

Abstract
Integrins are heterodimers, but recent in vitro and in vivo experiments suggest that they are also able to associate through their transmembrane domains to form homomeric interactions. Two fundamental questions are the biological relevance of these aggregates and their form of interaction in the membrane domain. Although in vitro experiments have shown the involvement of a GxxxG‐like motif, several crosslinking in vivo data are consistent with an almost opposite form of interaction between the transmembrane α‐helices. In the present work, we have explored these two questions using molecular dynamics simulations for all available integrin types. We have tested the hypothesis that homomeric interactions are evolutionary conserved, and essential for the cell, using conservative substitutions to filter out nonnative interactions. Our results show that two models, one involving a GxxxG‐like motif (model I) and an almost opposite form of interaction (model II) are conserved across all α and β integrin types, both in homodimers and homotrimers, with different specificities. No conserved interaction was found for homotetramers. Our results are completely independent from experimental data, both during molecular dynamics simulations and in the selection of the correct models. We rationalize previous seemingly conflicting findings regarding the nature of integrin interhelical homomeric interactions. Proteins 2006. © 2006 Wiley‐Liss, Inc.
Keywords: molecular dynamics, membrane, integrins, protein–protein interactions, homology
INTRODUCTION
Integrins are heterodimeric type I transmembrane proteins formed by noncovalent association of an α and a β‐subunit. Each subunit contains a large extracellular domain, a single transmembrane (TM) spanning α‐helix and a short cytoplasmic tail.1 Different types of α integrins can combine with different β counterparts, forming a variety of heterodimers. In humans, 18 α‐chains can interact with eight different β‐chains to form 24 different α/β heterodimers with varied functions.2 By spanning the membrane, the integrins serve as a dynamic linkage between cytoplasm and extracellular space, transducing signals across the membrane to mediate cell growth, differentiation, gene expression, motility, and apoptosis.3 Inside‐out signal transduction involves integrin cytoplasmic tails separation and subsequent ectodomain conformational changes, which alters the affinity of integrins for extracellular ligands.4, 5, 6
A number of experimental results suggest the existence of transmembrane α/β interactions.7, 8, 9 For example, electron cryomicroscopy and single particle analysis,10 cysteine scanning mutagenesis in the transmembrane domain11 and activation by disruption of transmembrane interactions of integrin αIIbβ3.12 Overall, for the α/β interaction, there seems to exist a general consensus on the type of interaction present in the inactive, low‐affinity form.11, 13, 14
But in addition to growing evidence indicating that α and β domains interact, there is also a strong tendency in vitro for α and β TM chains to form homooligomers, both in zwitterionic and acidic micelles15 and in biological membranes.16, 17 The latter authors examined the interaction of TM domains of the α2, αIIb, α4, β1, β3, and β7 integrins when expressed as chimeric proteins, showing that most TM domains homooligomerize to some extent. Also, a study of both cytoplasmic and transmembrane part of the αIIb/β3 integrin demonstrated only homooligomerization, but not formation of heterooligomers.18
The way in which TM integrin homomeric interactions take place may be similar to that of glycophorin A (GpA), an interaction that involves a GxxxG‐like motif, which was observed by Arkin and Brunger19 to be prevalent in TM sequences. Indeed, this motif can be found in multiple sequence alignments of predicted TM spanning regions of integrins (see Fig 1, residues highlighted in gray). The importance of this and other related motifs in α‐helical TM domains has been later been demostrated in exhaustive statistical analyses.20 Further, a selection of a random library of TM sequences for homodimerization clearly showed that the GxxxG motif is sufficient for strong helix–helix interactions.21 Using the TOXCAT assay,22 a test that measures the oligomerization of a chimeric protein containing a TM helix in the Escherichia coli inner membrane via transcriptional activation of the gene for chloramphenicol acetyltransferase, a sequence critical for integrin αIIb‐TM homodimerization that involved the GxxxG motif was suggested by Li et al.17
Figure 1.
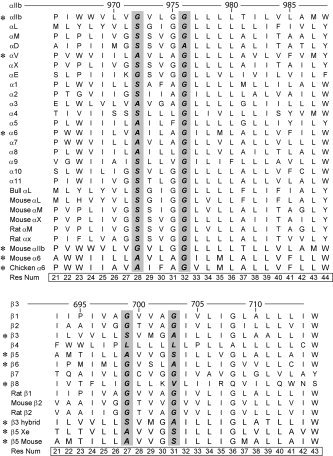
Alignment for the transmembrane sequences of integrin α (upper panel) or β (lower panel) used in this work. The specific numbering corresponding to human αIIβ (or β3) is indicated at the top of each respective panel. The residue numbering used in this work (common to all α or β sequences for convenience) is indicated at the bottom of each panel. The two black columns indicate the position of the small‐residue‐xxx‐small‐residue motif, or GxxxG‐like motif, for each sequence. The search for α (or β) homooligomers was started with a small subgroup of sequences indicated with an asterisk (*) (see Materials and Methods) referred to for convenience as “αIIb‐like” or “β3‐like” in the text.
