

Abstract
The previous epidemic of severe acute respiratory syndrome (SARS) has ended. However, many questions concerning how the aetiological agent, the novel SARS coronavirus (CoV), causes illness in humans remain unanswered. The pathology of fatal cases of SARS is dominated by diffuse alveolar damage. Specific histological changes are not detected in other organs. These contrast remarkably with the clinical picture, in which there are apparent manifestations in multiple organs. Both pathogen and host factors are important in the pathogenesis of SARS. The choice of specific receptors and the unique genome of the SARS‐CoV are important elements in understanding the biology of the pathogen. For the host cells, the outcome of SARS‐CoV infection, whether there are cytopathic effects or not, depends on the cell types that are infected. At the whole‐body level, immune‐mediated damage, due to activation of cytokines and/or chemokines and, perhaps, autoimmunity, may play key roles in the clinical and pathological features of SARS. Continued research is still required to determine the pathogenetic mechanisms involved and to combat this new emerging human infectious disease. Copyright © 2006 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: SARS, coronavirus, pathogenesis, cytokines, chemokines
Introduction
Severe acute respiratory syndrome (SARS) is a new viral disease caused by a novel coronavirus, SARS‐CoV (Figure 1) 1, 2. The saga of SARS has officially come to an end, as no more new cases have been reported since 2004. Many questions, particularly those related to how SARS‐CoV causes disease, however, remain unanswered.
Figure 1.
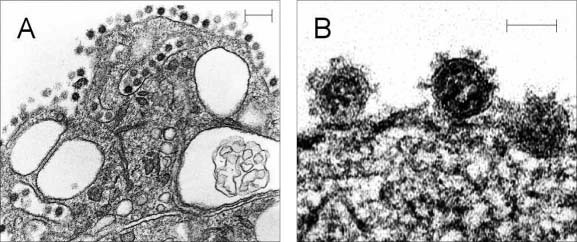
SARS‐CoV replicates in cultured Vero E6 cells and is produced in large numbers inside cytoplasmic vesicles (A). Virus particles can also be seen budding through the cytoplasmic membrane (B). Each virion particle is 60–90 nm in size by transmission electron microscopy and is characterized by the numerous club‐shaped projections on the outside, a ring beneath the envelope, and an electron‐lucent centre. Scale bars = 200 nm (A) and 50 nm (B)
The disease caused by SARS‐CoV differs from the diseases caused by the previously known human coronaviruses, 229E and OC43. SARS‐CoV infection results in severe and potentially fatal lung disease 1, 2. Although the majority of patients recovered after 1–2 weeks of debilitating febrile illness, a substantial proportion (up to one‐third) developed severe inflammation of the lung, requiring ventilator support and intensive care. Many patients in this group deteriorated into acute respiratory distress syndrome (ARDS). The mortality of this group of patients is high 3. Manifestations in other organ systems are characteristic. Lymphopenia 4, gastrointestinal symptoms 5, impaired liver function 6, 7, and impaired renal function 8 are common. The possibility of viral infection in multiple organs has been raised and viral replication in the lung, kidney, and gastrointestinal tract was reported 9, 10. In addition, prolonged shedding of virus was found in some convalescent patients 11. However, chronic infection by SARS‐CoV has not, to date, been documented in humans. Moreover, asymptomatic carriage of SARS‐CoV is rare 12.
There are significant age differences in the prognosis of SARS. Children have a good prognosis 13, while elderly patients with chronic illnesses fare badly. SARS is predominantly a lower respiratory tract disease, yet the most consistent and powerful prognostic indicator reported so far is blood lactate dehydrogenase (LDH) concentration 1, which is most likely a surrogate indicator and may reflect the extent of ongoing tissue damage.
Both pathogen and host factors are important for the progression of an infection. Here, we review the pathology of SARS infection. Specific features of the pathogen SARS‐CoV itself are then addressed. Finally, host factors, particularly an emerging understanding of immunological and inflammatory responses to SARS‐CoV infection, are discussed.
