

Abstract
abstract: Newly emergent infectious diseases are a global public health problem. The population dense regions of Southeast Asia are the epicenter of many emerging diseases, as evidenced by the outbreak of Nipah, SARS, avian influenza (H5N1), Dengue, and enterovirus 71 in this region in the past decade. Rapid identification, epidemiologic surveillance, and mitigation of transmission are major challenges in ensuring public health safety. Here we describe a powerful new approach for infectious disease surveillance that is based on polymerase chain reaction (PCR) to amplify nucleic acid targets from large groupings of organisms, electrospray ionization mass spectrometry (ESI‐MS) for accurate mass measurements of the PCR products, and base composition signature analysis to identify organisms in a sample. This approach is capable of automated analysis of more than 1,500 PCR reactions a day. It is applicable to the surveillance of bacterial, viral, fungal, or protozoal pathogens and will facilitate rapid characterization of known and emerging pathogens.
Keywords: PCR, RT‐PCR, ESI‐MS, mass spectrometry, infectious disease surveillance, viral diseases, broad detection, emerging infections
INTRODUCTION
Southeast Asia has been the source of several emerging zoonotic‐ and vector‐borne disease outbreaks over the years. 1 These include acute encephalitis caused by a novel paramyxovirus, Nipah virus; 2 , 3 hand, foot, and mouth disease (HFMD) caused by coxsackie A16 and enterovirus 71; 4 viral encephalitis and hemorrhagic fevers caused by arboviruses, such as Dengue and Japanese encephalitis viruses; 5 and episodic outbreaks of influenza A virus (H5N1, H9N2, etc.) and acute respiratory syndrome caused by the SARS coronavirus. 6 , 7 , 8 , 9
A fundamental problem in rapid identification of the etiology of infectious disease outbreaks is the sheer number of microbes, both known and novel, that could potentially be the causative agents. According to a recent review, 10 there are more than 1,400 species of microbes known to be pathogenic to humans. Of these, more than 60% were found to be zoonotic and more than 175 pathogenic species were associated with emerging infectious diseases. These numbers do not include numerous strain variants of each organism or classes of pathogens that infect plants or animals. Further, species variants of an otherwise benign group of microbes may prove infectious to humans and strain‐level genetic changes may confer resistance to antimicrobial agents.
Most of the new technologies being developed for detection of infectious agents incorporate a version of quantitative polymerase chain reaction (PCR), based upon the use of highly specific primers and probes and designed to selectively identify specific pathogenic organisms. Paradoxically, this approach requires assumptions about the type and strain of the infectious agent, limiting detection to a small number of known pathogens. It is both impractical and cost prohibitive to run a large number of individual tests in an attempt to identify every known infectious organism. Moreover, existing tests for known organisms will not identify newly emerging organisms.
Broad‐range PCR methods provide an alternative to single‐agent tests. By amplifying gene targets conserved across groups of organisms, broad‐range PCR has the potential to generate amplification products across entire genera, families, or, as with bacteria, an entire domain of life. This strategy has been successfully employed in the past for bacterial and viral detection. 11 , 12 , 13 , 14 , 15 , 16 The drawback of this approach for epidemiologic applications is that the analysis of PCR products for mixed amplified samples requires sequencing of hundreds of colonies per reaction, and thus it cannot be performed rapidly or cost effectively on large numbers of samples. New approaches to the parallel detection of multiple infectious agents include multiplexed PCR methods 17 , 18 and microarray strategies. 19 , 20 , 21 Microarray strategies are promising because undiscovered organisms might be detected by hybridization to probes designed to conserved regions of known families of bacteria and viruses. However, these methods are not well suited for the high‐throughput detection of a broad spectrum of microorganisms potentially present in complex biological mixtures.