Intriguingly, however, homomeric interactions that are not consistent with the involvement of a GxxxG‐like motif have been observed between α chains by crosslinking of the inactive αIIb/β3 dimer,11 and also between β chains23 induced by a G708N mutation in β3 TM. The functional relevance of these interactions has been discussed by these authors, and a role for integrin transmembrane homomeric interactions in integrin clustering when binding to multimeric ligands is possible.
Weak integrin TM interactions, however, make difficult the observation of in vitro mutagenesis effects. Also, some forms may be transient or not abundant, and they may be difficult to detect experimentally. One of the ways, although arguably indirect, to enquire on the existence and biological relevance of a given protein–protein interaction is to test its stability using evolutionary conservation data, using the idea that none of the mutations appeared during evolution, and present in homologous sequences, disrupt a native interaction. The general idea for this strategy is illustrated in Figure 2(A), which shows an example of results obtained in the present work for some of the α integrin sequences.
Figure 2.
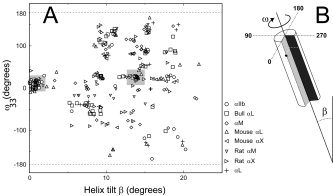
Example of results obtained to find evolutionary conserved structures as described in detail previously37 (only the results for a few α sequences are shown, for clarity). (A) Result obtained after exploring all possible conformational space of integrin transmembrane α sequences. For each sequence, similar low‐energy structures (typically, backbone RMSD lower than 1 Å) are grouped in clusters and averaged. These averaged low‐energy structures are indicated with symbols here, and are plotted on a plane described by helix tilt and rotational orientation for an arbitrarily chosen residue (here ω33). After considering the results from all sequences tested, a “complete set” is a group of these structures that contains representatives from all sequences. Hence, the backbone structure obtained by averaging converging structures for different homologs is not destabilized by conservative mutations. In this figure, the location of two “complete sets,” corresponding to α homodimeric models I and II (see Results), are indicated by gray squares. We stress that this plot is only a visual guide, and although the models (symbols) look close in the ω–β plane, the ultimate test of similarity is RMSD (see Materials and Methods). (B) Schematic representation of the rotational orientation ω and the helix tilt, β.
Using this method, we obtained previously24 the correct transmembrane homodimeric structure of glycophorin A at less than 1 Å RMSD from the structure determined by NMR.25 We later extended this method to the prediction of structures of various transmembrane α‐helical bundles, for example, phospholamban or Influenza A M2.26 Crucially, a picture emerged from that work that for some of these oligomers, for example, tetrameric M2, the correct structure was not found unless either the helix tilt was restrained to the experimentally obtained value or sampled at small intervals, so that all conformational space was fully explored. Indeed, only in these conditions the correct structure of M2 could be found.26 In the absence of experimentally determined helix tilt, the only alternative is to sample the helix tilt at small intervals, and we have recently followed this helix tilt sampling approach to find evolutionary conserved models of the transmembrane homooligomers of coronavirus envelope protein E.27
In the present work we have studied, without the help of any experimental restraint, the transmembrane homooligomeric interactions of integrins using an exhaustive global search,28 but sampling the helix tilt at 5° intervals, and using the integrin transmembrane sequence of 27 α subtypes and 14 β subtypes of integrin. We have explored the plausibility of dimeric, trimeric, and tetrameric homooligomers using a very stringent clustering protocol. The results obtained here are self‐consistent and the interpretation is unambiguous, that is, experimental data is not necessary to select the correct models.
We have found many models of interaction for homodimeric and homotrimeric oligomers that have been conserved through evolution and which can be used as reference for future mutagenesis studies.
MATERIALS AND METHODS
Global Search Molecular Dynamics (GSMD) Protocol
The simulations were performed using a Compaq Alpha Cluster SC45, which contains 44 nodes. All calculations were carried out using the parallel version of the Crystallography and NMR System (CNS Version 0.3), the Parallel Crystallography and NMR System (PCNS).29 The global search was carried out in vacuo with united atoms, explicitly describing only polar and aromatic hydrogen atoms as described elsewhere28 using CHI 1.1 (CNS Helical Interactions). As the models tested are homooligomers, the interaction between the helices was assumed to be symmetrical.
Trials were carried out starting from either left or right crossing angle configurations. The initial helix tilt, β, was restrained to 0° and the helices were rotated about their long helical axes in 10° increments until the rotation angle reached 350°. Henceforth, the simulation was repeated by increasing the helix tilt in discrete steps of 5°, up to 45°. We must note that the restraint for the helix tilt is not completely strict, that is, at the end of the simulation a drift of up to ±5° from the initial restrained value could be observed in some cases. Three trials were carried out for each starting configuration using different initial random velocities.
