

Periodontitis is one of the most complex infectious diseases of the human body. Individual periodontal lesions may harbor millions of genomic copies of herpesviruses (179) as well as papillomaviruses, human immunodeficiency virus (HIV), human T‐lymphotropic virus type 1, torquetenovirus, and hepatitis B and C viruses (190). Herpesvirus‐infected periodontal sites tend to exhibit more breakdown than herpesvirus‐free sites, and a herpesviral active infection is associated with an elevated risk of progressive periodontal disease (189). Furthermore, the oral cavity supports more than 700 bacterial species, and the periodontal pocket area harbors more than 400 bacterial species (148). Periodontopathic bacteria, such as Porphyromonas gingivalis and Tannerella forsythia, possess virulence factors involved in colonizing periodontal sites, neutralizing local host defenses and destroying periodontal tissues (78, 192).
The host immune response attempts to control both pathogenic viruses and bacteria in periodontal sites. However, it is unclear if various immune mediators, such as certain cytokines and chemokines, exert primarily a protective or a destructive role in periodontal disease. Also, some immune mechanisms that are active against viruses may diminish antibacterial immune responses, and vice versa. It may be that periodontitis is the result of extensive and partly opposing immune responses against viral–bacterial combined infections (179). Major advances in the diagnosis, prevention and treatment of periodontitis probably depend upon a better understanding of the pathogenic infections and the associated host responses.
This review article presents evidence that viruses and bacteria in aggregate produce a greater pathogenic effect than the sum of the individual agents, and discusses how the concept of a herpesviral–bacterial combined infection may change our understanding of the pathogenesis, and possibly the management, of destructive periodontal disease. Emphasis is placed on synergistic interactions between periodontal Epstein–Barr virus and cytomegalovirus, and major suspected periodontopathic bacteria.
Viral–bacterial synergy in medical infections
Respiratory disease represents the best‐known example of serious viral–bacterial co‐infections (113, 129). Worldwide, respiratory infections give rise to more disease and death than any other human infection and are one of the leading causes of morbidity and mortality (121). Seasonal influenza in the USA is estimated to result in more than 200,000 hospitalizations and 36,000 deaths annually (218). The great ‘Spanish flu’ influenza pandemic of 1918–1919 caused 20–50 million deaths worldwide and an estimated 675,000 deaths in the USA, affecting mostly young adults and not the usual children and old individuals. At the peak, half of the world’s population was clinically infected. During the influenza pandemic years of 1918–1919 (type A influenza, subtype H1N1) and 1957–1958 (type A influenza, subtype H2N2), the incidence of secondary bacterial pneumonia, the most common cause of excess mortality during influenza outbreaks, varied between 2 and 18% in different populations (123). The influenza A (swine flu) pandemic of 2009 involved a novel H1N1 strain. The main bacteria in influenza‐associated pneumonia are Streptococcus pneumoniae, alpha‐hemolytic streptococci, Haemophilus influenzae, Staphylococcus aureus and Moraxella (Branhamella) catarrhalis (123). A combined infection with influenza viruses and S. aureus causes particularly severe and fatal pneumonia in both children and adults (29). Respiratory viruses other than the influenza viruses can also interact pathogenically with bacterial pathogens, for example human parainfluenza virus with S. pneumoniae (50) and adenovirus with Bordetella pertussis (185). A study of community‐acquired childhood pneumonia revealed that mixed viral–bacterial infections, which comprised about 75% of the pneumonias studied, resulted in more severe inflammation and clinical illness than single viral or bacterial infections (87).
Viruses can induce alterations in the respiratory tract, which allow resident bacteria to multiply to levels capable of inducing inflammation (6). Respiratory tract viruses can inhibit the ciliary activity of the respiratory epithelium and thereby increase the risk for bacterial superinfection (6). Respiratory cells infected with influenza A virus, respiratory syncytial viruses or adenoviruses show enhanced bacterial adherence in both in vitro and in vivo model systems (69). The influenza virus may predispose to secondary bacterial pneumonia by denudating the respiratory epithelium and exposing basement membrane elements (e.g. fibrinogen) to which bacteria can attach (47, 123). An influenza virus infection can also cause prolonged desensitization of lung sentinel cells to toll‐like receptor ligands, resulting in a reduced presence of neutrophils and an increased secondary bacterial load (39). In addition, alteration of neutrophil functions by the influenza virus may decrease the clearance of pulmonary bacteria (97, 104).
Acute otitis media has been linked to synergistic interactions between viruses and bacteria (7). Otitis media is often associated with viruses, such as rhinovirus, respiratory syncytial virus, adenovirus, coronavirus, influenza virus (232), cytomegalovirus (238) and other herpesviruses (20), as well as with bacteria, including H. influenzae, S. pneumoniae, M. catarrhalis, and Prevotella and Peptostreptococcus species (19). Acute otitis media with a tympanostomy tube showed S. pneumoniae, S. aureus, H. influenzae, Pseudomonas aeruginosa and yeast (166). Viruses were detected in 65% of acute otitis media samples that were positive for H. influenzae, in 77% of samples that were positive for S. pneumoniae and in 73% of samples that were positive for M. catarrhalis (170). Tympanic membrane changes with acute otitis media are more severe in patients co‐infected with respiratory viruses and bacteria than in patients who have a single infection with either of the two types of infectious agents (237). Viruses interact with bacteria in acute otitis media to boost the local inflammatory process and also have a profound adverse effect on resolution of the disease (75).
Other human diseases seem also to have a combined viral–bacterial etiopathogeny. Recurrent bacterial sinusitis may develop as a complication to viral colds (5). Infectious mononucleosis caused by infection with Epstein–Barr virus can give rise to tonsillar upgrowth of Prevotella intermedia and Fusobacterium nucleatum, and subsequently pharyngotonsillitis (18). The Lemierre’s syndrome, which is characterized by severe pharyngitis and sepsis, can occur as a sequel to Epstein–Barr virus‐induced mononucleosis and is frequently associated with an overgrowth of Fusobacterium necrophorum (53). Gastroenteritis is often associated with mixed infections of viruses (rotavirus, adenovirus, norovirus, astrovirus) and bacteria (pathogenic Escherichia coli, Salmonella, Shigella, Campylobacter jejuni), and the combined viral–bacterial infection can lead to enhanced pathosis (117). Appendicitis has been associated with measles virus, adenovirus and herpesviruses, as well as with Bacteroides fragilis, E. coli, Yersinia, Salmonella and Shigella (91). In Helicobacter pylori‐associated gastritis, the activation of a latent Epstein–Barr virus infection by monochloramine from infiltrating neutrophils can aggravate the gastric disease (128). In organ transplant recipients, cytomegalovirus and human herpesviruses type 6 or type 7 can induce serious opportunistic infections with bacteria, fungi, protozoa and other viruses, probably as a result of immunomodulation by the herpesviruses (12, 137, 150). A renal transplant patient developed a subhepatic abscess as a result of infection with cytomegalovirus and P. gingivalis (103). Vascular diseases occur at a higher frequency as a result of a combined infection with herpes simplex virus and Chlamydia pneumoniae (165) or periodontopathic bacteria (224) than from a single infection with either of these agents. Rheumatic heart disease and autoimmune myocarditis may involve dual infection with coxsackievirus and group A streptococci, which have the potential to dysregulate immune mechanisms and trigger an autoimmune reaction (167). HIV infection causes a variety of severe bacterial, fungal and viral infections (46, 100, 174, 242). Moreover, P. gingivalis may up‐regulate expression of the HIV‐1 R5‐specific co‐receptor CCR5 in oral keratinocytes and thereby increase the risk for oral infection and dissemination of R5‐tropic HIV‐1 strains (58). Also, various gram‐positive and gram‐negative bacteria of the periodontitis biofilm have the capacity to re‐activate HIV in latently infected T cells, macrophages and dendritic cells (80), and the vaginosis bacteria Prevotella bivia and Peptostreptococcus asaccharolyticus, but no other vaginal bacteria studied, are potential activators of HIV expression in monocytoid cells and T‐lymphocytic cells (72).
Animal models have been used to study viral–bacterial interactions. Respiratory pathosis was enhanced in mice when the influenza virus or other viruses co‐infected with bacteria (198). In a mouse model of pathogenetic synergy, an influenza virus infection antecedent to an S. pneumoniae challenge caused pneumonia and led to 100% mortality, whereas an infection with S. pneumoniae prior to an influenza virus infection provided protection from influenza and improved survival rates (123). Immune mediators, including cytokines and chemokines, through toll‐like receptors/mitogen‐activated protein kinase pathways, seem to play important roles in the pathogenesis of the influenza virus–S. pneumoniae co‐infection (184). Calves dually infected with bovine respiratory syncytial virus and Haemophilus somnus, both bovine respiratory pathogens, contracted lung disease and harbored H. somnus in the lungs, whereas the lungs of monoinfected calves showed neither pathosis nor H. somnus infection (57). Acute otitis media was induced in 63% of mice that had been stably colonized by S. pneumoniae and then infected with the influenza virus, whereas all mock‐infected mice remained free of disease (122). Mice infected with herpes simplex virus, adenovirus or vaccinia virus revealed enhanced susceptibility to E. coli pyelonephritis (59). Mice inoculated intraperitoneally with murine cytomegalovirus together with P. aeruginosa, S. aureus or Candida albicans exhibited mortality rates of 80–100%, whereas immunization against murine cytomegalovirus abrogated the synergistic effect on mortality for all combinations of the infectious agents studied (70). In immunocompromised rats, a symptomatic infection with rat cytomegalovirus induced severe neutropenia and subsequently a high rate of enteric gram‐negative rod bacteremia (201). A markedly higher mortality rate was found in mice co‐infected with murine cytomegalovirus and P. gingivalis than in mice mono‐infected with either cytomegalovirus or P. gingivalis (203). A significantly lower level of systemic interferon‐gamma was detected in the dually infected mice than in the mono‐infected mice, suggesting that P. gingivalis was able to reduce the antiviral interferon‐gamma response and thus increase the pathogenicity of the co‐infecting cytomegalovirus (203).
For completeness, a latent herpesvirus infection may, in some experimental settings, up‐regulate the activation state of the innate immunity, thereby providing immune benefits for the host. Mice latently infected with murine Epstein–Barr virus or cytomegalovirus can show resistance to infection with the bacterial pathogens Listeria monocytogenes and Yersinia pestis (10). However, the herpesvirus‐induced protection against bacterial infection seems to be transient, lasting only a few months (236).
Collectively, numerous studies of natural diseases and experimental infections conclude that viral–bacterial co‐infections produce more disease than single infections with either of the two types of infectious agents. Early studies on viral–bacterial synergism emphasized a decreased clearance or an enhanced pathogenicity of the bacteria involved in the infectious complex, but more recent investigations also point to the possibility of an increased virulence of the virus. As millions of herpesviruses and bacteria can inhabit individual periodontitis sites (179), it is reasonable to assume that herpesviral–bacterial interactions also play a role in the development of human periodontitis.
Herpesviruses in periodontal diseases
Diagnostic considerations
Studies from various countries have found genomic copies of Epstein–Barr virus and cytomegalovirus to be present in most periodontitis lesions and to be less prevalent in gingivitis and healthy periodontal sites (Table 1). Study‐to‐study variation in the detection of periodontal herpesviruses may be a result of (i) the clinical status of the study subjects, (ii) the viral diagnostic methods utilized, or (iii) true geographical/ethnic differences in herpesviral occurrence.
Table 1.
Recent studies on the prevalence of subgingival genome‐copies of Epstein–Barr virus (EBV) and human cytomegalovirus (HCMV) in periodontal disease
| Study (country) | Virus | Aggressive periodontitis; percentage positive samples | Chronic periodontitis; percentage positive samples | Gingivitis; percentage positive samples | Healthy/normal periodontium; percentage positive samples |
|---|---|---|---|---|---|
| Imbronito et al. 2008 (85) (Brazil) | EBV‐1 | 33% | 47% | 20% | 0% |
| HCMV | 48% | 50% | 40% | 57% | |
| Combs et al. 2008 (28) (USA) | HCMV | No data | 4% | No data | 0% |
| Chalabi et al. 2008 (22) (Iran) | EBV‐1 + 2 | No data | 79% | No data | 7 |
| HCMV | No data | 59% | No data | 0 | |
| Grande et al. 2008 (61) (Brazil) | EBV | No data | 48% | No data | No data |
| HCMV | No data | 80% | No data | No data | |
| Rotola et al. 2008 (168) (Italy)* | EBV | 55% | 46% | No data | 8% |
| HCMV | 0% | 0% | No data | 8% | |
| Ding et al. 2008 (40) (China) | HCMV | 44% | No data | No data | 13% |
| Botero et al. 2008 (16) (Columbia) | HCMV | No data | 80% | No data | 25% |
| Saygun et al. 2008 (179) (Turkey) | EBV | 60% | No data | 13% | No data |
| HCMV | 53% | No data | 7% | No data | |
| Sunde et al. 2008 (207) (Norway)** | EBV | No data | 40% | No data | 7% |
| HCMV | No data | 12% | No data | 0% | |
| Imbronito et al. 2008 (84) (Brazil) | EBV | No data | 45% | No data | No data |
| HCMV | No data | 83% | No data | No data | |
| Moghim et al. 2007 (133) (Iran) | EBV | No data | 61% | No data | 3% |
| Wu et al. 2007 (235) (China) | EBV‐1 + 2 | No data | 38% | 20% | 21% |
| HCMV | No data | 63% | 49% | 42% | |
| Botero et al. 2007 (15) (Columbia) | HCMV | 40% | 60% | No data | 18% |
| Watanabe et al. 2007 (230) (Brazil) | EBV | 57% | No data | 30% | No data |
| HCMV | 7% | No data | 0% | No data | |
| Wu et al. 2006 (234) (China) | EBV‐1 + 2 | No data | 66% | 32% | 17% |
| Chen et al. 2006 (24) (China) | HCMV | No data | 59% | No data | 32% |
| Klemenc et al. 2005 (95) (Slovania) | EBV | No data | 44% | No data | 0% |
| HCMV | No data | 3% | No data | 0% | |
| Konstantinidis et al. 2005 (96) (Greece) | EBV | No data | 55% | No data | 9% |
| Kubar et al. 2005 (98) (Turkey) | EBV | 89% | 46% | No data | No data |
| HCMV | 78% | 27% | No data | No data | |
| Li et al. 2004 (107) (China) | EBV | 58% | 23% | 19% | No data |
| Tantivanich et al. 2004 (214) (Thailand) | HCMV | No data | 34% | No data | 3% |
| Percentage average (percentage median) positive samples | EBV HCMV | 65% (57%) 44% (44%) | 49% (46%) 44% (55%) | 22% (20%) 24% (24%) | 8% (7%) 17% (11%) |
*Gingival biopsies were studied. Patients received multiple sessions of nonsurgical periodontal therapy before virological sampling. Latent herpesvirus‐7 was identified in 90% of the periodontitis lesions studied.
**Patients received conventional periodontal therapy before virological sampling.
Clinical status of the study subjects
Identification of periodontal herpesviruses is influenced by the sample site and method utilized. Because pathogen loads typically peak during periods of active disease, herpesvirus recovery from periodontal sites depends greatly on the accuracy of the clinical diagnosis. The difficulty of defining periodontitis for the purpose of research was recently pointed out (156). The use of patient age as a main criterion for distinguishing between ‘aggressive’ and ‘chronic’ periodontitis may not be a reliable indicator of active or stable disease. The term ‘normal’ periodontal site is sometimes used to suggest absence of attachment loss rather than inflammation‐free gingiva. As herpesviruses may enter even slightly inflamed periodontal sites, sensitive polymerase chain reaction (PCR) techniques may show viruses in ‘unhealthy’ control sites. Conversely, periodontal therapy can markedly reduce herpesvirus subgingival counts and thus cause a significant underestimation of the viral load associated with untreated disease (177). Reliable data on the prevalence and quantity of periodontal herpesviruses may require study of virgin lesions with a well‐defined disease‐activity status.
