

Abstract
Human periodontitis is associated with a wide range of bacteria and viruses and with complex innate and adaptive immune responses. Porphyromonas gingivalis, Tannerella forsythia, Aggregatibacter actinomycetemcomitans, Treponema denticola, cytomegalovirus and other herpesviruses are major suspected pathogens of periodontitis, and a combined herpesvirus–bacterial periodontal infection can potentially explain major clinical features of the disease. Cytomegalovirus infects periodontal macrophages and T‐cells and elicits a release of interleukin‐1β and tumor necrosis factor‐α. These proinflammatory cytokines play an important role in the host defense against the virus, but they also have the potential to induce alveolar bone resorption and loss of periodontal ligament. Gingival fibroblasts infected with cytomegalovirus also exhibit diminished collagen production and release of an increased level of matrix metalloproteinases. This article reviews innate and adaptive immunity to cytomegalovirus and suggests that immune responses towards cytomegalovirus can play roles in controlling, as well as in exacerbating, destructive periodontal disease.
Our knowledge of viral infections has increased significantly in the past couple of decades. New viruses and their pathogenicity are constantly being identified and characterized. Norovirus has joined the rotaviruses as a major pathogen of gastroenteritis, and metapneumovirus, hantavirus, niphavirus, hendravirus, Ebola virus and severe acute respiratory syndrome (SARS) coronavirus were identified after fatal respiratory outbreaks (71, 83, 85, 92, 184). Human herpesvirus species have been related to a variety of oral and non‐oral diseases (149, 150), HIV to AIDS and the associated oral pathoses (23, 149), and papillomaviruses to genital and oropharyngeal cancers (110, 135). Vaccination against smallpox virus and poliovirus has become the most successful public health measure ever in preventive medicine.
During acute viral infections, large amounts of virions are aerosolized or shed into the respiratory tract, feces, saliva or other biological fluids, and pose a risk for individuals in close contact (149, 151). Paramyxoviruses, influenza viruses, respiratory syncytial virus, niphavirus and Ebola virus are spread by aerosols (33, 71, 92, 97, 184). Relatively few viruses are transmitted from lesions of the skin, but herpes simplex virus type 1 infection is commonly acquired through labial contact (4, 18, 60). HIV, papillomaviruses and herpes simplex type 2 are examples of sexually transmitted viruses. Cytomegalovirus can be transmitted transplacentally from mother to child and give rise to preterm birth and pre‐eclampsia (52, 183).
Viruses infecting oral, gastric, dermal, respiratory or genital sites encounter skin or mucosa as the first barrier for entrance (Fig. 1). The mild acidic and dry environment of the skin makes it difficult for most viruses to establish infection. The oral mucosa is thinner than the skin, and is wet but covered with mucins, immunglobulins and other protective factors in saliva. Saliva of cytomegalovirus‐positive subjects possesses a neutralizing activity compared with saliva of seronegative subjects (134). Herpesviruses usually enter the host through minor breaks or abrasions of the skin or mucosa and replicate productively in epidermis or dermis, fibroblasts, macrophages and neural ending cells (16, 37, 39, 59, 68, 87, 166, 168). Papillomaviruses can replicate in skin cells and in mucosal cells. Respiratory and enteric viruses can infect and replicate in epithelial cells without requiring a break of the mucosal barrier (16, 33, 92, 132).
Figure 1.
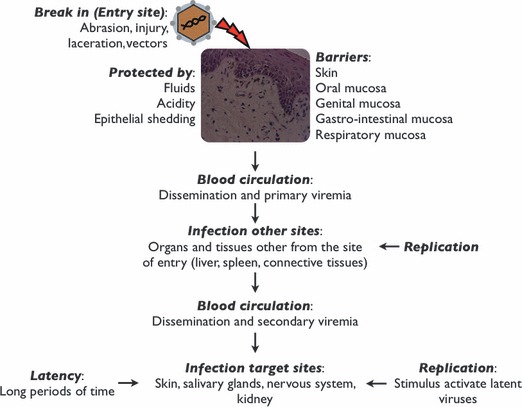
Viral infection steps: entry, replication, dissemination and infection of target cells/organs. Virions enter the host organism and spread to target tissues/organs where they can replicate and/or cause a persistent infection (latency). Latent viruses can become reactivated by several immune‐compromising events, such as smoking, inflammation, stress, trauma and immunosuppressive diseases.
Herpesviruses may cause illness by mechanisms that are direct, indirect or immune‐response linked, and illnesses range from subclinical or mild disease to encephalitis, pneumonia and other potentially lethal infections, and even to lymphoma, sarcoma and carcinoma (18, 40, 59, 60, 70, 104, 135, 150, 165). Several herpesvirus species are present in the saliva of most individuals, and are usually acquired through salivary contact early in life (151). Epstein–Barr virus can replicate in salivary glands and be released into saliva (5, 37, 149, 151, 166). Salivary Epstein–Barr virus and cytomegalovirus can also originate from periodontitis lesions (136, 150, 151). In the oral cavity, herpesviruses are involved in acute gingival infections, destructive periodontal disease, apical periodontitis, ulcerations of mucosa, odontogenic cyst, giant cell granuloma, autoimmune disease and various types of neoplasm (7, 41, 70, 104, 133, 135, 138, 150, 155, 165).
Periodontitis is associated with a wide range of bacteria and viruses and with complex humoral and cellular immune responses. Porphyromonas gingivalis, Tannerella forsythia, Aggregatibacter actinomycetemcomitans, Treponema denticola and some newly identified unculturable species are suspected pathogens of severe periodontitis (8, 64). However, the traditional concept of periodontitis being a bacterial disease seems unable to explain the site‐specificity and several other clinical characteristics of the disease, whereas the notion of a combined herpesvirus–bacterial periodontal infection can at least hypothetically account for major features of the disease (148). Close associations among herpesviruses, bacteria and periodontitis are consistent with a periodontopathic role of both types of the infectious agents. By infecting structural cells and host defense cells of the periodontium, herpesviruses may reduce the ability of periodontal tissues to withstand bacterial insults (24, 25). This review presents evidence for a role of herpesviruses in the pathogenesis of destructive periodontal disease, and because human cytomegalovirus shows a particularly strong association with disease‐active periodontitis (41, 99, 148, 150, 161), its pathobiology is evaluated in greater detail.
Characteristics of herpesviruses
Herpesvirus virions vary in size from 120 to 250 nm and consist of a double‐stranded linear DNA molecule surrounded by an icosahedral capsid, a proteinaceous tegument and a lipid‐containing envelope with embedded viral glycoproteins. Cytomegalovirus has the largest genome (∼230 kbp) of human herpesviruses, coding for more than 70 viral proteins. Herpesvirus virions acquire the envelope during the egress through the nuclear membrane. The Herpesviridae family is divided into the alpha subfamily (herpes simplex virus‐1, herpes simplex virus‐2 and varicella‐zoster virus), the beta subfamily (human cytomegalovirus, human herpesvirus‐6 and human herpesvirus‐7) and the gamma subfamily (Epstein–Barr virus and human herpesvirus‐8). Each herpesvirus subfamily maintains latent infection in specific cell population(s). Alpha herpesviruses exhibit a relatively short reproductive cycle, rapid lysis of infected cells and latency in sensory neural ganglia. Beta herpesviruses demonstrate a long reproductive cycle, slowly progressing infection and tropism for a large range of cells. Gamma herpesviruses are usually specific for B‐lymphocytes, and viral latency is typically found in lymphoid tissue (2, 5, 59, 67, 68, 87, 106, 115, 142, 146).
Following initial entry, herpesviruses produce a localized infection at the site of entry or enter the systemic circulation to infect distant tissues and organs (Fig. 1). The pattern of infection is partly determined by the mode of release of the virions from the infected cell (16, 167) (Fig. 2). If the release takes place from the apical part of the cell, the infection typically becomes localized. A release of virions from the basolateral side of the cell tends to produce a disseminating infection (16, 167, 168), or in the case of Epstein–Barr virus, an infection of adjacent epithelial cells (166, 168, 180).
Figure 2.
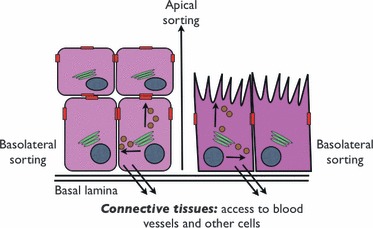
Different pathways of intracellular sorting of viruses. Virions released from stratified epithelium (left) or intestinal epithelium (right), by either apical sorting or basolateral sorting, can infect adjacent cells, enter connective tissue and gain access to blood vessels, or be dispersed into body lumens.
The outcome of the viral infection depends upon the relative efficacy of cellular components to block viral DNA replication and of viral transcriptional products to interfere with host defenses (74, 103, 131). Herpesvirus replication and production of infectious virions involves the sequential activation of three sets of genes, termed immediate‐early, early and late genes (32, 123, 154, 166, 173). The immediate‐early genes initiate viral DNA transcription and viral protein production. Early gene expression depends on the previous synthesis of viral proteins but not viral DNA synthesis. Herpesvirus immediate‐early and early genes employ the host cell RNA polymerase II for transcription. The expression of herpesvirus late genes depends on the presence of viral proteins and DNA synthesis (56, 173).
Herpesvirus dissemination occurs when the production of virions prevails over the ability of the host immune system to suppress viral replication (Fig. 1). Viruses entering the lymphatic circulation can reach regional lymph nodes through replication in the vascular endothelium (2) or through infected monocytes and lymphocytes (94, 168). Dissemination allows viruses to reach new permissive cells to create a productive infection or a state of latency. A secondary viremia may subsequently translocate progeny virus particles to additional body sites.
Virus glycoproteins mediate attachment to specific receptors on the cell surface, which forms the basis for cellular and tissue tropism (Table 1) (68, 142). For example, herpes simplex viruses replicate remarkably well in epithelial cells, fibroblasts and macrophages, and spread to adjacent cells to produce the typical ulcerative lesion (19). Herpes simplex virus infection of neuronal endings (128) causes spread of the virus to trigeminal and spinal ganglia (87), where it establishes latency and forms the main source for successive bouts of replication.
Table 1.
