

Abstract
Apart from direct detection of the infecting organisms or biomarker of the pathogen itself, surrogate host markers are also useful for sensitive and early diagnosis of pathogenic infections. Early detection of pathogenic infections, discrimination among closely related diseases with overlapping clinical manifestations, and monitoring of disease progression can be achieved by analyzing blood biomarkers. Therefore, over the last decade large numbers of proteomics studies have been conducted to identify differentially expressed human serum/plasma proteins in different infectious diseases with the intent of discovering novel potential diagnostic/prognostic biomarkers. However, in‐depth review of the literature indicates that many reported biomarkers are altered in the same way in multiple infectious diseases, regardless of the type of infection. This might be a consequence of generic acute phase reactions, while the uniquely modulated candidates in different pathogenic infections could be indicators of some specific responses. In this review article, we will provide a comprehensive analysis of differentially expressed serum/plasma proteins in various infectious diseases and categorize the protein markers associated with generic or specific responses. The challenges associated with the discovery, validation, and translational phases of serum/plasma biomarker establishment are also discussed.
Keywords: Acute phase proteins, Biomarkers, Infectious diseases, Plasma, Serum
Abbreviations
- AIDS
acquired immunodeficiency syndrome
- HUPO
Human Proteome Organisation
- PPP
plasma proteome project
- PSI
proteomics standards initiative
- SARS
severe acute respiratory syndrome
1. Introduction
The spectacular advancements in proteomics research, which have been achieved during the preceding decade have propelled its expansion into diverse fields of clinical research 1. While the field of personalized medicine and targeted therapeutics is gaining popularity, integrative personal omics profiling is becoming an attractive choice for molecular diagnostics and therapeutics 2. The comprehensive proteomic analyses of different biological fluids, particularly serum and plasma, have attracted substantial attention for the identification of protein biomarkers as early detection surrogates for human diseases 3. Infectious diseases directly contribute over 25% of the total annual deaths worldwide. The number increases when deaths that occur as a consequence of past infections or due to the complications associated with chronic infections are included 4. Existing, re‐emerging, and newly emerged infectious diseases are considered to be the leading global public health problems and one of the major causes of disease‐related casualties, particularly in developing countries 5, 6.
Even though detection of the presence of causative organisms or pathogen‐related markers and examination of clinical symptoms are generally used for routine diagnosis of infectious diseases, sensitive and early diagnosis of pathogenic infections is also achievable through the analysis of surrogate host markers 7. Additionally, investigation of pathogen‐induced alterations in the host proteome under diseased conditions can provide valuable information regarding disease pathogenesis and host immune responses 8. In order to sustain viability within the host system and ensure their replication, pathogens develop versatile mechanisms to exploit their host's cells and to induce new permeability pathways to allow the uptake of nutrients and the removal of waste products 9. This significantly affects various vital physiological processes and, as a consequence, modulates the host's proteome. Upon infection, a continuous interaction between the pathogen and the host immune system initiates a complex immune response to hold back the pathogenic infection and growth through multiple antiparasitic effector functions, including inhibition of invasion and cytoadherence, antibody‐dependent cytotoxicity, and cellular inhibition.
Serum/plasma proteins can be used for early diagnosis, differentiation between infections with very similar etiology and clinical manifestations, as well as for monitoring disease progression, since many components of the blood proteome often exhibit alterations in expression level as a consequence of external pathogenic infections and show excellent correlation with disease progression/severity 10, 11. Large numbers of serum/plasma proteomics studies aimed at the identification of novel potential diagnostic/prognostic biomarkers or understanding disease pathobiology have been reported during the past decade 3. However, altered expression levels of potential biomarkers have often been found to be nonspecific and are altered in multiple infectious diseases, irrespective of the type of infection. This might be an effect of inflammation‐mediated acute phase response signaling 12. Our research group is investigating the alterations in the human serum proteome in different infectious diseases, including falciparum and vivax malaria 13, 14, dengue fever 15, and leptospirosis 16, to identify the common and unique protein signatures. Moreover, we have compiled a list of differentially expressed serum proteins in ten different types of protozoan, viral, and bacterial infectious diseases that are often fatal, including malaria, dengue, meningitis, acquired immunodeficiency syndrome (AIDS), severe acute respiratory syndrome (SARS), diarrheas, hepatitis B and C, tuberculosis, pneumonia, and leptospirosis, from over 200 published studies conducted using proteomics and immunoassay‐based approaches. This review article intends to provide an inclusive representation of overlapping or specific protein alterations in human serum/plasma proteome due to various infectious diseases. The challenges and critical issues associated with different phases of serum/plasma biomarker discovery ventures are also discussed.
2. Serum/plasma biomarkers in different infectious diseases
The analysis of alterations in the human serum/plasma proteome as a consequence of pathogenic infections is certainly informative for researchers studying disease pathogenesis and host immune responses or interested in identifying diagnostic or prognostic host markers 3. Biomarkers are indicator biomolecules that assist in early diagnosis, discriminate between different diseases, and provide valuable tools for monitoring the progression/severity of disease. Early detection of infectious diseases could reduce the complications of secondary infections, fatalities due to disease severity, and also the unnecessary costs of improper and delayed diagnosis and treatment. Although existing diagnostic approaches (including analysis of clinical symptoms, microscopic detection of the causative pathogenic organism, molecular and immunologic diagnostic methods, and rapid diagnostic tests), which are usually implemented clinically, are adequately robust and sensitive for detection of the pathogen in symptomatic patients, the sensitivity is lower in asymptomatic subjects or in patients at a very early stage of infection (Table 1). Furthermore, existing routine detection techniques are unable to provide any prognostic information regarding the infection, or to clearly discriminate among infections that have overlapping clinical manifestations. To this end, protein markers are potential candidates for the development of alternative more‐sensitive diagnostic and prognostic approaches for infectious diseases. Identification of a panel of biomarkers might be attractive for detection of specific infections, as well as for discrimination among nearly similar identical manifestations.
Table 1.
Challenges associated with the routine diagnostic methods for different infectious diseases
| Disease (causative organism)a | Diagnostic methodsb | Challenges/limitations |
|---|---|---|
| 1. Malaria (Parasitic protozoan)P. falciparum and P. vivax |
|
|
| 2. Dengue and dengue hemorrhagic fever (DHF)(Viral)Serotypes of dengue virus (DENV 1–4) |
|
|
| 3. Meningitis (Bacterial)Neisseria meningitidis and Streptococcus pneumoniae(Viral)Enteroviruses, herpes simplex virus type 2/1, Varicella zoster virus |
|
|
| 4. Acquired immunodeficiency syndrome (AIDS)(Viral)Human immune deficiency virus (HIV) |
|
|
| 5. Severe acute respiratory syndrome (SARS)(Viral)Member of the Coronavirus family |
|
|
| 6. Diarrhea (bacterial)E. coli, Shigella, Salmonella, Campylobacter(Viral)Rotavirus, Norovirus(Protozoan)Giardia lamblia |
|
|
| 7. Hepatitis A, B, and C(Viral)Hepatitis A/B/C virus (HAV/HBV/HCV) |
|
|
| 8. Tuberculosis(Mycobacterial)M. tuberculosis |
|
|
| 9. Pneumonia(Bacterial)Streptococcus Haemophilus Chlamydophila(Viral)Rhinoviruses, Coronaviruses, Influenza virus |
|
|
| 10. Leptospirosis(Bacterial)Spirochete of genus Leptospira |
|
|
CT, computed tomography; MRI, magnetic resonance imaging; CSF, cerebrospinal fluid.
If an infection is caused by multiple different microorganisms, major causal pathogens are listed.
Clinical manifestations, that is, signs and symptoms of the diseases (not discussed here) are also studied for diagnosis.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The use of serum or plasma as biological fluid for studying disease pathobiology and identification of biomarkers have their own pros and cons. Plasma is more easily separable from the cellular components of the blood and a slightly higher volume (10–15%) of plasma is generally obtained from an equal quantity of whole blood compared to serum. Furthermore, a plasma sample can be obtained quickly since no coagulation time is required and coagulation‐induced interferences/variations can be avoided. In contrast, due to the absence of fibrinogen and other blood clotting factors in serum, the total protein content and complexity is reduced compared to plasma. According to a recent study by Zimmerman et al. involving paired comparison of plasma and serum samples from the same individuals, there were negligible differences in the numbers of peptide and protein identifications or in the overall percentages of semitryptic peptides or methionine oxidized peptides between these two biological fluids, although higher variability of semitryptic peptides were observed in serum 17. In particular, the variations in serum samples are introduced due to the inconsistency in the coagulation and separation processes. A comparative analysis of human serum and EDTA, heparin, and citrate‐anticoagulated plasma conducted by Human Proteome Organisation (HUPO) clearly indicated that plasma with EDTA (or citrate) anticoagulant provides more reproducible results than serum, and therefore plasma has been recommended as a more attractive biological fluid for proteomics research 18.
The different steps involved in the standard work‐flow of proteomics‐based serum/plasma biomarker discovery are shown in Fig. 1. After sample collection, protein extraction and processing involve multiple steps, often including depletion of high‐abundance proteins and prefractionation prior to the actual proteomic analysis due to the complexity of serum/plasma samples and the wide dynamic range of protein concentration 19, 20, but this can lead to unintended removal of proteins bound to the protein targeted for removal. Different gel‐, MS‐, and array‐based techniques can be applied for comparative proteomic profiling, while the results obtained in the discovery phase are usually validated using immunoassay‐based approaches. Readers are directed to the protocols in the specific articles referred to in Fig. 1 for the technological details and working principles of the different proteomic approaches shown 21, 22, 23, 24. Finally, the specificity, sensitivity, and predictive accuracies of the identified potential marker proteins are evaluated by receiver operating characteristic (ROC) curves and multivariate statistical analyses.
Figure 1.