Studies that examine a small number of periodontal sites in each subject will inevitably underestimate the prevalence of herpesviruses in the study population and the total number of herpesvirus genome‐copies in the entire periodontium. Studies of herpesviruses in subgingival sites will also not account for the substantial herpesvirus counts residing within inflamed gingival tissue (98, 168).
Viral diagnostic methods
The era of relying upon in vitro cell culture for routine laboratory diagnosis of viral infections has truly passed. Viral isolation indicates an active and possibly disease‐producing infection, but isolation of viruses is difficult, costly and time‐consuming. High‐fidelity PCR‐based techniques have become the standard for identification and quantification of periodontal herpesviruses (99, 146), and the transcription of late herpesvirus genes for structural proteins is understood to signify productive viral replication and is commonly used to indicate an active herpesvirus infection (60, 124). PCR identification of oral herpes simplex virus may yield two‐ to fourfold more positive samples than viral culture (42, 227). Nested PCR may unveil more periodontal sites that are positive for cytomegalovirus than viral culture or real‐time PCR (16) or end‐point detection PCR (23). Nested PCR technology is particularly efficient in detecting low viral loads (168). However, PCR primers that amplify templates of microbial communities at different efficiencies may yield biased results (86). Misamplification and false‐positive herpesvirus results may emerge when there are shared regions of nucleotide sequences between herpesvirus species and unknown infectious agents. Nevertheless, as periodontal Epstein–Barr virus and cytomegalovirus have been identified using a variety of PCR primers in platforms of end‐point detection PCR, nested PCR, reverse transcription PCR and real‐time PCR, the risk of a systematic misidentification of these viruses is small. However, different PCR primers and protocols may detect herpesviruses with a varying degree of proficiency (28), and technical expertise and quality assurance methods vary among laboratories (102, 144, 183). Preparation of the target nucleic acid constitutes a particularly vulnerable stage of PCR protocols.
Geographical/ethnic differences in herpesviral occurrence
The genotype distribution and seroprevalence of Epstein–Barr virus (82, 175) and cytomegalovirus (147, 168) differ among populations. Similarly to medical infections (157), some herpesvirus subtypes may exhibit increased periodontopathogenicity. Glycoprotein B (gB) homologues within the herpesvirus family participate in virus entry and cell‐to‐cell spread, and the encoding gene is commonly used in genotyping (145). The cytomegalovirus gB‐II genotype seems to predominate in chronic periodontitis, whereas the cytomegalovirus gB‐I genotype is more closely related to gingivitis and a healthy periodontium (235). In addition, herpesviruses occur at a higher prevalence in low‐income countries than in affluent countries (3), and the herpesvirus seroprevalence in high‐income countries exhibits significant racial, educational and socioeconomic disparities, starting at an early age and persisting into middle age (43, 44, 243).
Periodontal herpesviruses
Epstein–Barr virus and cytomegalovirus genomes have been identified in periodontitis lesions with prevalences ranging from a few per cent to more than 80% (Table 1). Cytomegalovirus seropositivity was identified in 95% of patients with periodontitis but in only 74% of patients with gingivitis (P = 0.057) (88). Epstein–Barr virus (105) and cytomegalovirus (240) are also common inhabitants of peri‐apical lesions of endodontic origin. Cytomegalovirus was detected in 32% of peri‐apical abscesses exhibiting spontaneous pain (23).
To assess, in more detail, the linkage between herpesviruses and periodontitis, patients have been grouped according to disease severity. Table 1 reveals the presence of Epstein–Barr virus in 65% of aggressive periodontitis lesions and in 49% of chronic periodontitis lesions and of cytomegalovirus in 44% of aggressive periodontitis lesions and in 44% of chronic periodontitis lesions. Although the prevalence of Epstein–Barr virus and cytomegalovirus was similar in aggressive and chronic periodontitis, the herpesvirus infection may differ qualitatively in the two diseases. An active (‘productive’) cytomegalovirus infection tends to be associated with periodontal sites showing no radiographic evidence of alveolar crestal lamina dura (211, 219), a finding consistent with disease‐active periodontitis (158). An active cytomegalovirus infection has also been linked to increased gingival inflammation in immunosuppressed organ‐transplant recipients (141). An acute herpes simplex virus‐type 1 infection in a 26‐year‐old patient caused, within a few hours, extensive gingival recession (155). By contrast, a latent (‘nonproductive’) herpesvirus infection is generally associated with chronic periodontitis sites exhibiting little propensity for disease progression (16).
Quantitatively, significantly more herpesvirus genomic copies inhabit progressive and untreated periodontitis lesions than stable or treated periodontitis sites (89, 90, 179). As many as 8.3 × 108 Epstein–Barr virus DNA copies and 4.6 × 105 cytomegalovirus DNA copies have been detected in samples of individual periodontal pockets (179), and even higher viral loads may reside within the gingival tissue of periodontitis lesions (98). Sunde et al. (208) identified up to one million genome‐copies of Epstein–Barr virus in periodontal sites of a 63 year‐old patient with refractory periodontitis. Herpesviruses other than Epstein–Barr virus and cytomegalovirus (110, 118, 138, 152, 168), as well as papillomavirus and other viruses (189), can also inhabit periodontitis lesions. Indeed, the total count of viruses approaches that of bacteria in some advanced periodontitis lesions. The abundance of herpesvirus genomic copies in individual periodontitis lesions translates into a heavy viral load for the entire periodontium of patients with severe and widespread periodontal disease. The high number of herpesviral copies in progressive periodontitis has implications for our understanding of the etiology of the disease.
Herpesviral–bacterial periodontal associations
Table 2 summarizes statistical relationships between periodontal herpesviruses and bacteria. A study in China found that 17% of Epstein–Barr virus‐positive periodontitis lesions, and 54% of cytomegalovirus‐positive periodontitis lesions, contained as many as six to eight species of major periodontopathic bacteria (40). Periodontal Epstein–Barr virus and cytomegalovirus seem to be most closely associated with P. gingivalis and T. forsythia (179), two bacteria with high periodontopathic potential (78). The link between cytomegalovirus and P. gingivalis appears to be particularly strong (193). Cytomegalovirus and P. gingivalis were linked to localized aggressive (juvenile) periodontitis in Jamaica with odds ratios of 4.6 and 7.8, respectively, but the cytomegalovirus–P. gingivalis dual infection was linked to the disease with an odds ratio as high as 51.4 (125). The significantly higher odds ratio of the cytomegalovirus‐P. gingivalis co‐infection than of the sum of the individual pathogens is suggestive of a pathogenetic synergy between the infectious agents. A study, in the USA, of 140 adults with gingivitis or periodontitis, linked Epstein–Barr virus‐1 and cytomegalovirus to elevated occurrence of the pathogens P. gingivalis, T. forsythia, P. intermedia, Prevotella nigrescens and Treponema denticola (Table 3). In a study from Japan, P. gingivalis comprised 0.25% of total salivary bacterial counts in Epstein–Barr virus‐positive periodontitis patients but only 0.02% (a 13‐fold difference) in Epstein–Barr virus‐negative patients (205). Periodontal Epstein–Barr virus (179, 207) and cytomegalovirus (85, 125, 140, 219) have also been related to the subgingival presence of the major periodontopathogen Aggregatibacter actinomycetemcomitans. An association between herpesviruses and anaerobic bacteria has also been demonstrated in periapical pathosis (171).
Table 2.
Statistically significant associations between subgingival Epstein–Barr virus and human cytomegalovirus and periodontopathic bacteria
| Herpesvirus | Bacterium | Study |
|---|---|---|
| Epstein–Barr virus | Porphyromonas gingivalis | Contreras et al. (31) |
| Imbronito et al. (85) | ||
| Saygun et al. (178) | ||
| Saygun et al. (179) | ||
| Sugano et al. (205) | ||
| Sunde et al. (207) | ||
| Tannerella forsythia | Contreras et al. (31) | |
| Saygun et al. (178) | ||
| Saygun et al. (179) | ||
| Prevotella intermedia | Contreras et al. (31) | |
| Imbronito et al. (85) | ||
| Prevotella nigrescens | Contreras et al. (31) | |
| Aggregatibacter actinomycetemcomitans | Michalowicz et al. (125) Sunde et al. (207) | |
| Campylobacter rectus | Saygun et al. (178) | |
| Treponema denticola | Contreras et al. (31) | |
| Cytomegalovirus | Porphyromonas gingivalis | Botero et al. (15) |
| Contreras et al. (31) | ||
| Michalowicz et al. (125) | ||
| Saygun et al. (178) | ||
| Saygun et al. (179) | ||
| Slots et al. (193) | ||
| Tannerella forsythia | Botero et al. (15) | |
| Contreras et al. (31) | ||
| Imbronito et al. (85) | ||
| Saygun et al. (178) | ||
| Saygun et al. (179) | ||
| Prevotella intermedia | Botero et al. (15) | |
| Saygun et al. (178) | ||
| Prevotella nigrescens | Contreras et al. (31) | |
| Aggregatibacter actinomycetemcomitans | Imbronito et al. (85) | |
| Michalowicz et al. (125) | ||
| Nowzari et al. (140) | ||
| Ting et al. (219) | ||
| Dialister pneumosintes | Slots et al. (196) | |
| Campylobacter rectus | Saygun et al. (179) | |
| Treponema denticola | Contreras et al. (31) |
Table 3.
Associations between Epstein–Barr virus‐1 and human cytomegalovirus and periodontopathic bacteria*
| Virus | Bacteria or disease | Odds ratio | P‐value |
|---|---|---|---|
| Epstein–Barr virus‐1 | Periodontitis severity | 5.1 | 0.05 |
| P. gingivalis | 3.4 | 0.01 | |
| P. gingivalis + P. intermedia | 4.4 | 0.005 | |
| P. gingivalis + T. denticola | 4.2 | 0.004 | |
| P. gingivalis + T. forsythia | 3.8 | 0.006 | |
| P. gingivalis + P. nigrescens | 2.7 | 0.05 | |
| P. gingivalis + T. forsythia + T. denticola | 4.1 | 0.005 | |
| P. gingivalis + P. nigrescens + T. denticola | 3.3 | 0.03 | |
| Cytomegalovirus | Periodontitis severity | 4.7 | 0.03 |
| P. gingivalis + P. nigrescens | 3.2 | 0.01 | |
| P. gingivalis + P. nigrescens + T. denticola | 2.6 | 0.05 | |
| P. gingivalis + T. forsythia + P. nigrescens | 3.2 | 0.01 |
*Adapted from Contreras et al. (31).
Herpes simplex virus may participate in periodontal disease in a subgroup of individuals (85, 89, 110, 138, 176, 178). Herpes simplex virus‐1, in combination with P. gingivalis, T. forsythia, P. intermedia or A. actinomycetemcomitans, has been linked to periodontitis (85), and, in combination with T. denticola, T. forsythia or Dialister pneumosintes, to periodontitis and necrotic dental pulp (138).
Despite a large body of supporting evidence, well‐designed studies of diverse populations are still needed to corroborate the findings of a relationship between various herpesviral–bacterial consortia and periodontal disease severity. Research is also needed to determine the extent to which herpesviral–bacterial co‐infections in periodontitis represent pathogenetically significant interactions or merely an independent etiologic importance of each of the infectious agents.
Immune responses to periodontal herpesviruses and bacteria
The periodontal immune response to herpesviral and bacterial infections is bifunctional with both tissue‐protective and tissue‐destructive effects, depending upon the type of the infectious agent and the immune competency of the individual. Features of herpesviral and bacterial infections of importance in the etiopathogeny of periodontitis are highlighted below.
General overview
Infectious agents causing periodontitis must be able to colonize periodontal sites, overcome local host defenses, proliferate in periodontal sites and participate in the breakdown of periodontal tissues (192). Also, periodontitis develops in a multistep process that reflects the dynamic interplay between the periodontal infectious agents and the host immune responses.
Successful immune control of a periodontal infection depends on a highly coordinated series of host defenses (36). The host identifies pathogens as nonself by recognizing pathogen‐associated molecular patterns (116). Pathogen recognition receptors and signaling pathways subsequently activate cells of the immune system. Cytokines mediate the interaction and regulation of immune cells. Optimally, the host executes immune responses sufficient to control the pathogens, but also ensures suppression of excessive immune reactions in order to limit the pathological consequences of inflammation. If uncoordinated, the host immune response by itself may cause pathosis.
Immune regulation is mediated by activated CD4 T cells, which, on the basis of characteristic cytokine profiles, are grouped into T helper (Th) type 1 (Th1), Th2, Th17 and induced T regulatory (iTreg) cells (52). Activated T helper cells secrete either pro‐ or anti‐inflammatory cytokines depending on the type and the mode of the immunologic stimulus. Th1 cells drive the type‐1 pathway (‘cellular immunity’) to fight enveloped viruses and intracellular bacterial pathogens, eliminate cancerous cells and stimulate delayed‐type hypersensitivity reactions (93). Activated Th1 cells are characterized as pro‐inflammatory and are a source of interferon‐gamma, interleukin (IL)‐1, IL‐2, IL‐6, IL‐12, IL‐18, IL‐23 and tumor necrosis factor‐alpha. Cytomegalovirus and other herpesviruses induce Th1‐type pro‐inflammatory cytokine responses (163). Pro‐inflammatory cytokines/chemokines promote leukocyte infiltration into the site of infection and play a crucial role in orchestrating the immune response against herpesvirus infections by enhancing the proliferation of T lymphocytes and facilitating cell–cell signaling.
Th2 cells induce the type‐2 pathway (‘humoral immunity’), which is linked to differentiation of B cells and antibody production (93). Antibodies are of critical importance in the defense against nonenveloped viruses and extracellular bacterial pathogens. The Th2 response evokes the secretion of the anti‐inflammatory and immunoregulatory cytokine IL‐10, and of IL‐4, IL‐5, IL‐6, IL‐9 and IL‐13.
However, the Th1/Th2 concept has inconsistencies, and several human and animal cytokine activities fail to fall into exclusive Th1 or Th2 patterns (93). IL‐6 can act as both a pro‐inflammatory and an anti‐inflammatory cytokine. Also, the various cytokines in inflammation have both unique and redundant roles, and can augment or inhibit the effect of other cytokines, complicating the assessment of the biological significance of individual cytokines. In addition, Th1 cells can inhibit Th2 cell differentiation by their expression of interferon‐gamma and IL‐12, while Th2 cells can inhibit Th1 cells by their expression of IL‐4, IL‐10 and transforming growth factor‐beta (233). The balance between Th1 and Th2 immune responses is regulated by iTreg cells and influenced by genetic and environmental factors. The feedback mechanisms among Th1 and Th2 type cytokines aim at amplifying or dampening local inflammatory responses in order to sustain an adequate host response and to safeguard the host against bursts of exaggerated inflammation.
Th1 and Th2 cell responses have received considerable interest in periodontal research (55, 71). As Th1 and Th2 immune responses are partly antagonistic to each other, the net effect of the combined Th1/Th2 immune response may determine whether a periodontal infection leads to limited gingival tissue reaction or to breakdown of periodontal attachment. Pro‐inflammatory cytokines occur at elevated levels in severe periodontitis lesions (62), where they exert immunomodulating activity as well as participate in collagen degradation and bone resorption (92). As periodontitis lesions concurrently harbor herpesviruses (which trigger Th1‐based cellular immunity) and pathogenic bacteria (which induce Th2‐based humoral immunity), the diseased periodontium may experience a changing dominance of either herpesviral or bacterial immunity.