Molecules involved in viral binding and entry into the host cell
| Virus | Binding | Entry | Tropism | Anatomic site of latency | ||
|---|---|---|---|---|---|---|
| Viral protein | Host molecule | Viral protein | Host receptor | |||
| Herpes simplex virus‐1 | Glycoprotein B, glycoprotein C | Heparan‐sulfate proteoglycans | Glycoprotein B | Paired immunoglobulin‐like type 2 receptor α | Epithelial, fibroblast, neurons | Sensory ganglia neurons, trigeminal and spinal ganglia |
| Glycoprotein D | Herpesvirus entry mediator, nectin 1, nectin 2,3‐O‐sulfotransferase‐modified heparan sulfate | |||||
| Herpes simplex virus‐2 | Glycoprotein B, glycoprotein C | Heparan‐sulfate proteoglycans | Glycoprotein D | Herpesvirus entry mediator, nectin 1, nectin 2,3‐O‐sulfotransferase‐modified heparan sulphate | Epithelial, fibroblast, neurons | Sensory ganglia neurons |
| Epstein–Barr virus | Glycoprotein 220, glycoprotein 350 | CD21 | Glycoprotein H, glycoprotein L, glycoprotein P42 | Major histocompatibility complex class II | Epithelial, fibroblasts, leucocytes Epstein–Barr virus can replicate in circulating B lymphocytes | B cells, pharyngeal epithelial cells |
| Cytomegalovirus | Glycoprotein B, glycoprotein M | Heparan‐sulfate proteoglycans | Glycoprotein B, glycoprotein H | Epidermal growth factor receptor, αvβ3‐integrin | Epithelial, fibroblasts, endothelial, leucocytes, immature dendritic cells Cytomegalovirus can replicate in circulating monocytes | Salivary glands, lymphocytes, macrophages, kidney, stromal cells |
Herpesvirus virions are released into respiratory‐tract aerosols, feces, saliva and other biological fluids, which constitute potential sources of viral transmission. Herpesvirus primary infection is followed by periods of latency and reactivation. In latency, the virus resides inside the cell without being identified by the immune system and can persist as a noninfectious form for considerable periods of time and even for a lifetime. Although the mechanisms of latency may be specific for a given virus, some general strategies include infection of nonpermissive cells, restriction of cytolytic effect, blocking of apoptosis and development of viral variants. Herpes simplex virus persistence in neuronal cells is genetically mediated by latency‐associated transcripts, which block the expression of lytic genes and prevent apoptosis (84). Cytomegalovirus latency involves a cellular intrinsic immunity mediated by nuclear body proteins that suppress the expression of viral immediate‐early proteins and subsequently the lytic infection (20, 57, 137, 154).
The virus genome is maintained during latency by integrating into the host cell genome or existing as an extrachromosomal plasmid (112). Retroviruses and some DNA viruses make use of genome integration. Epstein–Barr virus and herpes simplex viruses produce a nucleosome‐associated circular episomal DNA copy of the virus genome (84). Table 1 lists anatomic sites for herpesvirus latency and, accordingly, the most likely locations for recurrent clinical infections (5, 37, 59, 60, 84, 87, 101, 104, 111, 115, 146, 173).
Mechanisms of pathogenesis of human cytomegalovirus
Human cytomegalovirus disease can occur as a primary infection when the virus enters an immunonaive host, as an endogenous infection when a cytomegalovirus‐infected individual experiences reactivation from latency, and as an exogenous reinfection when a previously infected individual becomes infected with a new cytomegalovirus strain. The common form for cytomegalovirus transmission is through biological fluids, but transplacental (‘congenital’) transmission can take place from a mother experiencing a primary infection that is not controlled by neutralizing antibodies (26, 52, 88, 178, 183). Human cytomegalovirus can infect glial cells of the nervous system, monocytes, stromal cells, endothelial cells, epithelial cells, smooth‐muscle cells and fibroblasts, and may occur in saliva, serum, blood cells, gingival crevicular fluid, urine, maternal milk, tears, stool, vaginal and cervical secretions, and semen (111, 178). Peripheral monocytes and circulating endothelial cells infected with cytomegalovirus may carry the virus to distant sites in the body (2, 115). Cytomegalovirus‐infected cells can give rise to an enlarged rounded cell size termed ‘cytomegalic inclusion‐bearing cells’.
Glycoprotein B is the ligand for cellular attachment and penetration of the cytomegalovirus virion, as evidence by the failure of glycoprotein B‐deficient cytomegalovirus to propagate in culture (145). The predominant host defense against glycoprotein B is antibody mediated. Once the virus penetrates the cell, cytomegalovirus genes become activated in a cascade‐like manner. Like all viruses, cytomegalovirus relies upon the protein‐synthesis machinery of the host cell for replication. The cytomegalovirus immediate‐early genes (activated 0–2 h after primary infection) are regulators of the production of tegument phosphoproteins (pp65, pp72 and pp86) involved in cell cycle regulation, cell metabolism, and activation of later replication stages (152, 154, 164, 176). pp65 is involved in immune evasion, pp71 in gene expression, and pp150 and pp28 in virion assembly and egress. The tegument phosphoproteins are the immunodominant target of the cytomegalovirus‐specific cytotoxic T‐lymphocytes. Prime candidates for vaccine development are glycoprotein B and pp65, to be used in a combination.
Cytomegalovirus initial replication takes advantage of cellular transcriptional factors (102, 140), such as TATA‐binding protein, transcription factors IIB and IID, transcription factors cyclic adenosine monophosphate response element‐binding (CREB) and CREB‐binding protein (CBP), the histone acetyltransferase P300/CBP‐associated factor (P/CAF) (114, 140) and the cytomegalovirus immediate‐early proteins, which target the tumor suppressor proteins RB and p53 to induce the S‐phase for cellular quiescence (32). The immediate‐early genes also activate cytomegalovirus early genes (activated <24 h), which explains the slow replication process of the virus. Activation of the late genes (>24 h) completes the replication process and produce components necessary for genomic DNA duplication and for virion assembly and release. The late genes are also involved in the induction of latency by regulating cell‐cycle mechanisms, expression of class I major histocompatibility complex and production of interferon‐γ (55, 146).
Cytomegalovirus‐infected cells release chemokine‐like virokines, including a fully functional homolog of interleukin‐8 capable of promoting chemotaxis and degranulation of neutrophils (113). Also, cytomegalovirus transcripts of US27, US28 and US33 genes are homologous to CC chemokine G protein‐coupled receptors (20, 96, 107). The production of both virokines and their receptors may create a chemoattractant‐ and chemokine‐depleted environment, which may further contribute to immune evasion and tissue destruction.
Immune response against cytomegalovirus infection
Cytomegalovirus infections induce strong and diverse immune responses that nonetheless are incapable of eradicating the virus. The humoral immune response elaborates antibodies against viral proteinaceous surface molecules. The cellular immune response attempts to eliminate cytomegalovirus‐infected cells by means of cytotoxic CD8+ T‐lymphocytes that recognize viral peptides on the surface of infected cells in the context of class I major histocompatibility complex molecules. Perhaps, because of a herpesvirus periodontal infection, aggressive periodontitis lesions contain fewer overall viable cells, more T‐suppressor lymphocytes and more B‐lymphocytes (Epstein–Barr virus effect) than chronic periodontitis lesions or healthy periodontal sites (143). In addition to the innate and adaptive defenses against virus infection, a third defense mechanism that operates at the intracellular level was discovered recently. The intrinsic antiviral defense counteracts cytomegalovirus infections by impeding the expression of the viral immediate‐early gene (158).
The innate immune system plays roles in the early defense against cytomegalovirus and in the priming of the adaptive immune response. Cytomegalovirus is subject to innate sensing by toll‐like receptors and subsequent activation of toll‐like receptors. Activated toll‐like receptors trigger signal‐transduction pathways that induce dendritic cells and macrophages to release proinflammatory cytokines and interferon‐α/β capable of activating and recruiting polymorphonuclear leukocytes and natural killer cells to the site of infection (21, 39, 40, 177). Murine cytomegalovirus recognizes toll‐like receptors 3 and 9 (48, 69, 157), and stimulates interaction between toll‐like receptor 2 and glycoprotein B/glycoprotein H with release of proinflammatory cytokines (21, 40, 78). Of interest, the expression level of the toll‐like receptors 2, 7 and 9, which recognize viral DNA (and some bacterial pathogens), is significantly elevated in periodontitis lesions compared with gingivitis lesions (13, 14, 79).
Natural killer cells are key participants in the clearance of experimental murine cytomegalovirus infections. Evidence of this can be inferred by the ability of an adoptive transfer of natural killer cells to protect against murine cytomegalovirus (29, 117), and certain strains of mice, which are resistant to murine cytomegalovirus in vivo, become susceptible upon the depletion of natural killer cells (29). The resistance to cytomegalovirus infection is mediated by activation of murine natural killer cell receptor Ly‐49H in the natural killer gene complex (28, 29). In humans, less is known about the role of natural killer cells in the immune defense against cytomegalovirus. Renal transplant patients show an increased activity of natural killer cells during primary and recurrent cytomegalovirus infections, presumably in an attempt to control the cytomegalovirus infection (171).
In the adaptive immune system, the role of antibodies to cytomegalovirus is unclear, but effective humoral immunity against human and murine cytomegalovirus seems to be necessary to restrict viral dissemination and disease severity (22, 30, 66, 77). Passive immunization of guinea pigs with antibodies against cytomegalovirus, although unable to prevent infection, increased the survival rate of the litter (26). Also, passive immunization of guinea pigs with anti‐glycoprotein B serum and active immunization of pregnant guinea pigs with a recombinant glycoprotein B vaccine reduced cytomegalovirus fetal infection and disease (35).
In humans, neutralizing antibodies against cytomegalovirus surface protein glycoprotein B, which is involved in cell attachment and penetration, accounts for 50% of the neutralizing antibodies in cytomegalovirus‐infected individuals (27). The cytomegalovirus surface protein, glycoprotein H, fuses the viral envelope with the host cell membrane and is also a trigger of neutralizing antibodies (124). Cytomegalovirus antibodies of a mother protect the fetus against cytomegalovirus infection (88). Pregnant women, who showed previous immunity against cytomegalovirus, transmitted the virus to the fetus at a significantly lower rate than women who acquired a primary cytomegalovirus infection during pregnancy (52). Cytomegalovirus antibodies of low avidity and low affinity were associated with an increased rate of viral transmission from the mother to the fetus (22). Serum IgG against cytomegalovirus were demonstrated more frequently in patients with periodontitis than in patients with gingivitis, but their role in the periodontal disease process is not known (79).