Triangulation identification for genetic evaluation of risks (TIGER) is a universal pathogen detection platform developed under a Defense Advanced Research Projects Agency (DARPA) biodefense program that is capable of identifying an extraordinarily broad range of pathogens ( fig. 1). The process uses electrospray ionization mass spectrometry (ESI‐MS) and base composition analysis of PCR amplification products from highly conserved genomic regions to identify and determine the relative quantity of pathogenic bacteria, viruses, fungi, or protozoa present in a sample. The fundamental distinction between this PCR/ESI‐MS strategy and traditional (specific) nucleic acid tests is the nature of the question being answered. Traditional nucleic acid tests answer the question: “Is infectious organism X in my sample?” The PCR/ESI‐MS method answers the question: “What infectious organisms are in my sample?” In effect, this is equivalent to running many thousands of specific nucleic acid tests because the identity of the infectious organism being detected does not need to be anticipated. The premise of this technology is that it can provide rapid, sensitive, and cost‐effective detection of a broad range of “normal” pathogenic organisms and simultaneously identify unexpected or emerging infectious organisms. This broad function technology may be the only practical way to rapidly diagnose diseases caused by emerging infectious organisms that might otherwise be missed or mistaken for a more common infection. 22 The technology has been validated for a variety of applications, including emerging virus identification, 23 determination of the pathogens involved in acute pneumonia epidemics, 24 and identification of biological weapons agents in a variety of sample types. 25 , 26
Figure 1.

Automated PCR/ESI‐MS biosensor. Shown in this view are key modules including amplicon purification (desalting), plate stackers, sample injection, and the mass spectrometer. Precise molecular weight determinations of amplicons yield unambiguous base compositions that are used to uniquely “fingerprint” each pathogen. The automated system is capable of analyzing more than 1,500 PCR reactions in 24 h.
The basis of the above approach is the principle that, despite the enormous diversity of microbes, there are sets of essential common features in their biomolecules. Parallel detection of infectious agents takes advantage of highly conserved regions within genes that flank highly variable regions. These conserved genomic features serve as anchors for broad‐range PCR priming to amplify, without prejudice, nucleic acids from most microorganisms in a sample. Such broad‐range PCR reactions targeted across the broadest possible grouping of organisms typically result in a mixture of amplicons, reflecting the complexity of the starting sample. With no further separation, this mixture of PCR amplicons is desalted and analyzed by ESI‐MS for accurate mass measurement, enabling us to determine the base composition (numbers of A, G, C, and T nucleotides) of the PCR amplicons. 23 , 25 , 26 Amplification relies on highly conserved regions, but the variability in sequence between the two primer sites, reflected in the base composition, allows us to distinguish between organisms.
A key aspect of this approach is the concept of “triangulation.” In this context, triangulation refers to taking measurements from multiple loci distributed across the microbial genome and using the combination of the measured base composition signatures to identify the organism. While no single PCR target region might give definitive species resolution for all microbes, species can be clearly identified and differentiated from one another using the triangulation strategy across multiple loci. A database of expected base compositions for each primer region for all known organisms was generated using an in silico PCR search algorithm (electronic PCR or ePCR). An existing ribonucleic acid (RNA) structure search algorithm has been modified to include PCR parameters, such as hybridization conditions, mismatches, and thermodynamic calculations. 27 This also provides information on primer specificity of the selected primer pairs. The regions are selected so that the nucleic acid composition of the resulting amplicons has sufficient variability to maximally resolve the target organisms. Thus, the measured base compositions from strategically selected locations of the genome are used as a signature to identify and distinguish the organisms present in the original sample.
Detailed experimental protocols have been published elsewhere. 25 Microbes have been identified from human and animal samples (throat swabs, skin wipes, hair, tissue) and from samples obtained from the environment (food products, soil, water, surface wipes, air). Nucleic acids are extracted from bacteria using either a heat or mechanical lysis step. Viral genome isolation is performed using commercially available kits that have been optimized for use in the system. Samples are divided into wells of a microtiter plate that contain pairs of broad‐range PCR primers.
A key element of the PCR/ESI‐MS system is the ability to quantify the levels of genomic material present in the samples. To accomplish this, we include an internal calibrant in each PCR reaction performed. The calibrant consists of a specially designed nucleic acid sequence that is similar to, but distinguishable from, sequences of organisms we might detect. This allows for the simultaneous presence of an accurately quantified amount of a known calibrant deoxyribonucleic acid (DNA) with similar amplification properties to the target sequence during the PCR cycle. Further, this serves as an internal positive control for every PCR reaction. The relative abundances of the end products (as measured by the mass peak heights) can be used in conjunction with the starting concentration of the calibrant to accurately estimate the amount of target sequence in the starting sample ( fig. 2). These measurements are highly reproducible over a wide range of concentrations and provide improved confidence for the determination of the quantity of the infectious agents in the sample. 24 , 25
Figure 2.