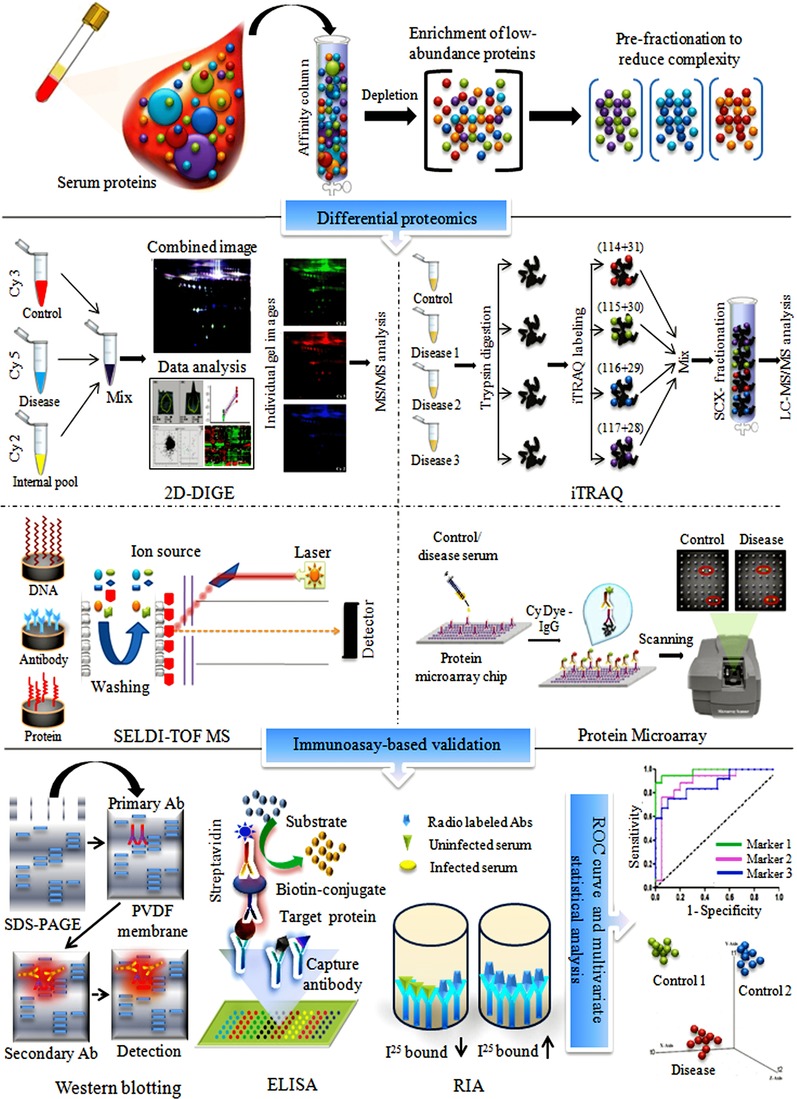
Standard work flow for different proteomics approaches commonly used in serum/plasma biomarker discovery. Prior to proteomic analysis, depletion of high‐abundance proteins, and prefractionation of the overall proteome are performed to reduce the complexity and dynamic range of protein concentration in serum/plasma samples. In order to perform comparative proteomic profiling of control and diseased samples, a variety of gel‐based, MS‐based, and array‐based techniques can be used. Results obtained in the initial discovery phase are usually validated with immunoassay‐based approaches, such as ELISA or Western blotting. Subsequently, ROC curve and multivariate statistical analysis are performed to determine the specificity and sensitivity and class prediction accuracy of the identified potential marker proteins.
We have performed a comprehensive analysis of the published literature in order to evaluate the overlapping and unique signatures of serum/plasma markers in ten often‐fatal protozoan, viral, and bacterial infectious diseases, including malaria, dengue, meningitis, AIDS, SARS, diarrheas, hepatitis B and C, tuberculosis, pneumonia, and leptospirosis. Experimental details (e.g., sample type, population size, type of controls, technologies employed in the discovery and validation phases of the study, and the important identified differentially expressed proteins) of many selected proteomics or immunoassay‐based studies on these infectious diseases have been provided in Table 2. Critical analysis of the published literature from various research groups clearly indicates that the expression levels of many reported potential biomarkers are modulated similarly in several infectious diseases, irrespective of the type of the pathogen, this might be a result of generic acute‐phase/stress reactions, while the specifically modulated candidates in different pathogenic infections could be indicators of some unique responses. Differential expression profiles of a few selected serum proteins in different infectious diseases are shown in Fig. 2. Serum/plasma proteins, which show opposite trends of differential expression in some infections compared to other types of infectious diseases, are promising candidates for diagnosis and discrimination analysis. Interestingly, serum levels of haptoglobin (Hp) were found to be significantly lower in malaria patients (both Plasmodium vivax and Plasmodium falciparum infection). Hp, being a positive acute phase protein, exhibits upregulation in other infectious diseases (Fig. 2; Supporting Information Table 1). Hp sequesters free hemoglobin (Hb), which is released during pathogen‐induced hemolysis as Hp–Hb complexes and leads to hypo‐ or ahaptoglobinemia. Hp is a promising inflammatory marker for evaluation of the severity of the Plasmodium infection and is a potential epidemiological marker for malaria 25, 26. Additionally, our comparative analysis of various infectious diseases indicates that even though some proteins, such as SAA or CRP, exhibit similar trends of differential expression (upregulation) in multiple infections, the levels of regulation (fold changes) are found to be quite different depending on the type of infectious agent, so these candidates could be effective classifiers for discriminating among different infectious manifestations (Fig. 2). It has been shown that establishment of a panel of serum biomarkers rather than a single candidate is much more effective for detection of specific infection and its discrimination from the other clinically related febrile illnesses 13. In our proteomic analyses, we have found that a panel of identified proteins consisting of six candidates (serum amyloid A, hemopexin, apolipoprotein E, haptoglobin, retinol‐binding protein, and apolipoprotein A‐I) can discriminate among malaria, dengue, and leptospirosis 13, 15, 16. Although these six proteins were not unique for malaria, dengue, or leptospirosis, the combination of their specific trends and levels of differential expression allowed the successful discrimination among these three infectious diseases having overlapping clinical manifestations. Within a panel, if the individual class prediction potential of the candidate markers is evaluated, it becomes apparent that each of them plays some specific contribution in discrimination of a particular infection from the healthy population and other related clinical manifestations. Therefore, we feel that the combination of clinicopathological parameters with serum/plasma markers can provide improved prediction accuracy for most diseases.
Table 2.
Differential expressions of serum/plasma proteins in different infectious diseasesa)
| Disease | Purpose of the study | Sample type and size | Technological details (discovery (1) and validation (2) phases) | Identified differentiallyexpressed candidates(Regulation)b) | Ref |
|---|---|---|---|---|---|
| 1. Malaria | Analysis of disease pathogenesis and host immune response and identification of protein markers for FM and VM | SerumFM: n = 20, VM: n = 17, HC: n = 20, FC (L): n = 6 |
|
Serum amyloid A (U), hemopexin (U), apolipoprotein E (U), haptoglobin (D), retinol‐binding protein (D), apolipoprotein A‐I (D) | 13, 14 |
| Identification of inflammation‐related biomarkers of FM | SerumSevere FM: n = 8, mild FM: n = 8, HC: n = 8 |
|
Serum amyloid A (U), apolipoprotein E (U), LPS‐binding protein (U), gelsolin (D), fibrinogen (U), clusterin (D) | 100 | |
| Proteomic analysis of haptoglobin and amyloid A protein levels in VM | PlasmaNAc) |
|
Serum amyloid A (U), Haptoglobin (D) | 101 | |
| Analysis of consequence of hemolysis in FM | PlasmaFM: n = 18, HC: n = 11, FC (pneumonia): n = 6 |
|
Gelsolin (D) | 102 | |
| 2. Dengue | Analysis of serum proteome and cytokine profiles in early febrile, defervescence, and convalescent stages of DF and DHF | SerumDF: n = 44, DHF: n = 18, HC: n = 50 |
|
Serum amyloid A2 (U), Haptoglobin (U), apolipoprotein E (U), hemopexin (U), plasma protease C1 inhibitor (U), clusterin (U), apolipoprotein CI (D), apolipoprotein CIV (D) | 47 |
| Comparative analysis of plasma from DF and HC | PlasmaDF: n = 13, HC: n = 13 |
|
C1 inhibitor (U), vitamin D‐binding protein (U), fibrinogen γ chain (U), apolipoprotein J (U), complement component C3c (U), prothrombin (D), histidine‐rich glycoprotein (D), apolipoprotein A‐IV & A‐I (D), transthyretin (D), complement C3b (D) | 103 | |
| Analysis of disease pathogenesis and identification of surrogate protein markers for DF | SerumDF: n = 6, HC: n = 8, FC (FM) : n = 8 |
|
Serum amyloid P (U), kininogen (D), complement C3 (D), C4 (U) & H (U), apolipoprotein A‐IV (D). hemopexin (D), protein C6 (U), clusterin (U) | 15 | |
| Comparative analysis of acute severe dengue (DHF) and acute nonsevere dengue (DF) | PlasmaDF: n = 5, DHF: n = 5 |
|
Leucine‐rich glycoprotein 1 (U), vitamin D binding protein (U), ferritin (U), peroxyredoxin‐2 (D), afamin (U), fibronectin (U), galectin 3 binding protein (U), C‐reactive protein (U) | 104 | |
| Identification of serum biomarkers of DF and DHF | SerumDF: n = 10, DHF: n = 10, HC: n = 8 |
|
α1‐Antitrypsin (U), NS1 protein (U) | 105 | |
| 3. Meningitis | Analysis of APPs in BM | Serum and CSFBM: n = 30,ViM: n = 30, HC: n = 100 | Immunoassay | C‐reactive protein (U), α‐1‐antitrypsin (U), α‐1‐acidgycoprotein (U), α‐2‐ceruloplasmin (U), α‐2‐haptoglobin (U) | 106 |