Some studies have suggested that Th1 cells are associated with stable periodontitis and that Th2 cells are associated with disease progression (56, 186). Other studies have reported a predominance of Th1 cells, or a reduced Th2 response, in diseased periodontal tissue (55, 56, 216). These findings are not necessarily in conflict with each other. A predominance of Th1 cells in disease‐stable sites may suggest a successful anti‐herpesvirus defense, and elevated levels of Th1 cells in progressing lesions may reflect an ongoing effort by the host to control an active herpesvirus infection. Similarly, the dispute about a dominance of either T cells or B cells in periodontitis sites may be partly unfounded (11, 71). A preponderance of T cells may reflect the attempt of the host to control an active herpesvirus infection. High B‐cell counts may be caused by specific antibody production or the result of an active Epstein–Barr virus infection, which, in the absence of sufficient cytotoxic T‐cell function, can induce B‐cell proliferation by polyclonal stimulation (21).
Herpesviruses
The eight human members of the herpesvirus family infect parenchymal cells, connective tissue cells, epithelial cells, various hematopoietic cells and other cell types, and can cause a variety of illnesses by mechanisms that are direct, indirect or immunoregulatory (151, 189, 190). The clinical outcome of herpesvirus infections ranges from subclinical or mild disease to encephalitis, pneumonia and other potentially lethal infections, and even to cancer, including lymphoma, sarcoma and carcinoma (190). The great majority of adults are carriers of Epstein–Barr virus and cytomegalovirus. Once infected, a person harbors the herpesvirus for life.
The herpesvirus replication‐cycle includes binding of viral envelope glycoproteins to cell‐membrane receptors, internalization and dismantling of the virus particle, migration of the viral DNA to the cell nucleus, transcription of viral genes, assembly of the virion and viral egress from the infected cell (Fig. 1). Herpesviruses destroy infected cells by active lytic replication. After primary infection, herpesviruses remain latent with limited expression of viral genes, albeit retaining the transcriptional and replicational capacity. Latency/persistence is maintained for Epstein–Barr virus in resting memory B lymphocytes, and for human cytomegalovirus in dendritic cells and in monocytes and their progenitors.
Figure 1.
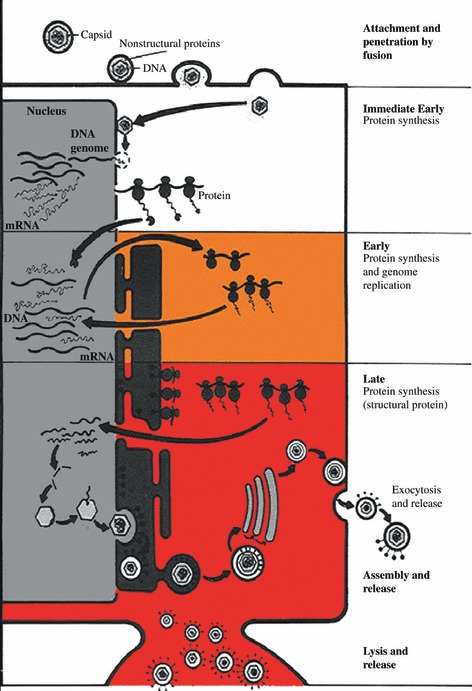
Schematic representation of herpesvirus replication. A herpesvirus virion initiates infection by fusing specific viral glycoproteins on the viral envelope with cellular receptors on the cell surface. After entering the cytoplasm, capsid is transported to the nuclear pore where viral DNA is released into the nucleus. Viral transcription and translation occur in 3 phases; immediate early, early, and late. Immediate early proteins are involved in viral transcriptional regulation and in mobilizing the cellular transcriptional machinery. Early proteins facilitate viral DNA replication. Late proteins are structural proteins of the virus that form empty capsids. Viral DNA is packaged into preformed capsids in the nucleus. Viral glycoproteins and tegument proteins are incorporated into the cellular membrane, and capsids become enveloped to form virions. Virions are transported via the endoplasmic reticulum, and infectious virions can either remain cell associated, spread to uninfected cells via virus‐induced fusion, or can be released from the cell by exocytosis or by cell lysis.
Active herpesvirus infections evoke strong innate and adaptive immune responses, which include both immune activation and immune suppression (34, 131, 164). Key effector cells of the innate immune system are dendritic cells, monocytes/macrophages and natural killer cells, whose function is to limit the viral burden until cells of the adaptive immunity become available to suppress the infection. The effector cells recognize viral proteins via toll‐like receptors, natural killer cell receptors or other pattern recognition receptors. Herpesvirus DNA reacts with toll‐like receptor 9, which is significantly up‐regulated in periodontitis lesions compared with gingivitis lesions (88). Cells of innate immunity employ cytokine secretion and cell‐mediated cytotoxicity as the primary anti‐herpesvirus effector mechanisms. Macrophages and polymorphonuclear leukocytes can destroy antibody‐coated virions or virus‐infected cells via reactive oxygen species, nitric oxide and activated caspases. Natural killer cells are an important source of interferon‐gamma and are able to kill herpesvirus‐infected cells via virus‐specific antibody‐dependent cell‐mediated cytotoxicity or via antibody‐independent mechanisms. In addition, natural killer cells share similarities with cytotoxic T cells and may play a role in the adaptive immunity (206).
The phagocytic cells internalize, process and express herpesvirus‐derived immunogenic peptides on their surface which, after linkage to major histocompatibility complex (MHC) molecules, can attach to and activate Th1 cells of the adaptive immune system. Cytomegalovirus can have a profound influence on the immune system of healthy individuals (25), and cytomegalovirus reactivity has been detected in as many as 30–50% (up to 80%) of the T cells of cytomegalovirus‐seropositive elderly individuals (200). Recognition of the herpesvirus proteins causes the induction of the pro‐inflammatory transcription factor nuclear factor‐kappaB (NF‐κB) with a subsequent release of cytokines and mobilization of CD8+ T cells (116). Activated CD8+ T cells differentiate into virus‐specific cytotoxic T cells capable of recognizing and killing cells carrying viral peptides. The release of CD8+ T‐cell‐associated antiviral cytokines, such as interferon‐gamma and tumor necrosis factor‐alpha, may also inhibit viral replication without killing the infected cell (66).
Herpesvirus infections in immunocompetent individuals also induce antibody production against herpesvirus proteins (209), and patients with periodontitis exhibit an elevated level of antibodies against herpesviruses (77, 88). However, antibodies against herpesviruses and against many other enveloped viruses do not ensure a favorable clinical outcome.
Herpesviruses have developed strategies to down‐regulate antiviral host defenses in order to persist in the midst of intense inflammation (154). Immunosuppression may thus take place, even in the presence of a substantial immune activation. Herpesviruses encode immunoevasive glycoproteins that interfere with antigen presentation, T‐cell immune surveillance and natural killer cell function (130). The viral proteins seek to alter or mimic MHC protein function, leukocyte activation and migration, induction and activity of cytokines and interferons, antibody‐based defense mechanisms, and host cell susceptibility to apoptosis (programmed cell death), which is the principal mode of physiologic elimination of cells (130).
Despite elaborate immune‐evasion efforts by herpesviruses, the host immune system generally prevails in immunocompetent subjects, perhaps because of the host immune‐sensing mechanisms that terminate viral gene expression before the assembly of infectious virions (161). However, herpesvirus disease incidence is elevated in immunologically immature subjects, and in individuals who are immunosuppressed as a result of advancing age (immunosenescence), diseases [e.g. HIV/acquired immune‐deficiency syndrome (AIDS)] or treatments (e.g. cancer chemotherapy, radiotherapy, pharmacological immunosuppression with organ transplantation, high‐dose corticosteroids).
Bacteria
Bacterial infections evoke functionalities of both the innate immune system and the adaptive immune system. Bacterial species attach to specific toll‐like receptors to establish some degree of specificity in the innate immune system and subsequently in the adaptive immune system (76, 94). Bacterial pathogens detected by toll‐like receptors on the surface of macrophages activate NF‐κB‐mediated transcription of cytokines and chemokines (136). Macrophages release cytokines and chemokines to recruit neutrophils in the innate immune system and serve as antigen‐presenting cells for lymphocytes in the adaptive immune system.
Neutrophils comprise more than 90% of the inflammatory cells in periodontal pockets (45). Their importance in periodontal defense is evidenced by the observation of severe periodontitis in most subjects exhibiting major neutrophil deficiencies (38). Neutrophils phagocytize and kill ingested bacteria by means of reactive oxygen species (e.g. the myeloperoxidase system and hypochlorite), antimicrobial proteins (e.g. defensins, lysozyme and lactoferrin), degradative enzymes (e.g. elastase and cathepsin G) and other microbicidal pathways (38).
Extracellular pathogenic bacteria activate Th2 cells, which release anti‐inflammatory cytokines and commit B cells to antibody production. P. gingivalis seems to evoke primarily a Th2‐type response in periodontitis patients (79). Antibacterial antibodies of the IgG1 subclass play important roles in opsonization and complement activation (38). Lipopolysaccharide of P. gingivalis and of other periodontopathic bacteria can also activate complement via the alternative pathway and induce the release of pro‐inflammatory cytokines (41). Complement assists antibodies by acting as an opsonin, by lysing bacterial cells and by attracting lymphocytes and neutrophils to the site of infection.
Intracellular bacteria trigger a Th1‐mediated immune response, which includes the release of interferon‐gamma, pro‐inflammatory cytokines and the IgG2 antibody isotype (48, 220). IgG2 serum antibody occurs at high levels in patients with localized aggressive (juvenile) periodontitis and, despite being a less efficient opsonin than IgG1 and IgG3, seems to protect against further tissue destruction (181). P. gingivalis (101, 106), A. actinomycetemcomitans (26, 169) and other periodontal species (27, 222, 225) have the ability to invade cells of the periodontium and thus may trigger a Th1, as well as a Th2, immune response. P. gingivalis‐specific T cells can produce both Th1 and Th2 cytokines irrespectively of the type of antigen‐presenting cells (54).
Pathogenic interactions among infectious agents in periodontal diseases
Epstein–Barr virus–cytomegalovirus co‐infection tends to be associated with severe types of oral disease, including aggressive periodontitis (89, 179, 219, 239), chronic periodontitis (22, 235), periodontal abscesses (180), acute necrotizing ulcerative gingivitis (30), symptomatic peri‐apical lesions (194) and oral ulcers (210). Cytomegalovirus–herpes simplex virus co‐infection has also been associated with increased gingival inflammation and periodontal attachment loss (110). Persons exhibiting a high rate of shedding of Epstein–Barr virus into saliva tend to reveal salivary cytomegalovirus DNA more often than those with a low rate of Epstein–Barr virus shedding, which may be suggestive of a cooperative relationship between the two viruses (64). Infants and children infected with both Epstein–Barr virus and cytomegalovirus have shown stronger T‐cell responses and more severe disease than children mono‐infected with either of the viruses (226). Feline calicivirus and feline herpesvirus were both shed in 88% of cats with chronic gingivostomatitis compared to 21% of cats without chronic oral inflammatory disease (112).
A concomitant infection with two herpesviruses may activate latent viral genomes by the mechanism of reciprocal transactivation, which connotes that gene products of one virus trigger the transcription of another virus (231). Two active viruses, each providing their own unique set of virulence factors, can result in a particularly severe disruption of the immune system and accelerate a disease process (213). Herpesvirus re‐activation causes a major spike in cytotoxic T cells and pro‐inflammatory cytokines (132), but also produces virus‐derived homologues of human IL‐10 (108) and other inhibitors of the antiviral Th1 cell‐mediated defense (195). Cytomegalovirus IL‐10 suppresses NF‐κB activation and subsequently transcription of tumor necrosis factor‐alpha and IL‐1beta (135). Specific genotypes of IL‐10 are correlated with an increased risk of progressive periodontitis (35). Other mechanisms of interaction between two viruses include the enhanced expression of viral receptors and co‐receptors in target cells as well as the production of superantigens (120).
Herpesviruses and specific bacteria in aggregate are also associated with enhanced severity of periodontal disease (179). Herpesviruses may aid in the initial colonization and upgrowth of periodontopathic bacteria. An active human cytomegalovirus infection of primary periodontal pocket epithelial cells or HeLa cells enhances the adherence of A. actinomycetemcomitans in a dose‐dependent mode (217); viral infections may destroy epithelial cells and expose new bacterial attachment sites in the basement membrane; and herpesvirus glycoproteins expressed on the surface of infected host cells may serve as receptors for bacteria. In addition, as A. actinomycetemcomitans can bind to the Fc part of the IgG molecule (221), the organism may attach to antibodies directed against the herpesvirus‐derived glycoproteins. Consistent with the concept of herpesviral–bacterial synergism, the initial phases of localized aggressive (juvenile) periodontitis demonstrate an active cytomegalovirus infection (219) and a remarkable increase in A. actinomycetemcomitans subgingival counts (197). Cytomegalovirus has, in several animal models, shown the ability to impair neutrophil chemotaxis, phagocytosis, oxidative burst and intracellular killing capacity (2); some of these neutrophil defects have also been described in localized aggressive periodontitis (182). Individuals who show an absence of herpesviral infection or of herpesviral re‐activation may exhibit a normal periodontium or minimal or nonprogressive disease, even in the presence of periodontopathic bacteria.
Herpesviruses can compromise the immune control of periodontopathic bacteria by a variety of mechanisms. Polymorphonuclear leukocytes seem to exhibit less efficient chemotaxis and bactericidal capacity in periodontitis patients with subgingival herpesviruses than in herpesvirus‐free subjects (142). Anti‐herpesvirus cytotoxic/suppressor T cells, which are present at elevated levels in aggressive periodontitis lesions (187), may adversely affect mammalian cells involved in the antibacterial periodontal defense. Th1 cytokines associated with active herpesvirus infections reduce Th2‐cell responses and thereby the antibody‐mediated control of pathogenic bacteria. Herpesviruses are capable of subverting macrophage (51, 63, 109), complement (111) and neutrophil (1) functions of importance in the antibacterial host defense. P. gingivalis and other exogenous‐like species, which may be primarily controlled by antibody‐mediated mechanisms (159, 215), may benefit most from a reduction in antibacterial immunity and outgrow co‐existing indigenous microorganisms. Taken together, an active herpesvirus infection has the potential to impair antibacterial host defenses and may trigger a microbial shift towards a more virulent subgingival microbiota.
The lipopolysaccharide of gram‐negative periodontopathic bacteria, most prominently P. gingivalis, can induce a Th1‐cell type release of pro‐inflammatory cytokines (41, 223), and may co‐operate with cytomegalovirus in stimulating IL‐1beta gene transcription (229). As Th1 cell activities can inhibit Th2 antibacterial immunity, the lipopolysaccharide‐induced Th1 cell response may partly constitute an immune‐evasive maneuver by periodontopathic bacteria in their efforts to survive in a hostile environment. On the other hand, the Th1 immune response may help to control P. gingivalis infections when they occur intracellularly (101, 106). Also, as herpesviruses may induce the overgrowth of P. gingivalis and other pathogenic species (179, 193, 196), the Th1‐mediated anti‐herpesvirus immune response may indirectly aid in containing the population of periodontopathic bacteria.