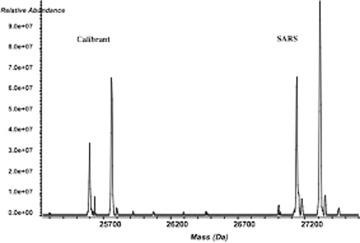
Mass spectra of calibrant and SARS coronavirus (SARS CoV) samples are shown. Both strands were amplified and thus there are two major peaks for both calibrant and SARS. The two templates were observed to be at a relative abundance ratio of approximately 1:1.5. From the input number of calibrant nucleic acid molecules (3 × 104), we estimated that SARS RNA was present in the sample at ∼4.5 × 104 genome copies.
DETECTION AND DISCRIMINATION OF KNOWN AND EMERGING VIRUSES
In this section, we describe two specific applications of the PCR/ESI‐MS method for identifying known and emerging viruses of particular relevance to Southeast Asia. These concepts are equally applicable to bacteria, 24 , 25 other viruses, fungi, and protozoa.
SARS Coronavirus
At the beginning of the SARS outbreak in early 2003, the etiology of the causative agent was not known. Several initial reports based on preliminary characterization of the isolated virus suggested it could be a member of one of several viral families, including Coronaviridae. We designed several broad‐range primers targeting the various genera within these families. For broad‐range detection of all coronaviruses, two PCR primer target regions in orf‐1b, one in the RNA‐dependent RNA polymerase (RdRp) and the other in Nsp14, were identified prior to publication of the SARS sequence. 23 Locations of primers within these regions were optimized both for sensitivity and broad‐range priming potential simultaneously by amplifying dilutions of multiple, diverse coronaviruses. In silico analysis of the final primer pairs against GenBank nucleotide database sequences showed that these primers would be expected to amplify all coronaviruses but no other viruses, bacteria, or human DNA. 27
Multiple isolates of a number of different coronavirus species were obtained from various sources (including the American Type Culture Collection [ATCC] and the Naval Health Research Center in San Diego). SARS isolates were obtained from the Centers for Disease Control and Prevention (CDC) and from Michael Buchmeier (The Scripps Research Institute, La Jolla, CA). PCR products for all isolates were generated, desalted, and analyzed by ESI‐MS by methods described previously. 28 The spectral signals were algorithmically processed to obtain base compositions of sense and antisense strands of each amplicon. 29 The results from the analysis of 14 coronavirus isolates are shown in table 1. For both target regions, the measured signals agreed with compositions expected from the known coronavirus sequences in GenBank. Several of the isolates used in this study did not have a genome sequence record in GenBank. Nevertheless, we were able to amplify all test viruses and experimentally determine their base compositions. These experimentally determined base compositions were later confirmed by sequencing and deposited in GenBank (AY874541, AY878317‐24). Thus the strategy described here enables identification of organisms without the need for prior knowledge of their sequence.
Table 1.
