaction buffer for 1 h at 25 °C before the reaction was initiated by the addition of the flourogenic peptide substrate Pyr‐Arg‐Thr‐Lys‐Arg‐AMC (Bachem [Bubendorf, Sweden], Switzerland) to a final concentration of 40 µ m. The components of the reaction were mixed with a 5‐s shake, and the progress of the reaction was monitored continuously at 37 °C by measuring the increase in fluorescence (λ ex = 355 nm and λ em = 460 nm) every 45 s for 1 h on a SpectraMax Gemini XS plate reader (Molecular Devices, Sunnyvale, CA, USA). The % inhibition was calculated by comparing it with wells with no inhibitor.
Measurement of IC50 Inhibition Constants
The 23 peptides identified as potential acetyl‐tripeptide‐aldehyde inhibitors against MVEV were re‐synthesised, and a 10‐m m stock was prepared. Varying concentrations of the inhibitor were incubated with 40 n m of the enzyme in 10‐m m Tris–HCl, pH 8.0, 1‐m m CHAPS and 30% glycerol buffer for 1 h at 25 °C. The reaction was initiated by the addition of the flourogenic peptide substrate Pyr‐Arg‐Thr‐Lys‐Arg‐AMC (Bachem) to a final concentration of 40 µ m. The components of the reaction were mixed with a 5‐s shake, and the progress of the reaction was monitored continuously at 37 °C by measuring the increase in fluorescence (λ ex = 355 nm and λ em = 460 nm) every 45 s for 1 h on a SpectraMax Gemini XS plate reader. The IC50 was derived by fitting the initial velocity against the log (inhibitor) with sigmoidal dose response curve in GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Results and Discussion
The solid‐phase synthesis of peptide aldehydes via the oxazolidine linker 4 is one of a number of reported methods, including the semicarbazone 10, the Weinreb amide 11, the ozoneolysis 12, 13, the thiazolidine 14, the backbone amide linker 15, synthesis via reduction of peptide thioesters 16 and oxidation of support‐bound peptide alcohols 17. The synthesis of peptide aldehydes has recently been reviewed 18. The oxazolidine linker satisfies the following criteria for a general linker for the immobilisation of aldehyde functionality: (i) it is low cost and easy to construct, (ii) it is chemically stable (long shelf life), (iii) it does not require preformation (i.e. the aldehyde couples directly to the solid phase), (iv) aldehyde attachment is generic and (v) it cleaves under mild conditions that leave no residue upon evaporation, allowing biological screening without further purification. The linker has been used to synthesise several peptide aldehydes including fellutamide B 19 and a library of reversible SARS coronavirus main protease inhibitors 20.
The method was developed and applied on the Multipin system, a technique developed for solid‐phase library synthesis. The Multipin method of peptide synthesis is an effective, low cost, simultaneous multiple synthesis technology, which gives researchers ready access to large numbers of peptides. The key to the method is that peptide synthesis and biological screening are totally integrated; no single peptide is given individual attention except at the coupling stages of the process. Synthesis has traditionally been performed on plastic pinsarranged in an 8 × 12 microtitre plate format, thus permitting the simultaneous preparation of sets of 96 peptides (or multiples thereof) 21. The fundamental advantage of this technology, which is an alternative to beaded cross‐linked resins, is the ease of handling large numbers in multiple parallel synthesis. Syntheses were performed on a hydrophilic graft polymer (polyamide), which is suited to aqueous chemistries and as described by Rademann et al. 20, is far superior to hydrophobic polymer matrices such as polystyrene resins and surfaces.
As shown in Scheme 1, the protected amino acid aldehyde is attached to the solid phase by condensation with a threonine residue 1 to form a support‐bound oxazolidine system 2. The oxazolidine is stable to non‐aqueous acid and to base, and as the secondary amine is difficult to acylate, it can tolerate subsequent peptide synthesis. Others have protected the secondary amine with a Boc group to eliminate possible acylation of this amine during peptide chain extension 20. Although this would be important for longer syntheses, and the synthesis of peptidomimetics requiring the use of more activated electrophiles, we found this extra protection step unnecessary for this short library synthesis. We do, however, recommend this advance made by Rademann 20 for future syntheses. Being stable to non‐aqueous acid, TFA may be used to remove acid‐labile protecting groups prior to release of the aldehyde function (i.e. cleavage). Importantly, the linker allows for side‐chain deprotection and subsequent removal of residual compounds such as TFA, organic solvents and scavengers before mild cleavage of the peptide aldehyde 4 using aqueous TFA (0.1%), containing acetonitrile as a cosolvent. The two‐step cleavage conditions allow for a shorter (30 min) TFA treatment because there is no concentration of product with no evidence of Pbf protecting group remaining on cleaved peptides. The mild cleavage conditions allowed for direct testing of all 400 unpurified peptide aldehydes in a protease assay.