Pathology of SARS in human and animal models
Diffuse alveolar damage is the most characteristic pathology in SARS
Most data on the human pathology of SARS come from autopsy studies of fatal cases 14, 15, 16, 17, 18, 19, 20, 21. These reports thus reflect the terminal stages and are likely to represent only the more severe end of the spectrum of SARS. Treatment and co‐morbid conditions might also modify the pathological changes. Diffuse alveolar damage at different stages of organization is the most consistent finding in the lungs of SARS patients in the terminal stage (Figures 2A–F). Multi‐nucleated syncytial cells (Figures 2G and 2H) are characteristic, although these cells are rare. Apart from when secondary infection occurs, the lack of a prominent inflammatory response is also distinctive. SARS‐CoV is explicitly detected in the alveolar lining cells (Figures 2I and 2J) 10, 22, 23, 24, 25, 26, 27. No specific pathology is identified in the gastrointestinal tract (Figure 3) 5, urinary system 8, or other organ systems 28, apart from that related to end‐stage multi‐organ failure or those changes secondary to treatment. It is important to note that in some organs such as the liver, while definitive and distinct morphological and functional changes are observed, SARS‐CoV may not be unequivocally demonstrable 29.
Figure 2.

Diffuse alveolar damage is the most consistent finding in the terminal stages of SARS. The lung may appear grossly consolidated (A) or have a honeycomb appearance (B). Although the latter finding may be related to pre‐morbid lung pathology, a correlation with interstitial fibrosis and disease duration has been demonstrated 21. Diffuse alveolar damage at different stages of organization, from fibrin deposition (C, H&E, original magnification ×200), to interstitial fibrosis (D, H&E, original magnification ×100) and cellular organization (E and F, H&E, original magnification ×400), can be detected. Atypical pneumocytes with enlarged nuclei and prominent nucleoli are often seen and some pneumocytes coalesce into syncytial multi‐nucleated cells (G, H&E, original magnification ×600). Multi‐nucleated histiocytes may also be found (H, H&E, original magnification ×600). SARS‐CoV can be detected in pneumocytes by in situ hybridization (I, using a DNA probe against the M gene, original magnification ×600 22). A large array of antibodies against the viral proteins including nucleocapsid N, spike S, membrane M, and SARS‐3a 23, has been developed for the detection of SARS‐CoV in formalin‐fixed, paraffin‐embedded tissue sections (J, showing immunohistochemical staining with an anti‐peptide antibody against N, original magnification ×600)
Figure 3.
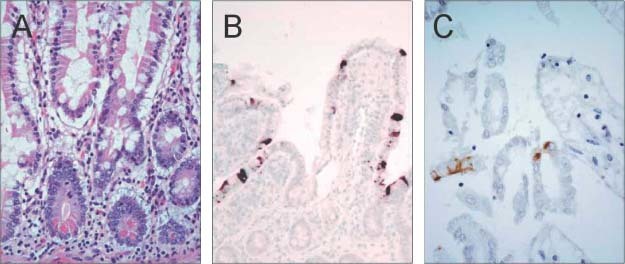
The small intestine shows no gross or microscopic pathology in terminal cases of SARS. Apart from autolytic changes, light microscopy reveals no specific abnormalities in the small bowel muocsa (A, original magnification ×400). However, SARS‐CoV can be detected on the surface enterocytes using in situ hybridization (B, with a DNA probe against the M gene, original magnification ×600 22) or immunohistochemical staining (C, using anti‐peptide antibody against SARS‐3a, original magnification ×600 23)
It is clear that our understanding of the pathology of SARS is incomplete. An obvious large gap is the lack of information on the early pathological changes of SARS. During the epidemic, very few biopsies were obtained from patients with clinically active SARS.
Animal models in the understanding of SARS
The study of animal models is important in a number of ways. It has allowed the establishment of SARS‐CoV as the aetiological agent 30. It also provides controlled conditions for the study of early changes in the disease. Initial studies of macaque models were promising. The histology of infected lung tissue is similar to that in humans 31, 32, 33. Both acute and organized stages of diffuse alveolar damage were seen when the macaques were sacrificed on the sixth day after a heavy dose of the virus. SARS‐CoV was detected in the alveolar epithelial cells and in the intra‐alveolar syncytial cells. However, detailed morphological studies and viral distribution in other organs in these animal studies are lacking. In studies involving longer observation times, the disease in macaque models appears self‐limiting and different from the genuine human disease. The usefulness of the macaque as a model of the disease remains to be established 32, 33.
Civet cats, domestic cats, and ferrets are thought to have been potential reservoirs of the virus during the epidemics and subsequent smaller outbreaks in mainland China 34. The animal coronavirus identified in civet cats shows high sequence identity with, but is distinct from, SARS‐CoV 2, 35. Recent evidence also suggests that wide Chinese horseshoe bats harbour a closely related bat‐SARS‐CoV which might also act as the animal reservoir 36. Again, details concerning the distribution of virus in different organs in these animals and the information on the pathology in the diseased or carrier animals are, surprisingly, sparse 32, 35.
