

Abstract
Nanotechnology is an expanding area of study with potentially pivotal applications in a discipline as medicine where new biomedical active molecules or strategies are continuously developing. One of the principal drawbacks for the application of new therapies is the difficulty to cross membranes that represent the main physiological barrier in our body and in all living cells. Membranes are selectively permeable and allow the selective internalization of substances; generally, they form a highly impermeable barrier to most polar and charged molecules, and represent an obstacle for drug delivery, limiting absorption to specific routes and mechanisms. Viruses provide attracting suggestions for the development of targeted drug carriers as they have evolved naturally to deliver their genomes to host cells with high fidelity.
A detailed understanding of virus structure and their mechanisms of entry into mammalian cells will facilitate the development and analysis of virus‐based materials for medical applications. Copyright © 2014 European Peptide Society and John Wiley & Sons, Ltd.
Keywords: virus, delivery, nanoparticle, peptide
Virus‐based nanoparticle for intracellular delivery.
Intracellular Delivery
Membranes are the most representative physiological barrier in our body and all living cells. Also, cellular organelles need membranes in order to maintain and to be protected by external microenvironmental damages. The plasma membrane is composed of tightly packed lipid molecules (including phospholipids, sphingolipids, and sterols), interdispersed with proteins that act as structural elements, transporters of nutrients, and environmental monitors. Membranes are selectively permeable, allowing the penetration of some substances but not others; in particular, they form a highly impermeable barrier to most polar and charged molecules and represent a physical barrier to drug absorption, limiting absorption to specific routes and mechanisms.
Therefore, one of the key requirements in both diagnosis and therapy of chronic diseases such as cancer is the efficient delivery of imaging agents and drugs to the target site. To do this, novel delivery tools are strongly required to increase their specificity and therapeutic index, respectively1. A variety of bio‐drugs, including peptides and proteins, are now produced on a commercial scale and should be intracellularly delivered to exert their therapeutic action inside the cytoplasm or onto specific organelles. Cells poorly internalize most drugs because they cross the membrane rather inefficiently2. Many treatments will not be active because drug concentration in the target disease site is not sufficient to generate a therapeutic effect. Moreover, a large amount of the drug is also delivered to normal tissues, which could result in severe side effects (low therapeutic index). Another important example is gene therapy. The latter is a method for the treatment or prevention of disease that uses genes to provide the patient with the genetic information necessary to produce specific therapeutic proteins required to correct or to modulate a disease using plasmids encoding for the proteins or down modulating the target through the use of interference methods, such as small interference RNAs (siRNA) or microRNAs 3. Key problems to overcome in gene therapy are both the delivery of the siRNA and the nucleic acid to specific locations within the body and the low plasma half‐life of nucleic acids.
Several methods exist for intracellular delivery of drugs or oligonucleotides and their choice strictly depends on the nature of the drug and on the specific place that has to be reached. Each method presents some advantages and disadvantages. For a delivery vector to be a promising tool for cellular uptake, both in vitro and in vivo have to be non‐toxic and non‐immunogenic. The uptake mechanism also plays a key role. Many vectors have been demonstrated to involve essentially endocytic mechanisms. However, endosomal entrapment can limit their activity; in fact, only a small quantity of the agent can escape this environment to achieve the specific desired biological site of action. Therefore, it is fundamental to exploit novel moieties and targeting systems that use different internalization mechanisms and represent alternative macromolecules of technological interest.
Plasmids may be incorporated into anionic or neutral liposomes to protect them against in vivo degradation and to enhance intracellular delivery. Different strategies have been developed in order to enhance the interaction and subsequent internalization of nanocarriers in the target cells.
In this light, pH‐sensitive liposomes are fusogenic at acidic pH and thus can be used to facilitate the endosomal disruption and subsequent release of plasmids in the cytoplasm. They usually consist of dioleoylphosphatidylethanolamine and a lipophilic anionic component. Another strategy is the development of proteoliposomes that incorporate viral proteins, fusogenic peptides, nuclear proteins, or nuclear localization peptides, which induce fusion of liposomes with the cell membranes and facilitate drug release and transport through the cytoplasm. In details, cationic lipids interact electrostatically with the negatively charged phosphate backbone of DNA, neutralizing the charges and promoting the condensation of DNA into a more compact structure. Usually, cationic lipids are mixed with a zwitterionic or neutral co‐lipid such as dioleoylphosphatidylethanolamine or cholesterol, respectively, to form liposomes or micelles. Cationic lipid‐based gene delivery systems lack target specificity, which results in low transfection efficiency in certain tissues due to the interference among cationic lipid‐binding macromolecules either in the circulation or in the extracellular matrix.
Another important concern in the treatment of human diseases is the crossing of physiological anatomic barriers such as blood–brain barrier (BBB). In fact, treatments of neurological disorders remain limited because of the inability of therapeutic agents to effectively cross the BBB that protects brain against invading organisms and unwanted substances and represents an obstacle for the penetration of drugs4. Most strategies to transport drugs inside the CNS determine the disruption of the anatomical texture of the BBB, thus impairing its natural function. In particular, effective delivery approaches should be cautiously assessed considering their impact on the overall protective function of the BBB. At the moment, delivery strategies across the BBB can be divided in invasive and non‐invasive approaches; invasive approaches involve the temporary disruption of the BBB, the intraventricular, or the intracerebral administration, whereas non‐invasive approaches involve the intravenous administration of the drug coupled to the carrier, which undergoes a receptor or adsorptive‐mediated transcytosis or the intranasal route. On these bases, targeted delivery of a drug to the intended site of action in the brain represents one of most promising non‐invasive approaches to overcome BBB, combining the advantages of brain targeting, high incorporation capacity, reduction of side effects, and circumvention of the multidrug efflux system5.
Viruses are Nanotechnological Device Examples of Intracellular Delivery
The example of nucleic acid delivery is illuminating in the fabrication of cell‐penetrating drug delivery systems. In this case, the efforts are addressed on how to introduce foreign nucleic acid or other pharmacological weapons into the cells. We can derive useful suggestions by understanding the virus infection process. In fact, viral carriers such as adenoviral vectors6 and retroviral/lentiviral vectors7, 8 were used for the first time in the past decades. They can bind their hosts and introduce their genetic or other organic materials into the target cells, thus mimicking an infectious process. Viral vectors are prepared removing the viral genome and substituting it with the therapeutic oligonucleotides into the viral husk9. These delivery systems showed relatively high transfection efficiency both in vitro and in vivo but raising several clinical safety and administration concerns. The latter pushed on the research on non‐viral carriers that have limited side effects if compared with attenuated viruses. In this light, the use of viral components that favor the intracellular uptake of the nanoparticles could be useful in drug delivery strategies. Therefore, sparked by the increasing knowledge on structures and life cycles of several viruses, many virus‐mimicking non‐viral carriers are being developed for therapeutic applications. In the present mini review, we will briefly describe how viral properties can be exploited for intracellular delivery using viral‐derived peptides10, modular viral‐derived protein assemblies11, and virus nanoparticles (VNPs)12.
Mechanism of Viral Entry into Host Cells
Once viruses, through one of the entrance doors, gain access into the body of a potential host, they immediately have to overcome the following challenges: (i) penetrating mucus layers, (ii) moving through the bloodstream, and (iii) replicating in living host cells. Replication further involves the delivery of the viral genome into the host cells and use of their intracellular machines. Once it has arrived in the proximity of cells to be infected, critical moments for viral infectious cycle are the following: (i) cellular receptor binding, (ii) internalization, and (iii) uncoating and releasing of viral nucleic acids at the proper site of replication (Figure 1).
Figure 1.
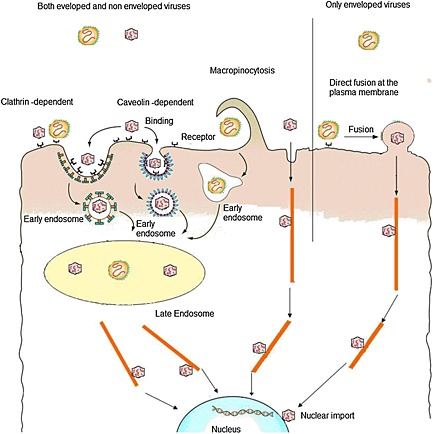
Viral entry and cellular trafficking.