Scheme 1.

Synthesis of 400‐member peptide aldehyde library.
Library Design Strategy
To demonstrate the powerful utility of this synthesis system, we chose to lock in the P1 amino acid, selected from a challenging but more importantly, a P1 residue that is common to many serine and cysteine proteases. Arginine is particularly well suited to this design criterion because it is the preferred substrate P1 cleavage amino acid for a number of proteases. To prepare for library synthesis, we synthesised the peptide aldehyde leupeptin as a control sequence. Leupeptin is a naturally occurring microbial peptide aldehyde derived from the culture filtrates of different streptomyces. Importantly, all C‐terminal arginine peptide aldehydes partially exist in the hydrated form as demonstrated by the [M + H + H2O] + peak in all MS run (Table 2). Lewis and Wolfenden 22 demonstrated this originally in their work on the active site of papain, whereas Gancsik et al. 23 extended this work using 13C NMR spectroscopy. The hydrated form also exists in N‐terminal aldehydes as demonstrated by Rademann, Meldal and Bock 24 and Groth and Meldal 25. Hence, in aqueous solutions, peptide aldehydes exist as an equilibrium state between the aldehyde and hydrated forms. For the library synthesis, we synthesised an extra two copies of leupeptin on each block as internal controls.
Table 2.
As‐synthesised mass spectrum of those inhibitors with an IC50 result under 15 µ m
| Peptide inhibitor | IC50 (μ m) | Mass spectrum |
|---|---|---|
| Ac‐CKR‐H | 8.5 ± 1.4 |

|
| Ac‐IWR‐H | 9.68 ± 2.2 |
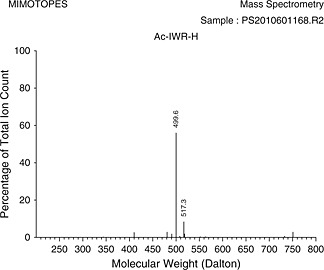
|
| Ac‐KKR‐H | 4.56 ± 1.6 |

|
| Ac‐LWR‐H | 8.8 ± 1.4 |

|
| Ac‐YWR‐H | 9.7 ± 2.8 |

|
| Ac‐WYR‐H | 10.90 ± 4.46 |

|
| Ac‐WRR‐H | 10.58 ± 1.24 |
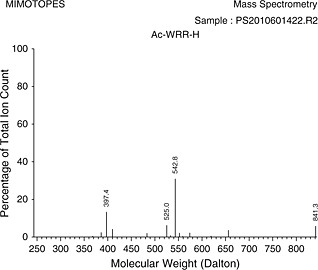
|
| Ac‐WLR‐H | 12.17 ± 3.12 |

|
| Ac‐CWR‐H | 12.53 ± 2.4 |
|
The modular nature of SynPhase lanterns allows for their use in traditional solution‐phase reaction vessels without the need for filtration, as is required with resins. This can be extended to the ‘split and mix’ technique if the lanterns are tagged with, e.g. coloured tags (SynPhase cogs and spindles) or radio frequency tags (Transtems). Both array and reaction vessel strategies can be used in combination to simplify the synthesis process. At the completion or during the synthesis, the SynPhase lanterns are sorted into the highly convenient 8 × 12 matrix format for multiple synthesis and cleavage of the target compounds. Hence, the following strategy was used to synthesise the 400‐member peptide aldehyde library resulting in a (20 × 20 × 1] combinatorial library (P3, P2 = 20 natural AAs, P1 is fixed as arginine).
Step 1: Attach Fmoc‐Arg(Pbf)‐H to 400 lanterns in a single reaction vessel (P1 position).