| Identification of surrogate markers for diagnosis of BM | Serum and CSFBM: n = 28, ViM: n = 25, HC: n = 27 | (1) 2D‐DIGEMALDI‐TOF/MS (2) WB, ELISA | Prostaglandin‐H2 d‐isomerase (U), haptoglobin (D), fibulin‐1 (U), fibrinogen beta chain (D), apolipoprotein E (U), GFAP (U) | 107 | |
| 4. AIDS | Analysis of APPs as systemic antiviral response in HIV‐1 infection | PlasmaAIDS: n = 19, HC: n = 5 (longitudinal study) |
|
Serum amyloid A (U), complement C3, apolipoproteins, C‐reactive protein, virus inhibitory peptide (VIRIP) (U) | 108 |
| Investigation of different isoforms of apolipoprotein AI in AIDS | PlasmaHIV: n = 10, HC: n = 10 |
|
ALB (U), haptoglobin β chain (U), immunoglobulin light chain (U), haptoglobin α 2 chain (U), transthyretin (U), apolipoprotein AI (D) | 109 | |
| Identification of serum markers of HIV‐1 latently infected LTNP AIDS | SerumHIV: n = 6, LTNP: n = 6, HC: n = 6 |
|
HIV‐1 enhancer binding protein 1 (U), ribonuclease III (U), heterochromatin protein 1 binding protein (U) | 110 | |
| 5. SARS | Identification of diagnostic and prognostic markers of SARS | SerumSARS: n = 39Non‐SARS: n = 39 | SELDI‐MS | Fibrinogen α‐E chain (D), platelet factor 4 (D), β‐thromboglobin (U), IgG Kappa light chain (U), N‐terminal fragment of complement C3c (U) | 111 |
| Analysis of inflammation inhibitors in SARS | PlasmaProgressive and convalescent SARS: n = 10 (each), HC: n = 9 |
|
α1‐Acid glycoprotein (U), haptoglobin (β and alpha‐2 chain) (U), fetuin (U), transthyretin (D), apolipoprotein A‐I (D), transferrin (D) | 112 | |
| Discovery of serum biomarkers for SARS | SerumSARS: n = 13, HC: n = 14, FC (pneumonia): n = 12 |
|
TF‐α 1‐AT (U), complement C4 fragments (U), serum amyloid A (U) | 113 | |
| Analysis of plasma proteome alterations in SARS | PlasmaSARS: n = 22, HC: n = 6 |
|
GSH peroxidise (U), Prx II (U), vitamin D binding protein (U), serum amyloid A (U), complement factor H‐related protein (U), haptoglobin β chain (U) | 114 | |
| 6. Diarrhea | Analysis of expression and release of leptin and proinflammatory cytokines | SerumDiarrhea: n = 30, FC (ulcerative colitis): n = 50 | WB, ELISA | Leptin (U), TNF‐α (U), IL‐1β (U), IL‐6 (U) | 115 |
| Analysis of serum TNF‐α in inflammatory bowel diseases | SerumDiarrhea: n = 46, FC (Crohn disease): n = 54 | ELISA | TNF‐α (U) | 116 | |
| 7. Hepatitis A and B | Analysis of α‐1 antitrypsin level in hepatitis B | SerumChronic HBV: n = 31, acute HBV: n = 10, HBV‐related HCC: n = 18, HC: n = 12 |
|
α‐1‐Antitrypsin (U) | 117 |
| Analysis of plasma gelsolin protein level in hepatitis‐B‐associated liver cirrhosis | PlasmaInactive HBV: n = 8HBV: n = 8 | (1) 2DE, LC‐ESIMS/MS | Gelsolin (D) | 118 | |
| 8.Tuberculosis | Identification of TB‐associated proteins in whole blood supernatant | PlasmaTB: n = 39, HC: n = 63 |
|
Retinol‐binding protein 4 (D), Fetuin‐A (α‐HS‐glycoprotein) (D) | 119 |
| Identification of diagnostic markers for TB | SerumTB: n = 129, FC (respiratory disease): n = 69, HC: n = 66 |
|
Orosomucoid (U) | 120 | |
| Identification of diagnostic markers for TB | SerumTB: n = 179, HC: n = 21 |
|
Serum amyloid A (U), transthyretin (D) C‐reactive protein (U) | 121 | |
| 9. Pneumonia | Analysis of systemic cytokine response in CAP | SerumP: n = 200, HC: n = 313 | ELISA | IL‐1RA, IL‐6, IL‐8, IL‐10 | 122 |
| Analysis of serum cytokines profile in P | SerumP: n = 14, HC: n = 5 | Immunoassay | IFN‐gama (D), IL‐12(U), C‐reactive protein (U) | 123 | |
| 10. Leptospirosis | Analysis of disease pathogenesis and host immune response and identification of surrogate protein markers for L | SerumL: n = 6, FC (FM): n = 8, HC: n = 18 |
|
Apolipoprotein A1 (D), apolipoprotein A‐IV (D), complement C4 (D), α‐1B‐glycoprotein precursor (U) | 16 |
| Analysis of induction of proinflammatory cytokines by leptospiral hemolysins | SerumL: n = 3, HC: n = 3 |
|
Proinflammatory factors (IL‐1b, IL‐6, IL‐17, and TNF‐a) (U), anti‐inflammatory factors (IL‐4, IL‐10, IL‐13, and sTNF RI) (U), immunoregulators (IL‐7, IL‐11, and IFN‐c) (U), colony‐stimulating factors (G‐CSF and GM‐CSF) (U) | 124 | |
| Analysis of serum nitrite levels in L | SerumL: n = 20, HC: n = 13 | ELISA | Serum nitrite (U) | 125 |
AM, aseptic meningitis; APP, acute phase proteins; BM, bacterial meningitis; CAP, community‐acquired pneumonia; DF, dengue fever; DHF, dengue hemorrhagic fever; FM, falciparum malaria; FC, febrile control; H, hepatitis; HC, healthy control; HCC, hepatocellular carcinoma; L, leptospirosis; LTNP, long‐term nonprogressor; P, pneumonia; TB, tuberculosis; ViM, viral meningitis; VM, vivax malaria; WB, Western blotting.
a) Representative studies are shown.
b) U, upregulation and D, downregulation.
c) NA, exact information not available.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Figure 2.
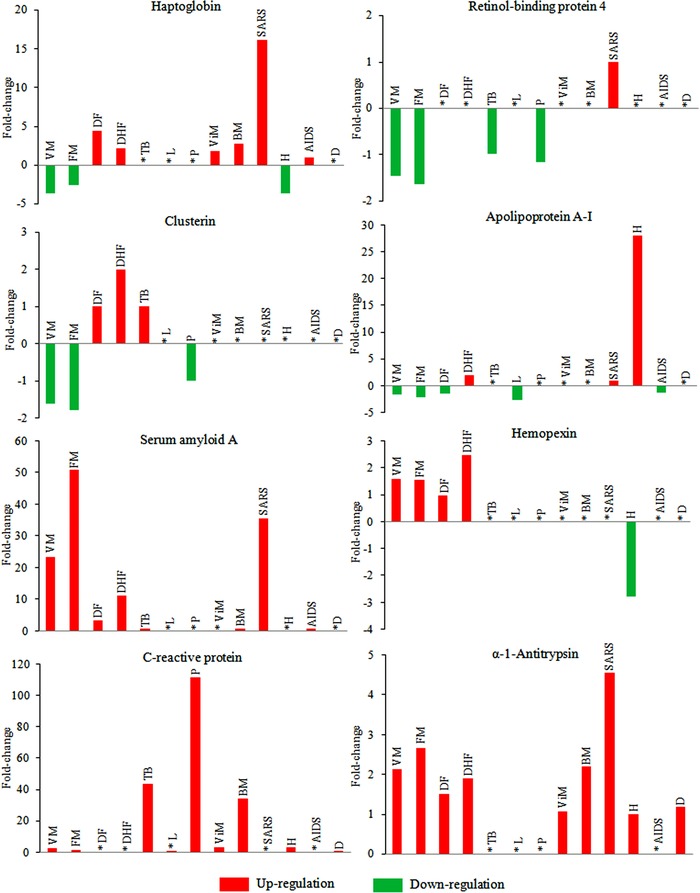
Differential expression of some selected serum/plasma proteins in different infectious diseases. Fold‐change values (up‐/downregulation) of the candidate proteins were obtained from different published studies. If differential expression of any particular protein is reported in multiple studies, representative data are shown. Exact differential expression values for each candidate are provided in the Supporting Information Table 1. Alterations in protein expression levels in different infectious diseases are determined using healthy subjects as controls. Fold‐change values are calculated by keeping the expression level of the proteins (mean value) in healthy population as baseline. *, indicates that the differential expression of that protein is not reported in that particular disease in humans. VM, vivax malaria; FM, falciparum malaria; DF, dengue fever; DHF, dengue hemorrhagic fever; TB, tuberculosis; L, leptospirosis; P, pneumonia; ViM, viral meningitis; BM, bacterial meningitis; SARS, severe acute respiratory syndrome; AIDS, acquired immunodeficiency syndrome; H, hepatitis, D, diarrhea.
Many serum/plasma proteins, such as α‐1‐antichymo‐trypsin, α‐1‐antitrypsin, serotransferrin, serum albumin, α‐2‐HS‐glycoprotein, etc., which exhibit similar trends of differential expression in multiple infections, are basically nonspecific indicators of inflammation or stress response, and are not promising from a diagnostic/prognostic point of view (Fig. 2). Very often, in isolated proteomics studies, where only disease versus healthy controls are compared, differentially expressed candidates are found, but many are simply a consequence of generic acute phase reactions, which are misclaimed as promising markers (Fig. 3A). In real‐life scenarios, biomarker candidates that are commonly altered in multiple infectious diseases cannot effectively discriminate between different clinical manifestations, unless the levels of differential expression of these candidate proteins are significantly different in other infections, or if they are used as part of a marker panel along with other more specific candidates. Other reviews have demonstrated the frequent appearance of HSP27, HSP60, ATP synthase, β‐actin, and enolase 1 as differentially expressed candidates in various published tissue proteomics analyses performed by using 2DE in combination with MS 12, 27. Such universal candidates should probably be excluded from the list of promising targets and may not actually be biomarkers of any specific disease state.
Figure 3.