Depending on the different complexity of the viral particles, various strategies can mediate an efficient infection as a result of the physical interactions between cells and viruses. The most critical barrier is represented by the complex membranous system surrounding and residing within the host cell and consisting in the following: (i) plasma membrane, (ii) a rather dense cytoplasm where molecular traffic is highly restricted and regulated13, and (iii) any other membranes that must be crossed in order to access the sites of viral replication or assembly. Viruses consist of an RNA or DNA genome surrounded by either multiple copies of capsid proteins (non‐enveloped viruses) or both capsid proteins and a lipid membrane (enveloped viruses). The size of animal viruses ranges from approximately 25 nm to over 300 nm, and depending on dimension and structure, viruses have acquired different strategies to use and control cell functions. Nowadays, the overall picture of entry of animal virus into the host cell is becoming increasingly complete. In fact, studies performed on several viruses and different cellular systems have shed light on the basic mechanisms of viral entry.
Although the fine molecular details at the interface of virus and cell surface interactions are quite complex and highly variable, the pathways allowing viruses to reach the sites of penetration seem to be limited. In fact, only a few endocytic mechanisms are involved. To bind cell surface, a wide variety of different proteins, lipids, and carbohydrates are used by viruses. These molecules are generally considered attachment factors that simply enable viruses to bind and to concentrate on the cell surface. Other interactions on the cell surface involve real receptors, which in addition to binding, actively promote their entry into cells, mediate conformational changes in the virus particle, and trigger signaling pathways that promote the overall infectious process. Thus, the first and highly specific step of viral infection is binding to the receptors present on the plasma membranes 14, 15, which may be either primary receptors or co‐receptors, the latter having lower binding affinities, and being involved only after primary receptors in order to enhance the binding strength16. Only few viruses are able of penetrating directly through the cell surface by fusing their envelope with the plasma membrane.
For some species of retroviridae, paramyxoviridae, and herpesviridae, the recognition of specific cell surface receptors triggers the fusion of their envelope with the plasma membrane. The best characterized fusion mechanism is that of HIV‐1. HIV‐1 cellular uptake is mediated by two glycoproteins, namely, gp120 and gp41. Binding of gp120 to the primary receptor, CD4, generates a conformational change of gp120, which determines a further interaction with the chemokine co‐receptors (CCR5 and CXCR4) and the exposure of a hydrophobic fusion peptide present on the N‐terminal region of gp41, which inserts into the plasma membrane and drives membrane fusion17.
Another example is represented by Herpes simplex virus type 1 (HSV‐1) of the α‐herpesvirinae subfamily, which undergoes a fusion event following the engagement of the glycoproteins gB and gD with cellular receptors18. Hence, fusion between viral envelopes and plasma membranes leads to the release of capsids into the cytoplasm.
Most viruses (both enveloped and non‐enveloped) undergo endocytosis, a fundamental cellular process involved in the uptake of many macromolecules. Viruses use several endocytic routes for cellular internalization: caveolar and clathrin‐dependent or caveolae‐independent. In clathrin‐mediated endocytosis, a ligand binds to a specific receptor and determines the clustering of the ligand–receptor complexes in coated pits on the plasma membrane, which then invaginates and pinches off from the membrane to form intracellular clathrin‐coated vesicles. Their depolymerization and fusion with each other result in formation of late endosomes that further fuse with lysosomes, where the cargo is degraded19. Although lysosomes represent a cul‐de‐sac for many molecules utilizing this cell internalization route, viruses have developed the ability to avoid lysosomal degradation escaping into the cytosol. Therefore, the penetration event follows endosomal internalization and is necessary for the delivery to the cytosol. In the case of enveloped viruses, penetration invariably involves membrane fusion, which, similarly to the already described fusion at the plasma membrane, is mediated by specific viral glycoproteins. The main difference between the two modes of fusion is that once inside the endosome, the viruses fuse their envelope with the limiting membrane of the endocytic vacuoles from the luminal side20.
Therefore, membrane fusion represents the common step in the entry of enveloped viruses, regardless of the chosen route and is also an essential and ubiquitous mechanism in most cellular events.
Description of the Structure of Viral Fusion Proteins
The viral fusion proteins undergo conformational changes as a consequence of either low endosomal pH or receptor binding and this leads to the exposure of hydrophobic peptides, loops, or patches, which then interact with and destabilize one or both the opposing membranes.
Viral fusion proteins undergo significant structural rearrangements from the pre‐fusion to the post‐fusion conformations which lead to the formation of a stable hairpin with the transmembrane and the fusion peptide domains at the same end of the trimeric elongated rod‐like structure. Three different classes of viral fusion proteins (Figure 2) have been identified to date based on their common post‐fusion structure [20, 21, 22, 23, 24, 25, 26, 27]. These are the following: (i) class I fusion proteins, characterized by trimers of hairpins containing a central α‐helical coiled‐coil structure (identified in orthomyxoviruses, paramyxoviruses, retroviruses, filoviruses, and coronaviruses), 28, 29, 30, 31, 32, (ii) class II fusion proteins, characterized by trimers of hairpins composed of β structures (main representatives are members of the Flaviviridae and Togaviridae families), 33, 34, and (iii) class III fusion proteins, with the central α‐helical trimeric core similar to class I and two fusion loops located at the tip of an enlongated β‐sheet similar to class II fusion proteins. The structures 35 of the G protein of vesicular stomatitis virus and of the gB protein of HSV‐1 have been described as class III fusion proteins 36, 37. Two more glycoproteins have lately been added to this class, namely, gp64 from baculovirus 38 and gB from Epstein–Barr virus 21.
Figure 2.
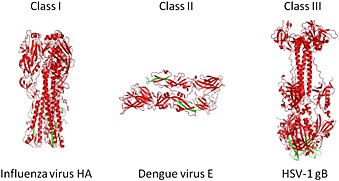
Structure of viral fusion proteins. An example for the three different classes is shown. The fusion pepride is shown in green.
Despite structural differences among the three classes, they induce membrane fusion in a similar manner through the formation of an analogous hairpin structure, which allows fusion peptides to insert into cell membranes and to drive membrane destabilization. Further awesome refolding steps result in the merging of the two lipid layers and the consequent release of the viral nucleocapsid inside the host cells.
Non‐enveloped viruses use a different mechanism for entry, which involves membrane disruption or pore formation to escape the endosomes. The ability to disrupt the endosomal membrane to allow release of the genetic material inside into the cytoplasm needs to be directly dependent on the capsid components, because the cellular membranes are facing not the viral envelope but simply the protein made capsids. The process of non‐enveloped viruses entry is generally started with a mechanism reminiscent of the entry of enveloped viruses, namely, triggering a conformational change of capsid proteins as a consequence of receptor binding or pH lowering. The conformational change allows the release of viral components with membrane lytic activity, which binds to the cell membrane, disrupt the lipid bilayer, and convey the viral particle across the membrane. Therefore, membrane penetration is mediated by short, membrane altering, amphipatic, or hydrophobic sequences contained in proteins, which have undergone a conformation transition, allowing such sequences to interact with membranes 39.
Despite several salient differences in the mechanism of entry among non‐enveloped viruses, similar events during entry collectively characterize these membrane lytic peptides: induced modifications of capsid proteins resulting in peptide exposure followed by outward projection of peptides able to interact with host membranes and disrupt them, resulting in the delivery of the viral genome inside the host cell. Several short sequences deriving from viral capsids and being able to mediate entry have been identified; for example, some peptides (as the lytic peptides of nodaviruses 40, picornaviruses, 41 and reoviruses42) can be generated by an autocatalytic cleavage step of a precursor, whereas other peptides are derived following a proteolytic activity of cellular enzymes such as in the case of rotavirus 43 and adenoviruses 44 or virally derived proteolysis such as pep46 and additional peptides of birnavirus45, 46.