It is equally conceivable that a bacterial infection can pave the way for a herpesvirus clinical infection. P. gingivalis sonicate has the potential to re‐activate Epstein–Barr virus (205). The etiopathogenic model for periodontitis, detailed below, proposes that a bacterial–viral–bacterial sequential infection gives rise to the disease. Dental biofilm bacteria initiate gingivitis with an influx of herpesvirus‐infected monocyte/macrophages, T cells or B cells (32). A subsequent immunosuppressive event may then re‐activate latent herpesviruses, resulting in the release of pro‐inflammatory cytokines and matrix metalloproteinases, and in the overgrowth of periodontopathic bacteria.
Proteolytic enzymes of periodontal bacteria can degrade pro‐inflammatory cytokines and other host‐defense systems (83, 212), thereby potentially activating a co‐existing latent herpesvirus infection. Human gingival epithelial cells challenged with P. gingivalis showed a fourfold increase in the primary cytokine IL‐1beta level, but the related secondary cytokines IL‐6 or IL‐8 were virtually undetectable as a result of direct degradation by P. gingivalis proteases, with lysine gingipain being the most effective (202). Thus, in addition to subverting host defenses against bacterial infections (153), gingipains may exert periodontopathogenicity by perturbing cytokine‐mediated antiviral host responses.
Lastly, herpesviruses and bacteria may interact bidirectionally, as shown in a study of infected wound chambers in mice (203), adding another level of complexity to the analysis of the pathophysiology of periodontitis. Mice co‐infected with murine cytomegalovirus and P. gingivalis in wound chambers exhibited significantly higher mortality than mice infected with either murine cytomegalovirus or P. gingivalis, or simultaneously with murine cytomegalovirus and E. coli (203). The increase in cytomegalovirus pathogenicity was related to an ability of P. gingivalis to suppress interferon‐gamma, perhaps because of the proteolytic activity of the organism. A low level of interferon‐gamma was found despite the capacity of cytomegalovirus to trigger the induction of interferon‐gamma, and despite the ability of bacterial lipopolysaccharide, via cytokine simulation, to induce interferon‐producing CD8+ T cells (160). Adding further to the complexity, cytomegalovirus encodes genes capable of inhibiting the interferon response and antiviral processes (37).
In teleological terms, periodontitis can perhaps be viewed as a by‐product of the efforts by the host to control periodontal herpesvirus infections and avoid systemic viral diseases. Periodontal pro‐inflammatory cytokines, in spite of being capable of inducing collagen degradation and alveolar bone resorption (92), may actually be overall beneficial by helping to prevent activation and systemic dissemination of virulent viruses (65). Similarly, although CD8+ T cells may impede antibacterial defenses, they also confer a critical antiviral function. Periodontitis may represent an excellent example of how host immune responses can exert simultaneously protective and destructive functionalities.
Herpesviral–bacterial model of periodontitis
The finding of abundant herpesviruses in periodontitis lesions redefines the pathogenic paradigm of the disease. Fig. 2 describes a model for the development of periodontitis, which as its core has a sequential infectious process that proceeds from bacteria to herpesvirus to bacteria (191). Initially, bacteria in the dental biofilm induce gingivitis, which permits latent herpesviruses, embedded in the DNA of macrophages, T lymphocytes and B lymphocytes, to infiltrate the periodontium (32). Cytomegalovirus can replicate in gingival tissue (68), which may help to sustain the periodontal infection. Re‐activation of the latent herpesviruses may occur spontaneously or during periods of decreased host defense, resulting from drug‐induced immunosuppression, concurrent infection, unusual and prolonged emotional stress, hormonal changes, physical trauma, etc. Probably not coincidentally, most herpesvirus‐activating factors are also suspected risk factors/indicators for periodontitis (162).
Figure 2.
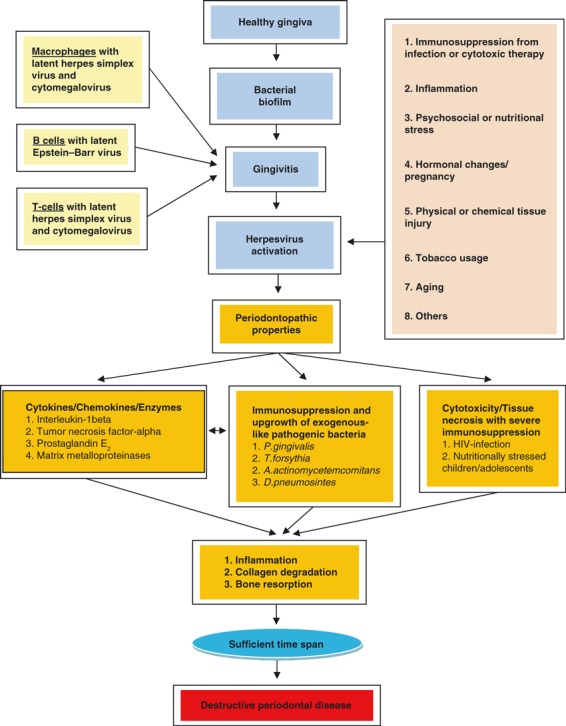
Herpesviral‐bacterial model of periodontitis.
In response to the active herpesvirus infection, the host elicits a robust T‐cell‐mediated immune response, comprised primarily of CD8+ T cells. To counteract the hostile host environment, herpesviruses in turn execute strategies to down‐regulate antiviral host defenses. Herpesviruses evade immune responses by disintegrating components of the MHC and interfering with antigen presentation, by silencing natural killer cells, by expressing a viral homolog of IL‐10, by diverting potent cytokine responses and by inhibiting apoptosis (189). The encounter between antiviral host defenses and virally mediated anti‐host responses results in a major release of pro‐inflammatory cytokines that have the potential to activate osteoclasts (13, 71) and to impair antibody‐mediated host defenses against exogenous‐like bacterial species, such as P. gingivalis and A. actinomycetemcomitans (179). The ensuing increase in pathogenic bacteria provides additional mechanisms of periodontal tissue destruction (78).
Cytomegalovirus or other herpesviruses can exert acute cytopathogenic effects on fibroblasts, epithelial cells, keratinocytes, endothelial cells, inflammatory cells and bone cells (17). An active herpesviral infection in severely immunocompromised patients may directly destroy periodontal cells and tissue by cytotoxic mechanisms, as seen in patients with necrotizing ulcerative gingivitis and noma. Herpesvirus infections may participate in oral collagen degradation, as suggested in in vivo (172) and in vitro (14) studies, and potentially interfere with periodontal tissue turnover and healing. Herpesvirus infections have also been related to diminished repair in periodontal guided tissue regeneration (199) and to dry socket formation after tooth extraction (73, 74).
In the herpesviral–bacterial model of periodontitis, herpesvirus‐related cytopathogenic effects, immune evasion, immunopathogenicity, latency, re‐activation from latency and tissue/site tropism comprise important aspects of periodontal pathosis. It is likely that the early stages of periodontitis in immunologically naïve hosts involve an active herpesviral infection that primarily causes cytopathogenic effects, whereas most clinical manifestations in immunocompetent individuals are secondary to cellular or humoral immune responses. The proposed model may help to clarify at least some of the clinical features of periodontitis (179, 189). The propensity for site tropism of herpesviruses may explain why periodontal tissue destruction can differ markedly from tooth‐to‐tooth in the same patient or from surface‐to‐surface in individual teeth. A vigorous anti‐herpesvirus host defense may ensure a prolonged period of periodontal stability, even in the presence of virulent bacteria. Herpesvirus re‐activation from the state of latency may trigger a burst of periodontal tissue damage and progressive disease. However, most immunocompetent individuals experience episodes of oral herpesvirus re‐activation lasting only a few hours or a few days (119), which is probably too short a time span to initiate or aggravate clinical periodontal disease.
Therapeutic implications
Conventional periodontal therapy can reduce the periodontal load of herpesviruses. Mechanical debridement has suppressed subgingival Epstein–Barr virus to undetectable levels in 12 of 21 patients (234), and has decreased subgingival Epstein–Barr virus genome‐copies by sixfold and subgingival cytomegalovirus genome‐copies by 38‐fold (177). After repeated debridement, 24 patients with periodontitis yielded no cytomegalovirus, but were found to have Epstein–Barr virus and herpesvirus‐7 (168), suggesting that cytomegalovirus is particularly susceptible to the effects of periodontal therapy. In a patient with Papillon–Lefèvre syndrome, mechanical debridement and systemic amoxicillin–metronidazole suppressed subgingival Epstein–Barr virus, cytomegalovirus and A. actinomycetemcomitans to undetectable levels and prevented further loss of periodontal attachment (143). The decrease in post‐treatment herpesvirus counts is probably caused by a reduction in gingivitis and thus in the numbers of virally infected inflammatory cells. Similarly, the low herpesvirus counts in healthy periodontal sites may be the result of a virtual absence of infected inflammatory cells. Cytomegalovirus was also not detected in healthy peri‐implant sites (139).
Saliva can contain high genome‐copy counts of herpesviruses. Medically healthy persons have been shown to contain 3.6 × 102 to 1.6 × 109 copies/ml of Epstein–Barr virus in saliva (173) and from 6 to 2.2 × 106 per 0.5 μg of DNA (228). Salivary cytomegalovirus DNA was detected in one‐half of periodontitis patients at levels of 3.3 × 103–4.2 × 104 copies/ml, whereas the virus was not detected in the saliva of periodontally normal individuals or in complete denture wearers (173), affirming the close relationship between cytomegalovirus and periodontitis. Herpesvirus‐6 and herpesvirus‐7 can occur in saliva with prevalences exceeding 90% and in concentrations of several million genome‐copies (126). Herpesvirus‐6 was detected in the whole saliva of 68% of healthy volunteers from Brazil (152). Herpesvirus‐8 reached salivary loads of 2.6–4.1 × 106 genome‐copies/ml in a renal allograft recipient (4). HIV‐infected individuals harbor higher salivary cytomegalovirus counts than non‐HIV‐infected persons (114, 115), and HIV‐infected individuals receiving antiretroviral therapy demonstrate higher salivary prevalence and copy counts of Epstein–Barr virus and other herpesviruses than normal control subjects (127). HIV‐infected individuals have revealed salivary median copy counts of 5.3 × 105/ml for Epstein–Barr virus and 3.3 × 103/ml for cytomegalovirus (64).
Conventional periodontal treatment has reduced salivary Epstein–Barr virus genome‐copy counts by 13‐fold and salivary cytomegalovirus copy counts by 65‐fold (177), salivary Epstein–Barr virus counts in two children with Kostmann syndrome by more than 100‐fold (241), and the average Epstein–Barr virus counts per ml of saliva from 946,000 to 9,010, and with six of 11 (54%) study patients with salivary Epstein–Barr virus pre‐pretreatment not showing the virus post‐treatment (81). Several patients in the above studies exhibited gingival inflammation post‐treatment; more efficacious anti‐gingivitis measures may have decreased the salivary herpesvirus counts even further (188). These treatment data suggest that diseased periodontal sites are an important source of salivary herpesviruses. The potential of periodontal therapy to decrease herpesvirus levels in saliva may reduce the risk of herpesvirus transmission and herpesvirus‐related diseases among close acquaintances.
The herpesviral–bacterial model of periodontitis provides a rationale for considering new approaches to disease prevention and treatment. Sunde et al. (208) treated a patient, who exhibited refractory periodontitis and high Epstein–Barr virus subgingival copy counts, with the anti‐herpesvirus drug, valacyclovir HCl, 500 mg twice a day for 10 days. The treatment suppressed subgingival Epstein–Barr virus to undetectable levels for at least 1 year and resulted in a ‘dramatic’ clinical improvement (208). Sunde et al. (208) proposed employing virus screening in periodontal treatment to determine when antiviral intervention is appropriate and if it has succeeded. Periodontal viruses may be identified successfully by using diagnostic DNA microarrays that are able to detect simultaneously herpes simplex virus types 1 and 2, Epstein–Barr virus, and cytomegalovirus as well as other viruses (134); or by using multiplex real‐time PCR techniques to quantify simultaneously the number of genome‐copies of herpes simplex virus, varicella–zoster virus, Epstein–Barr virus, cytomegalovirus and human herpesvirus‐6 (49, 67). Newer metagenomic pyrosequencing techniques may be even more proficient in identifying known and unknown periodontal viruses.
Anti‐herpesvirus chemotherapy can also decrease the salivary viral load. A short course of valacyclovir, 2 g twice on the day of treatment and 1 g twice the following day, resulted in a significant decrease in the salivary occurrence of Epstein–Barr virus compared with controls (126). Valacyclovir, 500 mg orally twice daily for 1 month, given to elite male distance runners, reduced the salivary load of Epstein–Barr virus by 82% compared with placebo (33). Valacyclovir therapy, 3 g per day for 14 days, resulted in a reduction, of more than 100‐fold, of Epstein–Barr virus genome‐copies in oral wash fluid of patients with acute infectious mononucleosis (9).
Chemotherapeutics are effective against viruses in the lytic phase, but not against viruses in the latent phase, limiting their potential use to disease‐active infections. Acyclovir types of drugs are acyclic nucleoside analogues that inhibit herpesviral DNA polymerase and replication of the viral genome. The orally administered acyclovir prodrug, valacyclovir, can reach serum concentrations similar to those of intravenously administered acyclovir and is prescribed for a variety of herpesviral diseases (149). Prolonged treatment with valacyclovir at dosages of 500–1000 mg/day is well tolerated, perhaps except in immunosuppressed individuals, and the adverse events are infrequent and generally mild, with headache being reported most often (6). To date, resistance to valacyclovir has not been clinically significant. However, randomized controlled trials are needed before anti‐herpesviral chemotherapy can be considered as standard clinical practice in the treatment of advanced periodontitis.
Future management of periodontal diseases may benefit from anti‐herpesviral immunotherapeutics: either prophylactic vaccines, which harness the immune system of healthy subjects to prevent infection with disease‐causing viruses; or therapeutic vaccines, which stimulate the immune system into combating existing viruses and disease. An efficacious vaccine against herpesviruses may also provide clinical proof‐of‐principle of the periodontopathic importance of the viruses. The notion of herpesviral–bacterial synergism in periodontitis implies that vaccination against herpesviruses can also contribute to the control of periodontopathic bacteria. One difficulty to overcome with prophylactic vaccination is declining immunity over time, which carries an increased risk of delayed‐onset primary herpesvirus diseases with increased morbidity. The US Institute of Medicine has assigned high priority to the development of vaccines against herpes simplex virus, Epstein–Barr virus and cytomegalovirus, to be given to 12‐year‐old children (204).
Summary
The etiopathogenesis of periodontitis includes virulence factors of herpesviruses and bacteria, host immune responses against viral and bacterial infections, and manipulation of host‐cell processes by the infectious agents. Herpesviruses may induce periodontitis by activating specific tissue‐destroying pathways of the immune system and by predisposing an individual to bacterial carriage or increased bacterial load. However, the molecular contribution of herpesviruses versus bacteria to periodontal pathosis remains little understood.
The inflamed periodontium appears to be a major site for Epstein–Barr virus and cytomegalovirus accumulation and re‐activation, especially in the progressive phase of periodontal disease. A single advanced periodontitis lesion may contain up to 100 million genomic copies of herpesviruses, approaching the total viable counts of bacteria. Immunosuppressive factors are potential triggers of herpesvirus re‐activation and, perhaps for that reason, are also major risk factors for periodontitis. An active herpesvirus infection in the periodontium may give rise to systemic viremia, but the magnitude and duration of this are unknown.