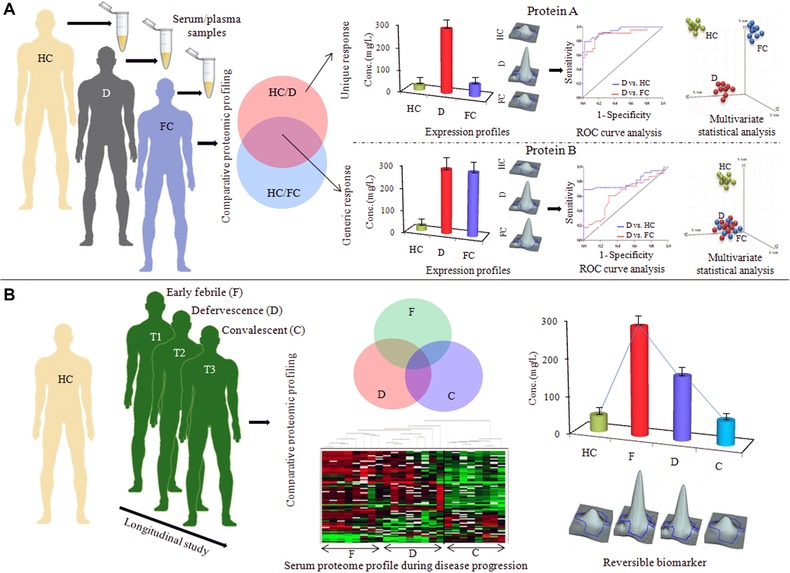
Crucial issues for designing clinical studies for serum/plasma biomarker discovery. (A) Selection of suitable febrile (diseased) controls for evaluating the specificity of the identified markers. Two potential markers (protein A and B) are significantly differentially expressed in an infectious disease population (D) compared to the healthy controls. Between those two candidates, differential expression of protein B is not specific for the disease population (D), it also shows an equal level of altered expression in another closely related infectious disease, which has been used as a febrile control (FC) for the disease D. While the expression level of protein A remained unaltered in the FC population, it showed some extent of specificity toward the disease population D. Downstream analysis of the specificities and sensitivities (ROC curve analysis) and class prediction capabilities of those two potential markers clearly indicates the superiority of the protein A as a potential marker for the disease state D, since it is not only useful in discrimination of disease D from healthy population, but also can successfully differentiate disease D from other closely related clinical manifestations. (B) Analysis of longitudinal cohorts for establishment of prognostic and disease monitoring marker proteins. Information about the reversibility and disease monitoring/prognostic capability of the identified disease surrogates can be obtained from multiple time point analysis (early febrile, defervescence, and convalescent stages of infection).
3. Challenges of biomarker discovery and crucial issues regarding experimental designing
3.1. Challenges associated with proteomics‐based serum/plasma biomarker discovery
Even though serum/plasma proteome analysis has gained considerable attention for investigation of pathogen‐induced alterations in the human host and for the identification of potential diagnostic and prognostic markers for various infectious diseases, there are quite a few basic challenges associated with the discovery, validation, and translational phases of serum/plasma biomarkers, mostly due to variations during the sample collection and processing steps (Fig. 4), PTMs, the presence of multiple isoforms of same proteins, and the sample complexity 3. Preanalytical variations introduced during sample collection, handling, and storage process are also challenging for the determination of true biomarkers 28. In addition, the complexity of biological samples, the very wide dynamic range of protein concentrations in plasma (1010–1012), the presence of high‐abundance proteins masking low‐abundance marker proteins, high levels of salts and other interfering compounds, insufficient sensitivity of the detection technology, and low throughput are the major obstacles for discovery of blood biomarkers. Despite recent technological advancements, there are several limitations of the current proteomics technologies, which are frequently used for the discovery of disease‐related marker proteins in serum/plasma samples 11. Details of the technological limitations associated with different types of proteomics approaches have been discussed elsewhere for gel‐based 29, 30, MS‐based 31, 32, 33, array‐based 34, 35, and label‐free proteomics 36.
Figure 4.
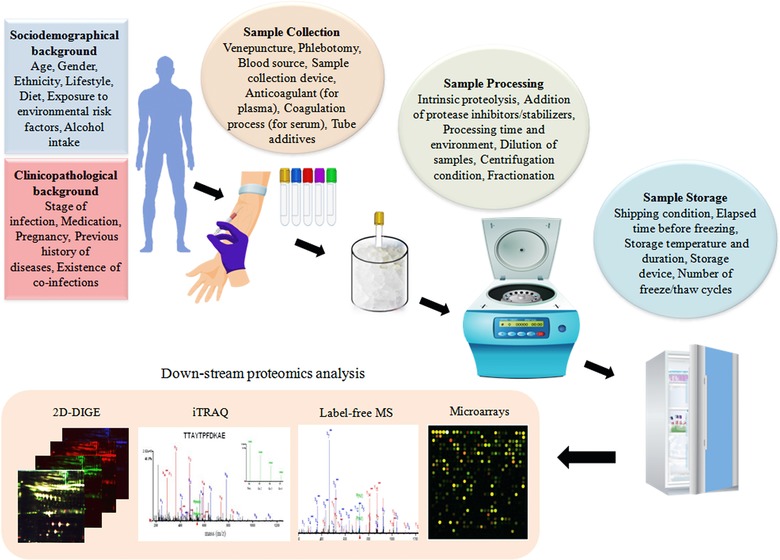
Different sources of preanalytical variability for proteomics biomarker discovery.
3.2. Crucial issues regarding experimental designing for clinical proteomic studies
It is often experienced in proteomic biomarker discovery studies that exciting and promising findings based on smaller populations sizes fail to live up to expectations in the validation stage and subsequent larger clinical trials, in part because of biological variability and lack of reproducibility of the techniques used. In a standard proteomic‐based blood biomarker discovery work‐flow, more than 1000 candidates are screened in the discovery phase often leading to the identification of nearly 100 differentially expressed candidates fulfilling the statistical/fold‐change criteria. Usually, about one tenth of these are confirmed in the validation/follow‐up phase, where targeted analysis is performed with bigger clinical cohorts using alternative technological approaches. Gradually, the number decreases even further during the clinical trials, and ultimately, only a few identified markers are found to have real clinical applicability and receive Food and Drug Administration (FDA) approval (Fig. 5). It should be pointed out that very few biomarkers or biomarker panels have actually been through the verification or validation stages. This is because the techniques used for biomarker discovery are generally not suitable for validation purpose. Antibody‐based methods, such as immunoassay‐based validation (Western blots and ELISA) are too expensive to be used on the huge number of potential candidates identified in the discovery phase and cannot be highly multiplexed 37. To this end, MS‐based targeted proteomic approaches, particularly, SRM (or MRM) are coming to the fore as alternative validation methods 38. In order to acquire results that can be extrapolated across the study population, consideration of a few critical factors at the beginning, when designing the clinical proteomic studies, for biomarker discovery is indispensible.
Figure 5.
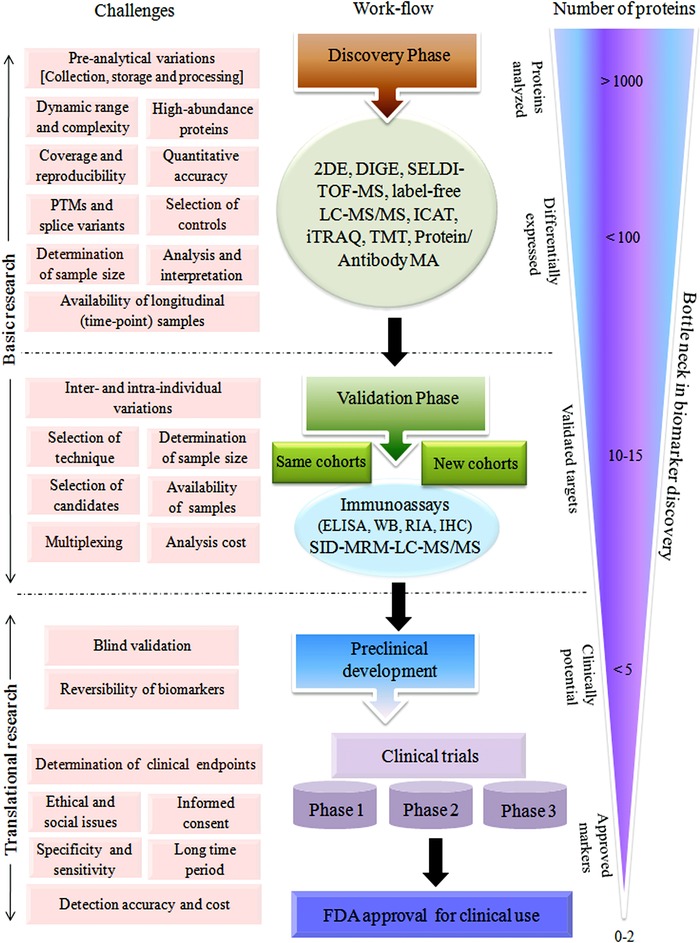
Different types of challenges associated with discovery, validation, and translational phases of serum/plasma biomarker establishment.
3.2.1. Selection of controls
Selection of appropriate controls is very crucial in the discovery phase for identification of indisputable biomarkers for any human disease (Fig. 3A). Frequently, researchers compare specific disease states with healthy subjects as controls, and whatever differentially expressed serum/plasma proteins satisfy, the statistical/fold‐change criteria have been reported as potential biomarkers, regardless of the specificity of the biomarker for that particular disease. Consequently, when various published articles are compared, it becomes evident that the differentially expressed serum/plasma proteins (compared to the healthy controls) identified in isolated studies from different research groups and reported as potential biomarkers for a specific infection might, in fact, be markers of multiple infectious diseases, and thereby cannot be used as reliable diagnostic markers for a specific disease.
3.2.2. Sociodemographical and clinicopathological background of the subjects
Diversities in socio‐epidemiological background, ethnicity, lifestyle, diet, exposure to various environmental risk factors and infectious agents, and hormonally related variables significantly affect the components of the serum/plasma proteome. Consequently, selection of control and diseased populations with comparable sex ratios, age ranges, demographic features, and dietary considerations is essential to minimize preanalytical variation. Clinicopathological details, particularly information regarding past history of infection, relapse cases, or presence of mixed/asymptomatic infection is crucial during the analysis of serum/plasma proteome of infectious diseases, since lack of detailed information may lead to misinterpretation of the obtained results. Therefore, meticulous tracking of all the variables is extremely important for obtaining reproducible results and for downstream analysis/interpretation of the findings. To this end, in developed countries stable biobanks/biorepositories for organized collection and storage of a huge number of biospecimens with comprehensive clinicopathological and socio‐epidemiological information can accelerate large‐scale clinical studies. In countries with low resource settings, due to the paucity of stable biobanking activities, obtaining well‐annotated biospecimens is often very challenging 39, 40.