After being released into the cytosol, viruses and viral capsids use the cytoplasmic transport systems of the cell, moving to sites of replication within the cytosol or the nucleus. Most viruses associate with microtubule‐based motors, such as dynein and dynactin, and move along microtubules toward the nucleus 13.
Consequently, many viruses, including most DNA viruses and some RNA viruses, need access to the nucleus of the host cell, because they depend on nuclear proteins for ensuing replication47, 48, 49 The nuclear envelope acts as a barrier between the cytoplasm and the nucleus, and transport of molecules into and out of the nucleus is tightly regulated. Therefore, viruses have developed several strategies to reach the nuclear milieu: (i) some viruses gain access to the nucleus during mitosis, when the limiting membrane is temporarily disassembled, (ii) HIV‐1, influenza A virus and others undergo extensive disassembly in the cytoplasm, but the cytoplasmic‐released components contain nuclear localization sequences and are thereby able to cross the nuclear pore using the host transport machinery, (iii) some viral capsids use importins or viral proteins to attach to the cytoplasmic side of the nuclear pore. Interaction with the nuclear pore promotes disassembly and the viral genome is released into the nucleus, (iv) some viral capsids (for example, those of hepatitis B virus) can cross intact nuclear pores for their small size, and (v) some viruses, such as parvoviruses, transiently disrupt the nuclear membrane and lamina and enter the nucleus 50.
Finally, after using the cellular machinery for genome synthesis and production of new viral proteins, progeny virions are assembled and then released from the cell.
Viral Peptides for Enhanced Intracellular Delivery
The successful clinical use of nanotechnology in the delivery of gene material is greatly because of the specific targeting of weapons to the diseased cells thus reducing the risk derived from the uptake by the normal tissue counterparts. Overcoming the physiological barriers of both cells and tissues can be allowed by the direct transfer across cell membrane involving transient permeabilization or alternatively after endocytosis, through transfer across vesicular membranes by lipid disruption and pore formation. These cell membrane changes can be part of a pathological process that characterizes the viral infections and the propagation of the viral genetic content in the host cells. In fact, the latter requires the permeabilization of cell membranes in order to allow viral penetration.
The alteration of intracellular vesicle trafficking is one of the major limits in the strategies based on the transfer of drugs or genes through the use of non‐viral carriers that have poor side effects and are also poorly efficient because of the endosomal vesicle sequestration 51. The subsequent fusion of these endosomes with lysosomes can induce the degradation of the pharmacologically active moiety with consequent reduced efficacy. In this light, viruses are a suitable tool in order to design strategies to stealth the endosomal‐lysosomal pathway and the consequent drug sequestration 52, 53. Peptide LAH4, from a family of His‐rich peptides, has also been shown to complex DNA and efficiently deliver nucleic acids into eukaryotic cells by pH‐triggered lysis of endosomes54, 55. There are many differences on the viral capsid and membrane proteins and peptides that drive the penetration and endosomal escape depending upon the different classes of viruses. However, the mechanisms involved in the regulation of these biological processes are common and depend upon similar structural characteristics of the viral moieties involved 56.
Recent work has identified three distinct classes of viral membrane fusion proteins based on the following structural criteria: (i) N‐terminal (class I), (ii) internal single loops (class II), and (iii) internal bipartite loops (class III). Fusion peptides strongly interact with the bilayer and undergo conformational changes, which are crucial for membrane fusion. It has been reported that the fusion peptide of influenza enhances the endosomal escape of polyplex 57, 58 or liposome‐encapsulated proteins59, 60, 61, 62 by mediating the insertion into the endosomal membrane. MPG is a chimeric peptide comprised of two independent domains. The first 17 amino acids of the N‐terminus are derived from glycine‐rich region of the viral gp41 and the hydrophilic C‐terminus from nuclear localization signal of the Simian Virus 40 (SV40) large T antigen 59, 60. The MPG hydrophobic domain allows the peptide insertion into the membrane; MPG does not enter cells by endocytosis but induces transient membrane destabilization.
Recently, it has been proved that gH625, a peptide derived from the glycoprotein H of HSV‐1, possesses the ability to carry cargo molecules across cell membranes63, 64, 65, 66. gH625 interacts with biological membranes, contributing to their merging and is able to traverse the membrane bilayer and transport a cargo into the cytoplasm. The peptide contains particular residues that are crucial for its capacity to interact with target lipid membranes. In particular, it is rich in hydrophobic residues that allow the peptide to enter into the bilayer and destabilize membranes. Fusion peptides are characterized by a typical amphipatic α‐helix structure, which mediates lipid–protein interactions during the binding of proteins to membranes, thereby triggering membrane fusion and translocation 67. Confocal microscopy studies on live cells showed the gH625 ability to cross membrane bilayers (Figure 3) and translocate into the cytosol as compared with (transactivator of transcription protein from HIV‐1) TAT peptide, which mainly enter cells by endocytosis 68. A shorter version of this peptide (missing the first histidine at position 625) was investigated in a study by Tu and Kim 69 and found to enhance the transfection efficiency of cationic liposomes more than 30‐fold in human cell lines improving the intracellular penetration of liposomes or lipoplexes. Another example of the suitability of gH625 is given by the effective delivery of intrinsically disordered proteins. A genetically modified recombinant gH625‐c‐prune was prepared through conjugation of c‐prune with gH625. C‐prune is the C‐terminal domain of h‐prune, overexpressed in breast, colorectal, and gastric cancers, interacting with multiple partners and representing an ideal target for inhibition of cancer development. Its C‐terminal domain results in an intrinsically disordered domain and is easily transduced through biomembranes due to net negative charge of gH625 70. gH625‐c‐prune fusion protein exhibited the ability to cross biomembranes, opening a new scenario for the use of gH625 as a novel multifunctional carrier 68. The successful use of fusogenic peptides in drug delivery systems is dependent on the specific targeting of drugs to tumor cells with minimal toxicity.
Figure 3.
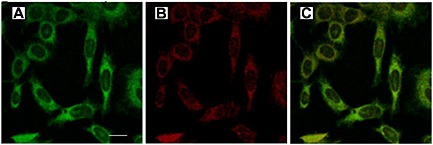
Fluorescence images of viral peptide labeled at 1 μM. In panel A, the peptide is shown in green, in panel B, the Lyso Tracker is shown in red, and panel C, the merge of the two images is shown. Bar = 10 µm.
Modular Viral‐based Vectors
The success of new strategies for treatment of diseases relies on the development of delivery devices capable of improving the therapeutic index of biologically active molecules as well as diagnosing the disease site of interest. A fundamental challenge of current diagnostics and therapeutics is the design of a single carrier system that has the potential to deliver therapeutics to the disease site with high fidelity, that is, targeted delivery, and allows both diagnosis and cell delivery, that is, cell penetration and uptake71. Nanotechnology has the potential to create platforms that combine targeting and delivery with imaging and targeted cell uptake (theranostics), 72, 73. Several classes of biomaterials can serve as platform for theranostics and are promising drug delivery carriers; they include nanocrystals, liposomes, virosomes, nanoemulsions, polymer protein conjugates, nanocomplexes, and nanoparticles71, 74, 75, 76, 77. According to the nanoparticle versatility, diagnostic and therapeutic drugs can be either physically encapsulated or covalently conjugated to nanoparticles (Figure 4). The carrier composition, shape, and surface decoration dictate its in vivo behavior and its actual translation into clinical use. The dimension of the nanosystem may be comprised between 1 and 1000 nm; but it is widely accepted that its diameter for cancer therapy should be in the range 10–100 nm, in order to make it easily penetrate the leaky tumor vasculature and accumulate within the tumor tissue.
Figure 4.
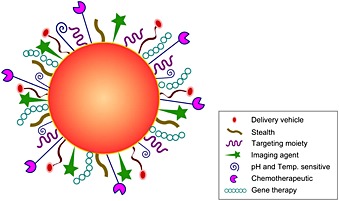
Multifunctional nanosystem with a choice of drug delivery vehicle, molecular imaging agent, tumor targeting ligand, synthetic and biological therapeutics with stealth properties.