Other common small laboratory animal models, such as the mouse, are not particularly useful. SARS‐CoV has a low virulence in ordinary laboratory mice and very high levels of inoculation are required to produce self‐limiting diseases. These features may be related to differences in the affinities of SARS‐CoV for human receptors and their murine homologues 37.
Pathogen factors: specific features of SARS‐CoV
SARS‐CoV uses a protector of lung damage, angiotensin‐converting enzyme 2, as a receptor
Characterization of the functional cellular receptor of SARS‐CoV provides important clues to the pathogenesis of SARS. Angiotensin‐converting enzyme 2 (ACE2) interacts directly with the Spike (S) proteins of the SARS‐CoV 38, 39, 40, 41, 42. The level of expression of ACE2 correlates with the efficiency of SARS‐CoV infection in cell culture models 42, 43, 44. ACE2 proteins are expressed by alveolar epithelial cells and by surface enterocytes of the small intestine 45, which are the primary target cells of SARS‐CoV. Studies in the intestine cell culture model, however, suggested that, in addition to ACE2, unknown co‐factors or co‐receptors are required to convey infectivity 46.
In addition to being a cellular receptor, ACE2 may contribute to the pathogenesis of DAD in SARS through its role in the tissue renin–angiotensin system. In a mouse model of alveolar damage induced by acid aspiration, the balance of the renin–angiotensin system appears to affect the development of DAD. ACE2, which acts as a negative regulator of the local renin–angiotensin system, protects the mouse lung against experimental damage 47, 48. SARS‐CoV co‐infection in these damaged animals down‐regulates ACE2 in the lungs of infected mice and the severity of lung damage can be alleviated by blocking the system 49. Exciting as these findings appear, the case of a new coronavirus, NL63, immediately provides an example that other factors are acting in the overall mechanism of lung damage. NL63 utilizes the same ACE2 protein as its receptor in the lung. However, infection with NL63 results in only minor cold symptoms and alveolar damage is rare 50. The insert/deletion genotype of the ACE gene was associated with DAD after SARS‐CoV infection in a small cohort of 44 patients 51. This association was, however, not replicated subsequently in a larger series 52. We also could not detect any association between the ACE2 genotype and disease severity in SARS‐CoV infection 53.
SARS‐CoV may also use the C‐type lectins as receptors for infecting immune cells
C‐type lectins, including CD209 and CD209L, are also SARS‐COV receptors: these were identified through the study of proteins that interact with the S (Spike) protein. CD209, also known as dendritic cell‐specific intercellular adhesion molecule‐grabbing non‐integrin (DC‐SIGN), was shown to mediate viral entry in a lentiviral pseudo‐type experimental model 54. In Chinese hamster ovary (CHO) cells expressing a human lung cDNA library, S protein and its fragments interacted directly with a second related cell surface glycoprotein, CD209L, also known as L‐SIGN or DC‐SIGNR 55. CD209L acts in conjunction with LSECtin (liver and lymph node sinusoidal endothelial cell C‐type lectin) and enhances viral infection 56. Tissue cultures expressing CD209 or CD209L were also susceptible to SARS‐CoV infection 54, 55, 57.
The possible involvement of dendritic cells is particularly interesting. Although SARS‐CoV does not replicate in dendritic cells, these cells may act as a reservoir and distribute the virus to other cell types 54, 58. This is an attractive concept and similar biological behaviours have been proposed for human immunodeficiency virus I (HIV I) 59. No SARS‐CoV has been detected in dendritic cells in autopsy and biopsy studies reported so far.
The unique 3′ end of the SARS‐CoV may hold the key to specific viral behaviours
The genome of SARS‐CoV consists of a single 27.69 kb positive‐strand RNA. The genomic sequences derived from different phases of the SARS epidemic revealed no association with sequence variation and virulence 60, 61.
There are two large open reading frames (ORFs) and 12 potential ORFs in the SARS‐CoV genome. The two large ORFs encode non‐structural proteins involved in replication. These proteins have relatively higher homologies to known coronaviruses. The remaining 12 ORFs are squeezed into the 3′ end of the genome. These ORFs include four genes encoding known structural proteins (envelope, membrane, nucleocapsid, and spike proteins, respectively). The remaining potential ORFs encode hypothetical SARS‐CoV‐specific proteins which lack obvious sequence similarity to known proteins 62, 63. The functions of these hypothetical proteins and their roles in SARS pathogenesis remain obscure 64, 65. Antibodies against some of these putative proteins, notably SARS3a and SARS6, can be detected in the serum of SARS patients 66. There is also evidence suggesting that a number of these proteins, including SARS3a, 3b, 7a, and 9b, were expressed in pneumocytes and enterocytes in deceased patients 23. However, differential expression patterns of these proteins in cell types showing different responses to SARS‐CoV infection have not been confirmed.