3.2.3. Effects of preanalytical variables
Understanding the possible sources of preanalytical variables and their subsequent impact on quantification of peptides and protein concentrations in serum/plasma samples is extremely crucial since these can adversely influence the ultimate findings and reproducibility of the entire analysis process 41. Gelfand and Omenn have provided a comprehensive systematic analysis of the diverse sources of preanalytical variability for plasma and serum proteome analysis and recommended experimental guidelines for reducing the undesirable variations that can be introduced from multiple sources during the sample collection, handling, and processing steps 42.
In addition to the sociodemographical and clinicopathological background of the patients, venepuncture and phlebotomy strategies, sample collection devices, intrinsic enzyme activity, duration and environment of sample processing, and shipping and storage conditions are the major sources of preanalytical variables (Fig. 4). A recent study by Zhao et al. has shown the influences of preanalytical variations of blood sampling and handling on the findings of quantitative immunoassays for rheumatoid arthritis 43. Findings obtained from this study indicate that while the effect of blood sample collection, processing, and handling processes is minimal on autoantibody biomarker measurements, these variables can substantially affect serum protein concentrations. Yi et al. have demonstrated the time‐dependent alterations in human plasma and serum proteins due to the proteolytic degradation by intrinsic plasma peptidases and proteases, and recommended the addition of protease inhibitors instantaneously with the blood samples during collection process to increase stabilization of the plasma proteins and prevent proteolysis‐related preanalytical variability 44. Repeated freeze‐thawing cycles also affect the serum/plasma protein stability and reproducibility of the obtained results, and should be avoided. Taken as a whole, the use of optimized blood collection devices, reduction of the time elapsed between blood collection and subsequent processing, control of the sample handling procedure and environment, stable storage temperature, and avoidance of multiple freeze/thaw cycles can cumulatively reduce the sample instability and preanalytical variations associated with serum/plasma proteomics biomarker studies 45.
3.2.4. Determination of sample size
Another vital issue is the determination of population size (diseased and control) during the discovery and validation phases of biomarker discovery 46 (Fig. 5). Definitely, the more the clinical cohorts studied, better the confidence level of the findings. Nevertheless, the analysis cost and the availability of suitable clinical samples often constrain the investigation of bigger clinical cohorts. Prior to the comparative proteomic analysis, power calculations should be performed to estimate the minimum number of biological replicates required from diseased and control populations. In a recent study, Gandhi et al. have shown through power calculation analysis that the DIGE experiment requires at least ten samples from each group to be confident of 1.5‐fold difference at the p < 0.05 significance level 47. However, the minimum sample size depends on the type of analysis, study population, and reproducibility of the experimental approaches implicated. It is generally advised to perform validation studies employing larger clinical cohorts than the sample size used in discovery phase 37. All in all, the design of appropriate proteomics experiments involving different types of febrile subjects as diseases controls, and validation of the initial findings in larger clinical cohorts using alternative technological approaches, is essential for obtaining conclusive protein markers for any specific infection that can subsequently be translated into real clinical applications.
3.2.5. Individual versus pooled sample
Another controversial issue for clinical proteomic profiling experiments is the analysis of pooled samples. Certainly, information obtained from analysis of individual samples is much more reliable than the analysis of pooled samples, since pooling of samples may not effectively reflect the real biological alterations. Moreover, actual differences might be masked due to the presence of some outliers with extreme biological variability within the pooled cohorts. Nevertheless, pooling of clinical samples is often practiced in quantitative proteomics analysis, particularly when larger numbers of samples need to be studied or there is not an adequate amount of each sample for individual analysis. In proteomic investigations using pooled samples, it is more acceptable if multiple small pools of control and diseased samples are analyzed, rather than preparation of a single pooled set containing all the samples to be analyzed. If sample pooling is performed during the discovery phase of the analysis, it is essential to validate the results in individual diseased and control samples selected randomly from the pooled populations.
3.2.6. Analysis of longitudinal cohort
The majority of published studies on alterations of the serum/plasma proteome in pathogenic infections exhibit a map of the differentially expressed proteome at a single time point during disease progression, generally in the febrile phase of the infection. Single time‐point analysis (case vs. control) involving the early febrile stage of infection may help to identify potential diagnostic markers. Longitudinal studies of infectious diseases, which involve repeated observations of the same subjects over long periods of time (early febrile, defervescence, and convalescent stages of infection), can provide additional valuable information regarding changes in the identified disease markers and their utility as disease monitoring or prognostic markers (Fig. 3B). To this end, a serum proteome and cytokine analysis in longitudinal cohorts of dengue fever and its life‐threatening complication dengue hemorrhagic fever has recently been reported by Kumar et al. 48. The authors have shown that an in‐depth investigation of serum proteome signatures at different time‐points during the infection and their correlation analysis with clinicopathological parameters can help to unravel mechanisms of dengue disease progression. However, collection of samples at multiple time points from patients with severe infection is often problematic due to ethical concerns.
4. Global initiatives, HUPO plasma proteome project (PPP) and proteomics standards initiative (PSI)
4.1. HUPO PPP
Due to various technological limitations, the complexity and the wide dynamic range of protein concentrations in plasma/serum samples, comprehensive coverage of the entire blood proteome, is extremely challenging, and often low‐abundance potential biomarkers are missed in individual studies conducted using any single proteomic profiling approach. With the goal of mapping the maximum number of proteins present in human plasma in 2002, HUPO introduced the Human PPP for advancement of biomarker discovery and validation processes 49 through multiple international collaborations (including a total of 55 laboratories across the globe) 50. The objectives of the HUPO PPP include comprehensive analysis of the entire protein component of human plasma and serum, identification of the dynamic biological and physiological variation in the plasma proteome within an individual, as well as an estimation of the extent of variations between individuals within the same and different populations under normal and diseased conditions, which would be extremely useful for the design of clinical studies 51. The pilot phase of HUPO PPP included the results from 35 collaborating laboratories and analytical groups, and generated a core dataset of over 3000 proteins (each with two or more high‐confidence peptides) 18, which was reduced to 889 proteins once very stringent selection criteria were implemented in a separate analysis of same dataset 52. In addition to the HUPO PPP, the Human Plasma Peptide Atlas, established by incorporating LC‐MS/MS shotgun proteomics results from several experiments conducted in various laboratories 53, is another resource for studying human plasma proteins 54.
As a further continuation of the HUPO PPP initiative, the subsequent phase of the HUPO PPP 2 was commenced in 2008 (at the 7th HUPO World Congress of Proteomics in Amsterdam, The Netherlands) to encourage the submission of high‐quality, large datasets of human plasma proteome analysis, the development of a comprehensive data repositories and integration of the PPP with other disease‐related initiatives of HUPO with regard to plasma/serum biomarker discovery 55. In recent years, omics‐based research has worked toward the in‐depth analysis of PTMs 56 and splice variants 57 to obtain information beyond differential expression analysis of candidate proteins/genes, which is often found to be inadequate for understanding complex biological processes or disease pathobiology 58, 59, 60. Recently, HUPO initiated a chromosome‐centric project HPP 61 to provide a more systematic catalog of all of the proteins related to each chromosome, in contrast to that provided by disease‐oriented approach (B/D‐HPP) 62.
4.2. HUPO PSI
Sharing scientific data among different research groups across the world is crucial for improvement of proteomics‐based biomarker discovery endeavors, since it allows different research communities to access, evaluate, and validate each other's findings and correlate the results with their own observations. Comparison and extrapolation of data generated in different research laboratories are possible only when uniform standards for proteomics procedures with negligible preanalytical and technological variations are practiced globally. To this end, in 2002, the HUPO PSI (http://psidev.sourceforge.net/) was established to develop universal reference standards for use in proteomics studies. These standard methods can be followed to introduce uniformity and reproducibility in data acquisition and analysis processes, which in turn can accelerate comparison, exchange, and interpretation of data 63, 64, and avoid the “fragmentation of proteomics data” generated in different research laboratories across the globe 65. The HUPO PSI committee is working on the development of standard operating procedures and quality assurance protocols for data handling and analysis at a global scale. Moreover, HUPO PSI's goal is to develop reporting requirements, data exchange formats, and controlled vocabularies to formalize the requirements of the proteomics community and to encourage participation in multidisciplinary collaborative projects, such as the Functional Genomics Experiment (FuGE) and Functional Genomics Investigation Ontology (FuGO), in order to accelerate the collation, comparison, and analysis of “multi‐omics” datasets 66. HUPO PSI's specified standards have already been successfully implemented in the field of MS and molecular interactions, including TraML format for exchange of SRM transition lists 67, mzIdentML data standard for MS‐based proteomics results 68, PSICQUIC and PSISCORE for accessing and scoring molecular interactions 69, and the International Molecular Exchange (IMEx) consortium for protein interaction data curation 70. According to a 2012 assessment, within 10 years of its establishment, the HUPO PSI has gained considerable popularity among the proteomics community worldwide and established itself as the foremost representative in the field of proteomics for facilitating data comparison, exchange, and verification 71.
While continuous introduction of new proteomic technologies with improved sensitivity and high‐throughput capability is overcoming some existing technological limitations, integration of proteomics research with other emerging research fields and collaborative research work at a global scale is also essential for translation of the existing knowledge into practical clinical applications.
5. Concluding remarks
The development of simple, accurate, low cost, and stable diagnostic tests and the implementation of these tests in developing countries are crucial since approximately 90% of infectious disease‐associated deaths occur in developing countries mainly due to the lack of early diagnosis and timely treatments 72. Certainly, one of the most promising applications of proteomics in clinics is the identification of next generation diagnostic, prognostic, or disease biomarkers that can effectively improve diagnostics and therapeutics. High‐throughput proteomic technologies suitable for the rapid screening and quantification of thousands of analytes with high accuracy are needed for the finding of authentic biomarkers. Ultimately, the primary objective of proteomics biomarker discovery research is to establish novel candidate markers that can be clinically useful, as well as to translate the existing findings into practical applications in diagnostics and therapeutics. However, in spite of having tremendous potential, actual bed‐side translation of the findings obtained from proteomic research has thus far been limited. The efforts of the proteomics community have started to show results, as is evident from success stories, such as the OVA1 test, which has obtained FDA clearance 73. Even though most of the biomarkers identified in various proteomic studies show promises in the discovery phases, they cannot get approval without passing the validation stage and clinical trials, which is a complex and long‐term procedure. While the global initiatives, such as the Human PPP, the Human Plasma Peptide Atlas, and the HUPO PSI, are designed to identify and characterize the inclusive proteomes of plasma, connect the existing plasma/serum proteomics studies across the world, and establish a high‐quality uniform standard for proteomics analysis, it is also the collective responsibility of individual research groups to integrate their research findings with these ongoing worldwide initiatives.