Liposomal aggregates are the oldest nanotherapeutic platform and have attracted great attention due to their success as in vivo carriers of both hydrophilic (entrapped in the aqueous core) and hydrophobic (entrapped within the lipid membrane) drugs 78, 79, 80, 81, 82. Liposomes, with size ranging in mean diameter from 50 to 300 nm, display unique pharmacokinetic properties and can be adapted to a wide range of therapeutic agents. Liposomes are non‐toxic, biodegradable, and non‐immunogenic; encapsulated drugs are protected from chemical or metabolic degradation after injection and their toxicity is reduced thanks to the decreased exposure of the drug to susceptible healthy tissues and increased antitumor activity resulting from a relatively long systemic circulation time (achieved through surface modification with polyethylene glycol or other polymers), an extended exposure, and tumor selective accumulation in sites of tumor growth. The association of a drug with liposomes markedly changes its pharmacokinetic and pharmacodynamic properties and lowers systemic toxicity; moreover, in order to enhance the antitumor efficacy, many research groups are also working to improve cellular internalization of liposomes through the addition of surface ligands such as penetratin, TAT, and membranotropic 83, 84.
Dendrimers are hyperbranched polymeric molecules with a nearly perfect 3D geometrical architecture and their unique branched topologies provide a platform for coupling of drugs and targeting moieties 85. As dendrimer generation increases, the end groups at their periphery become more closely packed, which allows to achieve concentrated payloads of drugs or spectroscopic labels for therapeutic and imaging applications 86. In order to enhance their cellular uptake, dendrimers have been coupled to cationic cell‐penetrating peptides as well as membranotropic peptides87.
Core nanoparticles are also a common platform for nano‐biosystem construction, and the core material may display unique properties for stability and or detection. Nanoparticles of very different shapes and compositions have been developed including synthetic or natural polymers and inorganic materials such as iron oxide, quantum dots, and gold68, 88, 89, 90, 91, 92.
Numerous groups are working on novel multicomponent carriers, which use packaging strategies to mimic viruses. Most of them are obtained by the mere linear fusion of their components. They include multifunctional fusion proteins using signaling and transport domains from diverse organisms assembled in a single protein [93]. Favaro et al., [94] recently reported the obtainment of the multifunctional protein derived from the fusion of the recombinant human dynein light chain Rp3, an N‐terminal DNA binding domain and the C‐terminal TAT sequence. This multifunctional fusion protein was intended to mimic viral vectors that take advantage of the microtubules for a faster intracellular movement toward the nucleus. Hatakeyama et al. 95 demonstrated that introduction of both pH‐sensitive fusogenic glutamic acid‐alanine‐leucine‐alanine repeat peptide (GALA) peptide and PEG‐Peptide‐DOPE conjugate (PPD) facilitates nanoparticle endosomal escape, thereby enhancing the efficiency of siRNA delivery and gene silencing; El‐Sayed et al. 96 described a novel stearylated derivative of the endosome‐disrupting peptide, INF7 derived from the influenza virus hemagglutinin protein, which allowed preparation of a multifunctional envelope‐type nano device that mimics the structure of the influenza virus.
Viral Nanoparticles as Delivery Vehicles
New nanocarrier platforms based on natural biological building blocks offer great promises for the development of variety of application in nanotechnology. A typical example of natural molecular assemblies and containers is represented by virus‐based nanoparticles, which are nanocages or nanorods assembled from capsid proteins of viruses obtained exploiting the recent and rapid advances in protein engineering and material science. Virus‐like particles (VLPs) are multi‐subunit protein complexes able to self‐assemble and form higher‐order structures mimicking native 3D conformation of viruses. The main difference with native viruses is that VLPs do not carry, hidden into the capsid protective shell, the viral genetic material but are empty cases or incorporate selected pieces of genetic material. Therefore, VLPs lacking a complete viral genome are unable to replicate and non‐infectious. Over the last three decades, VLPs have been demonstrated very useful for the production of safe vaccines 97, 98, and the first vaccine approved by the Food and Drug Administration in 1986 was the vaccine against hepatitis B virus. From many points of view, VLPs are indistinguishable from VNP, in fact, the basic strategy behind the production of VLP and VNP is exactly the same: genetic engineering of capsid protein and their expression in microbial factories (e.g., bacteria, yeast, insect cells, mammalian cells, and plants). The principal difference is that VNPs are intended for creating novel material to be applied to the broader field of nanotechnology to solve different material‐based problems and in the scope of the present review for drug delivery. Because of the appropriate size generally ranging from 20 to 200 nm, their homogeneity, possible functionalization, easy of modification through chemical and genetic routes, and possibility of convenient preparation and scale up in microbial factories VNP are emerging as the most promising platform for gene and drug delivery applications 12, 99, 100. Plant and animal viruses and also bacteriophages have been employed for producing VNPs. The principal plant viruses include Tobacco Mosaic Virus (TMV), Brome Mosaic Virus, Cowpea Chlorotic Mottle Virus (CCMV), and Red Clover Necrotic Mosaic Virus, whereas animal viruses are principally represented by SV40, Alphavirus, Polyoma JC virus, and adenovirus. Some phages such as bacteriophages M13 and MS2 and have also been explored for the construction of VNPs (Table 1).
Table 1.
Virus nanoparticles employed in biomedicine
| VIRUS (short name) | Symmetry | Size (nm) | Application in biomedicine | References | ||
|---|---|---|---|---|---|---|
| Outer | Inner | |||||
| Plant viruses | Cowpea Chlorotic Mottle Virus (CCMV) | Icosahedral | 28 | 18 | Biocompatible nanoplatforms | [104] |
| Cowpea Mosaic Virus (CPMV) | Icosahedral | 28 | 20 | Doxorubicin delivery | [101] | |
| Red Clover Necrotic Mosaic Virus (RCNMV) | Icosahedral | 35 | 17 | Multifunctional cell targeting VNP | [102] | |
| Animal viruses | Adenovirus | icosahedral | 90 | 62 | Anticancer bleomycin (BLM) delivery | [107] |
| Simian virus 40 (SV40) | icosahedral | 50 | 36 | Nanoplatform for therapeutic use | [106] | |
| Polyomavirus (JCV) | Icosahedral | 45 | 32 | Drug‐releasing VNP | [106] | |
| Bacteriophages | MS2 | Icosahedral | 27 | 21 | RNA, DNA‐based drugs delivery | [108] |
| P22 | Icosahedral | 64 | 54 | Nanoscale MRI contrast agent | [109] | |
| Qβ | Icosahedral | 27 | 21 | Imaging agents delivery | [110] | |
| M13 | Helical | 6.6 × 880 | — | Convection‐enhanced delivery | [111] | |
There are mainly two ways to design a VNP‐carrying pharmaceutical molecules or heterogeneous genetic material as a cargo: (i) the therapeutic cargo can be encapsulated inside the capsid, and (ii) the therapeutic cargo is attached on the surface of the nanoparticle (Figure 5). Several viruses have been proved to be amenable to both chemical and genetic manipulation of their inner cavity as well as the outer surface, so that the further attachment of drug molecules is rendered feasible. The tools of molecular biology allows for the engineering of capsid proteins subunits by modifying amino acid residues at key position within the assembled structure. This allows for the precise spatial and numerical control of reactive functional groups for the chemical attachment of ligands.
Figure 5.
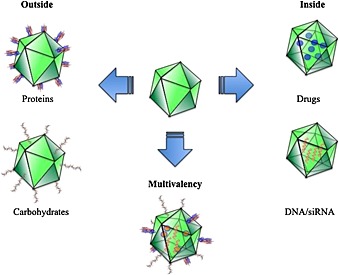
Viral capside nanocarriers that transport bioactive molecules on their surface or internalized in their core.