By expressing the hypothetical proteins individually in tissue culture, we are beginning to see data on the cellular functions of these proteins. SARS3a appears to be important in mediating apoptosis in some cell types 67. The SARS3a protein is incorporated into the viron particle and may also act as one of the structural proteins 68, 69, 70. Through an unknown mechanism, host cells overexpressing SARS3a have increased expression of fibrinogen mRNA 71. SARS7a has been implied in mediating apoptosis through the caspase‐dependent pathways 72.
Host responses are important in SARS‐CoV infection
The effect of SARS‐CoV infection varies in different cell types. Apoptosis and syncytial formation are seen in infected monkey renal epithelial cells (Vero E6) 67. Persistent infection with no change in cellular morphology or doubling time was detected in the colon cancer cell line LoVo 46. In clinical specimens, SARS‐CoV was detected in the lungs and small intestine. Severe cellular damage is characteristically detected in the lungs of SARS patients, while no morphological changes are observed in the small intestine. The basis of these differences in cellular responses is not clear. The tissue/cellular tropism may be partly related to differential expression of membrane receptors for the SARS‐CoV 22. These observations highlight the importance of host cell responses in SARS‐CoV infection. It is also clear from these observations that cytopathic damage alone cannot explain the pathogenesis of SARS.
The marked heterogeneity of the disease course and outcome after SARS infection suggests that host responses may play an important role in pathogenesis. DAD or ARDS appears to be a common pathway of lung parenchyma damage initiated by a variety of aetiologies, including SARS‐CoV infection itself, systemic sepsis, shock, and direct lung contusion. Once an inflammatory process reaches a certain intensity, it may self‐perpetuate. The cellular inflammatory infiltrate releases toxic metabolites and proteolytic enzymes, which may cause further damage to the lung parenchyma. The surrounding inflamed capillaries launch the coagulation cascade and recruit more immune cells 73, 74.
Immune‐mediated damage may be the main key to SARS pathology
Our previous investigation in the 1997 H5N1 influenza outbreak showed that patients who died of the disease had lymphoid depletion associated with marked elevation of circulating concentrations of cytokines, including interleukin‐6 (IL‐6), IL‐2 receptor, and interferon‐gamma 75. With the observation of characteristic lymphopenia in SARS, it has been postulated that the SARS‐CoV may similarly trigger an exaggerated hyper‐cytokinemic response in patients with DAD after viral infection 20. Current understanding indicates that patients with a more intense immune response are those at risk of a poor outcome, as the immune system also mounts a profound reaction to the bystander, the lung parenchyma, and causes DAD 76. SARS patients have variable humoral responses to individual epitopes 66. However, early sero‐conversion and high peak total SARS‐CoV IgG levels were associated with more severe disease in a cohort of 325 patients 77. Hence, particularly strong humoral responses to SARS‐CoV infection might not be protective but, perhaps, might be harmful to the host. The specific epitopes upon which these ‘damaging’ antibodies act await further characterization.
There is evidence that disarray of the immune system towards the host's own antigens may play a role in the pathology of SARS. In the early phase, within 1 week of SARS‐CoV infection, IgM and IgG autoantibodies against antigens located in the cytoplasm of lung epithelial cells (Figure 4) were detected in the sera of 36 Chinese SARS patients (Lo, unpublished observations). In another cohort of 22 SARS patients, immune activity against antigens from lung epithelial cell lines and endothelial cell lines was found in some patients' sera obtained approximately 1 month after infection 78. Moreover, high levels of these autoimmune activities in the sera were shown to be cytotoxic to lung epithelial cells and endothelial cells in culture. Autoimmune antibodies may be important in mediating tissue damage at certain stages of the disease. The cause of the autoimmunity is not fully understood. These autoantibodies may be the result of humoral responses to innate antigens exposed accidentally during direct damage of the lung and, perhaps, the endothelium by SARS‐CoV. Alternatively, autoimmunity may be due to cross‐reactivity of antibodies against some specific epitopes of the SARS‐CoV proteins.
Figure 4.