Coronaviruses and their experimentally determined base compositions. Sense and antisense strand masses are shown for each isolate
| Group | Abbreviation | Source | Measured masses (Da) | Derived Base compositions | Measured masses (Da) | Derived base compositions |
|---|---|---|---|---|---|---|
| CCoV 1‐71 | VR809 | 27486.514 | A24 G24 C8 T32 | 42475.955 | A33 G31 C19 T54 | |
| 26936.574 | A32 G8 C24 T24 | 42185.117 | A54 G19 C31 T33 | |||
| CCoV ‐TN449 | VR2068 | 27471.510 | A24 G24 C9 T31 | 42474.899 | A34 G30 C18 T55 | |
| 26952.548 | A31 G9 C24 T24 | 42184.072 | A55 G18 C30 T34 | |||
| FCoV WSU | VR‐989 | 27471.517 | A24 G24 C9 T31 | 42490.945 | A33 G31 C18 T55 | |
| 79‐1683 | 26952.556 | A31 G9 C24 T24 | 42169.118 | A55 G18 C31 T33 | ||
| 1 | FCoV DF2 | VR2004 | 27472.497 | A23 G25 C10 T30 | 42450.904 | A33 G30 C19 T55 |
| 26953.536 | A30 G10 C25 T23 | 42209.081 | A55 G19 C30 T33 | |||
| HCoV 229E | VR740 | 27450.532 | A25 G24 C11 T28 | 42462.994 | A36 G30 C20 T51 | |
| 26975.545 | A28 G11 C24 T25 | 42198.061 | A51 G20 C30 T36 | |||
| HCoV 229E | NHRC | 27450.506 | A25 G24 C11 T28 | 42462.930 | A36 G30 C20 T51 | |
| 26975.512 | A28 G11 C24 T25 | 42198.040 | A51 G20 C30 T36 | |||
| BCoV Calf | VR874 | 27358.452 | A22 G22 C12 T32 | 42606.039 | A38 G32 C15 T52 | |
| Diarrheal virus | 27066.586 | A32 G12 C22 T22 | 42052.897 | A52 G15 C32 T38 | ||
| HCoV OC43 | NHRC | 27328.473 | A22 G22 C14 T30 | 42580.959 | A38 G31 C15 T53 | |
| 27098.562 | A30 G14 C22 T22 | 42076.028 | A53 G15 C31 T38 | |||
| MHV MHV1 | VR261 | 27344.491 | A21 G23 C14 T30 | 42602.022 | A37 G34 C18 T48 | |
| 27083.564 | A30 G14 C23 T21 | 42061.016 | A48 G18 C34 T37 | |||
| 2 | MHV JHM‐thermostable | VR1426 | 27344.497 | A21 G23 C14 T30 | 42529.960 | A34 G34 C21 T48 |
| 27083.571 | A30 G14 C23 T21 | 42136.047 | A48 G21 C34 T34 | |||
| MHV MHV‐ | VR764 | 27344.503 | A21 G23 C14 T30 | 42599.989 | A34 G35 C18 T50 | |
| A59 | 27083.572 | A30 G14 C23 T21 | 42064.089 | A50 G18 C35 T34 | ||
| RtCoV 8190 | VR1410 | 27344.491 | A21 G23 C14 T30 | 42544.967 | A34 G34 C20 T49 | |
| 27083.567 | A30 G14 C23 T21 | 42120.041 | A49 G20 C34 T34 | |||
| 3 | IBV Egg‐adapted | VR22 | 27396.544 | A24 G24 C14 T26 | 42530.984 | A33 G32 C17 T55 |
| 27032.524 | A26 G14 C24 T24 | 42129.100 | A55 G17 C32 T33 | |||
| 4 | SCoV TOR2 | University of Manitoba | 27298.518 | A27 G19 C14 T28 | 42519.906 | A34 G33 C20 T50 |
| 27125.542 | A28 G14 C19 T27 | 42144.026 | A50 G20 C33 T34 | |||
| SCoV Urbani | CDC | 27298.518 | A27 G19 C14 T28 | 42519.906 | A34 G33 C20 T50 | |
| 27125.542 | A28 G14 C19 T27 | 42144.026 | A50 G20 C33 T34 | |||
We further demonstrated the potential to detect multiple viruses in the same sample, as might occur during a co‐infection. Pooled viral extracts from three human coronavirus isolates, HCoV‐229E, HCoV‐OC43, and SARS CoV, were amplified in a mixture and analyzed by PCR/ESI‐MS. Signals from all three viruses were clearly detected and resolved in the mass spectrum demonstrating that co‐infections of more than one coronavirus species could be identified. In addition to broad identification of all coronaviruses tested, including the SARS CoV, we were able to accurately classify the SARS isolates as unrelated but distant members of the group 2 coronaviruses, consistent with the findings of Snijder et al. 30 These results are described in detail in Sampath et al. 23
Vector‐Borne Viruses
Viral encephalitidis is caused by a number of different viruses from different viral families. Important among these are members of the genus Alphavirus (Togaviridae family) and the genus Flavivirus (Flaviviridae family); both are single‐strand, positive‐strand RNA viruses. These are major vector‐borne diseases in Southeast Asia and Pacific Rim countries. While the alphaviruses Venezuelan, and eastern and western equine encephalitis viruses (VEEV, EEEV, and WEEV) have received much attention in the Americas for their ability to infect humans and their potential to be used as biowarfare agents, Ross River, Chikungunya, Barmah Forest, and other alphaviruses are common and detrimental to public health in the Asia Pacific regions. 5 , 31 , 32 Genomic changes drive the emergence and reemergence of these viruses as high‐consequence pathogens by causing alterations in host range and virulence properties, 33 and the resulting diversity at the nucleotide and protein levels poses a great challenge for identification and diagnostic strategies. As an example, VEE complex viruses display 13 serologically distinct types belonging to 6 antigenic subtypes (I to VI), and the EEE virus antigenic complex has 4 antigenic subtypes (I–IV), spanning the North and South American isolates. 34