Clusters were identified with a minimum number of eight similar structures. Any structure belonging to a certain cluster was within 1.5 Å RMSD (root mean square deviation) from any other structure within the same cluster. Finally, the structures belonging to each cluster were averaged and subjected to energy minimization. These final averaged structures, described by a certain tilt and rotational orientation at a specified arbitrary residue, were taken as the representatives of the respective clusters [symbols in Fig. 2(A)].
The tilt angle of the models, β, was taken as the average of the angles between each helix axis in the bundle and the bundle axis. The bundle axis, coincident with the normal to the bilayer, was calculated by CHI. The helix axis was calculated as a vector with starting and end points above and below a defined residue, where the points correspond to the geometric mean of the coordinates of the five α carbons N‐terminal and the five α carbons C‐terminal to the defined residue. The rotational orientation angle ω of a residue is defined by the angle between a vector perpendicular to the helix axis, oriented towards the middle of the peptidic C=O bond of the residue, and a plane that contains both the helical axis and the normal to the bilayer. In this work, to compare the models, a residue was chosen arbitrarily, and the ω angle is always given for residue 33 (see common numbering in Fig. 1, lower row) both for α and β sequences. Intersequence comparisons between low‐energy clusters were performed by calculating the RMSD between their α‐carbon backbone. Fitting was performed using the program ProFit (http://www.bioinf.org.uk/software/profit). The energies calculated correspond to the total energy of the system, including both bonded, for example, bond, angle, dihedral, improper, and nonbonded, that is, Van der Waals and electrostatic terms.28 The interaction energy for the residues was calculated with the function chi_interaction implemented in CHI.
Homologous Sequences Used for Integrin α and β
Homologous sequences were obtained using ncbi homoloGene search (http://www.ncbi.nlm.nih.gov/). The definition of the 27 α sequences, the abbreviation (inside parentheses) used in Figure 1 and the RefSeq database accession numbers are: αL precursor [Homo sapiens] (Human_L), NP_002200; αM precursor [Homo sapiens] (Human_M), NP_000623; αIIb precursor [Homo sapiens] (αIIb) NP_000410; αD [Homo sapiens] (αD), XP_496142; αV precursor [Homo sapiens] (αV), NP_002201; αX [Mus musculus] (αX), NP_067309; αE [Homo sapiens] (αE), NP_002199; α1 precursor [Homo sapiens] (α1), NP_852478; α2 precursor [Homo sapiens] (α2), NP_002194; α3 isoform b precursor [Homo sapiens] (α3), NP_005492; α4 precursor [Homo sapiens] (α4), NP_000876; α5 precursor [Homo sapiens] (α5), NP_002196; α6 [Homo sapiens] (α6), NP_000201; α7 precursor [Homo sapiens] (α7), NP_002197; α8 [Homo sapiens] (α8), XP_167711; α9 [Homo sapiens] (α9), NP_002198; α10 precursor [Homo sapiens] (α10), NP_003628; α11 [Homo sapiens] (α11), NP_036343; Bos taurus integrin αL precursor (Bull αL), AY267467; αL [Mus musculus] (Mouse αL), NP_032426; αM [Mus musculus] (Mouse αM), NP_032427; αX [Mus musculus] (Mouse αX), NP_067309; αM [Rattus norvegicus] (Rat αM), NP_036843; αX [Rattus norvegicus] (Rat αX), NP_113879, αIIb [Mus musculus] (Mouse αIIb), NP_034705; α6 [Mus musculus] (Mouse α6), NP_032423; α6 [Gallus gallus] (Chicken α6), NP_990620.
Similarly, 14 sequences were used for the simulations of integrin β: β1 isoform 1D precursor [Homo sapiens] (β1), NP_391988; β2 precursor [Homo sapiens] (β2), NP_000202; β3 precursor [Homo sapiens] (β3), NP_000203; β4 [Homo sapiens] (β4), NP_000204; β5 [Homo sapiens] (β5), NP_002204; β6 [Homo sapiens] (β6), NP_000879; β7 [Homo sapiens] (β7), NP_000880; β8 [Homo sapiens] (β8), NP_002205; β1 [Rattus norvegicus] (Rat β1), NP_058718; β2 [Mus musculus] (Mouse β2), NP_032430; β2 precursor [Rattus norvegicus] (Rat β2), XP_228072; hybrid integrin β3 subunit precursor [synthetic construct] (β3 hybrid), AAF44692; Itgb5‐prov protein [Xenopus laevis] (β5 Xe), AAH76844 and β5 [Mus musculus] (β5 mouse), NP_034710.