Step 2: Divide 400 lanterns into sets of 20 and place in 20 reaction vessels for coupling 20 natural Fmoc‐protected amino acids (P2 position).
Step 3: Array ‘dipeptide’ aldehyde lanterns onto reaction backing plates and couple 20 natural Fmoc amino acids in microtitre plate format (P3 position).
Step 4: Acetylate the library by immersing plates in baths containing capping solution.
Step 5: Cleave 400 peptide aldehydes by first immersing backing plates with lanterns into baths containing TFA for concomitant side‐chain protecting group removal, and after several washes, cleave from the solid support into microtitre plates containing pre‐weighed tubes.
Step 6: Freeze dry in parallel, analyse and weigh, and screen in biological assay.
By utilising a strategy of positioning, the lanterns in five blocks of 8 × 10 (80 peptide aldehydes per block), the synthesis of the library becomes straightforward. After the second amino acid is coupled (step 2), the lanterns are arrayed on backing plates so as to orchestrate the third amino acid coupling in two side‐by‐side rows of 10 wells, using a multi‐channel pipette to dispense the activated amino acids.
The synthesis of the Fmoc‐Arg(Pbf)‐H followed the same protocol reported earlier 4. Synthesis of the protected amino acid aldehydes was achieved via reduction of the corresponding N,O‐dimethylhydroxamate with LiAlH4 by adapting the method of Fehrentz and Castro 26. The yield for attachment of Fmoc‐Arg(Pbf)‐H was 50% determined by quantitative Fmoc test. Standard peptide synthesis methodologies were used for the synthesis of 400 peptide aldehydes. Although there is potential to acylate the oxazolidine nitrogen during peptide synthesis, it is relatively unreactive 27, and no acylation occurs with DIC/HOBt activation. Acetylation with acetic anhydride is performed at 4 °C for 5 min to avoid reaction with the oxazolidine nitrogen.
The 400 isolated peptide aldehydes were weighed and analysed by mass spectrometry individually. The average mass obtained was 1.8 mg with a standard deviation of 0.2 mg (4.1 µmol, SD 0.5 µmol) that represents an average yield (based on an initial loading of Fmoc‐Arg(Pbf)‐H of 7 µmol) of 60%. All 400 peptide aldehydes showed target molecule peak by MS (Table 2 for representative MS). The mild cleavage conditions allowed for direct testing of unpurified peptide aldehyde library in a protease assay.
The 400 aldehydes were screened against the NS2B‐NS3 domain of MVEV using a protease assay (Figure 1). Among the 400 aldehyde peptide inhibitors tested at 10‐μ m concentration, 23 peptides showed >50% inhibition at 10.0 µ m. The remaining stock with these 23 peptides was further tested at 5, 1.0 and 0.1 µ m. The inhibition (%) for these peptides is represented in Figure 1. The acetyl‐tripeptide‐aldehyde inhibitors such as CKR and IWR showed inhibition even at 1.0 µ m.
Figure 1.

Inhibition (%) for the aldehyde peptides against MVEV at 10, 5.0, 1.0 and 0.1 u m.
The 23 peptides identified as potential acetyl‐tripeptide‐aldehyde inhibitors against MVEV were re‐synthesised and IC50 inhibition constants obtained (Table 1). The compounds were tested in duplicate with a goodness of fit (R 2) between 0.94 and 0.98. Table 2 presents the as‐synthesised mass spectrum of those inhibitors with an IC50 result under 15 µ m.
Table 1.