An active herpesvirus infection correlates with periodontitis disease activity and may be a major contributor to the periodontal immune response. Herpesviruses are potent inducers of pro‐inflammatory cytokines that have the potential to activate osteoclasts and matrix metalloproteinases. An active herpesvirus infection can also impair antibacterial immune mechanisms and potentially cause an upgrowth of periodontopathic bacteria. Some periodontopathic bacteria may re‐activate a latent herpesvirus infection. Synergism among herpesviruses and bacteria may play an important role in the onset and progression of periodontitis. The interplay of herpesviruses and bacteria in periodontitis may be compared to a marionette theater where the puppeteer is the virus and the puppets are the bacteria. Even if the puppeteer (virus) controls the performance (disease), both the puppeteer and the puppets are necessary for the marionette theater to function.
Obviously, it is difficult to link a herpesvirus infection that occurs in childhood to periodontitis that debuts several decades later. An important question is whether the herpesvirus–periodontitis association is etiologically based or is merely an epiphenomenon to gingival inflammation. Studies on herpesviral etiology of medical diseases face similar difficulties in delineating cause and effect. To distinguish between causality and correlation, information is needed on the extent to which an active herpesvirus infection gives rise to destructive periodontal disease, and the extent to which disease‐active periodontitis may activate a latent herpesvirus infection. A causal relationship may be inferred if the removal of herpesviruses by specific antiviral medication arrests, reverses or prevents periodontitis. Studies assessing temporal and histological correlations may also help to elucidate the role of herpesviruses in periodontal disease. Such studies may take advantage of molecular techniques able to quantify herpesvirus loads and relate either active or latent herpesvirus infection to periodontitis‐disease activity. However, the definitive answer to the periodontopathic significance of herpesviruses will probably have to await the development of efficacious anti‐herpesvirus vaccines.
The notion of a herpesviral–bacterial co‐infection in periodontitis may turn out to be the pathogenetic Rosetta stone that unlocks many of the intricacies of the disease. Herpesvirus‐induced periodontitis implies that anti‐herpesvirus immunity constitutes an important aspect of achieving a long‐lasting state of stable periodontal conditions. The development of herpesvirus vaccines in the not too distant future makes the topic of periodontopathic herpesviruses particularly intriguing. Control of herpesviruses by vaccination may foreshadow a future with a diminishing role for the traditional periodontal therapies of surgery and antibiotics. It is hoped that the issues raised in this review will help to steer periodontal research into new fertile fields of investigation.
References
- 1. Abramson JS, Mills EL. Depression of neutrophil function induced by viruses and its role in secondary microbial infections. Rev Infect Dis 1988: 10: 326–341. [DOI] [PubMed] [Google Scholar]
- 2. Abramson JS, Wheeler JG. Virus‐induced neutrophil dysfunction: role in the pathogenesis of bacterial infections. Pediatr Infect Dis J 1994: 13: 643–652. [PubMed] [Google Scholar]
- 3. Adjei AA, Armah HB, Gbagbo F, Boamah I, Adu‐Gyamfi C, Asare I. Seroprevalence of HHV‐8, CMV, and EBV among the general population in Ghana, West Africa. BMC Infect Dis 2008: 8: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al‐Otaibi LM, Al‐Sulaiman MH, Teo CG, Porter SR. Extensive oral shedding of human herpesvirus 8 in a renal allograft recipient. Oral Microbiol Immunol 2009: 24: 109–115. [DOI] [PubMed] [Google Scholar]
- 5. Alho OP. Viral infections and susceptibility to recurrent sinusitis. Curr Allergy Asthma Rep 2007: 5: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bakaletz LO. Viral potentiation of bacterial superinfection of the respiratory tract. Trends Microbiol 1995: 3: 110–114. [DOI] [PubMed] [Google Scholar]
- 7. Bakaletz LO. Otitis media In: Brogden KA, Guthmiller JM. editors. Polymicrobial diseases. Washington, DC: American Society for Microbiology, 2002: 259–298. [Google Scholar]
- 8. Baker DA, Blythe JG, Miller JM. Once‐daily valacyclovir hydrochloride for suppression of recurrent genital herpes. Obstet Gynecol 1999: 94: 103–106. [DOI] [PubMed] [Google Scholar]
- 9. Balfour HH Jr, Hokanson KM, Schacherer RM, Fietzer CM, Schmeling DO, Holman CJ, Vezina HE, Brundage RC. A virologic pilot study of valacyclovir in infectious mononucleosis. J Clin Virol 2007: 39: 16–21. [DOI] [PubMed] [Google Scholar]
- 10. Barton ES, White DW, Cathelyn JS, Brett‐McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW IV. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 2007: 447: 326–329. [DOI] [PubMed] [Google Scholar]
- 11. Berglundh T, Donati M, Zitzmann N. B cells in periodontitis: friends or enemies? Periodontol 2000 2007: 45: 51–66. [DOI] [PubMed] [Google Scholar]
- 12. Boeckh M, Nichols WG. Immunosuppressive effects of beta‐herpesviruses. Herpes 2003: 10: 12–16. [PubMed] [Google Scholar]
- 13. Botero JE, Contreras A, Parra B. Profiling of inflammatory cytokines produced by gingival fibroblasts after human cytomegalovirus infection. Oral Microbiol Immunol 2008: 23: 291–298. [DOI] [PubMed] [Google Scholar]
- 14. Botero JE, Contreras A, Parra B. Effects of cytomegalovirus infection on the mRNA expression of collagens and matrix metalloproteinases in gingival fibroblasts. J Periodontal Res 2008: 43: 649–657. [DOI] [PubMed] [Google Scholar]
- 15. Botero JE, Parra B, Jaramillo A, Contreras A. Subgingival human cytomegalovirus correlates with increased clinical periodontal parameters and bacterial coinfection in periodontitis. J Periodontol 2007: 78: 2303–2310. [DOI] [PubMed] [Google Scholar]
- 16. Botero JE, Vidal C, Contreras A, Parra B. Comparison of nested polymerase chain reaction (PCR), real‐time PCR and viral culture for the detection of cytomegalovirus in subgingival samples. Oral Microbiol Immunol 2008: 23: 239–244. [DOI] [PubMed] [Google Scholar]
- 17. Britt WJ, Alford CA. Cytomegalovirus In: Fields BN, Knipe DM, Howley PM. editors. Fields Virology, 3rd edn Philadelphia: Lippincott‐Raven, 1996: 2493–2524. [Google Scholar]
- 18. Brook I. The association of anaerobic bacteria with infectious mononucleosis. Anaerobe 2005: 11: 308–311. [DOI] [PubMed] [Google Scholar]
- 19. Brook I. The role of bacterial interference in otitis, sinusitis and tonsillitis. Otolaryngol Head Neck Surg 2005: 133: 139–146. [DOI] [PubMed] [Google Scholar]
- 20. Bulut Y, Karlidag T, Seyrek A, Keles E, Toraman ZA. Presence of herpesviruses in middle ear fluid of children with otitis media with effusion. Pediatr Int 2007: 49: 36–39. [DOI] [PubMed] [Google Scholar]
- 21. Cesarman E. Epstein‐Barr virus (EBV) and lymphomagenesis. Front Biosci 2002: 7: e58–65. [DOI] [PubMed] [Google Scholar]
- 22. Chalabi M, Moghim S, Mogharehabed A, Najafi F, Rezaie F. EBV and CMV in chronic periodontitis: a prevalence study. Arch Virol 2008: 153: 1917–1919. [DOI] [PubMed] [Google Scholar]
- 23. Chen V, Chen Y, Li H, Kent K, Baumgartner JC, Machida CA. Herpesviruses in abscesses and cellulitis of endodontic origin. J Endod 2009: 35: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen LL, Sun WL, Yan J, Yu ZS. [Correlation between infection of different glycoprotein B genotypes of human cytomegalovirus and human chronic periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi 2006: 41: 212–215 (Chinese). [PubMed] [Google Scholar]
- 25. Chidrawar S, Khan N, Wei W, McLarnon A, Smith N, Nayak L, Moss P. Cytomegalovirus‐seropositivity has a profound influence on the magnitude of major lymphoid subsets within healthy individuals. Clin Exp Immunol 2009: 155: 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christersson LA, Albini B, Zambon JJ, Wikesjö UM, Genco RJ. Tissue localization of Actinobacillus actinomycetemcomitans in human periodontitis. I. Light, immunofluorescence and electron microscopic studies. J Periodontol 1987: 58: 529–539. [DOI] [PubMed] [Google Scholar]
- 27. Colombo AV, Silva CM, Haffajee A, Colombo AP. Identification of oral bacteria associated with crevicular epithelial cells from chronic periodontitis lesions. J Med Microbiol 2006: 55: 609–615. [DOI] [PubMed] [Google Scholar]
- 28. Combs DR, Reilly EA, Dawson DR III, Avdiushko SA, Danaher RJ, Miller CS. Detection of human cytomegalovirus in dental plaque from individual periodontal sites by real‐time polymerase chain reaction. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008: 106: 840–844. [DOI] [PubMed] [Google Scholar]
- 29. Connor E, Powell K. Fulminant pneumonia caused by concomitant infection with influenza B virus and Staphylococcus aureus . J Pediatr 1985: 106: 447–550. [DOI] [PubMed] [Google Scholar]
- 30. Contreras A, Falkler WA Jr, Enwonwu CO, Idigbe EO, Savage KO, Afolabi MB, Onwujekwe D, Rams TE, Slots J. Human Herpesviridae in acute necrotizing ulcerative gingivitis in children in Nigeria. Oral Microbiol Immunol 1997: 12: 259–265. [DOI] [PubMed] [Google Scholar]
- 31. Contreras A, Umeda M, Chen C, Bakker I, Morrison JL, Slots J. Relationship between herpesviruses and adult periodontitis and periodontopathic bacteria. J Periodontol 1999: 70: 478–484. [DOI] [PubMed] [Google Scholar]
- 32. Contreras A, Zadeh HH, Nowzari H, Slots J. Herpesvirus infection of inflammatory cells in human periodontitis. Oral Microbiol Immunol 1999: 14: 206–212. [DOI] [PubMed] [Google Scholar]
- 33. Cox AJ, Gleeson M, Pyne DB, Saunders PU, Clancy RL, Fricker PA. Valtrex therapy for Epstein–Barr virus reactivation and upper respiratory symptoms in elite runners. Med Sci Sports Exerc 2004: 36: 1104–1110. [DOI] [PubMed] [Google Scholar]
- 34. Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 2009: 22: 76–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cullinan MP, Westerman B, Hamlet SM, Palmer JE, Faddy MJ, Seymour GJ, Middleton PG, Taylor JJ. Progression of periodontal disease and interleukin‐10 gene polymorphism. J Periodontal Res 2008: 43: 328–333. [DOI] [PubMed] [Google Scholar]
- 36. Cutler CW, Teng YT. Oral mucosal dendritic cells and periodontitis: many sides of the same coin with new twists. Periodontol 2000 2007: 45: 35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeFilippis VR. Induction and evasion of the type I interferon response by cytomegaloviruses. Adv Exp Med Biol 2007: 598: 309–324. [DOI] [PubMed] [Google Scholar]
- 38. Dennison DK, Van Dyke TE. The acute inflammatory response and the role of phagocytic cells in periodontal health and disease. Periodontol 2000 1997: 14: 54–78. [DOI] [PubMed] [Google Scholar]
- 39. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, Van Rijt LS, Lambrecht BN, Sirard JC, Hussell T. Sustained desensitization to bacterial Toll‐like receptor ligands after resolution of respiratory influenza infection. J Exp Med 2008: 205: 323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ding F, Feng XH, Meng HX, Zhao YB, Zhang L, Lu RF, Chen ZB. Relationship between herpesviruses and periodontal pathogenic bacteria in subgingival plaque. Beijing Da Xue Xue Bao 2008: 40: 318–322 (Chinese). [PubMed] [Google Scholar]
- 41. Dixon DR, Bainbridge BW, Darveau RP. Modulation of the innate immune response within the periodontium. Periodontol 2000 2004: 35: 53–74. [DOI] [PubMed] [Google Scholar]
- 42. Djuric M, Jankovic L, Jovanovic T, Pavlica D, Brkic S, Knezevic A, Markovic D, Milasin J. Prevalence of oral herpes simplex virus reactivation in cancer patients: a comparison of different techniques of viral detection. J Oral Pathol Med 2009: 38: 167–173. [DOI] [PubMed] [Google Scholar]
- 43. Dowd JB, Aiello AE, Alley DE. Socioeconomic disparities in the seroprevalence of cytomegalovirus infection in the US population: NHANES III. Epidemiol Infect 2008: 16: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dowd JB, Haan MN, Blythe L, Moore K, Aiello AE. Socioeconomic gradients in immune response to latent infection. Am J Epidemiol 2008: 167: 112–120. [DOI] [PubMed] [Google Scholar]
- 45. Ebersole JL. Humoral immune responses in gingival crevice fluid: local and systemic implications. Periodontol 2000 2003: 31: 135–166. [DOI] [PubMed] [Google Scholar]
- 46. Egusa H, Soysa NS, Ellepola AN, Yatani H, Samaranayake LP. Oral candidosis in HIV‐infected patients. Curr HIV Res 2008: 6: 485–499. [DOI] [PubMed] [Google Scholar]
- 47. El Ahmer OR, Raza MW, Ogilvie MM, Weir DM, Blackwell CC. Binding of bacteria to HEp‐2 cells infected with influenza A virus. FEMS Immunol Med Microbiol 1999: 23: 331–341. [DOI] [PubMed] [Google Scholar]
- 48. Elkins KL, Cowley SC, Bosio CM. Innate and adaptive immunity to Francisella . Ann N Y Acad Sci 2007: 1105: 284–324. [DOI] [PubMed] [Google Scholar]
- 49. Engelmann I, Petzold DR, Kosinska A, Hepkema BG, Schulz TF, Heim A. Rapid quantitative PCR assays for the simultaneous detection of herpes simplex virus, varicella zoster virus, cytomegalovirus, Epstein‐Barr virus, and human herpesvirus 6 DNA in blood and other clinical specimens. J Med Virol 2008: 80: 467–477. Erratae in: J Med Virol 2008; 80: 1505, J Med Virol 2008; 80: 2177. [DOI] [PubMed] [Google Scholar]
- 50. Fiore AE, Iverson C, Messmer T, Erdman D, Lett SM, Talkington DF, Anderson LJ, Fields B, Carlone GM, Breiman RF, Cetron MS. Outbreak of pneumonia in a long‐term care facility: antecedent human parainfluenza virus 1 infection may predispose to bacterial pneumonia. J Am Geriatr Soc 1998: 46: 1112–1117. [DOI] [PubMed] [Google Scholar]
- 51. Gafa V, Manches O, Pastor A, Drouet E, Ambroise‐Thomas P, Grillot R, Aldebert D. Human cytomegalovirus downregulates complement receptors (CR3, CR4) and decreases phagocytosis by macrophages. J Med Virol 2005: 76: 361–366. [DOI] [PubMed] [Google Scholar]