Cytomegalovirus replication is primarily controlled and restricted by cell‐mediated immunity, involving CD8+ T‐cells, CD4+ T‐cells and γδ T‐cells. Serious cytomegalovirus diseases occur almost exclusively in patients with deficient cellular immunity. The essential role of T‐cell immunity was first recognized in murine cytomegalovirus models, where the elimination of lymphocytes coincided with increased cytomegalovirus reactivation and dissemination, and the adoptive transfer of virus‐specific CD8+ cytotoxic T‐cells conferred protection from an otherwise lethal cytomegalovirus challenge (46). A selective depletion of lymphocyte subsets in mice showed CD8+ T‐cells to be the most important component of the immune control of cytomegalovirus (105, 125). Depletion of CD8+ T‐cells in simian immunodeficiency virus‐infected monkeys reactivated a latent cytomegalovirus infection (11).
In humans, a primary cytomegalovirus infection during pregnancy caused fetal CD8+ lymphocytes to expand into mature and functional T‐cells (95). In AIDS patients, interferon‐γ‐producing cytomegalovirus‐specific CD8+ T‐cells conferred protection against cytomegalovirus‐associated retinitis (73). In allogenic bone marrow transplant recipients, infusion of donor‐derived cytomegalovirus‐specific CD8+ T‐cells restored antigen‐specific cellular immunity and protected against cytomegalovirus‐associated clinical complications (129, 174). Analyses of virus‐specific T‐cell responses in renal transplant recipients demonstrated that CD8+ T‐cell responses were able to limit viremia and protect against cytomegalovirus disease. Studies have found that half of transplant patients lacking a detectable anti‐cytomegalovirus T‐cell response developed cytomegalovirus disease (31, 126, 141).
The cytomegalovirus CD8+ T‐cell response is characterized by an accumulation of a polyclonal T‐cell repertoire and a reduction in the naive T‐cell pool (47, 118). The cytomegalovirus‐specific CD8+ T‐cell response depends on the functional avidity of antigen‐specific CD8+ T‐cells, which is substantially lower during an acute infection than in the long‐term pool of memory T‐cells (47). The selection of the cytomegalovirus‐specific repertoire of T‐cells is also determined by the structural organization of the human leukocyte antigen–peptide complex and the efficiency of the endogenous antigen presentation by the virus‐infected cells (179). Clonotypes with a restriction in T‐cell receptors demonstrate a more efficient recognition of virus‐infected cells and a more terminally differentiated phenotype than T‐cells expressing a diverse repertoire of T‐cell receptors. Taken together, the hierarchy of host immune responses and the memory‐cell repertoire in cytomegalovirus infections depends on the avidity of the antigen‐specific CD8+ T‐cells, the efficiency of viral epitope presentation and characteristics of the HLA–peptide complex (98).
The memory T‐cell population in most viral infections expands during acute infection, contracts once the infection is cleared and finally survives as a stable pool of memory T cells (153). However, the latent herpesvirus infection displays continuous expansions and contractions in the memory T‐cell pool similar to that seen during the acute phase of infection (45). The cytomegalovirus‐specific memory CD8+ T‐cell population fluctuates both in function and in absolute numbers, despite an overall stable count of total T‐cells (49). The fluctuation of the memory CD8+ T‐cell population takes place in the absence of detectable cytomegalovirus virions, indicating that the T‐cell fluctuation is not caused by a periodic viral reactivation.
In addition to the CD8+ T‐cells, CD4+ T‐cells are critical in the control of cytomegalovirus infections (51). A selective depletion of CD4+ T‐cells increases the incidence of recurrent murine cytomegalovirus infection in mice (117). Also, CD4+ T‐cells contribute to the control of primary murine cytomegalovirus infection in mice depleted of CD8+ T‐cells (76). Deficiency in cytomegalovirus‐specific CD4+ T‐cell immunity has been linked to a prolonged urinary and salivary shedding of cytomegalovirus in otherwise healthy children (111). Similarly to the CD8+ cytomegalovirus‐specific T‐cell compartment, a high proportion of CD4+ T‐cells in healthy seropositive individuals are committed to anti‐cytomegalovirus immunity (46). The specificity of the cytomegalovirus‐specific CD4+ T‐cell response exhibits broad antigen recognition, with glycoprotein B‐specific CD4+ T‐cell responses predominating in healthy individuals (>30%), although UL14 and UL16 specific responses are prominent in a small number (<5%) of individuals (156).
Avoidance and subversion of host defenses by cytomegalovirus
Although cytomegalovirus can induce cytolysis in a wide range of cells, the viral‐associated pathology cannot be explained merely by direct cellular killing. Herpesviruses establish a lifelong latent infection by subverting the host’s innate and adaptive immune defenses and the intrinsic antiviral defense mechanisms (172). Cytomegalovirus suppresses the antiviral function of the cellular proteins involved in the intrinsic antiviral defense by elaborating viral regulatory proteins that either disrupt the subnuclear structure or induce a proteasomal degradation of the intrinsic antiviral proteins (158). It is of pathogenetic importance that viral proteins produced during replication can subvert critical functions of the host cell, such as modulation of the cell cycle and cellular gene expression, down‐regulation of class I major histocompatibility complex and inhibition of apoptosis (32, 46, 114, 139, 185).
A major immune‐evasion mechanism of cytomegalovirus is related to class I major histocompatibility complex‐restricted antigen presentation (12). Cytotoxic T‐lymphocytes recognize cytomegalovirus antigenic peptides, which are generated by means of an immediate‐early‐1 transcription factor and presented in complex with class I major histocompatibility complex molecules (63). However, the cytomegalovirus phosphoprotein pp65, which has kinase activity capable of phosphorylating and selectively blocking the processing and presentation of immediate‐early‐derived antigenic peptides (57), can prevent the cytotoxic T‐lymphocyte response (58). Also, the cytomegalovirus genome encodes five proteins – US2, US3, US6, US10 and US11 – that block the formation and/or export of class I major histocompatibility complex–peptide complexes and induce a rapid down‐regulation of class I major histocompatibility complex expression (3, 74, 75). Cytomegalovirus can also interfere with antigen presentation through the class II major histocompatibility complex pathway by means of US2, which can target the class II major histocompatibility complex molecules for proteasome degradation (163), and by proteins that hinder the interferon‐γ‐induced expression of class II major histocompatibility complex molecules (100).
Natural killer cells selectively recognize and kill cells lacking cell‐surface‐expressed major histocompatibility complex class I molecules (91), making cytomegalovirus‐infected cells with down‐regulated major histocompatibility complex molecules potentially vulnerable to natural killer cell‐mediated lysis. However, human cytomegalovirus produces proteins, such as pp65 and UL16, which can block natural killer cell activation (9, 44). Cytomegalovirus matrix protein pp65 phosphorylates the immediate‐early‐1 peptides, which selectively blocks the processing and presentation of the immediate‐early‐1 protein via the major histocompatibility complex class I pathway, thereby preventing the immediate‐early‐1 specific cytotoxic T‐lymphocyte immune response. This mechanism acts in conjunction with other human cytomegalovirus proteins (US2, US3, US6, US10 and US11) that block the production, the export and the expression of major histocompatibility complex class I molecules (3, 43, 46, 53, 57, 58, 74, 75, 176). Furthermore, the expression of the molecules CD40 and CD80, which serve as co‐stimulators for antigen presentation, are down‐regulated in cytomegalovirus‐infected monocytes (62).
Cytomegalovirus can also impede natural killer cell recognition by expressing virus‐encoded major histocompatibility complex class I homologues that act as decoy proteins (103). For example, the major histocompatibility complex class I molecule, human leukocyte antigen‐E, which depends on the binding of a signal peptide derived from other host major histocompatibility complex class I molecules, suppresses natural killer cell recognition by binding the inhibitory receptor CD94/NKG2A (46, 89, 162). However, the cytomegalovirus gene products UL40 and UL16 contain a sequence homologous to the signal peptides, which can substitute and up‐regulate cell‐surface expression of human leukocyte antigen‐E to protect virus‐infected cells from natural killer cell‐induced killing (89, 162).
The cytomegalovirus genome also encodes a variety of homologues that mimic the behavior of host proteins and divert the immune response. One such homologue is the human major histocompatibility complex class I homologue UL18, which, like major histocompatibility complex class I molecules, binds β2‐microglobulin but, in contrast to the human molecule, shows specific binding only to leukocyte immunoglobulin‐like receptor 1, a receptor prominently displayed on monocytes and B‐cells (43). The binding of leukocyte immunoglobulin‐like receptor 1 to UL18 resembles the binding to host major histocompatibility complex class I molecules (34), but the precise biological effects of UL18 activity during a cytomegalovirus infection are unknown (127). Four cytomegalovirus genes –UL33, UL78, US27 and US28– encode homologues of seven transmembrane G protein‐coupled receptors (36), and of these, US28 encodes a chemokine receptor that binds most human CC chemokines and the CX3C chemokine, fractalkine (54, 82). Cytomegalovirus also encodes a homologue (UL111a) of the immunosuppressive cytokine interleukin‐10 (86), a viral tumor necrosis factor receptor (UL144) (15), a potent interleukin‐8‐like chemokine, viral CXC‐1, which is capable of inducing chemotaxis of human peripheral blood neutrophils (UL146) (113), and various anti‐apoptotic gene products (UL36 and UL37) (61, 147).
Cytomegalovirus can inhibit apoptosis by synthesis of proteins that prevent killing of the infected cell. Inhibition of apoptosis is essential for the accumulation of molecular precursors necessary for virion assembly and release. Cytomegalovirus immediate‐early‐1 and immediate‐early‐2 proteins inhibit apoptosis induced by tumor necrosis factor‐α (185). Cytomegalovirus proteins also block caspase 8‐induced apoptosis, and cytomegalovirus UL36 and UL37 genes inhibit Fas ligation‐mediated apoptosis (61). Sustained viral activity in nonapoptotic cells can give rise to chronic inflammation.
Periodontopathogenicity of cytomegalovirus
Periodontitis may, at least in part, result from the attempt of the host to neutralize infecting viruses and bacteria. While specific bacteria are widely considered to be the main etiologic agents of periodontitis, various herpesviruses have been revealed to have a close relationship with the disease. Cytomegalovirus has been detected immunohistologically in biopsies from marginal (155) and apical (133) periodontitis lesions, in gingival monocytes and in T‐cells from periodontitis patients (42), and in periapical cysts (7). An active cytomegalovirus periodontal infection has been linked to disease‐active periodontitis (41, 161), and the virus may also play a role in other types of periodontal disease (149).