The assignment of the transmembrane domain for these sequences was based on the hydrophilicity/surface probability plots and the transmembrane predictions from the TMHMM server.30 According to these predictors, the transmembrane region of these sequences spans 24 residues for the α chain and 23 for the β chain. The alignment of these sequences in the TM domain is shown in Figure 1.
Because of the tremendous computational work needed, to optimize the search, we first limited the search to a certain subgroup of integrin types indicated in Figure 1 by a star. If one or more conserved models were found for this subgroup, other sequences were tested for the existence of these models. The rationale for starting with the sequences is the abundant experimental studies performed on the transmembrane domain of the major platelet integrin αIIb/β3. The initial selection of α sequences therefore included αIIb, the natural partner of β3, αV, which also associates to β3 (Fig. 3), and α6, which has the highest sequence similarity to αIIb when using BLAST (http://www.ncbi.nlm.nih.gov/BLAST/Blast.cgi). The initial selection of β sequences included β3 and other β sequences able to bind αV,31 that is, β5, β6, and β8 (Fig. 3).
Figure 3.
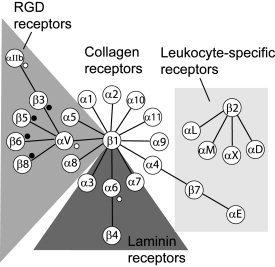
Mammalian types of integrin and types of heterodimeric associations (adapted from R.O. Hynes31). The α and β sequences used for the first group of simulations (“αIIb‐like” and “β3‐like,” see text) are indicated with white or black dots, respectively.
RESULTS AND DISCUSSION
TM Homodimer for α Integrin
When a homodimeric model was assumed for the “αIIb‐like” subgroup (see legend in Fig. 1), two “complete sets,” or conserved models, were found restraining the helix tilt to 15°, and only when the configuration was right handed. In one of these models (model I), with helix tilt β = 19° and rotational orientation ω33 = 28°, the residues located at the “G” position in the GxxxG‐like motif are involved in the interaction [Fig. 4(a), residues 972 and 976]. The other conserved model, with almost opposite orientation (model II), is shown in Figure 4(b), with β = 6° and ω33 = 16°. The opposite orientation of models I [Fig. 4(a)] and II [Fig. 4(b)] is shown clearly when comparing the orientation of W967 in these two models. When other helix tilts or left‐handed configurations were tested, no other complete sets were found across this subgroup of sequences. The RMSD between any pair of structures belonging to these two complete sets, either model I or II, was never higher than 0.8 Å.
Figure 4.
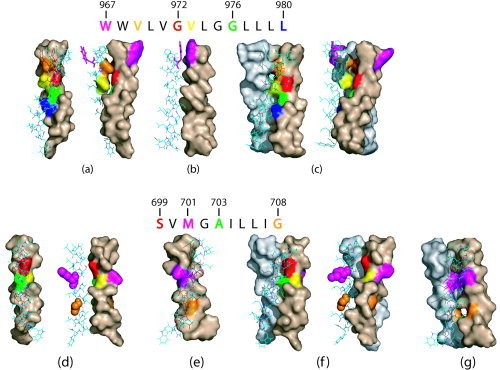
Homomeric interactions predicted here for α (upper panel) and β (lower panel) integrins in the TM domain. The sequence used corresponds to either αIIb (upper panel) or β3 (lower panel). (a) α homodimer, model I (front and side view); (b) α homodimer model II (side view); (c) α homotrimer model I (front and side view); (d) β homodimer model I (front view and side view); (e) β homodimer model II (side view); (f) β homotrimer model I (front and side view); (g): β homotrimer model II (side view).
When our search was extended to the remaining α sequences, model I was conserved in all α sequences tested. Model II was also conserved for the remaining sequences, but there were complex overlaps between different subtypes and, in contrast to model I, a common structure with RMSD less than 1 Å for all integrins types could not be found. Indeed, a virtually identical (RMSD less than 0.8 Å) model II was shared by all integrins, except for αM, α3, α2, and αX. We point out that this does not mean that a model similar to model II presented here is not present in the aforementioned sequences. For example, a group formed by αM and αX shared a “complete set” in which the helices were rotated approximately 50° (not shown) from the model II above. Related to this, we note that in previous reports we have used homologous sequences, found in different species, of a given protein. Here, in contrast, we have used different integrins types, which perform very different functions, in the same species (humans). This is clearly a more stringent condition for finding conserved interactions, as the variability in the sequences is potentially greater. The lack of a sufficient number of suitable homologous sequences of the same integrin type for different species, precluded the determination of a similar structure to model II in sequences α3 and α2, although it is possible that a similar interaction, that is, one that does not involve the GxxxG motif, is also present there.