The authors declare no competing interests.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Table S1. Differential expressions of selected serum/plasma proteins in different infectious diseases as reported in the published literature.
ACKNOWLEDGMENT
This work was supported by Board of Research in Nuclear Sciences (BRNS) DAE grant (2013/37B/24/BRNS) and a seed grant (young investigator award) 12IRAWD011 from the IIT Bombay to SS. SR was supported by the IIT Bombay fellowship.
Colour Online: See the article online to view Figs. 1–5 in colour.
6 References
- 1. Petricoin, E. , Wulfkuhle, J. , Espina, V. , Liotta, L. A. , Clinical proteomics: revolutionizing disease detection and patient tailoring therapy. J. Proteome. Res. 2004, 3, 209–217. [DOI] [PubMed] [Google Scholar]
- 2. Chen, R. , Mias, G. I. , Li‐Pook‐Than, J. , Jiang, L. et al., Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012, 148, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ray, S. , Reddy, P. J. , Jain, R. , Gollapalli, K. et al., Proteomic technologies for the identification of disease biomarkers in serum: advances and challenges ahead. Proteomics 2011, 11, 2139–2161. [DOI] [PubMed] [Google Scholar]
- 4. Morens, D. M. , Folkers, G. K. , Fauci, A. S. , The challenge of emerging and re‐emerging infectious diseases. Nature 2004, 430, 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daszak, P. , Cunningham, A. A. , Hyatt, A. D. , Emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 2000, 287, 443–449. [DOI] [PubMed] [Google Scholar]
- 6. Jones, K. E. , Patel, N. G. , Levy, M. A. , Storeygard, A. et al., Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banoo, S. , Bell, D. , Bossuyt, P. , Herring, A. et al., Evaluation of diagnostic tests for infectious diseases: general principles. Nat. Rev. Microbiol. 2010, 8, S17–S29. [PubMed] [Google Scholar]
- 8. Hanash, S. , Disease proteomics. Nature 2003, 422, 226–232. [DOI] [PubMed] [Google Scholar]
- 9. Olszewski, K. L. , Morrisey, J. M. , Wilinski, D. , Burns, J. M. et al., Host‐parasite interactions revealed by Plasmodium falciparum metabolomics. Cell Host Microbe 2009, 5, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson, N. L. , Anderson, N. G. , The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell Proteomics 2002, 1, 845–867. [DOI] [PubMed] [Google Scholar]
- 11. Issaq, H. J. , Xiao, Z. , Veenstra, T. D. , Serum and plasma proteomics. Chem. Rev. 2007, 107, 3601–3620. [DOI] [PubMed] [Google Scholar]
- 12. Wang, P. , Bouwman, F. G. , Mariman, E. C. , Generally detected proteins in comparative proteomics—a matter of cellular stress response? Proteomics 2009, 9, 2955–2966. [DOI] [PubMed] [Google Scholar]
- 13. Ray, S. , Renu, D. , Srivastava, R. , Gollapalli, K. et al., Proteomic investigation of falciparum and vivax malaria for identification of surrogate protein markers. PLoS One 2012, 7, e41751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ray, S. , Kamath, K. S. , Srivastava, R. , Raghu, D. et al., Serum proteome analysis of vivax malaria: an insight into the disease pathogenesis and host immune response. J. Proteomics 2012, 75, 3063–3080. [DOI] [PubMed] [Google Scholar]
- 15. Ray, S. , Srivastava, R. , Tripathi, K. , Vaibhav, V. et al., Serum proteome changes in dengue virus‐infected patients from a dengue‐endemic area of India: towards new molecular targets? OMICS 2012, 16, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srivastava, R. , Ray, S. , Vaibhav, V. , Gollapalli, K. et al., Serum profiling of leptospirosis patients to investigate proteomic alterations. J. Proteomics 2012, 76(Spec No), 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zimmerman, L. J. , Li, M. , Yarbrough, W. G. , Slebos, R. J. et al., Global stability of plasma proteomes for mass spectrometry‐based analyses. Mol. Cell Proteomics 2012. 11, M111, 014340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Omenn, G. S. , States, D. J. , Adamski, M. , Blackwell, T. W. et al., Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly‐available database. Proteomics 2005, 5, 3226–3245. [DOI] [PubMed] [Google Scholar]
- 19. Veenstra, T. D. , Global and targeted quantitative proteomics for biomarker discovery. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 847, 3–11. [DOI] [PubMed] [Google Scholar]
- 20. Simpson, K. L. , Whetton, A. D. , Dive, C. , Quantitative mass spectrometry‐based techniques for clinical use: biomarker identification and quantification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tonge, R. , Shaw, J. , Middleton, B. , Rowlinson, R. et al., Validation and development of fluorescence two‐dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [DOI] [PubMed] [Google Scholar]
- 22. Cristea, I. M. , Gaskell, S. J. , Whetton, A. D. , Proteomics techniques and their application to hematology. Blood 2004, 103, 3624–3634. [DOI] [PubMed] [Google Scholar]
- 23. Aebersold, R. , Mann, M. , Mass spectrometry‐based proteomics. Nature 2003, 422, 198–207. [DOI] [PubMed] [Google Scholar]
- 24. Ramachandran, N. , Srivastava, S. , Labaer, J. , Applications of protein microarrays for biomarker discovery. Proteomics Clin. Appl. 2008, 2, 1444–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rougemont, A. , Dumbo, O. , Bouvier, M. , Soula, G. et al., Hypohaptoglobinaemia as an epidemiological and clinical indicator for malaria. Results of two studies in a hyperendemic region in West Africa. Lancet 1988, 2, 709–712. [DOI] [PubMed] [Google Scholar]
- 26. Yerly, S. , Bouvier, M. , Rougemont, A. , Srivastava, I. et al., Development of a haptoglobin ELISA. Its use as an indicator for malaria. Acta Trop. 1990, 47, 237–244. [DOI] [PubMed] [Google Scholar]
- 27. Petrak, J. , Ivanek, R. , Toman, O. , Cmejla, R. et al., Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics 2008, 8, 1744–1749. [DOI] [PubMed] [Google Scholar]
- 28. Rai, A. J. , Gelfand, C. A. , Haywood, B. C. , Warunek, D. J. et al., HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. [DOI] [PubMed] [Google Scholar]
- 29. Gorg, A. , Weiss, W. , Dunn, M. J. , Current two‐dimensional electrophoresis technology for proteomics. Proteomics 2004, 4, 3665–3685. [DOI] [PubMed] [Google Scholar]
- 30. Miller, I. , Crawford, J. , Gianazza, E. , Protein stains for proteomic applications: which, when, why? Proteomics 2006, 6, 5385–5408. [DOI] [PubMed] [Google Scholar]
- 31. Patterson, S. D. , Data analysis—the Achilles heel of proteomics. Nat. Biotechnol. 2003, 21, 221–222. [DOI] [PubMed] [Google Scholar]
- 32. deVera, I. E. , Katz, J. E. , Agus, D. B. , Clinical proteomics: the promises and challenges of mass spectrometry‐based biomarker discovery. Clin. Adv. Hematol. Oncol. 2006, 4, 541–549. [PubMed] [Google Scholar]
- 33. Qian, W. J. , Jacobs, J. M. , Liu, T. , Camp, D. G. , Smith, R. D. , Advances and challenges in liquid chromatography‐mass spectrometry‐based proteomics profiling for clinical applications. Mol. Cell Proteomics 2006, 5, 1727–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kodadek, T. , Protein microarrays: prospects and problems. Chem. Biol. 2001, 8, 105–115. [DOI] [PubMed] [Google Scholar]
- 35. Talapatra, A. , Rouse, R. , Hardiman, G. , Protein microarrays: challenges and promises. Pharmacogenomics 2002, 3, 527–536. [DOI] [PubMed] [Google Scholar]
- 36. Ray, S. , Mehta, G. , Srivastava, S. , Label‐free detection techniques for protein microarrays: prospects, merits and challenges. Proteomics 2010, 10, 731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paulovich, A. G. , Whiteaker, J. R. , Hoofnagle, A. N. , Wang, P. , The interface between biomarker discovery and clinical validation: the tar pit of the protein biomarker pipeline. Proteomics Clin. Appl. 2008, 2, 1386–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aebersold, R. , Burlingame, A. L. , Bradshaw, R. A. , Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell Proteomics 2013, 12, 2381–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sgaier, S. K. , Jha, P. , Mony, P. , Kurpad, A. et al., Public health. Biobanks in developing countries: needs and feasibility. Science 2007, 318, 1074–1075. [DOI] [PubMed] [Google Scholar]
- 40. Ray, S. , Moiyadi, A. , Srivastava S., Epidemiology: Biorepositories for cancer research in developing countries. Nat. Rev. Clin. Oncol. 2013, 10, 434–436. [DOI] [PubMed] [Google Scholar]
- 41. Rai, A. J. , Vitzthum, F. , Effects of preanalytical variables on peptide and protein measurements in human serum and plasma: implications for clinical proteomics. Expert. Rev. Proteomics 2006, 3, 409–426. [DOI] [PubMed] [Google Scholar]
- 42. Gelfand, C. A. , Omenn, G. S. , in: Ivanov A. R., Lazarev A. V. (Eds.), Sample Preparation in Biological Mass Spectrometry, Springer, Heidelberg, London, New York: 2011, pp. 269–290. [Google Scholar]
- 43. Zhao, X. , Qureshi, F. , Eastman, P. S. , Manning, W. C. et al., Pre‐analytical effects of blood sampling and handling in quantitative immunoassays for rheumatoid arthritis. J. Immunol. Methods 2012, 378, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yi, J. , Kim, C. , Gelfand, C. A. , Inhibition of intrinsic proteolytic activities moderates preanalytical variability and instability of human plasma. J. Proteome Res. 2007, 6,1768–1781. [DOI] [PubMed] [Google Scholar]