The plant virus Cowpea mosaic virus (CPMV) with a covalent modification linking doxorubicin, an anticancer drug, to the capsid shell has been explored for its potential application in anticancer therapy 101. When doxorubicin was conjugated through a stable amide bond to the CPMV carrier system, the formulation induces time‐delayed, but enhanced, toxicity to HeLa cells compared with free drug. Other plant viruses used to create VNP carriers for doxorubicin are Red clover necrotic mosaic virus 102 and Hibiscus chlorotic ringspot virus 103. When VNPs loaded with doxorubicin and armed with a targeting peptide were delivered to HeLa cells, a cytotoxic effect was observed.
Investigations on biodistribution and clearance of non‐targeted CCMV 104, a member of the Bromoviridae family, showed a broad distribution and movement throughout most mice tissues and organs, rapid excretion, absence of long term persistence within tissue and organs, and no toxicity after a single injection, suggesting the possibility to use CCMV as a safe and biocompatible nanoplatforms for applications in biomedicine. For accurate diagnosis of this inflammatory disease, molecular imaging is becoming a necessity, and the plant viral nanoparticle platform based on TMV has been explored in order to target vascular cell adhesion molecule‐1105, which is highly expressed on activated endothelial cells at atherosclerotic plaques. TMV was modified to carry near‐infrared dyes and chelated Gd ions to build a multimodal probe in magnetic resonance and optical imaging to be used in an atherosclerotic ApoE‐/‐ mouse model. The obtained results indicated molecular targeting of atherosclerotic plaques.
In the case of animal viruses, the main examples of VNP construction have been provided by JC virus, SV40, and adenovirus. JC polyomavirus was employed to realize a drug‐releasing VNP in which the release mechanism is triggered by changes in pH 106. VNPs were composed of a major coat protein, VP1, and inner core protein, VP2, which was used to anchor the hexahistidine motif (His6) tags into the inside of the VNP. The His6 tag allowed a specific and reversible attachment for drug molecules that can enter through the 1 nm pores present on the VNP and be released when the pH is lowered as a consequence of the protonation of histidines. While a dodecahedron based on adenovirus composed of 12 copies of a pentameric viral protein responsible for virus penetration was used to deliver the lipophilic, non‐permeant, and labile anticancer antibiotic bleomycin (BLM)107. Successful BLM delivery by such a vector consisted in significantly improved drug bioavailability. Importantly, the adenovirus dodecahedron formed VNPs much smaller than the virion itself and with different intra‐particle interactions.
Finally, also, bacteriophages have been used for the production of delivery vectors. Bacteriophage MS2‐based VNPs were able to selectively deliver nanoparticles, chemotherapeutic drugs, siRNA cocktails, and protein toxins to human hepatocellular carcinoma (HCC), 108. MS2 VNPs had been modified with a peptide (SP94) that can bind to HCC and can deliver high concentrations of encapsidated cargo to the cytosol of HCC cells. SP94‐targeted VNPs loaded with doxorubicin, cisplatin, and 5‐fluorouracil selectively killed the HCC cell line, Hep3B, at nanomolar concentrations. VNPs that encapsidate a siRNA cocktail, silencing expression of cyclin family members, induced growth arrest and apoptosis of Hep3B at picomolar concentrations, showing that MS2 VNPs can be easily modified and can specifically encapsidate a variety of disparate cargos inducing selective cytotoxicity of cancer in vitro.
Concluding Remarks
The intracellular uptake of drugs or nucleic acid for the treatment of human diseases is an important challenge in medicine and requires multidisciplinary efforts in order to be successful. Nanotechnologies have been increasingly used in order to design and develop nanometric carriers for the delivery of therapeutic agents in the diseases, tissues, and cells. The teachings coming from the virus world could be enormously useful in order to enhance the delivery potential of such nanocarriers. Viruses are naturally evolved nanoparticles able to efficiently deliver genetic material through physiological barriers such as the cellular membranes. The mechanisms by which viruses infect cells and cross the membranes could be extremely useful in order to design strategies based upon both the use of the viral capsids as nanocarriers themselves and the decoration of nanocarriers with cell‐penetrating peptides of viral origin. These strategies could be successful in both favoring the delivery in diseased tissues and in favoring their penetration in diseased cells. The tight interaction between different biomedical scientists (microbiologists, biochemists, chemists, pharmacologists, and medical doctors) could represent the way to see the light at the end of the tunnel: how to efficiently deliver a drug to a diseases' tissue sparing the normal counterpart.
Biographies
Stefania Galdiero obtained her doctorate in Chemistry in the field of peptide's chemistry at the University of Naples ‘Federico II’ in 1998. She carried out research activities at the Columbia University of New York in 1996–1997. In 1999, she earned the position of lecturer in Inorganic Chemistry at the University of Naples, and since 2012, she is an adjunct professor at Loyola University Chicago. She is a member of the Department of Pharmacy of the University of Naples Federico II and of the Biostructures and Bioimages Institute of the National Research Council of Italy. Recent research activities focus on antimicrobial peptides and mechanisms of drug intracellular delivery. Elucidating the principles that govern the protein/membrane interactions and understanding the molecular forces underlying peptide–peptide interactions within the lipid environment of the cell membrane are major goals.

Annarita Falanga received the degree in Food Science and Technology at the University of Naples ‘Federico II’, in 2004. From 2004 to 2007, she attended the PhD course in Microbiological Science during which she was involved in the synthesis and characterization of fusogenic domains derived from membrane glycoproteins of herpes simplex virus type I. From 2007 to 2012, she earned several Research Contracts at Consiglio Nazionale delle Ricerche continuing her studies on peptides with antimicrobial activity. Her current research interests are in the development of stable formulation for radiopharmaceutical molecules.

Mariateresa Vitiello obtained her degree in Biological Science at the University of Naples ‘Federico II’ in 1991. She received her PhD in Microbiology in 1999 and held postdoctoral position in Microbiology since 2008 when Dr. Vitiello become Consultant in Clinical Microbiology and Virology. She has published more than 50 peer‐reviewed articles in the field of microbial pathogenesis and microbial‐induced signal transmission.

Paolo Grieco graduated in medicinal chemistry at the University of Naples ‘Federico II’, Italy, and obtained his PhD on 1993, working on solid‐phase peptide synthesis. He has been a postdoctoral fellow at the Department of Chemistry and Biochemistry, University of Arizona, working on the development of peptide and peptidomimetic ligands active on melanocortin receptors. In 2001, he was appointed an associate professor in medicinal chemistry at the University of Naples ‘Federico II’. Since 2012, he is a full professor in Medicinal Chemistry at the Department of Pharmacy at the University of Naples Federico II. He is the author of more than 160 scientific publications in international journals and several patents on discover of new molecules as potential therapeutic agents and for diagnostic applications.

Michele Caraglia graduated in Medicine in 1990 and achieved the PhD degree in Cellular Biochemistry in 2001. He is presently an associate professor in Biochemistry at the Second University of Naples. Prof. Caraglia has been involved in the study of the escape mechanisms of human cancer to the apoptosis induced by cytokines such as interferons alpha and beta and on the delivery of aminobisphosphonates and miRNAs by nanocarriers in tumor cells.

Giancarlo Morelli is a full professor of Chemistry at the University of Naples ‘Federico II’ (Napoli, Italy) and head of the Research Center on Bioactive Peptides. His research interest is devoted to the development of diagnostic and therapeutic agents based on bioactive peptides. He also focuses on the development of new nanovectors and peptide modified supramolecular aggregates for diagnosis and therapy in nanomedicine.

Massimiliano Galdiero obtained his MD degree from the University of Naples ‘Federico II’ in 1992. He earned his PhD in Virology from the University of Cambridge (UK) in 1998. He was appointed lecturer in Microbiology in 1994 at the Department of Animal Health of the Faculty of Veterinary Medicine of the University of Naples ‘Federico II’. He moved to the Faculty of Medicine of the Second University of Naples where he was appointed associate professor and then full professor of Microbiology in 2004. His more recent research interests are focused on the study of host–microbial interactions and antimicrobial development.