TM Homotrimer of α Integrin
When a homotrimeric model was assumed for the “αIIb‐like” subgroup, a complete set was found for a right handed configuration (β = 19° and ω = 6°), and only when the helix tilt was restrained to 15°. The structure representing this model (a model I type, i.e., where the GxxxG‐like motif is involved in the interaction) is shown in Figure 4(c). No other complete sets were found for other tilts or left‐handed configurations. The RMSD between any pair of structures belonging to this complete set was never higher than 1 Å. When the search was extended to other sequences, this model was still conserved. Model II of interaction was not found to be conserved in α homotrimers.
TM Homodimer of the β Integrin
Simulations were performed initially for the “β3‐like” subgroup formed by the star‐labeled sequences (Fig. 1, lower panel). Only two models or “complete sets” were found to be conserved, both right handed. A model equivalent to model I [Fig. 4(d), see residues 699 and 703 corresponding to the SxxxA motif participating in the interaction] was found when the helix tilt was restrained to 5° in a right‐handed configuration, with β = 5° and ω = 27°. Another model of almost opposite orientation (equivalent to model II) was found at β = 18° and ω = −21° [Fig. 4(e)]. The opposite orientation of models I [Fig. 4(d)] and II [Fig. 4(e)] is shown clearly when comparing the orientation of M701 and G708 in these two models. The RMSD between any pair of structures belonging to these complete sets was never higher than 1 Å. No complete sets were found for other tilts or left‐handed conformations. When the search was extended to the rest of the sequences, model I was conserved for all β sequences. A more complex overlap was found around model II, but with RMSD less than 1.5 Å, a common structure was conserved for all sequences, except for β4 and β7.
TM Homotrimer of the β Integrin
When a homotrimeric model was assumed for the “β3‐like” subgroup, two conserved models were found, both right handed. A model equivalent to model I was found when the helix tilt was restrained to 15° in a right‐handed configuration (β = 15° and ω = 97°) [see Fig 4(f)]. Another model, of opposite orientation, equivalent to model II, was found at β = 16° and ω = −43° [Fig. 4(g)]. No complete sets were found for other tilts or left‐handed configurations. The RMSD between any pair of structures belonging to this complete set was never higher than 1 Å. As for the homodimers, when search was extended to other β sequences, the first model (model I) was conserved in all instances. Model II showed also the same behavior than for the homodimer, and a common model was found for all sequences (if RMSD <1.5 Å), except for β4 and β7. In general, therefore, in contrast to the very stable backbone model I, homomeric model II seems to have drifted slightly during evolution both for α and β chain homomeric interactions.
Incidentally, we have also observed that these results also stand when using other sequences of β integrins, from coral and sponges,32 that are evolutionarily distant from the ones presented here. Despite the lack of close homology, these sequences still present a GxxxG‐like motif, and models equivalent to models I and II for β integrins were also found (not shown). As the sequences diverge, however, the structure representing a particular mode of interaction starts to diverge from a tight complete set and partial overlaps can be found. These results are difficult to interpret in terms of sequence similarity or function.
As mentioned before, a sharper, although more complex, picture would have emerged if we had been able to use different homologs of a single integrin subtype, which is equivalent to test different homologous sequences, for different species, of the same protein. However, the fact that we have been able to obtain these two models of interaction using integrins that perform totally different functions confirms the robustness of our findings and suggests that these two models are general forms of interaction across all the integrin family spectrum. The different interactions observed in our computational work are summarized schematically in Figure 5.
Figure 5.
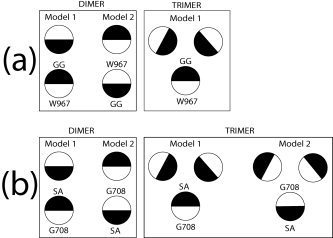
Summary of the transmembrane integrin homomeric interactions found in our computational work. All homooligomers are right handed. The scheme represents the models of interaction found for homodimers and homotrimers in αIIb (a) and β3 (b). Each helix is represented by two halves. For αIIb, the two Gly residues in the GxxxG‐like motif, G972 and G976, are located in one of the halves. Residue W967 is located at an opposite location, in the other half (see text). For β3, the two small (Gly) residues in the GxxxG‐like motif, residues S and A, are located in the same half, whereas G708 (and M701) is at an opposite orientation, in the other half.
DISCUSSION
Our computational results have been obtained independently from any previous experimental data, and clearly show that two right‐handed types of homomeric interaction in the transmembrane domain of α and β integrins (models I and II) are evolutionarily conserved. We also predict that these models are present in homodimers as well as in homotrimers, although with the exception of α homotrimers, where model II is not conserved.