- 52. Gaffen SL, Hajishengallis G. A new inflammatory cytokine on the block: Re‐thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL‐17. J Dent Res 2008: 87: 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garimorth K, Kountchev J, Bellmann R, Semenitz B, Weiss G, Joannidis M. Lemierre’s syndrome following infectious mononucleosis. Wien Klin Wochenschr 2008: 120: 181–183. [DOI] [PubMed] [Google Scholar]
- 54. Gemmell E, Carter CL, Grieco DA, Sugerman PB, Seymour GJ. P. gingivalis‐specific T‐cell lines produce Th1 and Th2 cytokines. J Dent Res 2002: 81: 303–307. [DOI] [PubMed] [Google Scholar]
- 55. Gemmell E, Seymour GJ. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontol 2000 2004: 35: 21–41. [DOI] [PubMed] [Google Scholar]
- 56. Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000 2007: 43: 14–40. [DOI] [PubMed] [Google Scholar]
- 57. Gershwin LJ, Berghaus LJ, Arnold K, Anderson ML, Corbeil LB. Immune mechanisms of pathogenetic synergy in concurrent bovine pulmonary infection with Haemophilus somnus and bovine respiratory syncytial virus. Vet Immunol Immunopathol 2005: 107: 119–130. [DOI] [PubMed] [Google Scholar]
- 58. Giacaman RA, Nobbs AH, Ross KF, Herzberg MC. Porphyromonas gingivalis selectively up‐regulates the HIV‐1 coreceptor CCR5 in oral keratinocytes. J Immunol 2007: 179: 2542–2550. [DOI] [PubMed] [Google Scholar]
- 59. Ginder DR. Increased susceptibility of mice infected with mouse adenovirus to Escherichia coli‐induced pyelonephritis. J Exp Med 1964: 120: 1117–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gozlan J, Salord JM, Chouaïd C, Duvivier C, Picard O, Meyohas MC, Petit JC. Human cytomegalovirus (HCMV) late‐mRNA detection in peripheral blood of AIDS patients: diagnostic value for HCMV disease compared with those of viral culture and HCMV DNA detection. J Clin Microbiol 1993: 31: 1943–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grande SR, Imbronito AV, Okuda OS, Lotufo RF, Magalhães MH, Nunes FD. Herpes viruses in periodontal compromised sites: comparison between HIV‐positive and ‐negative patients. J Clin Periodontol 2008: 35: 838–845. [DOI] [PubMed] [Google Scholar]
- 62. Graves DT, Cochran D. The contribution of interleukin‐1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol 2003: 74: 391–401. [DOI] [PubMed] [Google Scholar]
- 63. Gredmark S, Tilburgs T, Söderberg‐Nauclér C. Human cytomegalovirus inhibits cytokine‐induced macrophage differentiation. J Virol 2004: 78: 10378–10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Griffin E, Krantz E, Selke S, Huang ML, Wald A. Oral mucosal reactivation rates of herpesviruses among HIV‐1 seropositive persons. J Med Virol 2008: 80: 1153–1159. [DOI] [PubMed] [Google Scholar]
- 65. Guidotti LG, Chisari FV. Cytokine‐mediated control of viral infections. Virology 2000: 273: 221–227. [DOI] [PubMed] [Google Scholar]
- 66. Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol 2001: 19: 65–91. [DOI] [PubMed] [Google Scholar]
- 67. Gunson RN, Maclean AR, Shepherd SJ, Carman W. Simultaneous detection and quantitation of cytomegalovirus, Epstein‐Barr virus, and adenovirus by use of real‐time PCR and pooled standards. J Clin Microbiol 2009: 47: 765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hai R, Chu A, Li H, Umamoto S, Rider P, Liu F. Infection of human cytomegalovirus in cultured human gingival tissue. Virol J 2006: 3: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hament JM, Kimpen JL, Fleer A, Wolfs TF. Respiratory viral infection predisposing for bacterial disease: a concise review. FEMS Immunol Med Microbiol 1999: 26: 189–195. [DOI] [PubMed] [Google Scholar]
- 70. Hamilton JR, Overall JC, Glasgow LA. Synergistic effect on mortality in mice with murine cytomegalovirus and Pseudomonas aeruginosa, Staphylococcus aureus, or Candida albicans infections. Infect Immun 1976: 14: 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Han X, Kawai T, Taubman MA. Interference with immune‐cell‐mediated bone resorption in periodontal disease. Periodontol 2000 2007: 45: 76–94. [DOI] [PubMed] [Google Scholar]
- 72. Hashemi FB, Ghassemi M, Faro S, Aroutcheva A, Spear GT. Induction of human immunodeficiency virus type 1 expression by anaerobes associated with bacterial vaginosis. J Infect Dis 2000: 181: 1574–1580. [DOI] [PubMed] [Google Scholar]
- 73. Hedner E, Vahlne A, Hirsch JM. Primary herpes simplex virus (type 1) infection delays healing of oral excisional and extraction wounds in the rat. J Oral Pathol Med 1990: 19: 471–476. [DOI] [PubMed] [Google Scholar]
- 74. Hedner E, Vahlne A, Kahnberg KE, Hirsch JM. Reactivated herpes simplex virus infection as a possible cause of dry socket after tooth extraction. J Oral Maxillofac Surg 1993: 51: 370–376. discussion 377–378. [DOI] [PubMed] [Google Scholar]
- 75. Heikkinen T, Chonmaitree T. Viral‐bacterial synergy in otitis media: Implications for management. Curr Infect Dis Reports 2000: 2: 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hirschfeld M, Weis JJ, Toshchakov V, Salkowski CA, Cody MJ, Ward DC, Qureshi N, Michalek SM, Vogel SN. Signaling by Toll‐like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun 2001: 69: 1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hochman N, Zakay‐Rones Z, Shohat H, Ever‐Hadani P, Ehrlich J, Schlesinger M, Morag A. Antibodies to cytomegalo and Epstein–Barr viruses in human saliva and gingival fluid. New Microbiol 1998: 21: 131–139. [PubMed] [Google Scholar]
- 78. Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the ‘red complex’, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000 2005: 38: 72–122. [DOI] [PubMed] [Google Scholar]
- 79. Houri‐Haddad Y, Wilensky A, Shapira L. T‐cell phenotype as a risk factor for periodontal disease. Periodontol 2000 2007: 45: 67–75. [DOI] [PubMed] [Google Scholar]
- 80. Huang CB, Emerson KA, Gonzalez OA, Ebersole JL. Oral bacteria induce a differential activation of HIV‐1 promoter in T cells, macrophages, and dendritic cells. Oral Microbiol Immunol 2009: 24: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Idesawa M, Sugano N, Ikeda K, Oshikawa M, Takane M, Seki K, Ito K. Detection of Epstein–Barr virus in saliva by real‐time PCR. Oral Microbiol Immunol 2004: 19: 230–232. [DOI] [PubMed] [Google Scholar]
- 82. Ikegaya H, Motani H, Sakurada K, Sato K, Akutsu T, Yoshino M. Forensic application of Epstein–Barr virus genotype: correlation between viral genotype and geographical area. J Virol Methods 2008: 147: 78–85. [DOI] [PubMed] [Google Scholar]
- 83. Imamura T, Travis J, Potempa J. The biphasic virulence activities of gingipains: activation and inactivation of host proteins. Curr Protein Pept Sci 2003: 4: 443–450. [DOI] [PubMed] [Google Scholar]
- 84. Imbronito AV, Grande SR, Freitas NM, Okuda O, Lotufo RF, Nunes FD. Detection of Epstein–Barr virus and human cytomegalovirus in blood and oral samples: comparison of three sampling methods. J Oral Sci 2008: 50: 25–31. [DOI] [PubMed] [Google Scholar]
- 85. Imbronito AV, Okuda OS, Maria de Freitas N, Moreira Lotufo RF, Nunes FD. Detection of herpesviruses and periodontal pathogens in subgingival plaque of patients with chronic periodontitis, generalized aggressive periodontitis, or gingivitis. J Periodontol 2008: 79: 2313–2321. [DOI] [PubMed] [Google Scholar]
- 86. Ishii K, Fukui M. Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl Environ Microbiol 2001: 67: 3753–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Juvén T, Mertsola J, Waris M, Leinonen M, Ruuskanen O. Clinical response to antibiotic therapy for community‐acquired pneumonia. Eur J Pediatr 2004: 163: 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kajita K, Honda T, Amanuma R, Domon H, Okui T, Ito H, Yoshie H, Tabeta K, Nakajima T, Yamazaki K. Quantitative messenger RNA expression of Toll‐like receptors and interferon‐alpha1 in gingivitis and periodontitis. Oral Microbiol Immunol 2007: 22: 398–402. [DOI] [PubMed] [Google Scholar]
- 89. Kamma JJ, Contreras A, Slots J. Herpes viruses and periodontopathic bacteria in early‐onset periodontitis. J Clin Periodontol 2001: 28: 879–885. [DOI] [PubMed] [Google Scholar]
- 90. Kamma JJ, Slots J. Herpesviral‐bacterial interactions in aggressive periodontitis. J Clin Periodontol 2003: 30: 420–426. [DOI] [PubMed] [Google Scholar]
- 91. Katzoli P, Sakellaris G, Ergazaki M, Charissis G, Spandidos DA, Sourvinos G. Detection of herpes viruses in children with acute appendicitis. J Clin Virol 2009: 44: 282–286. [DOI] [PubMed] [Google Scholar]
- 92. Kawashima N, Stashenko P. Expression of bone‐resorptive and regulatory cytokines in murine periapical inflammation. Arch Oral Biol 1999: 44: 55–66. [DOI] [PubMed] [Google Scholar]
- 93. Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev 2003: 8: 223–246. [PubMed] [Google Scholar]
- 94. Kinane DF, Demuth DR, Gorr SU, Hajishengallis GN, Martin MH. Human variability in innate immunity. Periodontol 2000 2007: 45: 14–34. [DOI] [PubMed] [Google Scholar]
- 95. Klemenc P, Skaleric U, Artnik B, Nograsek P, Marin J. Prevalence of some herpesviruses in gingival crevicular fluid. J Clin Virol 2005: 34: 147–152. [DOI] [PubMed] [Google Scholar]
- 96. Konstantinidis A, Sakellari D, Papa A, Antoniadis A. Real‐time polymerase chain reaction quantification of Epstein–Barr virus in chronic periodontitis patients. J Periodontal Res 2005: 40: 294–298. [DOI] [PubMed] [Google Scholar]
- 97. Kosai K, Seki M, Yanagihara K, Nakamura S, Kurihara S, Imamura Y, Izumikawa K, Kakeya H, Yamamoto Y, Tashiro T, Kohno S. Two‐dimensional gel electrophoresis analysis in simultaneous influenza pneumonia and bacterial infection in mice. Clin Exp Immunol 2008: 152: 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kubar A, Saygun I, Özdemir A, Yapar M, Slots J. Real‐time polymerase chain reaction quantification of human cytomegalovirus and Epstein–Barr virus in periodontal pockets and the adjacent gingiva of periodontitis lesions. J Periodontal Res 2005: 40: 97–104. [DOI] [PubMed] [Google Scholar]
- 99. Kubar A, Saygun I, Yapar M, Özdemir A, Slots J. Real‐time PCR quantification of cytomegalovirus in aggressive periodontitis lesions using TaqMan technology. J Periodontal Res 2004: 39: 81–86. [DOI] [PubMed] [Google Scholar]
- 100. Kuritzkes DR, Walker BD. HIV‐1 pathogenesis, clinical manifestations, and treatment In: Knipe DM, Howley PM. editors. Fields Virology, 5th edn Philadelphia: Lippincott, Williams & Wilkins, 2007: 2187–2214. [Google Scholar]
- 101. Lamont RJ, Yilmaz O. In or out: the invasiveness of oral bacteria. Periodontol 2000 2002: 30: 61–69. [DOI] [PubMed] [Google Scholar]
- 102. Landry ML, Eid T, Bannykh S, Major E. False negative PCR despite high levels of JC virus DNA in spinal fluid: Implications for diagnostic testing. J Clin Virol 2008: 43: 247–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee SC, Fung CP, Lin CC, Tsai CJ, Chen KS. Porphyromonas gingivalis bacteremia and subhepatic abscess after renal transplantation: a case report. J Microbiol Immunol Infect 1999: 32: 213–216. [PubMed] [Google Scholar]
- 104. LeVine AM, Koeningsknecht V, Stark JM. Decreased pulmonary clearance of S. pneumoniae following influenza A infection in mice. J Virol Methods 2001: 94: 173–186. [DOI] [PubMed] [Google Scholar]
- 105. Li H, Chen V, Chen Y, Baumgartner JC, Machida CA. Herpesviruses in endodontic pathoses: association of Epstein–Barr virus with irreversible pulpitis and apical periodontitis. J Endod 2009: 35: 23–29. [DOI] [PubMed] [Google Scholar]
- 106. Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular survival and vascular cell‐to‐cell transmission of Porphyromonas gingivalis . BMC Microbiol 2008: 8: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li Y, Zhang JC, Zhang YH. The association between infection of Epstein–Barr virus and chronic periodontitis. Zhonghua Kou Qiang Yi Xue Za Zhi 2004: 39: 146–148. (Chinese). [PubMed] [Google Scholar]
- 108. Lin YL, Chang PC, Wang Y, Li M. Identification of novel viral interleukin‐10 isoforms of human cytomegalovirus AD169. Virus Res 2008: 131: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lin YL, Li M. Human cytomegalovirus and Epstein–Barr virus inhibit oral bacteria‐induced macrophage activation and phagocytosis. Oral Microbiol Immunol 2009: 24: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ling LJ, Ho CC, Wu CY, Chen YT, Hung SL. Association between human herpesviruses and the severity of periodontitis. J Periodontol 2004: 75: 1479–1485. [DOI] [PubMed] [Google Scholar]
- 111. Loenen WA, Bruggeman CA, Wiertz EJ. Immune evasion by human cytomegalovirus: lessons in immunology and cell biology. Semin Immunol 2001: 13: 41–49. [DOI] [PubMed] [Google Scholar]
- 112. Lommer MJ, Verstraete FJ. Concurrent oral shedding of feline calicivirus and feline herpesvirus 1 in cats with chronic gingivostomatitis. Oral Microbiol Immunol 2003: 18: 131–134. [DOI] [PubMed] [Google Scholar]
- 113. Lossli CG. Influenza and the interaction of viruses and bacteria in respiratory infections. Medicine (Baltimore) 1973: 52: 369–384. [DOI] [PubMed] [Google Scholar]
- 114. Lucht E, Albert J, Linde A, Xu W, Brytting M, Lundeberg J, Uhlén M, Bratt G, Sandström E, Heimdahl A, Nord CE, Wahren B. Human immunodeficiency virus type 1 and cytomegalovirus in saliva. J Med Virol 1993: 39: 156–162. [DOI] [PubMed] [Google Scholar]
- 115. Lucht E, Brytting M, Bjerregaard L, Julander I, Linde A. Shedding of cytomegalovirus and herpesviruses 6, 7, and 8 in saliva of human immunodeficiency virus type 1‐infected patients and healthy controls. Clin Infect Dis 1998: 27: 137–141. [DOI] [PubMed] [Google Scholar]
- 116. Mahanonda R, Pichyangkul S. Toll‐like receptors and their role in periodontal health and disease. Periodontol 2000 2007: 43: 41–55. [DOI] [PubMed] [Google Scholar]
- 117. Marchall JA. Mixed infections of intestinal viruses and bacteria in humans In: Brogden KA, Guthmiller JM. editors. Polymicrobial diseases. Washington, DC: American Society for Microbiology, 2002: 299–316. [Google Scholar]