Several mechanisms exist by which cytomegalovirus may contribute to periodontitis. As depicted in Fig. 3, the herpesviral–bacterial interactive model for destructive periodontal disease starts with dental‐plaque bacteria inducing inflammation of gingiva. The first cells to respond to the bacterial challenge are sulcular and junctional epithelial cells, which release defensins and cytokines, notably interleukin‐8 and interleukin‐1β (6, 8, 50). The gingival connective tissue reacts by recruiting monocytes, macrophages and neutrophils (50, 81, 170), followed by CD4 cells in a T helper‐1 and T helper‐2 combined response (14, 181). The cytomegalovirus latent genome is carried into the periodontium by infected macrophages and T‐cells (42), and cytomegalovirus activation may subsequently give rise to infection of additional cell types. An active cytomegalovirus infection in macrophages and T‐cells triggers a significant release of interleukin‐1β and tumor necrosis factor‐α (40, 72, 80, 94, 119, 121). These proinflammatory mediators recruit antiviral inflammatory cells to the site of infection but also induce osteoclast differentiation and the release of matrix metalloproteinases (109). Cytomegalovirus activation takes place with decreased cellular immunity and the activation process itself can further reduce the host immunity. Macrophages infected with cytomegalovirus or Epstein–Barr virus exhibit a decreased host response, with inhibition of phagocytic activity, tumor necrosis factor‐α production and toll‐like receptor‐9 expression (90). If the duration of diminished immunity is sufficiently long, an upgrowth of specific periodontopathic bacteria and destructive periodontal disease may ensue (1, 150). Cytomegalovirus can replicate in cultured gingival tissue (65) and enhance the adherence of A. actinomycetemcomitans to such cells (160), thereby providing an additional mechanism for increasing the pathogen load.
Figure 3.
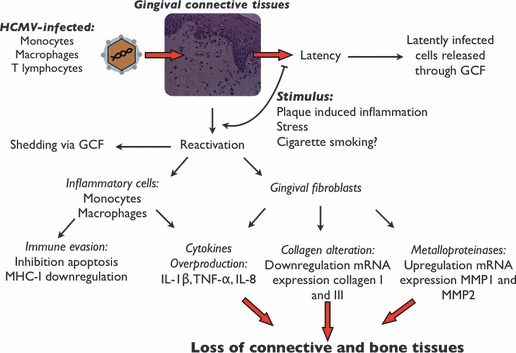
Proposed model linking cytomegalovirus to periodontal breakdown. Cytomegalovirus‐infected cells reach periodontal connective tissues, and reactivation or latency occur within the connective tissues. Virions infect other cells (gingival fibroblasts and inflammatory cells), which results in the release of proinflammatory cytokines and increased susceptibility to tissue breakdown, including loss of aveolar bone and periodontal attachment. GCF, gingival crevicular fluid; IL, interleukin; MHC‐I, major histocompatibility complex class I; MMP, matrix metalloproteinase; TNF‐α, tumor necrosis factor‐α.
Cytomegalovirus‐induced inflammation may either aggravate or control the cytomegalovirus infection. Tumor necrosis factor‐α can activate the cytomegalovirus immediate‐early gene promoter (120, 121), a necessary step in the replication of the virus. Interleukin‐1β and interleukin‐4 have been shown to up‐regulate the activity of the cytomegalovirus immediate‐early gene promoter in human umbilical vein endothelial cells (130). It is of interest that positive correlations have been found between the levels of tumor necrosis factor‐α and interleukin‐1β, and the severity of periodontal disease (144, 159). However, tumor necrosis factor‐α and interleukin‐1β serve ultimately an anti‐cytomegalovirus function by stimulating the cellular immunity and promoting an influx of cytotoxic lymphocytes. The complex profile of cytokines in the periodontal disease microenvironment may be supportive for activation of cytomegalovirus, at least for a limited period until cellular immune responses reverse the viral activation (Fig. 3).
Psychosomatic stress can significantly affects the body’s immune systems, and chronic stress has been associated with an enhanced risk of periodontal disease (38, 122). Stress responses serve as a replication stimulatory signal for herpesviruses. Catecholamines [epinephrine (adrenaline), norepinephrine (noradrenaline) and dopamine] released during stressful events directly stimulate the cytomegalovirus immediate‐early gene enhancer/promoter in monocytic cells via β2 adrenergic receptors (119, 120). In addition, catecholamines are involved in the regulation of immunoresponsiveness through the interleukin‐10 promoter/enhancer via the cyclic adenosine monophosphate (cAMP)/protein kinase A‐dependent pathway (86, 116) (Fig. 3).
Cytomegalovirus DNA can be detected in neutrophilic leukocytes during a productive infection, probably as a result of phagocytosis (102). Infection of neutrophils with cytomegalovirus may induce abnormalities in adherence, chemotactic, phagocytic, oxidative, secretory and bactericidal activities of polymorphonuclear neutrophils (1). Neutrophils are key cells in controlling periodontal bacterial infections (169, 170), and the phagocytic and bactericidal capacities of periodontal neutrophils seem to be significantly impaired in subjects carrying herpesviruses in oral lymphocytes and epithelial cells, compared with virus‐free subjects (108). The functional defects of neutrophils identified in patients with localized aggressive periodontitis (170) may partly be caused by an ongoing cytomegalovirus‐active infection of these patients (161) (Fig. 3).
Cytomegalovirus can infect and establish latency in gingival fibroblasts. Primary cultured gingival fibroblasts are permissive for cytomegalovirus infection (Fig. 4), and the pp72 cytomegalovirus antigen identified by immunofluorescence staining is expressed in fibroblasts within 24 h of replication (Fig. 5). Botero et al. (25) demonstrated that in vitro cultured gingival fibroblasts infected with cytomegalovirus resulted in a dose‐dependent down‐regulation of mRNA expression for collagens I and III, compared with UV‐light‐inactivated inoculated cells. mRNA analysis of gingival specimens showed that the expression of collagens was lower in cytomegalovirus‐positive periodontitis samples than in healthy gingiva samples (25). Cytomegalovirus can also affect the production of matrix metalloproteinases. An up‐regulation of the expression of mRNA for matrix metalloproteinases 1 and 2 was detected in cytomegalovirus‐positive gingival biopsies (25) (Fig. 3). Studies on guided tissue regeneration have found an interesting relationship between periodontal herpesviruses and impairment of clinical attachment gain (17, 93).
Figure 4.
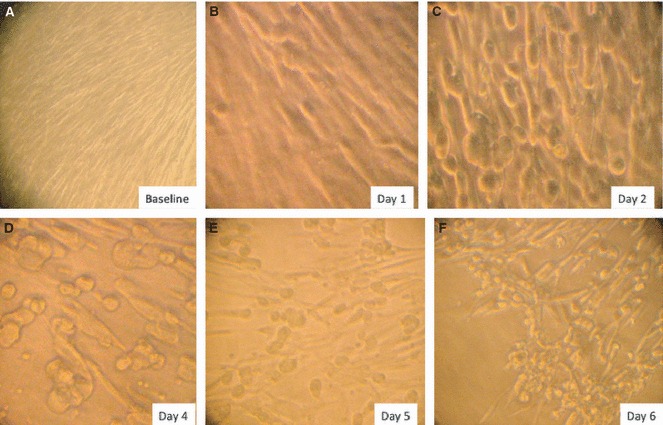
Primary culture of gingival fibroblasts infected with cytomegalovirus Towne strain. Gingival fibroblasts are shown before infection (A), 1 day after infection (B), 2 days after infection (C), 4 days after infection (D), 5 days after infection (E) and 6 days after infection (F). Note the characteristic cytopathic effects and the increasing number of infected cells over time.
Figure 5.
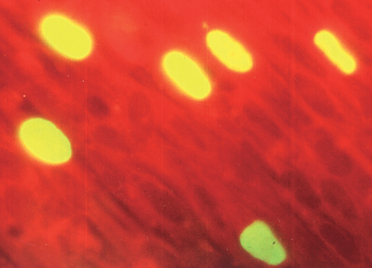
Indirect immunofluorescence staining of cytomegalovirus‐infected gingival fibroblasts with the primary antibody against the immediate‐early pp72 protein.
Gingival fibroblasts, although not inflammatory cells per se, are able to produce a wide assortment of proinflammatory cytokines (24). Cytomegalovirus‐infected gingival fibroblasts produced more interleukin‐1β, tumor necrosis factor‐α, interleukin‐1α, interleukin‐12p40, interleukin‐12p70, interleukin‐6, interleukin‐8 and interferon‐γ than noninfected control fibroblasts (24). Gingival specimens from cytomegalovirus‐positive periodontitis lesions showed an up‐regulation of mRNAs for interleukin‐1β and tumor necrosis factor‐α (24). The proinflammatory cytokines produced in response to a cytomegalovirus infection aim to attract antiviral cytotoxic T cells and natural killer cells to the site of infection. However, at high levels, proinflammatory cytokines may interfere with collagen production by fibroblasts (24) and stimulate bone‐resorbing osteoclasts (24). Also, as proinflammatory cytokines suppress the release of anti‐inflammatory cytokines, which are involved in antibody production and in the containment of bacterial pathogens, an active cytomegalovirus infection may result in the overgrowth of periodontopathic bacteria (41, 148).
Cytomegalovirus stimulates cytokine release from infected monocytes. Contreras et al. (42) found the cytomegalovirus genome in gingival mononuclear cells (55%) and T‐cells (20%) from periodontitis patients. Cytomegalovirus‐infected monocytes produce more interleukin‐1β than noninfected monocytes, perhaps because of interactions between viral proteins and gene promoters within the infected cell (72, 175). Cytomegalovirus proteins bG and glycoprotein H bind to the monocyte toll‐like receptor‐2 that leads to nuclear factor‐κB, and probably C/EBPβ, activation via a p38‐dependent phosphorylation event (10, 39). In addition, direct transactivation of the interleukin‐1β promoter by immediate‐early proteins 1 and 2 results in a high and sustained production of interleukin‐1β (40, 72, 140, 175, 182) (Fig. 3).