Transmembrane α Oligomers
Our results for the α homodimer are in contrast with a recent study33 that studied α homodimeric interactions using a computational method similar to the one used here, and where two models, with a left‐ and right‐handed configuration were proposed. In the aforementioned study, however, only 10 integrin subtypes were used. In addition, the helix tilt was not restrained, and hence, the conformational space searched was not complete, leading to ambiguous results,33 the interpretation of which ultimately required the consideration of previous experimental data. In contrast, in the present work, we have used 27 α sequences, and even under our stringent RMSD and clustering parameters (cf. previous report33), we are able to detect two conserved models of interaction for all these sequences without the need to take into account any experimental data.
In addition, we also show that an evolutionarily conserved mode of interaction (model I) also exists for α homotrimers, which suggests that α homotrimers observed in vitro for constructs involving both TM and cytoplasmic tail of αIIb integrins (TM‐CYTO) in dodecylphosphocholine (DPC) micelles15 are probably not artifacts. The fact that these homotrimers were only observed at high peptide concentration suggests that their stability is lower than that of α homodimers. However, results derived from calculation of energy (see Material and Methods) and packing efficiency34 (http://www.molmovdb.org/cgi-bin/voronoi.cgi) (unpublished results) of these α homotrimers relative to the model I or II α homodimers do not explain this hypothetical lower stability for the homotrimer.
Our independent predictions are nevertheless consistent with previous findings. For example, the helix tilt for our α homodimeric model I [see Fig. 4(a)] is 19°, which corresponds to a crossing angle of 38° for a symmetric dimer. This is remarkably consistent with a “model I‐like” αIIb homodimer proposed by W.F. DeGrado and coworkers based on an exhaustive search of rigid‐helix interactions and mutagenesis data17 where a modified motif, VGxxGG instead of GVxxG for GpA, was proposed. In the model I we report, residues G972 and G976, that pertain to the GxxxG motif (see residues 28 and 32 in Fig. 1, top panel) interact with V969 and V973 of the other helix (number 25 and 29 in Fig. 1, top panel). Also, mutations at L980, L980A, and L980V, which have been reported to greatly increase homodimerization17 are involved in the interaction. In contrast to this latter report,17 however, calculations of the interaction energy per residue (Fig. 6) in the αIIb dimer model I do not show that the residues preceeding G in the GxxxG motif (i.e, residues V971 and G975) are important for the interaction. Our results for model I are therefore more consistent with a typical GpA type mode of interaction in all integrins. The discrepancy between these results may be due to the different strategy used and the ambiguity in mutagenesis studies; for example, G975L or G975V was found to impair dimerization, but G975A was as dimerizing as the native residue.17 Small details aside, because we find that this homodimeric model I has been conserved through all integrin types, the model described for αIIb17 is just a particular instance of a more general form of interaction that includes all α representatives, as has been also suggested by experiments involving other integrin TM domains.16
Figure 6.
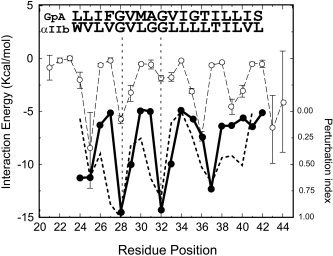
Average interaction energy (left axis) of individual residues corresponding to the α integrin homodimer model I using all available α sequences (broken line), with average values (○) and standard deviation (vertical bars). For comparison, a perturbation index (right axis) obtained from a mutation sensitivity experiment17 for the αIIb homodimer (thick broken line) and glycophorin (thick solid line) is also shown (1, maximum sensitivity; 0, lowest sensitivity). Residues 28 and 32 (vertical broken lines) correspond to the two interface glycine residues in the GxxxG‐like motif.
The mode of interaction of αIIb in the α/β heterodimer (αIIb/β3) has been described for the inactive state,11 with the residues at the G position in the GxxxG‐like motif participating in helix–helix contacts; in contrast, residue W967 points away from the α/β interface. Interestingly, mutation W967C resulted in the formation of a (αIIb/β3)2 species, a dimer of dimers, through formation of a disulfide bond.11 This suggests that in this particular case, interaction between αIIb chains is GxxxG‐like motif independent [similar to our model II of interaction, Fig. 4(b)]. Our computational results show that this form of interaction is neither accidental nor specific to αIIb, because we have found it to be evolutionarily conserved across all integrins. Coexistence of heterodimer and α homodimer is therefore possible taking model II into account, as it has also been suggested earlier.33
Transmembrane β Oligomers
Two conformations for the β3 homotrimer, of opposite handedness, have been proposed previously33 based on restraints from mutagenesis data. In contrast, our results are independent from experimental data, and using 14 β sequences we show that two models are evolutionarily conserved, but they are both right handed. In addition, we predict that these models are not only present in homotrimers, as previous experimental data suggests,23 but also in homodimers.