- 118. Mardirossian A, Contreras A, Navazesh M, Nowzari H, Slots J. Herpesviruses 6, 7 and 8 in HIV‐ and non‐HIV‐associated periodontitis. J Periodontal Res 2000: 35: 278–284. [DOI] [PubMed] [Google Scholar]
- 119. Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, Corey L. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis 2008: 198: 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mbopi‐Kéou FX, Bélec L, Teo CG, Scully C, Porter SR. Synergism between HIV and other viruses in the mouth. Lancet Infect Dis 2002: 2: 416–424. [DOI] [PubMed] [Google Scholar]
- 121. McChlery S, Ramage G, Bagg J. Respiratory tract infections and pneumonia. Periodontol 2000 2009: 49: 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. McCullers JA, Karlström A, Iverson AR, Loeffler JM, Fischetti VA. Novel strategy to prevent otitis media caused by colonizing Streptococcus pneumoniae . PLoS Pathog 2007: 3: e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet‐activating factor receptor. J Infect Dis 2002: 186: 341–350. [DOI] [PubMed] [Google Scholar]
- 124. Meyer‐König U, Serr A, Von Laer D, Kirste G, Wolff C, Haller O, Neumann‐Haefelin D, Hufert FT. Human cytomegalovirus immediate early and late transcripts in peripheral blood leukocytes: diagnostic value in renal transplant recipients. J Infect Dis 1995: 171: 705–709. [DOI] [PubMed] [Google Scholar]
- 125. Michalowicz BS, Ronderos M, Camara‐Silva R, Contreras A, Slots J. Human herpesviruses and Porphyromonas gingivalis are associated with juvenile periodontitis. J Periodontol 2000: 71: 981–988. [DOI] [PubMed] [Google Scholar]
- 126. Miller CS, Avdiushko SA, Kryscio RJ, Danaher RJ, Jacob RJ. Effect of prophylactic valacyclovir on the presence of human herpesvirus DNA in saliva of healthy individuals after dental treatment. J Clin Microbiol 2005: 43: 2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Miller CS, Berger JR, Mootoor Y, Avdiushko SA, Zhu H, Kryscio RJ. High prevalence of multiple human herpesviruses in saliva from human immunodeficiency virus‐infected persons in the era of highly active antiretroviral therapy. J Clin Microbiol 2006: 44: 2409–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Minoura‐Etoh J, Gotoh K, Sato R, Ogata M, Kaku N, Fujioka T, Nishizono A. Helicobacter pylori‐associated oxidant monochloramine induces reactivation of Epstein–Barr virus (EBV) in gastric epithelial cells latently infected with EBV. J Med Microbiol 2006: 55: 905–911. [DOI] [PubMed] [Google Scholar]
- 129. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med 2008: 358: 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mocarski ES Jr. Immune escape and exploitation strategies of cytomegaloviruses: impact on and imitation of the major histocompatibility system. Cell Microbiol 2004: 6: 707–717. [DOI] [PubMed] [Google Scholar]
- 131. Mocarski ES Jr, Shenk T, Pass RF. Cytomegaloviruses In: Knipe DM, Howley PM. editors. Fields Virology, 5th edn Philadelphia: Lippincott, Williams & Wilkins, 2007: 2702–2772. [Google Scholar]
- 132. Mogensen TH, Paludan SR. Molecular pathways in virus‐induced cytokine production. Microbiol Mol Biol Rev 2001: 65: 131–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Moghim SH, Chalabi M, Abed AM, Rezaei F, Tamizifar H. Prevalence of Epstein–Barr virus type 1 in patients with chronic periodontitis by nested‐PCR. Pak J Biol Sci 2007: 15;10: 4547–4550. [DOI] [PubMed] [Google Scholar]
- 134. Müller R, Ditzen A, Hille K, Stichling M, Ehricht R, Illmer T, Ehninger G, Rohayem J. Detection of herpesvirus and adenovirus co‐infections with diagnostic DNA‐microarrays. J Virol Methods 2009: 155: 161–166. [DOI] [PubMed] [Google Scholar]
- 135. Nachtwey J, Spencer JV. HCMV IL‐10 suppresses cytokine expression in monocytes through inhibition of Nuclear Factor‐kappaB. Viral Immunol 2008: 21: 477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nagasawa T, Kiji M, Yashiro R, Hormdee D, Lu H, Kunze M, Suda T, Koshy G, Kobayashi H, Oda S, Nitta H, Ishikawa I. Roles of receptor activator of nuclear factor‐kappaB ligand (RANKL) and osteoprotegerin in periodontal health and disease. Periodontol 2000 2007: 43: 65–84. [DOI] [PubMed] [Google Scholar]
- 137. Nichols WG, Corey L, Gooley T, Davis C, Boeckh M. High risk of death due to bacterial and fungal infection among cytomegalovirus (CMV)–seronegative recipients of stem cell transplants from seropositive donors: evidence for indirect effects of primary CMV infection. J Infect Dis 2002: 185: 273–282. [DOI] [PubMed] [Google Scholar]
- 138. Nishiyama SA, Nakano V, Velásquez‐Melendez G, Avila‐Campos MJ. Occurrence of herpes simplex virus 1 and three periodontal bacteria in patients with chronic periodontitis and necrotic pulp. Can J Microbiol 2008: 54: 326–330. [DOI] [PubMed] [Google Scholar]
- 139. Nowzari H, Botero JE, DeGiacomo M, Villacres MC, Rich SK. Microbiology and cytokine levels around healthy dental implants and teeth. Clin Implant Dent Relat Res 2008: 10: 166–173. [DOI] [PubMed] [Google Scholar]
- 140. Nowzari H, Jorgensen MG, Ta TT, Contreras A, Slots J. Aggressive periodontitis associated with Fanconi’s anemia. A case report. J Periodontol 2001: 72: 1601–1606. [DOI] [PubMed] [Google Scholar]
- 141. Olczak‐Kowalczyk D, Pawłowska J, Cukrowska B, Kluge P, Witkowska‐Vogtt E, Dzierzanowska‐Fangrat K, Wrześniewska D, Smirska E, Grenda R. Local presence of cytomegalovirus and Candida species vs oral lesions in liver and kidney transplant recipients. Ann Transplant 2008: 13: 28–33. [PubMed] [Google Scholar]
- 142. Ongrádi J, Sallay K, Kulcsár G. The decreased antibacterial activity of oral polymorphonuclear leukocytes coincides with the occurrence of virus‐carrying oral lymphocytes and epithelial cells. Folia Microbiol (Praha) 1987: 32: 438–447. [DOI] [PubMed] [Google Scholar]
- 143. Pacheco JJ, Coelho C, Salazar F, Contreras A, Slots J, Velazco CH. Treatment of Papillon‐Lefèvre syndrome periodontitis. J Clin Periodontol 2002: 29: 370–374. [DOI] [PubMed] [Google Scholar]
- 144. Pang XL, Fox JD, Fenton JM, Miller GG, Caliendo AM, Preiksaitis JK. American Society of Transplantation Infectious Diseases Community of Practice; Canadian Society of Transplantation. Interlaboratory comparison of cytomegalovirus viral load assays. Am J Transplant 2009: 9: 258–268. [DOI] [PubMed] [Google Scholar]
- 145. Pang X, Humar A, Preiksaitis JK. Concurrent genotyping and quantitation of cytomegalovirus gB genotypes in solid‐organ‐transplant recipients by use of a real‐time PCR assay. J Clin Microbiol 2008: 46: 4004–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Parra B, Slots J. Detection of human viruses in periodontal pockets using polymerase chain reaction. Oral Microbiol Immunol 1996: 11: 289–293. [DOI] [PubMed] [Google Scholar]
- 147. Pass RF. Epidemiology and transmission of cytomegalovirus infection. J Infect Dis 1985: 152: 243–248. [DOI] [PubMed] [Google Scholar]
- 148. Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000 2006: 42: 80–87. [DOI] [PubMed] [Google Scholar]
- 149. Patel R. Valaciclovir: development, clinical utility and potential. Expert Opin Investig Drugs 1997: 6: 173–189. [DOI] [PubMed] [Google Scholar]
- 150. Peleg AY, Husain S, Qureshi ZA, Silveira FP, Sarumi M, Shutt KA, Kwak EJ, Paterson DL. Risk factors, clinical characteristics, and outcome of Nocardia infection in organ transplant recipients: a matched case‐control study. Clin Infect Dis 2007: 44: 1307–1314. [DOI] [PubMed] [Google Scholar]
- 151. Pellett PE, Roizman B. The family Herpesviridae: A brief introduction In: Knipe DM, Howley PM. editors. Fields Virology, 5th edn Philadelphia: Lippincott, Williams & Wilkins, 2007: 2479–2499. [Google Scholar]
- 152. Pereira CM, Gasparetto PF, Corrêa ME, Costa FF, De Almeida OP, Barjas‐Castro ML. Human herpesvirus 6 in oral fluids from healthy individuals. Arch Oral Biol 2004: 49: 1043–1046. [DOI] [PubMed] [Google Scholar]
- 153. Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000 2000: 24: 153–192. [DOI] [PubMed] [Google Scholar]
- 154. Powers C, DeFilippis V, Malouli D, Früh K. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 2008: 325: 333–359. [DOI] [PubMed] [Google Scholar]
- 155. Prato GP, Rotundo R, Magnani C, Ficarra G. Viral etiology of gingival recession. A case report. J Periodontol 2002: 73: 110–114. [DOI] [PubMed] [Google Scholar]
- 156. Preshaw PM. Definitions of periodontal disease in research. J Clin Periodontol 2009: 36: 1–2. [DOI] [PubMed] [Google Scholar]
- 157. Puchhammer‐Stöckl E, Görzer I. Cytomegalovirus and Epstein–Barr virus subtypes – the search for clinical significance. J Clin Virol 2006: 36: 239–248. [DOI] [PubMed] [Google Scholar]
- 158. Rams TE, Listgarten MA, Slots J. Utility of radiographic crestal lamina dura for predicting periodontitis disease‐activity. J Clin Periodontol 1994: 21: 571–576. [DOI] [PubMed] [Google Scholar]
- 159. Rams TE, Listgarten MA, Slots J. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis subgingival presence, species‐specific serum immunoglobulin G antibody levels, and periodontitis disease recurrence. J Periodontal Res 2006: 41: 228–234. [DOI] [PubMed] [Google Scholar]
- 160. Raué HP, Brien JD, Hammarlund E, Slifka MK. Activation of virus‐specific CD8+ T cells by lipopolysaccharide‐induced IL‐12 and IL‐18. J Immunol 2004: 173: 6873–6881. [DOI] [PubMed] [Google Scholar]
- 161. Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. Murine model of cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol 2008: 325: 315–331. [DOI] [PubMed] [Google Scholar]
- 162. Reddy MS. Reaching a better understanding of non‐oral disease and the implication of periodontal infections. Periodontol 2000 2007: 44: 9–14. [DOI] [PubMed] [Google Scholar]
- 163. Rentenaar RJ, Gamadia LE, Van DerHoek N, Van Diepen FN, Boom R, Weel JF, Wertheim‐van Dillen PM, Van Lier RA, Ten Berge IJ. Development of virus‐specific CD4(+) T cells during primary cytomegalovirus infection. J Clin Invest 2000: 105: 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Rickinson AB, Kieff E. Epstein–Barr virus In: Knipe DM, Howley PM. editors. Fields Virology, 5th edn Philadelphia: Lippincott, Williams & Wilkins, 2007: 2656–2700. [Google Scholar]
- 165. Roivainen M, Viik‐Kajander M, Palosuo T, Toivanen P, Leinonen M, Saikku P, Tenkanen L, Manninen V, Hovi T, Mänttäri M. Infections, inflammation, and the risk of coronary heart disease. Circulation 2000: 101: 252–257. [DOI] [PubMed] [Google Scholar]
- 166. Roland PS, Parry DA, Stroman DW. Microbiology of acute otitis media with tympanostomy tubes. Otolaryngol Head Neck Surg 2005: 133: 585–595. [DOI] [PubMed] [Google Scholar]
- 167. Root‐Bernstein R, Vonck J, Podufaly A. Antigenic complementarity between coxsackie virus and streptococcus in the induction of rheumatic heart disease and autoimmune myocarditis. Autoimmunity 2009: 42: 1–16. [DOI] [PubMed] [Google Scholar]
- 168. Rotola A, Cassai E, Farina R, Caselli E, Gentili V, Lazzarotto T, Trombelli L. Human herpesvirus 7, Epstein–Barr virus and human cytomegalovirus in periodontal tissues of periodontally diseased and healthy subjects. J Clin Periodontol 2008: 35: 831–837. [DOI] [PubMed] [Google Scholar]
- 169. Rudney JD, Chen R, Sedgewick GJ. Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial intracellular flora within human buccal cells. J Dent Res 2005: 84: 59–63. [DOI] [PubMed] [Google Scholar]
- 170. Ruohola A, Meurman O, Nikkari S, Skottman T, Salmi A, Waris M, Osterback R, Eerola E, Allander T, Niesters H, Heikkinen T, Ruuskanen O. Microbiology of acute otitis media in children with tympanostomy tubes: prevalences of bacteria and viruses. Clin Infect Dis 2006: 43: 1417–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Sabeti M, Slots J. Herpesviral‐bacterial coinfection in periapical pathosis. J Endod 2004: 30: 69–72. [DOI] [PubMed] [Google Scholar]
- 172. Saboia‐Dantas CJ, Coutrin de Toledo LF, Siqueira JF Jr, Sampaio‐Filho HR, Carvalho JJ, Pereira MJ. Natural killer cells and alterations in collagen density: signs of periradicular herpesvirus infection? Clin Oral Investig 2008: 12: 129–135. [DOI] [PubMed] [Google Scholar]
- 173. Şahin S, Saygun I, Kubar A, Slots J. Periodontitis lesions are the main source of salivary cytomegalovirus. Oral Microbiol Immunol 2009: 24: 340–342. [DOI] [PubMed] [Google Scholar]
- 174. Samarayanake LP, Leung WK, Jin L. Oral mucosal fungal infections. Periodontol 2000 2009: 49: 39–59. [DOI] [PubMed] [Google Scholar]
- 175. Sandvej K, Zhou XG, Hamilton‐Dutoit S. EBNA‐1 sequence variation in Danish and Chinese EBV‐associated tumours: evidence for geographical polymorphism but not for tumour‐specific subtype restriction. J Pathol 2000: 191: 127–131. [DOI] [PubMed] [Google Scholar]
- 176. Santangelo R, D’Ercole S, Graffeo R, Marchetti S, Deli G, Nacci A, Piccolomini R, Cattani P, Fadda G. Bacterial and viral DNA in periodontal disease: a study using multiplex PCR. New Microbiol 2004: 27: 133–137. [PubMed] [Google Scholar]
- 177. Saygun I, Kubar A, Özdemir A, Slots J. Periodontitis lesions are a source of salivary cytomegalovirus and Epstein–Barr virus. J Periodontal Res 2005: 40: 187–191. [DOI] [PubMed] [Google Scholar]
- 178. Saygun I, Kubar A, Özdemir A, Yapar M, Slots J. Herpesviral‐bacterial interrelationships in aggressive periodontitis. J Periodontal Res 2004: 39: 207–212. [DOI] [PubMed] [Google Scholar]
- 179. Saygun I, Kubar A, Şahin S, Şener K, Slots J. Quantitative analysis of association between herpesviruses and bacterial pathogens in periodontitis. J Periodontal Res 2008: 43: 352–359. [DOI] [PubMed] [Google Scholar]
- 180. Saygun I, Yapar M, Özdemir A, Kubar A, Slots J. Human cytomegalovirus and Epstein–Barr virus type 1 in periodontal abscesses. Oral Microbiol Immunol 2004: 19: 83–87. [DOI] [PubMed] [Google Scholar]
- 181. Schenkein HA, Barbour SE, Tew JG. Cytokines and inflammatory factors regulating immunoglobulin production in aggressive periodontitis. Periodontol 2000 2007: 45: 113–127. [DOI] [PubMed] [Google Scholar]