This article is published in Journal of Peptide Science as part of the Special Issue devoted to contributions presented at the 1st International Conference on Peptide Materials for Biomedicine and Nanotechnology, Sorrento, October 28–31, 2013, edited by Professor Giancarlo Morelli, Professor Claudio Toniolo and Professor Mariano Venanzi.
References
- 1. Moros M, Mitchell SG, Grazu V , de la Fuente JM . The fate of nanocarriers as nanomedicines in vivo: important considerations and biological barriers to overcome. Curr. Med. Chem. 2013; 20: 2759–2778. [DOI] [PubMed] [Google Scholar]
- 2. Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007; 59: 748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin L, Zeng X, Liu M, Deng Y, He N. Current progress in gene delivery technology based on chemical methods and nano‐carriers. Theranostics 2014; 4: 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alyautdin R, Khalin I, Nafeeza MI, Haron MH, Kuznetsov D. Nanoscale drug delivery systems and the blood‐brain barrier. Int. J. Nanomedicine 2014; 9: 795–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinzon‐Daza ML, Campia I, Kopecka J, Garzon R, Ghigo D, Riganti C. Nanoparticle‐ and liposome‐carried drugs: new strategies for active targeting and drug delivery across blood‐brain barrier. Curr. Drug Metab. 2013; 14: 625–640. [DOI] [PubMed] [Google Scholar]
- 6. Hartman ZC, Appledorn DM, Serra D, Glass O, Mendelson TB, Clay TM, Amalfitano A. Replication‐attenuated human adenoviral type 4 vectors elicit capsid dependent enhanced innate immune responses that are partially dependent upon interactions with the complement system. Virology 2008; 374: 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cooray S, Howe SJ, Thrasher AJ. Retrovirus and lentivirus vector design and methods of cell conditioning. Methods Enzymol. 2012; 507: 29–57. [DOI] [PubMed] [Google Scholar]
- 8. Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005; 16: 1241–1246. [DOI] [PubMed] [Google Scholar]
- 9. Selkirk SM. Gene therapy in clinical medicine. Postgrad. Med. J. 2004; 80: 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galdiero S, Vitiello M, Falanga A, Cantisani M, Incoronato N, Galdiero M. Intracellular delivery: exploiting viral membranotropic peptides. Curr. Drug Metab. 2012; 13: 93–104. [DOI] [PubMed] [Google Scholar]
- 11. Aris A, Villaverde A. Modular protein engineering for non‐viral gene therapy. Trends Biotechnol. 2004; 22: 371–377. [DOI] [PubMed] [Google Scholar]
- 12. Bronstein LM. Virus‐based nanoparticles with inorganic cargo: what does the future hold? SMALL 2011; 7: 1609–1618. [DOI] [PubMed] [Google Scholar]
- 13. Radtke K, Dohner K, Sodeik B. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell. Microbiol. 2006; 8: 387–400. [DOI] [PubMed] [Google Scholar]
- 14. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000; 69: 531–569. [DOI] [PubMed] [Google Scholar]
- 15. Marsh M, Helenius A. Virus entry: open sesame. Cell 2006; 124: 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greber UF. Signalling in viral entry. Cell. Mol. Life Sci. 2002; 59: 608–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klasse PJ. The molecular basis of HIV entry. Cell. Microbiol. 2012; 14: 1183–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. Herpes virus fusion and entry: a story with many characters. Viruses 2012; 4: 800–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maxfield FR, McGraw TE. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004; 5: 121–132. [DOI] [PubMed] [Google Scholar]
- 20. Harrison SC. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res. 2005; 64: 231–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Backovic M, Longnecker R, Jardetzky TS. Structure of a trimeric variant of the Epstein–Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 2009; 106: 2880–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harrison SC. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008; 15: 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lamb RA, Paterson RG, Jardetzky TS. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 2006; 344: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stiasny K, Heinz FX. Flavivirus membrane fusion. J. Gen. Virol. 2006; 87: 2755–2766. [DOI] [PubMed] [Google Scholar]
- 25. Kielian M. Class II virus membrane fusion proteins. Virology 2006; 344: 38–47. [DOI] [PubMed] [Google Scholar]
- 26. Kielian M, Rey FA. Virus membrane‐fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 2006; 4: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weissenhorn W, Hinz A, Gaudin Y. Virus membrane fusion. FEBS Lett. 2007; 581: 2150–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981; 289: 366–373. [DOI] [PubMed] [Google Scholar]
- 29. Fass D, Harrison SC, Kim PS. Retrovirus envelope domain at 1.7 angstrom resolution. Nat. Struct. Biol. 1996; 3: 465–469. [DOI] [PubMed] [Google Scholar]
- 30. Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 1998; 2: 605–616. [DOI] [PubMed] [Google Scholar]
- 31. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006; 439: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu Y, Liu Y, Lou Z, Qin L, Li X, Bai Z, Pang H, Tien P, Gao GF, Rao Z. Structural basis for coronavirus‐mediated membrane fusion. Crystal structure of mouse hepatitis virus spike protein fusion core. J. Biol. Chem. 2004; 279: 30514–30522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick‐borne encephalitis virus at 2 Å resolution. Nature 1995; 375: 291–298. [DOI] [PubMed] [Google Scholar]
- 34. Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Rey FA. The Fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001; 105: 137–148. [DOI] [PubMed] [Google Scholar]
- 35. Modis Y, Ogata S, Clements D, Harrison SC. A ligand‐binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2003; 100: 6986–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low‐pH form of the vesicular stomatitis virus glycoprotein G. Science 2006; 313: 187–191. [DOI] [PubMed] [Google Scholar]
- 37. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006; 313: 217–220. [DOI] [PubMed] [Google Scholar]
- 38. Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008; 15: 1024–1030. [DOI] [PubMed] [Google Scholar]
- 39. Banerjee M, Johnson JE. Activation, exposure and penetration of virally encoded, membrane‐active polypeptides during non‐enveloped virus entry. Curr. Protein Pept. Sci. 2008; 9: 16–27. [DOI] [PubMed] [Google Scholar]
- 40. Zlotnick A, Reddy VS, Dasgupta R, Schneemann A, Ray WJ, Jr , Rueckert RR, Johnson JE. Capsid assembly in a family of animal viruses primes an autoproteolytic maturation that depends on a single aspartic acid residue. J. Biol. Chem. 1994; 269: 13680–13684. [PubMed] [Google Scholar]
- 41. Arnold E, Luo M, Vriend G, Rossmann MG, Palmenberg AC, Parks GD, Nicklin MJ, Wimmer E. Implications of the picornavirus capsid structure for polyprotein processing. Proc. Natl. Acad. Sci. U. S. A. 1987; 84: 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008; 27: 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arias CF, Romero P, Alvarez V, Lopez S. Trypsin activation pathway of rotavirus infectivity. J. Virol. 1996; 70: 5832–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greber UF, Webster P, Weber J, Helenius A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996; 15: 1766–1777. [PMC free article] [PubMed] [Google Scholar]