Transmembrane β homodimers have been observed in β1, β3, and β7,16, 35 and the importance of the GxxxG motif (interaction equivalent to our model I) has been confirmed experimentally by mutagenesis using GALLEX, a two‐hybrid system that follows heterodimerization of membrane proteins in the E. coli inner membrane. This seems to suggest that model I of interaction is more stable than model II for β homodimers or homotrimers. This would also suggest that polypeptides encompassing the transmembrane domain and cytoplasmic tail (TM‐CYTO) of β3 that have been found to form homotrimers in DPC15 probably correspond to model I. In contrast, only when the model II form of interaction is stabilized, for example, the mutant G708N in β3 which promoted homotrimerization,23 a model II form of interaction [Fig. 4(g)] would be detected (see position of G708 in β3). As for the α homooligomers, however, calculation of energy and packing efficiency for β2 and β3 (not shown) show consistently model II of interaction being more stable and well packed than model I. Also, among the homotrimers, the β homotrimer model II seems to be the most stable and well‐packed homooligomeric form. More detailed analyses are needed to explain this discrepancies.
But have these homomeric interactions any functional relevance? The putative coexistence of integrin hetero‐ and homo‐oligomers has been rationalized in a context where heteromeric interactions would stabilize the transmembrane region in a low‐affinity and/or intermediate affinity state,11 whereas homooligomers would be present in the active state, crosslinking individual molecules, and stabilizing focal adhesions.33 Consistent with this hypothesis, β3 TM homotrimerization induced constitutive activation and integrin clustering, suggesting a push–pull mechanism,14 although other studies failed to detect homomeric interactions after αIIbβ3 integrin activation.11 Nevertheless, a role for integrin transmembrane homomeric interactions in integrin clustering when binding to multimeric ligands36 is possible. The fact that several homomeric interactions are evolutionary conserved strongly support this possibility.
CONCLUSION
Our results provide an explanation for seemingly conflicting reports in in vivo and in vitro transmembrane homomeric interaction of integrins. We have found that two modes of interaction are evolutionarily conserved. One of these interactions (model I) involves the GxxxG‐like motif, which has been proposed previously on the basis of mutagenesis data in the transmembrane domains. The other model (model II) involves the opposite face of the helix, which is consistent with previous experimental data. Because our models have been obtained with independence of any previous experimental restraint and only using the perturbing effect of evolutionary conservation data as a filtering parameter, we suggest that these interactions are present in vivo. The present studies provide a fertile ground for experimentation. We are presently studying the in vivo effects of these potentially disruptive mutations in the integrin transmembrane domain.
Acknowledgements
J.T. thanks the financial support of Biomedical Research Council (BMRC) of Singapore and the facilities at the Bioinformatics Research Center (BIRC) of Nanyang Technological University. We are also grateful to Paul D. Adams for kindly providing CHI.
REFERENCES
- 1. Carman CV, Springer TA. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol 2003; 15: 547–556. [DOI] [PubMed] [Google Scholar]
- 2. Hemler M. Integrins. In: Kreis T, Vale R, editors. Extracellular matrix, anchor, and adhesion proteins. Oxford: Oxford University Press; 1999. [Google Scholar]
- 3. Watt FM. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J 2002; 21: 3919–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 2003; 301: 1720–1725. [DOI] [PubMed] [Google Scholar]
- 5. Hantgan RR, Paumi C, Rocco M, Weisel JW. Effects of ligand‐mimetic peptides Arg‐Gly‐Asp‐X (X = Phe, Trp, Ser) on alpha IIb beta 3 integrin conformation and oligomerization. Biochemistry 1999; 38: 14461–14474. [DOI] [PubMed] [Google Scholar]
- 6. Travis MA, Humphries JD, Humphries MJ. An unraveling tale of how integrins are activated from within. Trends Pharmacol Sci 2003; 24: 192–197. [DOI] [PubMed] [Google Scholar]
- 7. Xiong JP, Stehle T, Diefenbach B, Zhang RG, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alpha V beta 3. Science 2001; 294: 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adair BD, Xiong JP, Maddock C, Goodman SL, Arnaout MA, Yeager M. Three‐dimensional EM structure of the ectodomain of integrin {alpha}V{beta}3 in a complex with fibronectin. J Cell Biol 2005; 168: 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hughes PE, Diaz‐Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem 1996; 271: 6571–6574. [DOI] [PubMed] [Google Scholar]