IC50 inhibition constants for 23 peptides that showed >50% inhibition at 10.0 µ m
| Peptide inhibitor | IC50 (μ m) | Peptide inhibitor | IC50 (μ m) |
|---|---|---|---|
| Ac‐CMR‐H | >50 | Ac‐NWR‐H | 32.89 ± 7.0 |
| Ac‐DAR‐H | >50 | Ac‐SCR‐H | >50 |
| Ac‐EMR‐H | >50 | Ac‐RQR‐H | >50 |
| Ac‐ENR‐H | >50 | Ac‐RRR‐H | 18.86 ± 6.86 |
| Ac‐CRR‐H | 22.26 ± 4.0 | Ac‐QGR‐H | >50 |
| Ac‐CKR‐H | 8.5 ± 1.4 | Ac‐WRR‐H | 10.58 ±1.24 |
| Ac‐CWR‐H | 12.53 ± 2.4 | Ac‐WVR‐H | >50 |
| Ac‐GWR‐H | >50 | Ac‐WKR‐H | 19.17 ± 3.18 |
| Ac‐IWR‐H | 9.68 ± 2.2 | Ac‐YWR‐H | 9.7 ± 2.8 |
| Ac‐KKR‐H | 4.56 ± 1.6 | Ac‐WLR‐H | 12.17 ± 3.12 |
| Ac‐KWR‐H | 15.5 ± 2.47 | Ac‐WYR‐H | 10.90 ± 4.46 |
| Ac‐LWR‐H | 8.8 ± 1.4 |
Conclusion
A 400‐member library of C‐terminal arginine peptide aldehydes was prepared by the straightforward approach described here and was demonstrated to be an effective tool in expediting rapid SAR analysis of serine proteases, such as MVEV.
References
- 1. Otto H‐H, Schirmeister T. Cysteine proteases and their inhibitors. Chem. Rev. 1997; 97: 133–171. [DOI] [PubMed] [Google Scholar]
- 2. Malcolm BA, Lowe C, Shechosky S, McKay RT, Yang CC, Shah VJ, Simon RJ, Vederas JC, Santi DV. Peptide aldehyde inhibitors of hepatitis A virus 3C proteinase. Biochemistry 1995; 34: 8172–8179. [DOI] [PubMed] [Google Scholar]
- 3. Scheidt KA, Roush WR, McKerrow JH, Selzer PM, Hansell E, Rosenthal PJ. Structure‐based design, synthesis and evaluation of conformationally constrained cysteine protease inhibitors. Bioorg. Med. Chem. 1998; 6: 2477–2494. [DOI] [PubMed] [Google Scholar]
- 4. Ede NJ, Eagle SN, Wickham G, Bray AM, Warne B, Shoemaker K, Rosenberg S. Solid phase synthesis of peptide aldehyde protease inhibitors. Probing the proteolytic sites of hepatitis C virus polyprotein. J. Peptide Sci. 2000; 6: 11–18. [DOI] [PubMed] [Google Scholar]
- 5. Ray D, Shi PY. Recent advances in flavivirus antiviral drug discovery and vaccine development. Recent Pat. Anti‐Infect. Drug Discov. 2006; 1: 45–55. [DOI] [PubMed] [Google Scholar]
- 6. Burrow JN, Whelan PI, Kilburn CJ, Fisher DA, Currie BJ, Smith DW. Australian encephalitis in the Northern Territory: clinical and epidemiological features, 1987–1996. Aust. N. Z. J. Med. 1998; 28: 590–596. [DOI] [PubMed] [Google Scholar]
- 7. Joy JK, Mee NF, Kuan WL, Perlyn KZ, Wen TS, Hill J. Biochemical characterisation of Murray Valley encephalitis virus proteinase. FEBS Letts. 2010; 584: 3149–3152. [DOI] [PubMed] [Google Scholar]
- 8. Yin Z, Patel SJ, Wang W‐L, Chan W‐L, Ranga Rao KR, Wang G, Ngew X, Patel V, Beer D, Knox JE, Ling Ma N, Ehrhardt C, Pheng Lim S, Vasudevan SG, Keller TH. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Letts. 2006; 16: 40–43. [DOI] [PubMed] [Google Scholar]
- 9. Smart SS, Mason TJ, Bennell PS, Maeji NJ, Geysen HM. High‐throughput purity estimation and characterization of synthetic peptides by electrospray mass spectrometry. Int. J. Peptide Protein Res. 1996; 47: 47–55. [DOI] [PubMed] [Google Scholar]
- 10. Murphy AM, Dagnino R, Jr. , Vallar PL, Trippe AJ, Sherman SL, Lumpkin RH, Tamura SY, Webb TR. Automated synthesis of peptide C‐terminal aldehydes. J. Am. Chem. Soc. 1992; 114: 3156–3157. [Google Scholar]
- 11. Fehrentz J‐A, Paris M, Heitz A, Velek J, Winternitz F, Martinez J. Solid phase synthesis of C‐terminal peptide aldehydes. J. Org. Chem. 1997; 62: 6792–6796. [Google Scholar]