Our approach for identifying and discriminating these viruses was to design several primer pairs that would amplify all members of the Alphavirus genus, and then design primers that amplify distinct regions of the Alphavirus isolates to distinguish subtypes. 25 Broad‐range primer pairs targeting the 5′‐end of the RNA virus genome (nonstructural protein, nsP1 gene) were designed and tested against a number of different viral isolates, including both old world and new world alphaviruses. All of the test isolates showed a perfect match to the expected base compositions where prior data were known, and the tested viral isolates were readily distinguished from one other ( table 2). Importantly, the base composition data for some of the isolates represented new measurements from regions that have not been sequenced, showing the potential of this strategy to identify previously unknown or newly emerging viruses. Additional confirmation primer pairs targeting the old and new world alphaviruses have been developed and together they provide complete resolution among all known alphavirus species (Eshoo et al., Personal Observation).
Table 2.
Alphavirus test isolates and their expected/observed base compositions.ab
| Virus | Strain | Sequence available | Expected base composition | Observed base composition |
|---|---|---|---|---|
| VEE | 3908 (subtype IC, 1995) | Yes | [21 23 23 19] | [21 23 23 19] |
| VEE | 66637 (subtype ID, 1981) | Yes | [21 23 23 19] | [21 23 23 19] |
| VEE | 68U201 (subtype IE, 1968) | Yes | [22 25 19 20] | [22 25 19 20] |
| VEE | 243937 (subtype IC, 1992) | Yes | [21 23 23 19] | [21 23 23 19] |
| WEE | OR71 (71V1658) | Yes | [22 26 19 19] | [22 26 19 19] |
| WEE | SD83 (R‐43738) | No | Unknown | [22 26 19 19] |
| WEE | ON41 (McMillan) | No | Unknown | [22 27 18 19] |
| WEE | Fleming (Fleming) | No | Unknown | [22 25 19 20] |
| EEE | (Parker strain) | Yes | [23 25 19 19] | [23 25 19 19] |
aThree of the WEE isolates shown in grey represent de novo measurements.
bAll isolates were obtained from Dr. Scott Weaver, UTHSC, Galveston, TX.
Influenza Virus Surveillance
The biosensor system described here is uniquely suited to influenza virus detection and surveillance in avian, human, or other animal reservoirs. Broad surveillance capability for all influenza viruses is complemented by simultaneous type and subtype level resolution of various identified influenza viruses. This will be useful in monitoring the emergence of new influenza viruses. Based upon the analysis of multiple influenza sequence alignments, pan‐influenza virus PCR primer sets have been developed that are capable of amplifying all three influenza virus species (A, B, and C) and subtypes (HxNy) from different animal hosts (human, avian, swine, etc.) and to distinguish their essential molecular features using base composition signatures (Sampath et al., Personal Observation). To measure the breadth of coverage and resolution offered by this panel we tested 50 different influenza virus isolates, including 22 avian isolates (representing 20 different H/N types, collected from 9 different species of wild birds), 18 human influenza A isolates (8 H1N1, 10 H3N2), 4 swine isolates (including 1 novel type), 1 equine isolate, and 6 human influenza B isolates. The avian influenza samples represented a variety of host species, influenza virus types, geographic locations, and dates of isolation. Despite the diversity of this sample set, the broad‐range primers generated amplicons from all isolates, while the base composition signatures from amplicons obtained with these primers distinguished the isolates (Sampath et al., manuscript submitted). The assay is high throughput (∼300 samples/24 h) and rapid (sample to answer in <6 h). In silico analysis of >1,400 influenza virus sequences from the GenBank database showed that the primer panel would detect all known influenza viruses and distinguish >90% of all species, with 100% resolution between avian H5N1 and any human isolate. Several hundred human respiratory specimens have been screened using this assay and have shown >97% sensitivity and specificity of detection compared to traditional characterization.