Conclusions and perspectives
Cytomegalovirus infections of the periodontium may explain several features of periodontitis, such as the insidious disease onset, the site‐specificity of the periodontal attachment loss, the mirror‐like disease pattern of localized aggressive periodontitis, and the overgrowth of specific bacterial species in the periodontal pocket. The dynamic interaction between infectious cytomegalovirus and host immune responses may partly account for the discontinuous pattern of periodontal disease progression and for the refractory state of disease in the case of inadequate immunity. Cytomegalovirus may participate in the development of periodontitis by causing macrophages and T‐cells to release osteoclast‐inducing interleukin‐1β and tumor necrosis factor‐α. Also, gingival fibroblasts infected with cytomegalovirus exhibit diminished production of collagens I and III and enhanced generation of matrix metalloproteinases 1 and 2. However, although biologically plausible, the extent to which cytomegalovirus participates in the destruction of the human periodontium is still a matter of research. Studies are needed to identify the environmental events and pathogenic pathways that trigger activation of cytomegalovirus in the peridontium, the possible link between cytomegalovirus reactivation and periodontitis disease activity, and the importance of anti‐cytomegalovirus immunity in controlling periodontal disease. Such information may help to explain why cytomegalovirus and other ubiquitous herpesviruses may cause periodontitis only in a relatively small subset of individuals and teeth. Hopefully, increased knowledge of the immunovirology of cytomegalovirus and other herpesviruses in periodontitis may lead to a greater understanding of periodontal host responses and to more effective preventive and therapeutic interventions, including future vaccination against periodontopathic herpesviruses.
References
- 1. Abramson JS, Mills EL. Depression of neutrophil function induced by viruses and its role in secondary microbial infections. Rev Infect Dis 1988: 10: 326–341. [DOI] [PubMed] [Google Scholar]
- 2. Adler B, Sinzger C. Endothelial cells in human cytomegalovirus infection: one host cell out of many or a crucial target for virus spread? Thromb Haemost 2009: 102: 1057–1063. [DOI] [PubMed] [Google Scholar]
- 3. Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJ, Ploegh HL, Peterson PA, Yang Y, Früh K. The ER‐luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997: 6: 613–621. [DOI] [PubMed] [Google Scholar]
- 4. Akhtar J, Shukla D. Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J 2009: 276: 7228–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amon W, Farrell PJ. Reactivation of Epstein‐Barr virus from latency. Rev Med Virol 2005: 15: 149–156. [DOI] [PubMed] [Google Scholar]
- 6. Andrew J, van Dyke D, van Dyke T. Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000 2003: 31: 55–76. [DOI] [PubMed] [Google Scholar]
- 7. Andric M, Milasin J, Jovanovic T, Todorovic L. Human cytomegalovirus is present in odontogenic cysts. Oral Microbiol Immunol 2007: 22: 347–351. [DOI] [PubMed] [Google Scholar]
- 8. Armitage GC. Comparison of the microbiologic features of chronic and aggressive periodontitis. Periodontol 2000 2010: 53: 70–88. [DOI] [PubMed] [Google Scholar]
- 9. Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, Gazit R, Gonen‐Gross T, Hanna J, Nahari E, Porgador A, Honigman A, Plachter B, Mevorach D, Wolf DG, Mandelboim O. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 2005: 6: 515–523. [DOI] [PubMed] [Google Scholar]
- 10. Baldassare JJ, Bi Y, Bellone CJ. The role of p38 mitogen‐activated protein kinase in IL‐1 beta transcription. J Immunol 1999: 162: 5367–5373. [PubMed] [Google Scholar]
- 11. Barry AP, Silvestri G, Safrit JT, Sumpter B, Kozyr N, McClure HM, Staprans SI, Feinberg MB. Depletion of CD8+ cells in sooty mangabey monkeys naturally infected with simian immunodeficiency virus reveals limited role for immune control of virus replication in a natural host species. J Immunol 2007: 178: 8002–8012. [DOI] [PubMed] [Google Scholar]
- 12. Basta S, Bennink JR. A survival game of hide and seek: cytomegaloviruses and MHC class I antigen presentation pathways. Viral Immunol 2003: 16: 231–242. [DOI] [PubMed] [Google Scholar]
- 13. Beklen A, Hukkanen M, Konttinen YT. Immunohistochemical localization of Toll‐like receptors 1–10 in periodontitis. Oral Microbiol Immunol 2008: 23: 425–431. [DOI] [PubMed] [Google Scholar]
- 14. Beklen A, Sorsa T, Konttinen YT. Toll‐like receptors 2 and 5 in human gingival epithelial cells co‐operate with T‐cell cytokine interleukin‐17. Oral Microbiol Immunol 2009: 24: 38–42. [DOI] [PubMed] [Google Scholar]
- 15. Benedict CA, Butrovich KD, Lurain NS, Corbeil J, Rooney I, Schneider P, Tschopp T, Ware CF. Cutting edge: a novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J Immunol 1999: 162: 6967–6970. [PubMed] [Google Scholar]
- 16. Bergelson JM. Intercellular junctional proteins as receptors and barriers to virus infection and spread. Cell Host Microbe 2009: 5: 517–521. [DOI] [PubMed] [Google Scholar]
- 17. Bertoldi C, Pellacani C, Lalla M, Consolo U, Pinti M, Cortellini P, Cossarizza A. Herpes simplex I virus impairs regenerative outcomes of periodontal regenerative therapy in intrabony defects. A pilot study. J Clin Periodontol 2012: 39: 385–392. [DOI] [PubMed] [Google Scholar]
- 18. Birek C, Ficarra G. The diagnosis and management of oral herpes simplex infection. Curr Infect Dis Rep 2006: 8: 181–188. [DOI] [PubMed] [Google Scholar]
- 19. Blank H, Burgoon CF, Baldridge GD, McCarthy PL, Urbach F. Cytologic smears in diagnosis of herpes simplex, herpes zoster, and varicella. J Am Med Assoc 1951: 146: 1410–1412. [DOI] [PubMed] [Google Scholar]
- 20. Bodaghi B, Jones TR, Zipeto D, Vita C, Sun L, Laurent L, Arenzana‐Seisdedos F, Virelizier JL, Michelson S. Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: withdrawal of chemokines from the environment of cytomegalovirus‐infected cells. J Exp Med 1998: 188: 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boehme KW, Compton T. Innate sensing of viruses by Toll‐like receptors. J Virol 2004: 78: 7867–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boppana SB, Britt WJ. Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J Infect Dis 1995: 171: 1115–1121. [DOI] [PubMed] [Google Scholar]
- 23. Botero JE, Arce RM, Escudero M, Betancourth M, Jaramillo A, Contreras A. Frequency of detection of periodontopathic and superinfecting bacteria in HIV‐positive patients with periodontitis. J Int Acad Periodontol 2007: 9: 13–18. [PubMed] [Google Scholar]
- 24. Botero JE, Contreras A, Parra B. Profiling of inflammatory cytokines produced by gingival fibroblasts after human cytomegalovirus infection. Oral Microbiol Immunol 2008: 23: 291–298. [DOI] [PubMed] [Google Scholar]
- 25. Botero JE, Contreras A, Parra B. Effects of cytomegalovirus infection on the mRNA expression of collagens and matrix metalloproteinases in gingival fibroblasts. J Periodontal Res 2008: 43: 649–657. [DOI] [PubMed] [Google Scholar]
- 26. Bratcher DF, Bourne N, Bravo FJ, Schleiss MR, Slaoui M, Myers MG, Bernstein DI. Effect of passive antibody on congenital cytomegalovirus infection in guinea pigs. J Infect Dis 1995: 172: 944–950. [DOI] [PubMed] [Google Scholar]
- 27. Britt WJ, Vugler L, Butfiloski EJ, Stephens EB. Cell surface expression of human cytomegalovirus (HCMV) gp55‐116 (gB): use of HCMV‐recombinant vaccinia virus‐infected cells in analysis of the human neutralizing antibody response. J Virol 1990: 64: 1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 2001: 292: 934–937. [DOI] [PubMed] [Google Scholar]
- 29. Bukowski JF, Warner JF, Dennert G, Welsh RM. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo . J Exp Med 1985: 161: 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bukowski JF, Woda BA, Habu S, Okumura K, Welsh RM. Natural killer cell depletion enhances virus synthesis and virus‐induced hepatitis in vivo. J Immunol 1983: 131: 1531–1538. [PubMed] [Google Scholar]
- 31. Bunde T, Kirchner A, Hoffmeister B, Habedank D, Hetzer R, Cherepnev G, Proesch S, Reinke P, Volk HD, Lehmkuhl H, Kern F. Protection from cytomegalovirus after transplantation is correlated with immediate early 1‐specific CD8 T cells. J Exp Med 2005: 201: 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Castillo JP, Kowalik TF. Human cytomegalovirus immediate early proteins and cell growth control. Gene 2002: 290: 19–34. [DOI] [PubMed] [Google Scholar]
- 33. Chan MC, Chan RW, Yu WC, Ho CC, Chui WH, Lo CK, Yuen KM, Guan YI, Nicholls JM, Peiris JS. Influenza H5N1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Respir Res 2009: 10: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR‐1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity 1999: 11: 603–613. [DOI] [PubMed] [Google Scholar]
- 35. Chatterjee A, Harrison CJ, Britt WJ, Bewtra C. Modification of maternal and congenital cytomegalovirus infection by anti‐glycoprotein b antibody transfer in guinea pigs. J Infect Dis 2001: 183: 1547–1553. [DOI] [PubMed] [Google Scholar]
- 36. Chee MS, Satchwell SC, Preddie E, Weston KM, Barrell BG. Human cytomegalovirus encodes three G protein‐coupled receptor homologues. Nature 1990: 344: 774–777. [DOI] [PubMed] [Google Scholar]
- 37. Chen T, Hudnall SD. Anatomical mapping of human herpesvirus reservoirs of infection. Mod Pathol 2006: 19: 726–737. [DOI] [PubMed] [Google Scholar]
- 38. Chiou LJ, Yang YH, Hung HC, Tsai CC, Shieh TY, Wu YM, Wang WC, Hsu TC. The association of psychosocial factors and smoking with periodontal health in a community population. J Periodontal Res 2010: 45: 16–22. [DOI] [PubMed] [Google Scholar]
- 39. Compton T. Receptors and immune sensors: the complex entry path of human cytomegalovirus. Trends Cell Biol 2004: 14: 5–8. [DOI] [PubMed] [Google Scholar]