- 12. Pothion C, Paris M, Heitz A, Rocheblave L, Rouch F, Fehrentz J‐A, Martinez J. Use of ozonolysis in the synthesis of C‐terminal peptide aldehydes on solid support. Tetrahedron Lett. 1997; 44: 7749–7752. [Google Scholar]
- 13. Hall BJ, Sutherland JD. A practical method for the combinatorial synthesis of peptide aldehydes. Tetrahedron Letts. 1998; 39: 6593–6596. [Google Scholar]
- 14. Galeotti N, Giraud M, Jouin P Solid phase synthesis of peptidyl aldehydes from C‐terminal thiazolidinyl peptides. Letts. Pept. Sci. 1997; 4: 437–440. [Google Scholar]
- 15. Jensen KJ, Alsina J, Songster MF, Vagner J, Albericio F, Barany G. Backbone amide linker (BAL) strategy for solid‐phase synthesis of C‐terminal‐modified and cyclic peptides. J. Am. Chem. Soc. 1998; 120: 5441–5452. [Google Scholar]
- 16. Wyslouch‐Cieszynska A, Tam PT. Peptides: Frontiers of Peptide Science, Proceedings of the Fifteenth American Peptide Symposium, Tam PJ, Kaumaya PTP. (eds). Kluwer Academic Publishers: The Netherlands, Dordrecht, 1999; 263–264. [Google Scholar]
- 17. Page P, Bradley M, Walters I, Teague S. Solid phase synthesis of tyrosine peptide aldehydes. Analogues of (S)‐MAPI. J. Org. Chem. 1999; 64: 794–799. [DOI] [PubMed] [Google Scholar]
- 18. Moulin A, Martinez J, Fehrentz J‐A. Synthesis of peptide aldehydes. J. Peptide Sci. 2007; 13: 1–15. [DOI] [PubMed] [Google Scholar]
- 19. Schneekloth JS, Jr. , Sanders JL, Hines J, Crews CM. Neurotrophic peptide aldehydes: solid phase synthesis of fellutamide B and a simplified analog. Bioorg. Med. Chem. Lett. 2006; 16: 3855–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Al‐Gharabli SI, Ali Shah ST, Weik S, Schmidt MF, Mesters JR, Kuhn D, Klebe G, Hilgenfeld R, Rademann J. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS‐CoV Mpro) inhibitors. Chem. Bio. Chem. 2006; 7: 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ede NJ. Multiple parallel synthesis of peptides on SynPhaseTM grafted supports. J. Immunol. Methods 2002; 267: 3–11. [DOI] [PubMed] [Google Scholar]
- 22. Lewis CA, Jr. , Wolfenden R. Thiohemiacetal formation by inhibitory aldehydes at the active site of papain. Biochemistry 1977; 16: 4890–4895. [DOI] [PubMed] [Google Scholar]
- 23. Gamcsik MP, Malthouse JPG, Primrose WU, Mackenzie NE, Boyd ASF, Russell RA, Scott AI. Structure and stereochemistry of tetrahedral inhibitor complexes of papain by direct NMR observation. J. Am. Chem. Soc. 1983; 105: 6324–6325. [Google Scholar]
- 24. Rademann J, Meldal M, Bock K. Solid‐phase synthesis of peptide isosteres by nucleophilic reactions with N‐terminal peptide aldehydes on a polar support tailored for solid‐phase organic chemistry. Chem. Eur. J. 1999; 5: 1218–1225. [Google Scholar]
- 25. Groth T, Meldal M. Synthesis of aldehyde building blocks protected as acid labile N‐Boc N,O‐acetals: toward combinatorial solid phase synthesis of novel peptide isosteres. J. Comb. Chem. 2001; 3: 34–44. [DOI] [PubMed] [Google Scholar]
- 26. Fehrentz J‐A, Castro B. An efficient synthesis of optically active α‐(t‐butoxycarbonylamino)‐aldehydes from α‐amino acids. Synthesis 1983; 676–678. [Google Scholar]
- 27. Ede NJ, Bray AM. A simple linker for the attachment of aldehydes to the solid phase. Application to solid phase synthesis by the MultipinTM method. Tetrahedron Lett. 1997; 38: 7119–7122. [Google Scholar]