SUMMARY
TIGER strategy uses a paradigm that allows simultaneous identification of multiple pathogens, including all bacteria, viruses, fungi, and other microorganisms present in a sample without the need for prior knowledge of a specific nucleotide target sequence. Thus previously unanticipated, newly emerging, or potentially unknown organisms can be identified using this platform. The strategy described here uses integrated ESI‐MS and base composition analysis of broad‐range PCR products. Broad‐range PCR reactions are capable of producing products from groups of organisms, rather than single species, and the information content of each PCR reaction is very high. In many cases, including the SARS‐CoV detection described above, priming across broadly conserved regions provides taxonomic resolution at the species level. Where additional subspecies level classification becomes important, broad primers can be supplemented with species‐specific primers that can detect even single nucleotide changes (SNPs) at highly variable loci. As a genotypic method for microbial identification, PCR/ESI‐MS can provide strain‐ or isolate‐level information. 35 These data may be important in epidemiological monitoring to determine the origin of a particular outbreak.
Due to its high throughput capabilities, the methods described will have great utility in a routine survey and detection setting. The mass spectrometer is capable of analyzing complex PCR products at a rate of approximately 1 min per sample. Because the process is performed in an automated, microtiter plate format, large numbers of samples can be examined (>1,400 PCR reactions/day/instrument), making this process practical for large‐scale analysis of clinical or environmental surveillance samples in public health laboratory settings. This approach can be extended to other viral, bacterial, fungal, or protozoal pathogen groups and is a powerful new paradigm for timely identification of previously unknown organisms that cause disease in humans or animals and for monitoring the progress of epidemics.
CONFLICT OF INTEREST
All authors are full‐time employees and stockholders of Ibis Biosciences, Inc., a wholly owned subsidiary of Isis Pharmaceuticals, Inc.
ACKNOWLEDGMENTS
TIGER technology was jointly developed by the Ibis Biosciences a subsidiary of Isis Pharmaceuticals and SAIC (San Diego, USA). We acknowledge DARPA, CDC, Department of Homeland Security (DHS), and National Institute of Allergy and Infectious Diseases (NIAID) for financial support for development of the technology and Dr. Jacqueline R. Wyatt, Ph.D. (http://www.JL-SciEdit.com) for carefully reviewing the manuscript; patents are currently pending in the United States and internationally. Several key participants contributed significantly to the development and implementation of various aspects of the technology but are too numerous to be listed individually by name.
REFERENCES
- 1. Lam, S.K. 1998. Emerging infectious diseases–Southeast Asia. Emerg. Infect. Dis. 4: 145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong, K.T. et al 2002. Nipah virus infection, an emerging paramyxoviral zoonosis. Springer Semin. Immunopathol. 24: 215–228. [DOI] [PubMed] [Google Scholar]
- 3. Wong, K.T. et al 2002. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol. 161: 2153–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cardosa, M.J. et al 2003. Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia‐Pacific region: comparative analysis of the VP1 and VP4 genes. Emerg. Infect. Dis. 9: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mackenzie, J.S. et al 2001. Emerging viral diseases of Southeast Asia and the Western Pacific. Emerg. Infect. Dis. 7: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poutanen, S.M. et al 2003. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 348: 1995–2005. [DOI] [PubMed] [Google Scholar]
- 7. Peiris, J. et al 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361: 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Falsey, A.R. & Walsh E.E.. 2003. Novel coronavirus and severe acute respiratory syndrome. Lancet 361: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ksiazek, T.G. et al 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348: 1953–1966. [DOI] [PubMed] [Google Scholar]