- 10. Adair BD, Yeager M. Three‐dimensional model of the human platelet integrin alpha(llb)beta(3) based on electron cryomicroscopy and x‐ray crystallography. Proc Natl Acad Sci USA 2002; 99: 14059–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol 2004; 2: e153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo BH, Carman CV, Takagi J, Springer TA. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc Natl Acad Sci USA 2005; 102: 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gottschalk KE, Adams PD, Brunger AT, Kessler H. Transmembrane signal transduction of the alpha(IIb)beta(3) integrin. Protein Sci 2002; 11: 1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W, Metcalf DG, Gorelik R, Li R, Mitra N, Nanda V, Law PB, Lear JD, Degrado WF, Bennett JS. A push–pull mechanism for regulating integrin function. Proc Natl Acad Sci USA 2005; 102: 1424–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li R, Babu CR, Lear JD, Wand AJ, Bennett JS, DeGrado WF. Oligomerization of the integrin alphaIIbbeta3: roles of the transmembrane and cytoplasmic domains. Proc Natl Acad Sci USA 2001; 98: 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schneider D, Engelman DM. Involvement of transmembrane domain interactions in signal transduction by alpha/beta integrins. J Biol Chem 2004; 279: 9840–9846. [DOI] [PubMed] [Google Scholar]
- 17. Li RH, Gorelik R, Nanda V, Law PB, Lear JD, DeGrado WF, Bennett JS. Dimerization of the transmembrane domain of integrin alpha(IIb) subunit in cell membranes. J Biol Chem 2004; 279: 26666–26673. [DOI] [PubMed] [Google Scholar]
- 18. Lu CF, Takagi J, Springer TA. Association of the membrane proximal regions of the alpha and beta subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem 2001; 276: 14642–14648. [DOI] [PubMed] [Google Scholar]
- 19. Arkin IT, Brunger AT. Statistical analysis of predicted transmembrane alpha‐helices. Biochim Biophys Acta 1998; 1429: 113–128. [DOI] [PubMed] [Google Scholar]
- 20. Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta‐branched residues at neighboring positions. J Mol Biol 2000; 296: 921–936. [DOI] [PubMed] [Google Scholar]
- 21. Russ WP, Engelman DM. The GxxxG motif: A framework for transmembrane helix–helix association. J Mol Biol 2000; 296: 911–919. [DOI] [PubMed] [Google Scholar]
- 22. Russ WP, Engelman DM. TOXCAT: a measure of transmembrane helix association in a biological membrane. 1999; 96: 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li RH, Mitra N, Gratkowski H, Vilaire G, Litvinov R, Nagasami C, Weisel JW, Lear JD, DeGrado WF, Bennett JS. Activation of integrin alpha IIb beta 3 by modulation of transmembrane helix associations. Science 2003; 300: 795–798. [DOI] [PubMed] [Google Scholar]
- 24. Briggs JA, Torres J, Arkin IT. A new method to model membrane protein structure based on silent amino acid substitutions. 2001; 44: 370–375. [DOI] [PubMed] [Google Scholar]
- 25. MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: Structure and implications. Science 1997; 276: 131–133. [DOI] [PubMed] [Google Scholar]
- 26. Torres J, Briggs JAG, Arkin IT. Contribution of energy values to the analysis of global searching molecular dynamics simulations of transmembrane helical bundles. Biophys J 2002; 82: 3063–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Torres J, Wang J, Parthasarathy K, Liu DX. The transmembrane oligomers of coronavirus protein E. Biophys J 2005; 88: 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams PD, Arkin IT, Engelman DM, Brunger AT. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat Struct Biol 1995; 2: 154–162. [DOI] [PubMed] [Google Scholar]
- 29. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse‐Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 1998; 54: 905–921. [DOI] [PubMed] [Google Scholar]
- 30. Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305: 567–580. [DOI] [PubMed] [Google Scholar]
- 31. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110: 673–687. [DOI] [PubMed] [Google Scholar]
- 32. Brower DL, Brower SM, Hayward DC, Ball EE. Molecular evolution of integrins: Genes encoding integrin beta subunits from a coral and a sponge. Proc Natl Acad Sci USA 1997; 94: 9182–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gottschalk KE, Kessler H. A computational model of transmembrane integrin clustering. Structure 2004; 12: 1109–1116. [DOI] [PubMed] [Google Scholar]
- 34. Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: standard radii and volumes. J Mol Biol 1999; 290: 253–266. [DOI] [PubMed] [Google Scholar]
- 35. Schneider D, Engelman DM. GALLEX, a measurement of heterologous association of transmembrane helices in a biological membrane. J Biol Chem 2003; 278: 3105–3111. [DOI] [PubMed] [Google Scholar]
- 36. Buensuceso C, de Virgilio M, Shattil SJ. Detection of integrin alpha IIbbeta 3 clustering in living cells. J Biol Chem 2003; 278: 15217–15224. [DOI] [PubMed] [Google Scholar]
- 37. Briggs JAG, Torres J, Arkin IT. A new method to model membrane protein structure based on silent amino acid substitutions. Proteins Struct Funct Genet 2001; 44: 370–375. [DOI] [PubMed] [Google Scholar]