- 40. Compton T, Kurt‐Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll‐like receptor 2. J Virol 2003: 77: 4588–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Contreras A, Slots J. Herpesviruses in human periodontal disease. J Periodontal Res 2000: 35: 3–16. [DOI] [PubMed] [Google Scholar]
- 42. Contreras A, Zadeh HH, Nowzari H, Slots J. Herpesvirus infection of inflammatory cells in human periodontitis. Oral Microbiol Immunol 1999: 14: 206–212. [DOI] [PubMed] [Google Scholar]
- 43. Cosman D, Fanger N, Borges L. Human cytomegalovirus, MHC class I and inhibitory signalling receptors: more questions than answers. Immunol Rev 1999: 168: 177–185. [DOI] [PubMed] [Google Scholar]
- 44. Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I‐related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001: 14: 123–133. [DOI] [PubMed] [Google Scholar]
- 45. Crough T, Burrows JM, Fazou C, Walker S, Davenport MP, Khanna R. Contemporaneous fluctuations in T cell responses to persistent herpes virus infections. Eur J Immunol 2005: 35: 139–149. [DOI] [PubMed] [Google Scholar]
- 46. Crough T, Khanna R. Immunobiology of human cytomegalovirus: from bench to bedsite. Clin Microbiol Rev 2009: 22: 76–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Day EK, Carmichael AJ, ten Berge IJ, Waller EC, Sissons JG, Wills MR. Rapid CD8+ T cell repertoire focusing and selection of high‐affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol 2007: 179: 3203–3213. [DOI] [PubMed] [Google Scholar]
- 48. Delale T, Paquin A, Asselin‐Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. MyD88‐dependent and ‐independent murine cytomegalovirus sensing for IFN‐alpha release and initiation of immune responses in vivo . J Immunol 2005: 175: 6723–6732. [DOI] [PubMed] [Google Scholar]
- 49. Dunn HS, Haney DJ, Ghanekar SA, Stepick‐Biek P, Lewis DB, Maecker HT. Dynamics of CD4 and CD8 T cell responses to cytomegalovirus in healthy human donors. J Infect Dis 2002: 186: 15–22. [DOI] [PubMed] [Google Scholar]
- 50. Ebersole JL. Humoral immune responses in gingival crevice fluid: local and systemic implications. Periodontol 2000 2003: 31: 135–166. [DOI] [PubMed] [Google Scholar]
- 51. Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Loffler J, Grigoleit U, Moris A, Rammensee HG, Kanz L, Kleihauer A, Frank F, Jahn G, Hebart H. Infusion of cytomegalovirus (CMV)‐specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002: 99: 3916–3922. [DOI] [PubMed] [Google Scholar]
- 52. Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med 1992: 326: 663–667. [DOI] [PubMed] [Google Scholar]
- 53. Furman MH, Dey N, Tortorella D, Ploegh HL. The human cytomegalovirus US10 gene product delays trafficking of major histocompatibility complex class I molecules. J Virol 2002: 76: 11753–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao JL, Murphy PM. Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J Biol Chem 1994: 269: 28539–28542. [PubMed] [Google Scholar]
- 55. Gawn JM, Greaves RF. Absence of IE1 p72 protein function during low‐multiplicity infection by human cytomegalovirus results in a broad block to viral delayed‐early gene expression. J Virol 2002: 76: 4441–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gibson W. Human cytomegalovirus: molecular biology In: Mahy BWJ, van Regenmortel MH. editors. Desk encyclopedia of general virology. San Diego, CA: Academic Press, 2010: 469–474. [Google Scholar]
- 57. Gilbert MJ, Riddell SR, Li CR, Greenberg PD. Selective interference with class I major histocompatibility complex presentation of the major immediate‐early protein following infection with human cytomegalovirus. J Virol 1993: 67: 3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gilbert MJ, Riddell SR, Plachter B, Greenberg PD. Cytomegalovirus selectively blocks antigen processing and presentation of its immediate‐early gene product. Nature 1996: 383: 720–722. [DOI] [PubMed] [Google Scholar]
- 59. Gilden DH, Mahalingam R, Cohrs R, Tyler KL. Herpesvirus infection of the nervous system. Nat Clin Pract Neurol 2007: 3: 82–94. [DOI] [PubMed] [Google Scholar]
- 60. Glick M. Clinical aspects of recurrent oral herpes simplex virus infection. Compend Contin Educ Dent 2002: 23(7 Suppl. 2): 4–8. [PubMed] [Google Scholar]
- 61. Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, Han JW, Lutz RJ, Watanabe S, Cahir McFarland ED, Kieff ED, Mocarski ES, Chittenden T. A cytomegalovirus‐encoded mitochondria‐localized inhibitor of apoptosis structurally unrelated to Bcl‐2. Proc Natl Acad Sci USA 1999: 96: 12536–12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grigoleit U, Riegler S, Einsele H, Laib Sampaio K, Jahn G, Hebart H, Brossart P, Frank F, Sinzger C. Human cytomegalovirus induces a direct inhibitory effect on antigen presentation by monocyte‐derived immature dendritic cells. Br J Haematol 2002: 119: 189–198. [DOI] [PubMed] [Google Scholar]
- 63. Groothuis TA, Griekspoor AC, Neijssen JJ, Herberts CA, Neefjes JJ. MHC class I alleles and their exploration of the antigen‐processing machinery. Immunol Rev 2005: 207: 60–76. [DOI] [PubMed] [Google Scholar]
- 64. Haffajee AD, Socransky SS. Microbial etiological agents of destructive periodontal diseases. Periodontol 2000 1994: 5: 78–111. [DOI] [PubMed] [Google Scholar]
- 65. Hai R, Chu A, Li H, Umamoto S, Rider P, Liu F. Infection of human cytomegalovirus in cultured human gingival tissue. J Virol 2006: 3: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Halary F, Pitard V, Dlubek D, Krzysiek R, de la Salle H, Merville P, Dromer C, Emilie D, Moreau JF, Dechanet‐Merville J. Shared reactivity of Vδ2(−) γδT cells against cytomegalovirus‐infected cells and tumor intestinal epithelial cells. J Exp Med 2005: 201: 1567–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hayward GS, Ambinder R, Ciufo D, Hayward SD, LaFemina RL. Structural organization of human herpesvirus DNA molecules. J Invest Dermatol 1984: 83(1 Suppl.): 29s–41s. [DOI] [PubMed] [Google Scholar]
- 68. Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 2008: 65: 1653–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hokeness‐Antonelli KL, Crane MJ, Dragoi AM, Chu WM, Salazar‐Mather TP. IFN‐alpha beta‐mediated inflammatory responses and antiviral defense in liver is TLR9‐independent but MyD88‐dependent during murine cytomegalovirus infection. J Immunol 2007: 179: 6176–6183. [DOI] [PubMed] [Google Scholar]
- 70. Hsu YC, Lu HF, Huang CC, Hsu RF, Su CY. Malignant lymphoepithelial lesions of the salivary gland. Otolaryngol Head Neck Surg 2006: 134: 661–666. [DOI] [PubMed] [Google Scholar]
- 71. Hui DS, Chan PK. Severe acute respiratory syndrome and coronavirus. Infect Dis Clin North Am 2010: 24: 619–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Iwamoto GK, Monick MM, Clark BD, Auron PE, Stinski MF, Hunninghake GW. Modulation of interleukin 1 beta gene expression by the immediate early genes of human cytomegalovirus. J Clin Invest 1990: 85: 1853–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jacobson MA, Maecker HT, Orr PL, D’Amico R, Van Natta M, Li XD, Pollard RB, Bredt BM. Results of a cytomegalovirus (CMV)‐specific CD8+/interferon‐gamma+ cytokine flow cytometry assay correlate with clinical evidence of protective immunity in patients with AIDS with CMV retinitis. J Infect Dis 2004: 189: 1362–1373. [DOI] [PubMed] [Google Scholar]
- 74. Jones TR, Sun L. Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. J Virol 1997: 71: 2970–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jones TR, Wiertz EJ, Sun L, Fish KN, Nelson JA, Ploegh HL. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc Natl Acad Sci USA 1996: 93: 11327–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jonjic S, Pavic I, Lucin P, Rukavina D, Koszinowski HU. Efficacious control of cytomegalovirus infection after long‐term depletion of CD8+ T lymphocytes. J Virol 1990: 64: 5457–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jonjic S, Pavic I, Polic B, Crnkovic I, Lucin P, Koszinowski UH. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med 1994: 179: 1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Juckem LK, Boehme KW, Feire AL, Compton T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 2008: 180: 4965–4977. [DOI] [PubMed] [Google Scholar]
- 79. Kajita K, Honda T, Amanuma R, Domon H, Okui T, Ito H, Yoshie H, Tabeta K, Nakajima T, Yamazaki K. Quantitative messenger RNA expression of Toll‐like receptors and interferon‐alpha1 in gingivitis and periodontitis. Oral Microbiol Immunol 2007: 22: 398–402. [DOI] [PubMed] [Google Scholar]
- 80. Kasprzak A, Zabel M, Wysocki J, Seidel J, Surdyk‐Zasada J, Filipiak B. Expression of mRNA for cytokines (TNF‐alpha and IL‐1alpha) in human cytomegalovirus (HCMV) and hepatitis B virus (HBV) infections. Folia Histochem Cytobiol 2002: 40: 63–68. [PubMed] [Google Scholar]
- 81. Kinane D, Lappin DF. Immune processes in periodontal disease: a review. Ann Periodontol 2002: 7: 62–71. [DOI] [PubMed] [Google Scholar]
- 82. Kledal TN, Rosenkilde MM, Schwartz TW. Selective recognition of the membrane‐bound CX3C chemokine, fractalkine, by the human cytomegalovirus‐encoded broad‐spectrum receptor US28. FEBS Lett 1998: 441: 209–214. [DOI] [PubMed] [Google Scholar]
- 83. Klempa B. Hantaviruses and the climate change. Clin Microbiol Infect 2009: 15: 518–523. [DOI] [PubMed] [Google Scholar]
- 84. Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 2008: 6: 211–221. [DOI] [PubMed] [Google Scholar]
- 85. Koo HL, Ajami N, Atmar RL, DuPont HL. Noroviruses: the leading cause of gastroenteritis worldwide. Discov Med 2010: 10: 1–70. [PMC free article] [PubMed] [Google Scholar]
- 86. Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL‐10 homolog (cmvIL‐10). Proc Natl Acad Sci USA 2000: 97: 1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kristensson K, Nennesmo L, Persson L, Lycke E. Neuron to neuron transmission of herpes simplex virus. Transport of virus from skin to brainstem nuclei. J Neurol Sci 1982: 54: 149–156. [DOI] [PubMed] [Google Scholar]
- 88. Lazzarotto T, Guerra B, Lanari M, Gabrielli L, Landini M. New advances in the diagnosis of congenital cytomegalovirus infection. J Clin Virol 2008: 41: 192–197. [DOI] [PubMed] [Google Scholar]