- 45. Yi, J. , Craft, D. , Gelfand, C. A. , Minimizing preanalytical variation of plasma samples by proper blood collection and handling. Methods Mol. Biol. 2011, 728, 137–149. [DOI] [PubMed] [Google Scholar]
- 46. Cairns, D. A. , Barrett, J. H. , Billingham, L. J. , Stanley, A. J. et al., Sample size determination in clinical proteomic profiling experiments using mass spectrometry for class comparison. Proteomics 2009, 9, 74–86. [DOI] [PubMed] [Google Scholar]
- 47. Gandhi, K. S. , McKay, F. C. , Diefenbach, E. , Crossett, B. et al., Novel approaches to detect serum biomarkers for clinical response to interferon‐beta treatment in multiple sclerosis. PLoS One 2010, 5, e10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumar, Y. , Liang, C. , Bo, Z. , Rajapakse, J. C. et al., Serum proteome and cytokine analysis in a longitudinal cohort of adults with primary dengue infection reveals predictive markers of DHF. PLoS Negl. Trop. Dis. 2012, 6, e1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Omenn, G. S. , Advancement of biomarker discovery and validation through the HUPO plasma proteome project. Dis. Markers 2004, 20, 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Omenn, G. S. , International collaboration in clinical chemistry and laboratory medicine: the Human Proteome Organization (HUPO) Plasma Proteome Project. Clin. Chem. Lab Med. 2004, 42, 1–2. [DOI] [PubMed] [Google Scholar]
- 51. Omenn, G. S. , The HUPO Human Plasma Proteome Project. Proteomics Clin. Appl. 2007, 1, 769–779. [DOI] [PubMed] [Google Scholar]
- 52. States, D. J. , Omenn, G. S. , Blackwell, T. W. , Fermin, D. et al., Challenges in deriving high‐confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat. Biotechnol. 2006, 24, 333–338. [DOI] [PubMed] [Google Scholar]
- 53. Deutsch, E. W. , Eng, J. K. , Zhang, H. , King, N. L. et al., Human Plasma Peptide Atlas. Proteomics 2005, 5, 3497–3500. [DOI] [PubMed] [Google Scholar]
- 54. Farrah, T. , Deutsch, E. W. , Aebersold, R. , Using the Human Plasma Peptide Atlas to study human plasma proteins. Methods Mol. Biol. 2011, 728, 349–374. [DOI] [PubMed] [Google Scholar]
- 55. Omenn, G. S. , Aebersold, R. , Paik, Y. K. , 7(th) HUPO World Congress of Proteomics: launching the second phase of the HUPO Plasma Proteome Project (PPP‐2) 16–20 August 2008, Amsterdam, The Netherlands. Proteomics 2009, 9, 4–6. [DOI] [PubMed] [Google Scholar]
- 56. Nesvizhskii, A. I. , Roos, F. F. , Grossmann, J. , Vogelzang, M. et al., Dynamic spectrum quality assessment and iterative computational analysis of shotgun proteomic data: toward more efficient identification of post‐translational modifications, sequence polymorphisms, and novel peptides. Mol. Cell Proteomics 2006, 5, 652–670. [DOI] [PubMed] [Google Scholar]
- 57. Ning, K. , Nesvizhskii, A. I. , The utility of mass spectrometry‐based proteomic data for validation of novel alternative splice forms reconstructed from RNA‐Seq data: a preliminary assessment. BMC Bioinform. 2010, 11(Suppl 11), S14 DOI: 10.1186/1471-2105-11-S11-S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang, J. Y. , Lee, J. C. , Hou, M. F. , Wang, C. L. et al., Alternative splicing for diseases, cancers, drugs, and databases. Sci. World J. 2013, 2013, 703568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Luo, Y. B. , Mitrpant, C. , Johnsen, R. , Fabian, V. et al., Investigation of splicing changes and post‐translational processing of LMNA in sporadic inclusion body myositis. Int. J. Clin. Exp. Pathol. 2013, 6, 1723–1733. [PMC free article] [PubMed] [Google Scholar]
- 60. Mann, M. , Jensen, O. N. , Proteomic analysis of post‐translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [DOI] [PubMed] [Google Scholar]
- 61. Paik, Y. K. , Jeong, S. K. , Omenn, G. S. , Uhlen, M. et al., The Chromosome‐Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol. 2012, 30, 221–223. [DOI] [PubMed] [Google Scholar]
- 62. Aebersold, R. , Bader, G. D. , Edwards, A. M. , van Eyk, J. E. et al., The biology/disease‐driven human proteome project (B/D‐HPP): enabling protein research for the life sciences community. J. Proteome Res. 2013, 12, 23–27. [DOI] [PubMed] [Google Scholar]
- 63. Orchard, S. , Hermjakob, H. , Apweiler, R. , The proteomics standards initiative. Proteomics 2003, 3, 1374–1376. [DOI] [PubMed] [Google Scholar]
- 64. Hermjakob, H. , Montecchi‐Palazzi, L. , Bader, G. , Wojcik, J. et al., The HUPO PSI's molecular interaction format—a community standard for the representation of protein interaction data. Nat. Biotechnol. 2004, 22, 177–183. [DOI] [PubMed] [Google Scholar]
- 65. Hermjakob, H. , The HUPO proteomics standards initiative—overcoming the fragmentation of proteomics data. Proteomics 2006, 6(Suppl 2), 34–38. [DOI] [PubMed] [Google Scholar]
- 66. Taylor, C. F. , Hermjakob, H. , Julian, R. K. Jr. , Garavelli, J. S. et al., The work of the Human Proteome Organisation's Proteomics Standards Initiative (HUPO PSI). OMICS 2006, 10, 145–151. [DOI] [PubMed] [Google Scholar]
- 67. Deutsch, E. W. , Chambers, M. , Neumann, S. , Levander, F. et al., TraML—a standard format for exchange of selected reaction monitoring transition lists. Mol. Cell Proteomics 2012, 11, R111, 015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jones, A. R. , Eisenacher, M. , Mayer, G. , Kohlbacher, O. et al., The mzIdentML data standard for mass spectrometry‐based proteomics results. Mol. Cell Proteomics 2012, 11, M111, 014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aranda, B. , Blankenburg, H. , Kerrien, S. , Brinkman, F. S. et al., PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat. Methods 2011, 8, 528–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Orchard, S. , Kerrien, S. , Abbani, S. , Aranda, B. et al., Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat. Methods 2012, 9, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Orchard, S. , Binz, P. A. , Borchers, C. , Gilson, M. K. et al., Ten years of standardizing proteomic data: a report on the HUPO‐PSI Spring Workshop: April 12–14th, 2012, San Diego, USA. Proteomics 2012, 12, 2767–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mabey, D. , Peeling, R. W. , Ustianowski, A. , Perkins, M. D. , Diagnostics for the developing world. Nat. Rev. Microbiol. 2004, 2, 231–240. [DOI] [PubMed] [Google Scholar]
- 73. Fung, E. T. , A recipe for proteomics diagnostic test development: the OVA1 test, from biomarker discovery to FDA clearance. Clin. Chem. 2010, 56, 327–329. [DOI] [PubMed] [Google Scholar]
- 74. A research agenda for malaria eradication: diagnoses and diagnostics. PLoS Med. 2011, 8, e1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Payne, D. , Use and limitations of light microscopy for diagnosing malaria at the primary health care level. Bull. World Health Organ. 1988, 66, 621–626. [PMC free article] [PubMed] [Google Scholar]
- 76. Tangpukdee, N. , Duangdee, C. , Wilairatana, P. , Krudsood, S. , Malaria diagnosis: a brief review. Korean J. Parasitol. 2009, 47, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Singh, M. P. , Majumdar, M. , Singh, G. , Goyal, K. et al., NS1 antigen as an early diagnostic marker in dengue: report from India. Diagn. Microbiol. Infect. Dis. 2010, 68, 50–54. [DOI] [PubMed] [Google Scholar]
- 78. Peeling, R. W. , Artsob, H. , Pelegrino, J. L. , Buchy, P. et al., Evaluation of diagnostic tests: dengue. Nat. Rev. Microbiol. 2010, 8, S30–S38. [DOI] [PubMed] [Google Scholar]
- 79. Gubler, D. J. , Meltzer, M. , Impact of dengue/dengue hemorrhagic fever on the developing world. Adv. Virus Res. 1999, 53, 35–70. [DOI] [PubMed] [Google Scholar]
- 80. Poppert, S. , Essig, A. , Stoehr, B. , Steingruber, A. et al., Rapid diagnosis of bacterial meningitis by real‐time PCR and fluorescence in situ hybridization. J. Clin. Microbiol. 2005, 43, 3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ghotaslou, R. , Farajnia, S. , Yeganeh, F. , Abdoli‐Oskouei, S. et al., Detection of acute childhood meningitis by PCR, culture and agglutination tests in Tabriz, Iran. Acta Med. Iran 2012, 50, 192–196. [PubMed] [Google Scholar]
- 82. Marcos, M. A. , Martinez, E. , Almela, M. , Mensa, J. , Jimenez de Anta, M. T. , New rapid antigen test for diagnosis of pneumococcal meningitis. Lancet 2001, 357, 1499–1500. [DOI] [PubMed] [Google Scholar]
- 83. Greenwald, J. L. , Burstein, G. R. , Pincus, J. , Branson, B. , A rapid review of rapid HIV antibody tests. Curr. Infect. Dis. Rep. 2006, 8, 125–131. [DOI] [PubMed] [Google Scholar]
- 84. Kvinesdal, B. B. , Nielsen, C. M. , Poulsen, A. G. , Hojlyng, N. , Immunofluorescence assay for detection of antibodies to human immunodeficiency virus type 2. J. Clin. Microbiol. 1989, 27, 2502–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dong, J. , Olano, J. P. , McBride, J. W. , Walker, D. H. , Emerging pathogens: challenges and successes of molecular diagnostics. J. Mol. Diagn. 2008, 10, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mahony, J. B. , Richardson, S. , Molecular diagnosis of severe acute respiratory syndrome: the state of the art. J. Mol. Diagn. 2005, 7, 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hines, J. , Nachamkin, I. , Effective use of the clinical microbiology laboratory for diagnosing diarrheal diseases. Clin. Infect. Dis. 1996, 23, 1292–1301. [DOI] [PubMed] [Google Scholar]