- 45. Da Costa B, Chevalier C, Henry C, Huet JC, Petit S, Lepault J, Boot H, Delmas B. The capsid of infectious bursal disease virus contains several small peptides arising from the maturation process of pVP2. J. Virol. 2002; 76: 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Galloux M, Libersou S, Alves ID, Marquant R, Salgado GF, Rezaei H, Lepault J, Delmas B, Bouaziz S, Morellet N. NMR structure of a viral peptide inserted in artificial membranes: a view on the early steps of the birnavirus entry process. J. Biol. Chem. 2010; 285: 19409–19421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greber UF, Fornerod M. Nuclear import in viral infections. Curr. Top. Microbiol. Immunol. 2005; 285: 109–138. [DOI] [PubMed] [Google Scholar]
- 48. Greber UF, Puntener D. DNA‐tumor virus entry‐‐from plasma membrane to the nucleus. Semin. Cell Dev. Biol. 2009; 20: 631–642. [DOI] [PubMed] [Google Scholar]
- 49. Whittaker GR. Virus nuclear import. Adv. Drug Deliv. Rev. 2003; 55: 733–747. [DOI] [PubMed] [Google Scholar]
- 50. Cohen S, Au S, Pante N. How viruses access the nucleus. Biochim. Biophys. Acta 1813; 2011: 1634–1645. [DOI] [PubMed] [Google Scholar]
- 51. Fominaya J, Wels W. Target cell‐specific DNA transfer mediated by a chimeric multidomain protein. Novel non‐viral gene delivery system. J. Biol. Chem. 1996; 271: 10560–10568. [DOI] [PubMed] [Google Scholar]
- 52. Leopold PL, Ferris B, Grinberg I, Worgall S, Hackett NR, Crystal RG. Fluorescent virions: dynamic tracking of the pathway of adenoviral gene transfer vectors in living cells. Hum. Gene Ther. 1998; 9: 367–378. [DOI] [PubMed] [Google Scholar]
- 53. Greber UF, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993; 75: 477–486. [DOI] [PubMed] [Google Scholar]
- 54. Prongidi‐Fix L, Sugawara M, Bertani P, Raya J, Leborgne C, Kichler A, Bechinger B. Self‐promoted cellular uptake of peptide/DNA transfection complexes. Biochemistry 2007; 46: 11253–11262. [DOI] [PubMed] [Google Scholar]
- 55. Bechinger B, Vidovic V, Bertani P, Kichler A. A new family of peptide‐nucleic acid nanostructures with potent transfection activities. J. Pept. Sci. 2011; 17: 88–93. [DOI] [PubMed] [Google Scholar]
- 56. Plank C, Zauner W, Wagner E. Application of membrane‐active peptides for drug and gene delivery across cellular membranes. Adv. Drug Deliv. Rev. 1998; 34: 21–35. [DOI] [PubMed] [Google Scholar]
- 57. Funhoff AM, van Nostrum CF, Lok MC, Fretz MM, Crommelin DJ, Hennink WE. Poly(3‐guanidinopropyl methacrylate): a novel cationic polymer for gene delivery. Bioconjug. Chem. 2004; 15: 1212–1220. [DOI] [PubMed] [Google Scholar]
- 58. Jiang X, Lok MC, Hennink WE. Degradable‐brushed pHEMA‐pDMAEMA synthesized via ATRP and click chemistry for gene delivery. Bioconjug. Chem. 2007; 18: 2077–2084. [DOI] [PubMed] [Google Scholar]
- 59. Deshayes S, Gerbal‐Chaloin S, Morris MC, Aldrian‐Herrada G, Charnet P, Divita G, Heitz F. On the mechanism of non‐endosomial peptide‐mediated cellular delivery of nucleic acids. Biochim. Biophys. Acta 2004; 1667: 141–147. [DOI] [PubMed] [Google Scholar]
- 60. Deshayes S, Morris M, Heitz F, Divita G. Delivery of proteins and nucleic acids using a non‐covalent peptide‐based strategy. Adv. Drug Deliv. Rev. 2008; 60: 537–547. [DOI] [PubMed] [Google Scholar]
- 61. Mastrobattista E, Koning GA, van Bloois L, Filipe AC, Jiskoot W, Storm G. Functional characterization of an endosome‐disruptive peptide and its application in cytosolic delivery of immunoliposome‐entrapped proteins. J. Biol. Chem. 2002; 277: 27135–27143. [DOI] [PubMed] [Google Scholar]
- 62. Fretz MM, Mastrobattista E, Koning GA, Jiskoot W, Storm G. Strategies for cytosolic delivery of liposomal macromolecules. Int. J. Pharm. 2005; 298: 305–309. [DOI] [PubMed] [Google Scholar]
- 63. Galdiero S, Falanga A, Vitiello G, Vitiello M, Pedone C, D'Errico G, Galdiero M. Role of membranotropic sequences from herpes simplex virus type I glycoproteins B and H in the fusion process. Biochim. Biophys. Acta 2010; 1798: 579–591. [DOI] [PubMed] [Google Scholar]
- 64. Galdiero S, Falanga A, Vitiello M, Browne H, Pedone C, Galdiero M. Fusogenic domains in herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 2005; 280: 28632–28643. [DOI] [PubMed] [Google Scholar]
- 65. Galdiero S, Falanga A, Vitiello M, Raiola L, Fattorusso R, Browne H, Pedone C, Isernia C, Galdiero M. Analysis of a membrane interacting region of herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 2008; 283: 29993–30009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Galdiero S, Falanga A, Vitiello M, Raiola L, Russo L, Pedone C, Isernia C, Galdiero M. The presence of a single N‐terminal histidine residue enhances the fusogenic properties of a membranotropic peptide derived from herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 2010; 285: 17123–17136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Falanga A, Vitiello MT, Cantisani M, Tarallo R, Guarnieri D, Mignogna E, Netti P, Pedone C, Galdiero M, Galdiero S. A peptide derived from herpes simplex virus type 1 glycoprotein H: membrane translocation and applications to the delivery of quantum dots. Nanomedicine 2011; 7: 925–934. [DOI] [PubMed] [Google Scholar]
- 68. Falanga A, Vitiello MT, Cantisani M, Tarallo R, Guarnieri D, Mignogna E, Netti P, Pedone C, Galdiero M, Galdiero S. A peptide derived from herpes simplex virus type 1 glycoprotein H: membrane translocation and applications to the delivery of quantum dots. Nanomed. Nanotechnol. 2011; 7: 925–934. [DOI] [PubMed] [Google Scholar]
- 69. Tu Y, Kim JS. A fusogenic segment of glycoprotein H from herpes simplex virus enhances transfection efficiency of cationic liposomes. J. Gene Med. 2008; 10: 646–654. [DOI] [PubMed] [Google Scholar]
- 70. Smaldone G, Falanga A, Capasso D, Guarnieri D, Correale S, Galdiero M, Netti PA, Zollo M, Galdiero S, Di Gaetano S, Pedone E. gH625 is a viral derived peptide for effective delivery of intrinsically disordered proteins. Int. J. Nanomedicine 2013; 8: 2555–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bao G, Mitragotri S, Tong S. Multifunctional nanoparticles for drug delivery and molecular imaging. Annu. Rev. Biomed. Eng. 2013; 15: 253–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wong IY, Bhatia SN, Toner M. Nanotechnology: emerging tools for biology and medicine. Genes Dev. 2013; 27: 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kiriyama A, Iga K, Shibata N. Availability of polymeric nanoparticles for specific enhanced and targeted drug delivery. Ther. Deliv. 2013; 4: 1261–1278. [DOI] [PubMed] [Google Scholar]
- 74. Ng KK, Lovell JF, Zheng G. Lipoprotein‐inspired nanoparticles for cancer theranostics. Acc. Chem. Res. 2011; 44: 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ryu JH, Koo H, Sun IC, Yuk SH, Choi K, Kim K, Kwon IC. Tumor‐targeting multi‐functional nanoparticles for theragnosis: new paradigm for cancer therapy. Adv. Drug Deliv. Rev. 2012; 64: 1447–1458. [DOI] [PubMed] [Google Scholar]
- 76. Arias JL. Advanced methodologies to formulate nanotheragnostic agents for combined drug delivery and imaging. Expert Opin. Drug Deliv. 2011; 8: 1589–1608. [DOI] [PubMed] [Google Scholar]
- 77. Falanga A, Tarallo R, Galdiero E, Cantisani M, Galdiero M, Galdiero S. Review of a viral peptide nanosystem for intracellular delivery. J. Nanophotonics 2013; 7: 071599–071599. [Google Scholar]