- 89. Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, Lanier LL. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med 1998: 187: 1681–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lin Y‐L, Li M. Human cytomegalovirus and Epstein–Barr virus inhibit oral bacteria‐induced macrophage activation and phagocytosis. Oral Microbiol Immunol 2009: 24: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 1990: 11: 237–244. [DOI] [PubMed] [Google Scholar]
- 92. Lo MK, Rota PA. The emergence of Nipha virus, a highly pathogenic paramyxovirus. J Clin Virol 2008: 43: 396–400. [DOI] [PubMed] [Google Scholar]
- 93. MacDonald ES, Nowzari H, Contreras A, Flynn J, Morrison J, Slots J. Clinical and microbiological evaluation of a bioabsorbable and a nonresorbable barrier membrane in the treatment of periodontal intraosseous lesions. J Periodontol 1998: 69: 445–453. [DOI] [PubMed] [Google Scholar]
- 94. Maciejewski JP, Bruening EE, Donahue RE, Sellers SE, Carter C, Young NS, St Jeor S. Infection of mononucleated phagocytes with human cytomegalovirus. Virology 1993: 195: 327–336. [DOI] [PubMed] [Google Scholar]
- 95. Marchant A, Appay V, van der Sande M, Dulphy N, Liesnard C, Kidd M, Kaye S, Ojuola O, Gillespie GM, Vargas Cuero AL, Cerundolo V, Callan M, McAdam KP, Rowland‐Jones SL, Donner C, McMichael AJ, Whittle H. Mature CD8(+) T lymphocyte response to viral infection during fetal life. J Clin Invest 2003: 111: 1747–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Margulies BJ, Browne H, Gibson W. Identification of the human cytomegalovirus G protein‐coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology 1996: 225: 111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Martinez MJ, Bray MP, Huggins JW. A mouse model of aerosol‐transmitted orthopoxviral disease: morphology of experimental aerosol‐transmitted orthopoxviral disease in a cowpox virus‐BALB/c mouse system. Arch Pathol Lab Med 2000: 124: 362–377. [DOI] [PubMed] [Google Scholar]
- 98. Mathew P, Hudnall SD, Elghetany MT, Payne DA. T‐gamma gene rearrangement and CMV mononucleosis. Am J Hematol 2001: 66: 64–66. [DOI] [PubMed] [Google Scholar]
- 99. Mehanonda R, Sa‐Ard‐Iam N, Rerkyen P, Champaiboon C, Vanavit N, Pichyangkul S. Innate antiviral immunity of periodontal tissue. Periodontol 2000 2011: 56: 143–153. [DOI] [PubMed] [Google Scholar]
- 100. Miller DM, Rahill BM, Boss JM, Lairmore MD, Durbin JE, Waldman JW, Sedmak DD. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J Exp Med 1998: 187: 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mitchell BM, Bloom DC, Corhrs RJ, Gilden DH, Kennedy PE. Herpes simplex virus 1 and varicella zoster virus latency in ganglia. J Neurol Virol 2003: 9: 194–204. [DOI] [PubMed] [Google Scholar]
- 102. Mocarski ES Jr. Cytomegalovirus and their replication In: Fields BN, Knipe DM, Howley PM, editors. Fields virology, 3rd edn Philadelphia: Lippincott‐Raven Publishers, 1996: 2447–2492. [Google Scholar]
- 103. Mocarski ES Jr. Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol 2002: 10: 332–339. [DOI] [PubMed] [Google Scholar]
- 104. Mueller NH, Gilden DH, Cohrs RJ, Mahalingam R, Nagel MA. Varicella zoster virus infection: clinical features, molecular pathogenesis of disease and latency. Neurol Clin 2008: 26: 675–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mutter W, Reddehase MJ, Busch FW, Buhring HJ, Koszinowski UH. Failure in generating hemopoietic stem cells is the primary cause of death from cytomegalovirus disease in the immunocompromised host. J Exp Med 1988: 167: 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Neipel F, Albrecht JC, Fleckenstein B. Human herpesvirus 8 – the first human Rhadinovirus. J Natl Cancer Inst Monogr 1998: 23: 73–77. [DOI] [PubMed] [Google Scholar]
- 107. Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C‐C chemokine receptor. Cell 1993: 72: 415–425. [DOI] [PubMed] [Google Scholar]
- 108. Ongradi J, Sallay K, Kulcsar G. The decreased antibacterial activity of oral polymorphonuclear leukocytes coincides with the occurrence of virus‐carrying oral lymphocytes and epithelial cells. Folia Microbiol (Praha) 1987: 32: 438–447. [DOI] [PubMed] [Google Scholar]
- 109. Page RC, Offenbacher S, Schroeder HE, Seymour GJ, Kornman KS. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontol 2000 1997: 14: 216–248. [DOI] [PubMed] [Google Scholar]
- 110. Pannone G, Santoro A, Papagerakis S, Lo Muzio L, De Rosa G, Bufo P. The role of human papillomavirus in the pathogenesis of head and neck squamous cell carcinoma: an overview. Infect Agent Cancer 2011: 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pass RF. Cytomegalovirus infection. Pediatr Rev 2002: 23: 163–170. [DOI] [PubMed] [Google Scholar]
- 112. Paulus C, Nitzsche A, Nevels M. Chromatinization of herpesvirus genomes. Rev Med Virol 2010: 20: 34–50. [DOI] [PubMed] [Google Scholar]
- 113. Penfold ME, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ. Cytomegalovirus encodes a potent alpha chemokine. Proc Natl Acad Sci USA 1999: 96: 9839–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Petrik DT, Schmitt KP, Stinski MF. Inhibition of cellular DNA synthesis by the human cytomegalovirus IE86 protein is necessary for efficient virus replication. J Virol 2006: 80: 3872–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Plachter B, Sinzger C, Jahn G. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res 1996: 46: 195–261. [DOI] [PubMed] [Google Scholar]
- 116. Platzer C, Döcke W, Volk H, Prösch S. Catecholamines trigger IL‐10 release in acute systemic stress reaction by direct stimulation of its promoter/enhancer activity in monocytic cells. J Neuroimmunol 2000: 105: 31–38. [DOI] [PubMed] [Google Scholar]
- 117. Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Luccaronin P, Jonjic S, Koszinowski UH. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med 1998: 188: 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Price DA, Brenchley JM, Ruff LE, Betts MR, Hill BJ, Roederer M, Koup RA, Migueles SA, Gostick E, Wooldridge L, Sewell AK, Connors M, Douek DC. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med 2005: 202: 1349–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Prosch S, Heine AK, Volk HD, Kruger DH. CCAAT/enhancer‐binding proteins alpha and beta negatively influence the capacity of tumor necrosis factor alpha to up‐regulate the human cytomegalovirus IE1/2 enhancer/promoter by nuclear factor kappa B during monocyte differentiation. J Biol Chem 2001: 276: 40712–40720. [DOI] [PubMed] [Google Scholar]
- 120. Prosch S, Staak K, Stein J, Liebenthal C, Stamminger T, Volk HD, Krüger DH. Stimulation of the human cytomegalovirus IE enhancer/promoter in HL‐60 cells by TNF alpha is mediated via induction of NF‐kappaB. Virology 1995: 208: 197–206. [DOI] [PubMed] [Google Scholar]
- 121. Pulliam L, Moore D, West DC. Human cytomegalovirus induces IL‐6 and TNF alpha from macrophages and microglial cells: possible role in neurotoxicity. J Neurovirol 1995: 1: 219–227. [DOI] [PubMed] [Google Scholar]
- 122. Rai B, Kaur J, Anand SC, Jacobs R. Salivary stress markers, stress and periodontitis: a pilot study. J Periodontol 2010: 82: 287–292. [DOI] [PubMed] [Google Scholar]
- 123. Rajcani J, Andrea V, Ingeborg R. Peculiarities of herpes simplex virus (HSV) transcription: an overview. Virus Genes 2004: 28: 293–310. [DOI] [PubMed] [Google Scholar]
- 124. Rasmussen L, Matkin C, Spaete R, Pachl C, Merigan TC. Antibody response to human cytomegalovirus glycoproteins gB and gH after natural infection in humans. J Infect Dis 1991: 164: 835–842. [DOI] [PubMed] [Google Scholar]
- 125. Reddehase MJ, Weiland F, Munch K, Jonjic S, Luske A, Koszinowski UH. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J Virol 1985: 55: 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Reusser P, Cathomas G, Attenhofer R, Tamm M, Thiel G. Cytomegalovirus (CMV)‐specific T cell immunity after renal transplantation mediates protection from CMV disease by limiting the systemic virus load. J Infect Dis 1999: 180: 247–253. [DOI] [PubMed] [Google Scholar]
- 127. Reyburn HT, Mandelboim O, Vales‐Gomez M, Davis DM, Pazmany L, Strominger JL. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature 1997: 386: 514–517. [DOI] [PubMed] [Google Scholar]
- 128. Richart SM, Simpson SA, Krummenacher C, Whitbeck JC, Pizer LI, Cohen GH, Eisenberg RJ, Wilcox CL. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin‐1/HveC. J Virol 2003: 77: 3307–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Riddell SR, Watanabe KS, Goodrich JM, Li CR, Agha ME, Greenberg PD. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992: 257: 238–241. [DOI] [PubMed] [Google Scholar]
- 130. Ritter T, Brandt C, Prösch S, Vergopoulos A, Vogt K, Kolls J, Volk HD. Stimulatory and inhibitory action of cytokines on the regulation of hCMV‐IE promoter activity in human endothelial cells. Cytokine 2000: 12: 1163–1170. [DOI] [PubMed] [Google Scholar]
- 131. Roizman B, Gu H, Mandel G. The first 30 minutes in the life of a virus: unrest in the nucleus. Cell Cycle 2005: 4: 1019–1021. [DOI] [PubMed] [Google Scholar]
- 132. Rossen JW, Horzinek MC, Rottier PJ. Coronavirus infection of polarized epithelial cells. Trends Microbiol 1995: 3: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Saboia‐Dantas CJ, Coutrin de Toledo LF, Sampaio‐Filho HR, Siqueira JF Jr. Herpesviruses in asymptomatic apical periodontitis lesions: an immunohistochemical approach. Oral Microbiol Immunol 2007: 22: 320–325. [DOI] [PubMed] [Google Scholar]
- 134. Saccoccio FM, Gallagher MK, Adler SP, McVoy MA. Neutralizing activity of saliva against cytomegalovirus. Clin Vaccine Immunol 2011: 18: 1536–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Saha A, Kaul R, Murakami M, Robertson ES. Tumor viruses and cancer biology: modulating signaling pathways for therapeutic intervention. Cancer Biol Ther 2010: 10: 961–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sahin S, Saygun I, Kubar A, Slots J. Periodontitis lesions are the main source of salivary cytomegalovirus. Oral Microbiol Immunol 2009: 24: 340–342. [DOI] [PubMed] [Google Scholar]