- 182. Schenkein HA, Van Dyke TE. Early‐onset periodontitis: systemic aspects of etiology and pathogenesis. Periodontol 2000 1994: 6: 7–25. [DOI] [PubMed] [Google Scholar]
- 183. Schloss L, Van Loon AM, Cinque P, Cleator G, Echevarria JM, Falk KI, Klapper P, Schirm J, Vestergaard BF, Niesters H, Popow‐Kraupp T, Quint W, Linde A. An international external quality assessment of nucleic acid amplification of herpes simplex virus. J Clin Virol 2003: 28: 175–185. [DOI] [PubMed] [Google Scholar]
- 184. Seki M, Yanagihara K, Higashiyama Y, Fukuda Y, Kaneko Y, Ohno H, Miyazaki Y, Hirakata Y, Tomono K, Kadota J, Tashiro T, Kohno S. Immunokinetics in severe pneumonia due to influenza virus and bacteria coinfection in mice. Eur Respir J 2004: 24: 143–149. [DOI] [PubMed] [Google Scholar]
- 185. Severien C, Teig N, Riedel F, Hohendahl J, Rieger C. Severe pneumonia and chronic lung disease in a young child with adenovirus and Bordetella pertussis infection. Pediatr Infect Dis J 1995: 14: 400–401. [DOI] [PubMed] [Google Scholar]
- 186. Seymour GJ, Gemmell E, Reinhardt RA, Eastcott J, Taubman MA. Immunopathogenesis of chronic inflammatory periodontal diseases: cellular and molecular mechanisms. J Periodontal Res 1993: 28: 478–486. [DOI] [PubMed] [Google Scholar]
- 187. Sigusch BW, Wutzler A, Nietzsch T, Glockmann E. Evidence for a specific crevicular lymphocyte profile in aggressive periodontitis. J Periodontal Res 2006: 41: 391–396. [DOI] [PubMed] [Google Scholar]
- 188. Slots J. Selection of antimicrobial agents in periodontal therapy. J Periodontal Res 2002: 37: 389–398. [DOI] [PubMed] [Google Scholar]
- 189. Slots J. Herpesviruses in periodontal diseases. Periodontol 2000 2005: 38: 33–62. [DOI] [PubMed] [Google Scholar]
- 190. Slots J. Oral viral infections of adults. Periodontol 2000 2009: 49: 60–86. [DOI] [PubMed] [Google Scholar]
- 191. Slots J, Contreras A. Herpesviruses: a unifying causative factor in periodontitis? Oral Microbiol Immunol 2000: 15: 277–280. [DOI] [PubMed] [Google Scholar]
- 192. Slots J, Genco RJ. Black‐pigmented Bacteroides species, Capnocytophaga species, and Actinobacillus actinomycetemcomitans in human periodontal disease: virulence factors in colonization, survival, and tissue destruction. J Dent Res 1984: 63: 412–421. [DOI] [PubMed] [Google Scholar]
- 193. Slots J, Kamma JJ, Sugar C. The herpesvirus‐Porphyromonas gingivalis‐periodontitis axis. J Periodontal Res 2003: 38: 318–323. [DOI] [PubMed] [Google Scholar]
- 194. Slots J, Nowzari H, Sabeti M. Cytomegalovirus infection in symptomatic periapical pathosis. Int Endod J 2004: 37: 519–524. Erratum in: Int Endod J 2005; 38: 854. [DOI] [PubMed] [Google Scholar]
- 195. Slots J, Saygun I, Sabeti M, Kubar A. Epstein‐Barr virus in oral diseases. J Periodontal Res 2006: 41: 235–244. [DOI] [PubMed] [Google Scholar]
- 196. Slots J, Sugar C, Kamma JJ. Cytomegalovirus periodontal presence is associated with subgingival Dialister pneumosintes and alveolar bone loss. Oral Microbiol Immunol 2002: 17: 369–374. [DOI] [PubMed] [Google Scholar]
- 197. Slots J, Ting M. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in human periodontal disease: occurrence and treatment. Periodontol 2000 1999: 20: 82–121. [DOI] [PubMed] [Google Scholar]
- 198. Smith H, Sweet C. Cooperation between viral and bacterial pathogens in causing human respiratory disease In: Brogden KA, Guthmiller JM. editors. Polymicrobial diseases. Washington, DC: American Society for Microbiology, 2002: 201–112. [Google Scholar]
- 199. Smith MacDonald E, Nowzari H, Contreras A, Flynn J, Morrison J, Slots J. Clinical and microbiological evaluation of a bioabsorbable and a nonresorbable barrier membrane in the treatment of periodontal intraosseous lesions. J Periodontol 1998: 69: 445–453. [DOI] [PubMed] [Google Scholar]
- 200. Söderberg‐Nauclér C. HCMV microinfections in inflammatory diseases and cancer. J Clin Virol 2008: 41: 218–223. [DOI] [PubMed] [Google Scholar]
- 201. Stals FS, Vd Bogaard AE, Bruggeman CA. Prevention of bacteraemia by systemic ciprofloxacin treatment in rat cytomegalovirus‐infected immunocompromised rats. J Antimicrob Chemother 1994: 34: 101–110. [DOI] [PubMed] [Google Scholar]
- 202. Stathopoulou PG, Benakanakere MR, Galicia JC, Kinane DF. The host cytokine response to Porphyromonas gingivalis is modified by gingipains. Oral Microbiol Immunol 2009: 24: 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Stern J, Shai E, Zaks B, Halabi A, Houri‐Haddad Y, Shapira L, Palmon A. Reduced expression of gamma interferon in serum and marked lymphoid depletion induced by Porphyromonas gingivalis increase murine morbidity and mortality due to cytomegalovirus infection. Infect Immun 2004: 72: 5791–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Stratton KR, Durch J, Lawrence RS. Institute of Medicine (US) Committee to Study Priorities for Vaccine Development Vaccines for the 21st century: a tool for decision making. Washington, DC: National Academies Press, 2000. [PubMed] [Google Scholar]
- 205. Sugano N, Ikeda K, Oshikawa M, Idesawa M, Tanaka H, Sato S, Ito K. Relationship between Porphyromonas gingivalis, Epstein‐Barr virus infection and reactivation in periodontitis. J Oral Sci 2004: 46: 203–206. [DOI] [PubMed] [Google Scholar]
- 206. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature 2009: 457: 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Sunde PT, Olsen I, Enersen M, Beiske K, Grinde B. Human cytomegalovirus and Epstein–Barr virus in apical and marginal periodontitis: A role in pathology? J Med Virol 2008: 80: 1007–1011. [DOI] [PubMed] [Google Scholar]
- 208. Sunde PT, Olsen I, Enersen M, Grinde B. Patient with severe periodontitis and subgingival Epstein–Barr virus treated with antiviral therapy. J Clin Virol 2008: 42: 176–178. [DOI] [PubMed] [Google Scholar]
- 209. Svahn A, Berggren J, Parke A, Storsaeter J, Thorstensson R, Linde A. Changes in seroprevalence to four herpesviruses over 30 years in Swedish children aged 9–12 years. J Clin Virol 2006: 37: 118–123. [DOI] [PubMed] [Google Scholar]
- 210. Syrjänen S, Leimola‐Virtanen R, Schmidt‐Westhausen A, Reichart PA. Oral ulcers in AIDS patients frequently associated with cytomegalovirus (CMV) and Epstein–Barr virus (EBV) infections. J Oral Pathol Med 1999: 28: 204–209. [DOI] [PubMed] [Google Scholar]
- 211. Tabanella G, Nowzari H. Cytomegalovirus‐associated periodontitis and Guillain‐Barré syndrome. J Periodontol 2005: 76: 2306–2311. [DOI] [PubMed] [Google Scholar]
- 212. Tam V, O’Brien‐Simpson NM, Chen YY, Sanderson CJ, Kinnear B, Reynolds EC. The RgpA‐Kgp proteinase‐adhesin complexes of Porphyromonas gingivalis inactivate the Th2 cytokines interleukin‐4 and interleukin‐5. Infect Immun 2009: 77: 1451–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Tang YW, Espy MJ, Persing DH, Smith TF. Molecular evidence and clinical significance of herpesvirus coinfection in the central nervous system. J Clin Microbiol 1997: 35: 2869–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Tantivanich S, Laohapand P, Thaweeboon S, Desakorn V, Wuthinuntiwong P, Chalermtaranukul S, Pansri P, Amarapal P, Balachandra K, Chantratita W, Dhepakson P. Prevalence of cytomegalovirus, human herpesvirus‐6, and Epstein–Barr virus in periodontitis patients and healthy subjects in the Thai population. Southeast Asian J Trop Med Public Health 2004: 35: 635–640. [PubMed] [Google Scholar]
- 215. Taubman MA, Haffajee AD, Socransky SS, Smith DJ, Ebersole JL. Longitudinal monitoring of human antibody in subjects with destructive periodontal diseases. J Periodontal Res 1992: 27: 511–521. [DOI] [PubMed] [Google Scholar]
- 216. Taubman MA, Kawai T. Involvement of T‐lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit Rev Oral Biol Med 2001: 12: 125–135. [DOI] [PubMed] [Google Scholar]
- 217. Teughels W, Sliepen I, Quirynen M, Haake SK, Van Eldere J, Fives‐Taylor P, Van Ranst M. Human cytomegalovirus enhances A. actinomycetemcomitans adherence to cells. J Dent Res 2007: 86: 175–180. [DOI] [PubMed] [Google Scholar]
- 218. Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. Influenza‐associated hospitalizations in the United States. JAMA 2004: 292: 1333–1340. [DOI] [PubMed] [Google Scholar]
- 219. Ting M, Contreras A, Slots J. Herpesviruses in localized juvenile periodontitis. J Periodontal Res 2000: 35: 17–25. [DOI] [PubMed] [Google Scholar]
- 220. Titball RW. Vaccines against intracellular bacterial pathogens. Drug Discov Today 2008: 13: 596–600. [DOI] [PubMed] [Google Scholar]
- 221. Tolo K, Helgeland K. Fc‐binding components: a virulence factor in Actinobacillus actinomycetemcomitans? Oral Microbiol Immunol 1991: 6: 373–377. [DOI] [PubMed] [Google Scholar]
- 222. Tribble GD, Lamont RJ. Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontol 2000 2010: 52: 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Udagawa N, Sato N, Yang S, Nakamura M, Yamashita T, Nakamura H, Noguchi T. Signal transduction of lipopolysaccharide‐induced osteoclast differentiation. Periodontol 2000 2007: 43: 56–64. [DOI] [PubMed] [Google Scholar]
- 224. Vilkuna‐Rautiainen T, Pussinen PJ, Roivainen M, Petäys T, Jousilahti P, Hovi T, Vartiainen E, Asikainen S. Serum antibody response to periodontal pathogens and herpes simplex virus in relation to classic risk factors of cardiovascular disease. Int J Epidemiol 2006: 35: 1486–1494. [DOI] [PubMed] [Google Scholar]
- 225. Vitkov L, Krautgartner WD, Hannig M. Bacterial internalization in periodontitis. Oral Microbiol Immunol 2005: 20: 317–321. [DOI] [PubMed] [Google Scholar]
- 226. Wakiguchi H, Hisakawa H, Kubota H, Kurashige T. Strong response of T cells in infants with dual infection by Epstein–Barr virus and cytomegalovirus. Pediatr Int 1999: 41: 484–489. [DOI] [PubMed] [Google Scholar]
- 227. Wald A, Huang ML, Carrell D, Selke S, Corey L. Polymerase chain reaction for detection of herpes simplex (HSV) DNA on mucosal surfaces: comparison with HSV isolation in cell culture. J Infect Dis 2003: 188: 1345–1351. [DOI] [PubMed] [Google Scholar]
- 228. Walling DM, Brown AL, Etienne W, Keitel WA, Ling PD. Multiple Epstein–Barr virus infections in healthy individuals. J Virol 2003: 77: 6546–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Wara‐Aswapati N, Boch JA, Auron PE. Activation of interleukin 1beta gene transcription by human cytomegalovirus: molecular mechanisms and relevance to periodontitis. Oral Microbiol Immunol 2003: 18: 67–71. [DOI] [PubMed] [Google Scholar]
- 230. Watanabe SA, De Fátima Correia‐Silva J, Horta MC, Da Costa JE, Gomez RS. EBV‐1 and HCMV in aggressive periodontitis in Brazilian patients. Braz Oral Res 2007: 21: 336–341. [DOI] [PubMed] [Google Scholar]
- 231. White MK, Gorrill TS, Khalili K. Reciprocal transactivation between HIV‐1 and other human viruses. Virology 2006: 352: 1–13. [DOI] [PubMed] [Google Scholar]
- 232. Winther B, Alper CM, Mandel EM, Doyle WJ, Hendley JO. Temporal relationships between colds, upper respiratory viruses detected by polymerase chain reaction, and otitis media in young children followed through a typical cold season. Pediatrics 2007: 119: 1069–1075. [DOI] [PubMed] [Google Scholar]
- 233. Wohlleben G, Erb KJ. Immune stimulatory strategies for the prevention and treatment of asthma. Curr Pharm Des 2006: 12: 3281–3292. [DOI] [PubMed] [Google Scholar]
- 234. Wu YM, Yan J, Chen LL, Sun WL, Gu ZY. Infection frequency of Epstein–Barr virus in subgingival samples from patients with different periodontal status and its correlation with clinical parameters. J Zhejiang Univ Sci B 2006: 7: 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Wu YM, Yan J, Ojcius DM, Chen LL, Gu ZY, Pan JP. Correlation between infections with different genotypes of human cytomegalovirus and Epstein–Barr virus in subgingival samples and periodontal status of patients. J Clin Microbiol 2007: 45: 3665–3670. Erratum in: J Clin Microbiol 2008; 46: 836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Yager EJ, Szaba FM, Kummer LW, Lanzer KG, Burkum CE, Smiley ST, Blackman MA. Gamma‐herpesvirus‐induced protection against bacterial infection is transient. Viral Immunol 2009: 22: 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Yano H, Okitsu N, Hori T, Watanabe O, Kisu T, Hatagishi E, Suzuki A, Okamoto M, Ohmi A, Suetake M, Sagai S, Kobayashi T, Nishimura H. Detection of respiratory viruses in nasopharyngeal secretions and middle ear fluid from children with acute otitis media. Acta Otolaryngol 2008: 13: 1–6. [DOI] [PubMed] [Google Scholar]
- 238. Yano H, Okitsu N, Watanabe O, Kisu T, Hori T, Hatagishi E, Okamoto M, Ohmi A, Yamada K, Sagai S, Suetake M, Kobayashi T, Nishimura H. Acute otitis media associated with cytomegalovirus infection in infants and children. Int J Pediatr Otorhinolaryngol 2007: 71: 1443–1447. [DOI] [PubMed] [Google Scholar]
- 239. Yapar M, Saygun I, Özdemir A, Kubar A, Şahin S. Prevalence of human herpesviruses in patients with aggressive periodontitis. J Periodontol 2003: 74: 1634–1640. [DOI] [PubMed] [Google Scholar]
- 240. Yazdi KA, Sabeti M, Jabalameli F, Eman eini M, Kolahdouzan SA, Slots J. Relationship between human cytomegalovirus transcription and symptomatic apical periodontitis in Iran. Oral Microbiol Immunol 2008: 23: 510–514. [DOI] [PubMed] [Google Scholar]
- 241. Yildirim S, Yapar M, Kubar A. Detection and quantification of herpesviruses in Kostmann syndrome periodontitis using real‐time polymerase chain reaction: a case report. Oral Microbiol Immunol 2006: 21: 73–78. [DOI] [PubMed] [Google Scholar]
- 242. Yin MT, Dobkin JF, Grbic JT. Epidemiology, pathogenesis, and management of human immunodeficiency virus infection in patients with periodontal disease. Periodontol 2000 2007: 44: 55–81. [DOI] [PubMed] [Google Scholar]
- 243. Zajacova A, Dowd JB, Aiello AE. Socioeconomic and race/ethnic patterns in persistent infection burden among U.S. adults. J Gerontol A Biol Sci Med Sci 2009: 64: 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]