- 10. Taylor, L.H. , Latham S.M. & Woolhouse M.E.. 2001. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356: 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidt, T.M. , DeLong E.F. & Pace N.R.. 1991. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bact. 173: 4371–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kroes, I. , Lepp P.W. & Relman D.A.. 1999. Bacterial diversity within the human subgingival crevice. Proc. Natl. Acad. Sci. (USA) 96: 14547–14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stephensen, C.B. , Casebolt D.B. & Gangopadhyay N.N.. 1999. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Vir. Res. 60: 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oberste, M.S. , Maher K. & Pallansch M.A.. 2002. Molecular phylogeny and proposed classification of the simian picornaviruses. J. Virol. 76: 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mack, D.H. & Sninsky J.J.. 1988. A sensitive method for the identification of uncharacterized viruses related to known virus groups: hepadnavirus model system. Proc. Natl. Acad. Sci. USA 85: 6977–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Echavarria, M. et al 1998. PCR method for detection of adenovirus in urine of healthy and human immunodeficiency virus‐infected individuals. J. Clin. Micro. 36: 3323–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fout, G.S. et al 2003. A multiplex reverse transcription‐PCR method for detection of human enteric viruses in groundwater. Appl. Environ. Microbiol. 69: 3158–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brito, D.A. , Ramirez M. & De Lencastre H.. 2003. Serotyping Streptococcus pneumoniae by multiplex PCR. J. Clin. Microbiol. 41: 2378–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson, K.H. et al 2002. High‐density microarray of small‐subunit ribosomal DNA probes. Appl. Environ. Microbiol. 68: 2535–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang, D. et al 2002. Microarray‐based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. USA 99: 15687–15692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang, D. et al 2003. Viral discovery and sequence recovery using DNA microarrays. PLOS Biol. 1: 257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sampath, R. & Ecker D.J.. 2004. Novel biosensor for infectious disease diagnostics In Forum on Microbial Threats: Learning from SARS: Preparing for the Next Disease Outbreak—Workshop Summary. Knobler S.E., Mahmoud A. & Lemon S., Eds.: 181–185. The National Academies Press. Washington , DC . [PubMed] [Google Scholar]
- 23. Sampath, R. et al 2005. Rapid identification of emerging pathogens: coronavirus. Emerg. Infect. Dis. 11: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ecker, D.J. et al 2005. Rapid identification and strain‐typing of respiratory pathogens for epidemic surveillance. Proc. Natl. Acad. Sci. USA 102: 8012–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hofstadler, S.A. et al 2005. TIGER: the universal biosensor. Int. J. Mass Spectrom. 242: 23–41. [Google Scholar]
- 26. Van Ert, M. et al 2004. Mass spectrometry provides accurate characterization of two genetic marker types in Bacillus anthracis . Biotechniques 37: 642–651. [DOI] [PubMed] [Google Scholar]
- 27. Macke, T. et al 2001. RNAMotif–An RNA secondary structure definition and search algorithm. Nuc. Acids Res. 29: 4724–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang, Y. & Hofstadler S.A.. 2003. A highly efficient and automated method of purifying and desalting PCR products for analysis by electrospray ionization mass spectrometry. Anal. Biochem. 316: 50–57. [DOI] [PubMed] [Google Scholar]
- 29. Muddiman, D.C. et al 1997. Length and base composition of PCR‐amplified nucleic acids using mass measurements from electrospray ionization mass spectrometry. Anal. Chem. 69: 1543–1549. [DOI] [PubMed] [Google Scholar]
- 30. Snijder, E.J. et al 2003. Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. J. Mol. Biol. 331: 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Linssen, B. et al 2000. Development of reverse transcription‐PCR assays specific for detection of equine encephalitis viruses. J. Clin. Microbiol. 38: 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sidwell, R.W. & Smee D.F.. 2003. Viruses of the Bunya‐Togaviridae families: potential as bioterrorism agents and means of control. Antiviral Res. 57: 101–111. [DOI] [PubMed] [Google Scholar]
- 33. Brault, A.C. et al 2004. Venezuelan equine encephalitis emergence: enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc. Natl. Acad. Sci. USA 101: 11344–11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brault, A.C. et al 1999. Genetic and antigenic diversity among eastern equine encephalitis viruses from North, Central and South America. Am. J. Trop. Med. Hyg. 6: 579–586. [DOI] [PubMed] [Google Scholar]
- 35. Ecker, J.E. et al 2006. High throughput genotyping of Acinetobacter baumannii . 44: 2921–2932. [Google Scholar]