- 137. Saffert RT, Penkert RR, Kalejta RF. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 2010: 84: 5594–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Saygun I, Sahin S, Muşabak U, Enhoş S, Kubar A, Günhan O, Slots J. Human cytomegalovirus in peripheral giant cell granuloma. Oral Microbiol Immunol 2009: 24: 408–410. [DOI] [PubMed] [Google Scholar]
- 139. Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. Cmv‐1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med 1990: 171: 1469–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Schwartz R, Helmich B, Spector DH. CREB and CREB‐binding proteins play an important role in the IE2 86‐kilodalton protein‐mediated transactivation of the human cytomegalovirus 2.2‐kilobase RNA promoter. J Virol 1996: 70: 6955–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sester U, Gartner BC, Wilkens H, Schwaab B, Wossner R, Kindermann I, Girndt M, Meyerhans A, Mueller‐Lantzsch N, Schafers HJ, Sybrecht GW, Kohler H, Sester M. Differences in CMV‐specific T‐cell levels and long‐term susceptibility to CMV infection after kidney, heart and lung transplantation. Am J Transplant 2005: 5: 1483–1489. [DOI] [PubMed] [Google Scholar]
- 142. Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest 2001: 108: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sigusch BW, Wutzler A, Nietzsch T, Glockmann E. Evidence for a specific crevicular lymphocyte profile in aggressive periodontitis. J Periodontal Res 2006: 41: 391–396. [DOI] [PubMed] [Google Scholar]
- 144. Silva N, Dutzan N, Hernandez M, Dezerega A, Rivera O, Aguillon JC, Aravena O, Lastres P, Pozo P, Vernal R, Gamonal J. Characterization of progressive periodontal lesions in chronic periodontitis patients: levels of chemokines, cytokines, matrix metalloproteinase‐13, periodontal pathogens and inflammatory cells. J Clin Periodontol 2008: 35: 206–214. [DOI] [PubMed] [Google Scholar]
- 145. Simmen KA, Singh J, Luukkonen BG, Lopper M, Bittner A, Miller NE, Jackson MR, Compton T, Früh K. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc Natl Acad Sci USA 2001: 98: 7140–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sinclair J, Sissons P. Latency and reactivation of human cytomegalovirus. J Gen Virol 2006: 87: 1763–1779. [DOI] [PubMed] [Google Scholar]
- 147. Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus‐encoded inhibitor of apoptosis that suppresses caspase‐8 activation. Proc Natl Acad Sci USA 2001: 98: 7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Slots J. Herpesviruses in periodontal diseases. Periodontol 2000 2005: 38: 33–62. [DOI] [PubMed] [Google Scholar]
- 149. Slots J. Oral viral infections of adults. Periodontol 2000 2009: 49: 60–86. [DOI] [PubMed] [Google Scholar]
- 150. Slots J. Human viruses in periodontitis. Periodontol 2000 2010: 53: 89–110. [DOI] [PubMed] [Google Scholar]
- 151. Slots J, Slots H. Bacterial and viral pathogens in saliva: disease relationship and infectious risk. Periodontol 2000 2011: 55: 48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Song YJ, Stinski MF. Effect of the human cytomegalovirus IE86 protein on expression of E2F‐responsive genes: a DNA microarray analysis. Proc Natl Acad Sci USA 2002: 99: 2836–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sprent J, Tough DF. T cell death and memory. Science 2001: 293: 245–248. [DOI] [PubMed] [Google Scholar]
- 154. Stinski MF. Cytomegalovirus and its replication In: Fields BN, Knipe DM, Hirsch MS, Melnick JL, Monath TP, Roizman B, editors. Virology. New York: Raven Press, 1990: 69:1959–1980. [Google Scholar]
- 155. Sunde PT, Olsen I, Enersen M, Beiske K, Grinde B. Human cytomegalovirus and Epstein–Barr virus in apical and marginal periodontitis: a role in pathology? J Med Virol 2008: 80: 1007–1011. [DOI] [PubMed] [Google Scholar]
- 156. Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern NF, Nelson JA, Picker LJ. Broadly targeted human cytomegalovirus‐specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 2005: 202: 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B. Toll‐like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA 2004: 101: 3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tavalai N, Stamminger T. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res 2011: 157: 128–133. [DOI] [PubMed] [Google Scholar]
- 159. Teles R, Sakellari D, Teles F, Konstantinidis A, Kent R, Socransky S, Haffajee A. Relationships among gingival crevicular fluid biomarkers, clinical parameters of periodontal disease, and the subgingival microbiota. J Periodontol 2010: 81: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Teughels W, Sliepen I, Quirynen M, Haake SK, van Eldere J, Fives‐Taylor P, van Ranst M. Human cytomegalovirus enhances A. actinomycetemcomitans adherence to cells. J Dent Res 2007: 86: 175–180. [DOI] [PubMed] [Google Scholar]
- 161. Ting M, Contreras A, Slots J. Herpesviruses in localized juvenile periodontitis. J Periodontal Res 2000: 35: 17–25. [DOI] [PubMed] [Google Scholar]
- 162. Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GW. Surface expression of HLA‐E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000: 287: 1031. [DOI] [PubMed] [Google Scholar]
- 163. Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, Cresswell P, Nelson JA, Riddell SR, Johnson DC. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 1999: 5: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 164. Tomtishen JP 3rd. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol J 2012: 9: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Toussirot E, Roudier J. Epstein–Barr virus in autoimmune diseases. Best Pract Res Clin Rheumatol 2008: 22: 883–896. [DOI] [PubMed] [Google Scholar]
- 166. Tsurumi T, Fujita M, Kudoh A. Latent and lytic Epstein Barr virus replication strategies. Rev Med Virol 2005: 15: 3–15. [DOI] [PubMed] [Google Scholar]
- 167. Tucker SP, Compams RW. Virus infections of polarized epithelial cells. Adv Virus Res 1992: 42: 187–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Tugizov S, Herrera R, Veluppillai P, Greenspan J, Greenspan D, Palefsky JM. Epstein–Barr virus (EBV)‐infected monocytes facilitate dissemination of EBV within the oral mucosal epithelium. J Virol 2007: 81: 5484–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. van Dyke TE, Levine MJ, Genco RJ. Neutrophil function and oral disease. J Oral Pathol 1985: 14: 95–120. [DOI] [PubMed] [Google Scholar]
- 170. van Dyke TE, Vaikuntam J. Neutrophil function and dysfunction in periodontal disease. Curr Opin Periodontol 1994: 1: 19–27. [PubMed] [Google Scholar]
- 171. Venema H, van den Berg AP, van Zanten C, van Son WJ, van der Giessen M, The TH. Natural killer cell responses in renal transplant patients with cytomegalovirus infection. J Med Virol 1994: 42: 188–192. [DOI] [PubMed] [Google Scholar]
- 172. Wagner CS, Walther‐Jallow L, Buentke E, Ljunggren HG, Achour A, Chambers BJ. Human cytomegalovirus‐derived protein UL18 alters the phenotype and function of monocyte‐derived dendritic cells. J Leukoc Biol 2008: 83: 56–63. [DOI] [PubMed] [Google Scholar]
- 173. Wagner EK, Sandri‐Goldin RM. Herpes simplex viruses: molecular biology In: Mahy BWJ, van Regenmortel MH. editors. Desk encyclopedia of general virology. San Diego, CA: Academic Press, 2010: 460–469. [Google Scholar]
- 174. Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T‐cell clones from the donor. N Engl J Med 1995: 333: 1038–1044. [DOI] [PubMed] [Google Scholar]
- 175. Wara‐aswapati N, Yang Z, Waterman WR, Koyama Y, Tetradis S, Choy BK, Webb AC, Auron PE. Cytomegalovirus IE2 protein stimulates interleukin 1beta gene transcription via tethering to Spi‐1/PU.1. Mol Cell Biol 1999: 19: 6803–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Wiebusch L, Asmar J, Uecker R, Hagemeier C. Human cytomegalovirus immediate‐early protein 2 (IE2)‐mediated activation of cyclin E is cell‐cycle‐independent and forces S‐phase entry in IE2‐arrested cells. J Gen Virol 2003: 84: 51–60. [DOI] [PubMed] [Google Scholar]
- 177. Wong KF, Yip SF, So CC, Lau GT, Yeung YM. Cytomegalovirus infection associated with clonal proliferation of T‐cell large granular lymphocytes: causal or casual? Cancer Genet Cytogenet 2003: 142: 77–79. [DOI] [PubMed] [Google Scholar]
- 178. Wu CA, Paveglio SA, Lingenheld EG, Zhu L, Lefrançois L, Puddington L. Transmission of murine cytomegalovirus in breast milk: a model of natural infection in neonates. J Virol 2011: 85: 5115–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wynn KK, Fulton Z, Cooper L, Silins SL, Gras S, Archbold JK, Tynan FE, Miles JJ, McCluskey J, Burrows SR, Rossjohn J, Khanna R. Impact of clonal competition for peptide‐MHC complexes on the CD8+ T‐cell repertoire selection in a persistent viral infection. Blood 2008: 111: 4283–4292. [DOI] [PubMed] [Google Scholar]
- 180. Xiao J, Palefsky JM, Herrera R, Berline J, Tugizov SM. EBV BMRF‐2 facilitates cell‐to‐cell spread of virus within polarized oral epithelial cells. Virology 2009: 388: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Yamazaki K, Nakajima T, Gemmel E, Polar B, Seymour GJ, Hara K. IL‐4 and IL6 producing cells in human periodontal disease tissue. J Oral Pathol Med 1994: 23: 347–353. [DOI] [PubMed] [Google Scholar]
- 182. Yang Z, Wara‐aswapati N, Yoshida Y, Walker N, Galson DL, Listman J, Auron PE. Dual regulatory role of human cytomegalovirus immediate‐early protein in IL1B transcription is dependent upon Spi‐1/PU.1. Biochem Biophys Res Commun 2002: 294: 854–863. [DOI] [PubMed] [Google Scholar]
- 183. Yinon Y, Farine D, Yudin MH. Screening, diagnosis, and management of cytomegalovirus infection in pregnancy. Obstet Gynecol Surv 2010: 65: 736–743. [DOI] [PubMed] [Google Scholar]
- 184. Zanetti AR, Zappa A. Emerging and re‐emerging infections at the turn of the millennium. Haemophilia 2010: 16: 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zhu H, Shen Y, Shenk T. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 1995: 69: 7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]