- 88. Nainan, O. V. , Xia, G. , Vaughan, G. , Margolis, H. S. , Diagnosis of hepatitis a virus infection: a molecular approach. Clin. Microbiol. Rev. 2006, 19, 63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Valsamakis, A. , Molecular testing in the diagnosis and management of chronic hepatitis B. Clin. Microbiol. Rev. 2007, 20, 426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Herrera, V. , Perry, S. , Parsonnet, J. , Banaei, N. , Clinical application and limitations of interferon‐gamma release assays for the diagnosis of latent tuberculosis infection. Clin. Infect. Dis. 2011, 52, 1031–1037. [DOI] [PubMed] [Google Scholar]
- 91. Dorman, S. E. , New diagnostic tests for tuberculosis: bench, bedside, and beyond. Clin. Infect. Dis. 2010, 50(Suppl 3), S173–S177. [DOI] [PubMed] [Google Scholar]
- 92. Syed Ahamed, K. B. , Raman, B. , Thomas, A. , Perumal, V. , Raja, A. , Role of QuantiFERON‐TB gold, interferon gamma inducible protein‐10 and tuberculin skin test in active tuberculosis diagnosis. PLoS One 2010, 5, e9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Al‐Zamel, F. A. , Detection and diagnosis of Mycobacterium tuberculosis. Expert. Rev. Anti Infect. Ther. 2009, 7, 1099–1108. [DOI] [PubMed] [Google Scholar]
- 94. Vernet, G. , Saha, S. , Satzke, C. , Burgess, D. H. et al., Laboratory‐based diagnosis of pneumococcal pneumonia: state of the art and unmet needs. Clin. Microbiol. Infect. 2011, 17(Suppl 3), 1–13. [DOI] [PubMed] [Google Scholar]
- 95. Siegel, M. O. , Fedorko, D. P. , Drake, S. K. , Calhoun, L. B. , Holland, S. M. , Legionella feeleii serotype 2 pneumonia in a man with chronic lymphocytic leukemia: a challenging diagnosis. J. Clin. Microbiol. 2010, 48, 2294–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kandi, S. , Diagnosis of community acquired pneumonia. J. Assoc. Physicians India 2012, 60(Suppl), 17–20. [PubMed] [Google Scholar]
- 97. Cumberland, P. , Everard, C. O. , Levett, P. N. , Assessment of the efficacy of an IgM‐elisa and microscopic agglutination test (MAT) in the diagnosis of acute leptospirosis. Am. J. Trop. Med. Hyg. 1999, 61, 731–734. [DOI] [PubMed] [Google Scholar]
- 98. Brandao, A. P. , Camargo, E. D. , da Silva, E. D. , Silva, M. V. , Abrao, R. V. , Macroscopic agglutination test for rapid diagnosis of human leptospirosis. J. Clin. Microbiol. 1998, 36, 3138–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Toyokawa, T. , Ohnishi, M. , Koizumi, N. , Diagnosis of acute leptospirosis. Expert. Rev. Anti Infect. Ther. 2011, 9, 111–121. [DOI] [PubMed] [Google Scholar]
- 100. Kassa, F. A. , Shio, M. T. , Bellemare, M. J. , Faye, B. et al., New inflammation‐related biomarkers during malaria infection. PLoS One 2011, 6, e26495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bahk, Y. Y. , Na, B. K. , Cho, S. H. , Kim, J. Y. et al., Proteomic analysis of haptoglobin and amyloid A protein levels in patients with vivax malaria. Korean J. Parasitol. 2010, 48, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Smith, D. B. , Janmey, P. A. , Sherwood, J. A. , Howard, R. J. , Lind, S. E. , Decreased plasma gelsolin levels in patients with Plasmodium falciparum malaria: a consequence of hemolysis? Blood 1988, 72, 214–218. [PubMed] [Google Scholar]
- 103. Albuquerque, L. M. , Trugilho, M. R. , Chapeaurouge, A. , Jurgilas, P. B. et al., Two‐dimensional difference gel electrophoresis (DiGE) analysis of plasmas from dengue fever patients. J. Proteome Res. 2009, 8, 5431–5441. [DOI] [PubMed] [Google Scholar]
- 104. Fragnoud, R. , Yugueros‐Marcos, J. , Pachot, A. , Bedin, F. , Isotope coded protein labeling analysis of plasma specimens from acute severe dengue fever patients. Proteome Sci. 2012, 10, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Thayan, R. , Huat, T. L. , See, L. L. , Tan, C. P. et al., The use of two‐dimension electrophoresis to identify serum biomarkers from patients with dengue haemorrhagic fever. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 413–419. [DOI] [PubMed] [Google Scholar]
- 106. Paradowski, M. , Lobos, M. , Kuydowicz, J. , Krakowiak, M. , Kubasiewicz‐Ujma, B. , Acute phase proteins in serum and cerebrospinal fluid in the course of bacterial meningitis. Clin. Biochem. 1995, 28, 459–466. [DOI] [PubMed] [Google Scholar]
- 107. Jesse, S. , Steinacker, P. , Lehnert, S. , Sdzuj, M. et al., A proteomic approach for the diagnosis of bacterial meningitis. PLoS One 2010, 5, e10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kramer, H. B. , Lavender, K. J. , Qin, L. , Stacey, A. R. et al., Elevation of intact and proteolytic fragments of acute phase proteins constitutes the earliest systemic antiviral response in HIV‐1 infection. PLoS Pathog. 2010, 6, e1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kim, S. S. , Kim, M. H. , Shin, B. K. , Na, H. J. et al., Different isoforms of apolipoprotein AI present heterologous post‐translational expression in HIV infected patients. J. Proteome Res. 2007, 6, 180–184. [DOI] [PubMed] [Google Scholar]
- 110. Van, D. R. , Guendel, I. , Kehn‐Hall, K. , Easley, R. et al., The identification of unique serum proteins of HIV‐1 latently infected long‐term non‐progressor patients. AIDS Res. Ther. 2010, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Poon, T. C. , Pang, R. T. , Chan, K. C. , Lee, N. L. et al., Proteomic profiling in SARS: diagnostic and prognostic applications. Hong Kong Med. J. 2009, 15(Suppl 8), 15–18. [PubMed] [Google Scholar]
- 112. Wan, J. , Sun, W. , Li, X. , Ying, W. et al., Inflammation inhibitors were remarkably up‐regulated in plasma of severe acute respiratory syndrome patients at progressive phase. Proteomics 2006, 6, 2886–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ren, Y. , He, Q. Y. , Fan, J. , Jones, B. et al., The use of proteomics in the discovery of serum biomarkers from patients with severe acute respiratory syndrome. Proteomics 2004, 4, 3477–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen, J. H. , Chang, Y. W. , Yao, C. W. , Chiueh, T. S. et al., Plasma proteome of severe acute respiratory syndrome analyzed by two‐dimensional gel electrophoresis and mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 17039–17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Biesiada, G. , Czepiel, J. , Ptak‐Belowska, A. , Targosz, A. et al., Expression and release of leptin and proinflammatory cytokines in patients with ulcerative colitis and infectious diarrhea. J. Physiol. Pharmacol. 2012, 63, 471–481. [PubMed] [Google Scholar]
- 116. Komatsu, M. , Kobayashi, D. , Saito, K. , Furuya, D. et al., Tumor necrosis factor‐alpha in serum of patients with inflammatory bowel disease as measured by a highly sensitive immuno‐PCR. Clin. Chem. 2001, 47, 1297–1301. [PubMed] [Google Scholar]
- 117. Tan, X. F. , Wu, S. S. , Li, S. P. , Chen, Z. , Chen, F. , Alpha‐1 antitrypsin is a potential biomarker for hepatitis B. Virol. J. 2011, 8, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Marrocco, C. , Rinalducci, S. , Mohamadkhani, A. , D'Amici, G. M. , Zolla, L. , Plasma gelsolin protein: a candidate biomarker for hepatitis B‐associated liver cirrhosis identified by proteomic approach. Blood Transfus. 2010, 8(Suppl 3), S105–S112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tanaka, T. , Sakurada, S. , Kano, K. , Takahashi, E. et al., Identification of tuberculosis‐associated proteins in whole blood supernatant. BMC Infect. Dis. 2011, 11, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhang, J. , Wu, X. , Shi, L. , Liang, Y. et al., Diagnostic serum proteomic analysis in patients with active tuberculosis. Clin. Chim. Acta 2012, 413, 883–887. [DOI] [PubMed] [Google Scholar]
- 121. Agranoff, D. , Fernandez‐Reyes, D. , Papadopoulos, M. C. , Rojas, S. A. et al., Identification of diagnostic markers for tuberculosis by proteomic fingerprinting of serum. Lancet 2006, 368, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Endeman, H. , Meijvis, S. C. , Rijkers, G. T. , van Velzen‐Blad, H. et al., Systemic cytokine response in patients with community‐acquired pneumonia. Eur. Respir. J. 2011, 37, 1431–1438. [DOI] [PubMed] [Google Scholar]
- 123. Tateda, K. , Matsumoto, T. , Ishii, Y. , Furuya, N. et al., Serum cytokines in patients with Legionella pneumonia: relative predominance of Th1‐type cytokines. Clin. Diagn. Lab Immunol. 1998, 5, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang, H. , Wu, Y. , Ojcius, D. M. , Yang, X. F. et al., Leptospiral hemolysins induce proinflammatory cytokines through Toll‐like receptor 2‐and 4‐mediated JNK and NF‐kappa B signaling pathways. PLoS One 2012, 7, e42266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gunaratna, R. I. , Handunnetti, S. M. , Bulathsinghalage, M. R. , Somaratne, P. et al., Serum nitrite levels in Sri Lankan patients with leptospirosis. Asian Pac. J. Trop. Med. 2012, 5, 75–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Table S1. Differential expressions of selected serum/plasma proteins in different infectious diseases as reported in the published literature.