- 78. Gregoriadis G, Ryman BE. Lysosomal localization of ‐fructofuranosidase‐containing liposomes injected into rats. Biochem. J. 1972; 129: 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cukierman E, Khan DR. The benefits and challenges associated with the use of drug delivery systems in cancer therapy. Biochem. Pharmacol. 2010; 80: 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Papisov MI. Theoretical considerations of RES‐avoiding liposomes: molecular mechanics and chemistry of liposome interactions. Adv. Drug Deliv. Rev. 1998; 32: 119–138. [DOI] [PubMed] [Google Scholar]
- 81. Miller CR, Bondurant B, McLean SD, McGovern KA, O'Brien DF. Liposome‐cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry 1998; 37: 12875–12883. [DOI] [PubMed] [Google Scholar]
- 82. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 2000; 65: 271–284. [DOI] [PubMed] [Google Scholar]
- 83. Tarallo R, Accardo A, Falanga A, Guarnieri D, Vitiello G, Netti P, D'Errico G, Morelli G, Galdiero S. Clickable functionalization of liposomes with the gH625 peptide from Herpes simplex virus type I for intracellular drug delivery. Chemistry 2011; 17: 12659–12668. [DOI] [PubMed] [Google Scholar]
- 84. Albrizio S, Giusti L, D'Errico G, Esposito C, Porchia F, Caliendo G, Novellino E, Mazzoni MR, Rovero P, D'Ursi AM. Driving forces in the delivery of penetratin conjugated G protein fragment. J. Med. Chem. 2007; 50: 1458–1464. [DOI] [PubMed] [Google Scholar]
- 85. Ma N, Ma C, Deng Y, Wang T, He N. Advances in applications of dendritic compounds. J. Nanosci. Nanotechnol. 2013; 13: 33–39. [DOI] [PubMed] [Google Scholar]
- 86. Lo ST, Kumar A, Hsieh JT, Sun X. Dendrimer nanoscaffolds for potential theranostics of prostate cancer with a focus on radiochemistry. Mol. Pharm. 2013; 10: 793–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Carberry TP, Tarallo R, Falanga A, Finamore E, Galdiero M, Weck M, Galdiero S. Dendrimer functionalization with a membrane‐interacting domain of herpes simplex virus type 1: towards intracellular delivery. Chemistry 2012; 18: 13678–13685. [DOI] [PubMed] [Google Scholar]
- 88. Guarnieri D, Falanga A, Muscetti O, Tarallo R, Fusco S, Galdiero M, Galdiero S, Netti PA. Shuttle‐mediated nanoparticle delivery to the blood‐brain barrier. Small 2013; 9: 853–862. [DOI] [PubMed] [Google Scholar]
- 89. Acharya A. Luminescent magnetic quantum dots for in vitro/in vivo imaging and applications in therapeutics. J. Nanosci. Nanotechnol. 2013; 13: 3753–3768. [DOI] [PubMed] [Google Scholar]
- 90. Loomba L, Scarabelli T. Metallic nanoparticles and their medicinal potential. Part II: aluminosilicates, nanobiomagnets, quantum dots and cochleates. Ther. Deliv. 2013; 4: 1179–1196. [DOI] [PubMed] [Google Scholar]
- 91. Geszke‐Moritz M, Moritz M. Quantum dots as versatile probes in medical sciences: synthesis, modification and properties. Mater. Sci. Eng. C, Mater. Biol. Appl. 2013; 33: 1008–1021. [DOI] [PubMed] [Google Scholar]
- 92. Cheki M, Moslehi M, Assadi M. Marvelous applications of quantum dots. Eur. Rev. Med. Pharmacol. Sci. 2013; 17: 1141–1148. [PubMed] [Google Scholar]
- 93. Glover DJ, Ng SM, Mechler A, Martin LL, Jans DA. Multifunctional protein nanocarriers for targeted nuclear gene delivery in nondividing cells. FASEB J. 2009; 23: 2996–3006. [DOI] [PubMed] [Google Scholar]
- 94. Favaro MT, de Toledo MA, Alves RF, Santos CA, Beloti LL, Janissen R, de la Torre LG, Souza AP, Azzoni AR. Development of a non‐viral gene delivery vector based on the dynein light chain Rp3 and the TAT peptide. J. Biotechnol. 2014; 173: 10–18. [DOI] [PubMed] [Google Scholar]
- 95. Hatakeyama H, Ito E, Akita H, Oishi M, Nagasaki Y, Futaki S, Harashima H. A pH‐sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA‐containing nanoparticles in vitro and in vivo . J. Control. Release 2009; 139: 127–132. [DOI] [PubMed] [Google Scholar]
- 96. El‐Sayed A, Masuda T, Khalil I, Akita H, Harashima H. Enhanced gene expression by a novel stearylated INF7 peptide derivative through fusion independent endosomal escape. J. Control. Release 2009; 138: 160–167. [DOI] [PubMed] [Google Scholar]
- 97. Zhao Q, Li S, Yu H, Xia N, Modis Y. Virus‐like particle‐based human vaccines: quality assessment based on structural and functional properties. Trends Biotechnol. 2013; 31: 654–663. [DOI] [PubMed] [Google Scholar]
- 98. Rodriguez‐Limas WA, Sekar K, Tyo KE. Virus‐like particles: the future of microbial factories and cell‐free systems as platforms for vaccine development. Curr. Opin. Biotechnol. 2013; 24: 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li F, Wang Q. Fabrication of nanoarchitectures templated by virus‐based nanoparticles: strategies and applications. SMALL 2014; 10: 230–245. [DOI] [PubMed] [Google Scholar]
- 100. Ma Y, Nolte RJ, Cornelissen JJ. Virus‐based nanocarriers for drug delivery. Adv. Drug Deliv. Rev. 2012; 64: 811–825. [DOI] [PubMed] [Google Scholar]
- 101. Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV‐DOX delivers. Mol. Pharm. 2013; 10: 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lockney DM, Guenther RN, Loo L, Overton W, Antonelli R, Clark J, Hu M, Luft C, Lommel SA, Franzen S. The Red clover necrotic mosaic virus capsid as a multifunctional cell targeting plant viral nanoparticle. Bioconjug. Chem. 2011; 22: 67–73. [DOI] [PubMed] [Google Scholar]
- 103. Ren Y, Wong SM, Lim LY. Folic acid‐conjugated protein cages of a plant virus: a novel delivery platform for doxorubicin. Bioconjug. Chem. 2007; 18: 836–843. [DOI] [PubMed] [Google Scholar]
- 104. Kaiser CR, Flenniken ML, Gillitzer E, Harmsen AL, Harmsen AG, Jutila MA, Douglas T, Young MJ. Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo . Int. J. Nanomedicine 2007; 2: 715–733. [PMC free article] [PubMed] [Google Scholar]
- 105. Lee D‐E, Kim A, Saravanakumar G, Koo H, Kwon I, Choi K, Park J, Kim K. Hyaluronidase‐sensitive SPIONs for MR/optical dual imaging nanoprobes. Macromol. Res. 2011; 19: 861–867. [Google Scholar]
- 106. Ohtake N, Niikura K, Suzuki T, Nagakawa K, Mikuni S, Matsuo Y, Kinjo M, Sawa H, Ijiro K. Low pH‐triggered model drug molecule release from virus‐like particles. ChemBioChem 2010; 11: 959–962. [DOI] [PubMed] [Google Scholar]
- 107. Zochowska M, Paca A, Schoehn G, Andrieu JP, Chroboczek J, Dublet B, Szolajska E. Adenovirus dodecahedron, as a drug delivery vector. PLoS One 2009; 4: e5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ashley CE, Carnes EC, Phillips GK, Durfee PN, Buley MD, Lino CA, Padilla DP, Phillips B, Carter MB, Willman CL, Brinker CJ, Caldeira Jdo C, Chackerian B, Wharton W, Peabody DS. Cell‐specific delivery of diverse cargos by bacteriophage MS2 virus‐like particles. ACS Nano 2011; 5: 5729–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Qazi S, Liepold LO, Abedin MJ, Johnson B, Prevelige P, Frank JA, Douglas T. P22 viral capsids as nanocomposite high‐relaxivity MRI contrast agents. Mol. Pharm. 2013; 10: 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pokorski JK, Hovlid ML, Finn MG. Cell targeting with hybrid Qβ virus‐like particles displaying epidermal growth factor. ChemBioChem 2011; 12: 2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ksendzovsky A, Walbridge S, Saunders RC, Asthagiri AR, Heiss JD, Lonser RR. Convection‐enhanced delivery of M13 bacteriophage to the brain. J. Neurosurg. 2012; 117: 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]