Serum taken from SARS patients during the acute phase of the disease contains IgG against cytoplasmic antigens of pneumocytes. Application of acute phase serum as a primary antibody lights up the cytoplasm of pneumocytes of autopsy adult lung sections (A, original magnification ×1000) as well as fetal lung sections (B, original magnification ×1000)
Chemokines are important immune mediators for lung pathology in SARS
The chemokines are a family of small proteins that play important roles in intercellular signalling and chemotaxis. Based on their protein sequences, they are broadly divided into α‐chemokines with a common C–X–C (cysteine–other–cysteine) structure of amino acid residues near the amino‐terminus which interacts predominantly with neutrophils, and β‐chemokines with a C–C (cysteine–cysteine) structure interacting with mononuclear cells. Recently, chemokines have been recognized for their roles in integrating the innate and adaptive immune responses to viral infection through a cytokine‐to‐chemokine‐to‐cytokine signalling cascade 79, 80, 81.
A global view of the spectrum of expression of the immune mediators was studied in SARS by measuring the circulating concentrations of these mediators at different stages of the disease. Most cytokines showed only transient and short‐lived activation in patients after SARS‐CoV infection 82. Even in patients who developed DAD, most cytokine concentrations were not significantly increased 83. In contrast, circulating concentrations of several chemokines, including CXCL9 (chemokine C–X–C motif ligand 9 or monokine induced by γ‐interferon), CXCL10 (chemokine C–X–C motif ligand 10 or interferon‐inducible protein‐10), and CCL2 (C–C motif ligand 2 or monocyte chemoattractant protein‐1), were markedly increased in SARS patients 82, 84, 85. Remarkably, the circulating concentration of CXCL10 measured early after infection is an independent prognostic indicator of disease outcome 86. These chemokines therefore appear to be important elements of the pathogenesis of SARS.
In the lung tissues obtained from seven SARS patients who died 86, chemokines CXCL10 (Figure 5) and IL‐18 were markedly activated (25‐ and 40‐fold compared with controls, respectively). The important roles of chemokines are underscored by the findings in an experimental mouse model of SARS‐CoV infection in which CXCL10 and a neutrophil chemokine, CXCL8 (chemokine C–X–C motif ligand 8), were also markedly activated 87. These findings in SARS compare favourably with the specific situation in HIV patients with lung allograft rejection and interstitial alveolitis, in which similar activation of the chemokine CXCL10 and its receptor CXCR3 (chemokine C–X–C motif receptor 3) was also found 88, 89.
Figure 5.

Chemokines are aberrantly expressed in terminal cases of SARS. Immunohistochemical staining using a monoclonal antibody against CXCL10 (IP‐10) demonstrated overexpression of CXCL10 in the pneumocytes of SARS patients (A, original magnification ×600) but not in control autopsy lung (B, original magnification ×600)
Other than pneumocytes, chemokines are also expressed and secreted by various different cell types. Global gene expression profiles, generated by cDNA microarray analysis of peripheral blood mononuclear cells (PBMCs) after in vitro exposure to SARS‐CoV, also reveal the importance of chemokine activation. Within 1 day after exposure to the virus, a number of chemokines (including CXCL10, CXCL9, and CCL2) were activated 90. PBMCs and macrophages do not support productive infection as viral replication is abortive and no infectious virus is produced. The roles of these cell types in the pathogenesis of SARS remain to be clarified. Nonetheless, these easily obtainable cell types provide convenient experimental models and allow some insight into the patterns of host responses to the infection to be studied. Similar findings were also reported in other cell types, such as dendritic cells, where the cytokine expression profiles are predominantly of inflammatory chemokines CCL3 (chemokine C–C motif ligand 3), CCL5 (chemokine C–C motif ligand 5), CXCL10, and CCL2. Unlike the usual response of dendritic cells to viral infection, anti‐viral cytokines, including IFN‐α (interferon‐alpha), IFN‐β, IFN‐γ, and IL‐12B, were not activated 58.
Immunogenetics of the host may affect the severity of SARS
Other than using serum inflammatory mediators to reflect the different degree of host inflammatory reaction during an infection, the intensity of the immune response is also genetically determined. The difference in genetic makeup between individuals is mostly accounted for by single base differences (single nucleotide polymorphisms, SNPs). Many studies have shown an association between SNPs and predisposition to ARDS, and survival after sepsis or other insults 91, 92. In the context of predisposition to ARDS after trauma, among parameters such as circulating concentrations of IL‐1, tumour necrosis factor and plasminogen activator inhibitor‐1 (PAI‐1), and the genotype of PAI‐1, insertion alleles at the promoter of PAI‐1 were associated with high concentrations of PAI‐1 in the plasma and a poor survival rate 93. In addition to PAI‐1, other genetic polymorphisms, such as angiotensin‐converting enzyme (ACE) 94, CD14 95, surfactant protein 96, and HLA genotypes 97, are also associated with predisposition to, severity, and outcome of ARDS. Although SASR‐CoV utilizes ACE2 as its receptor and ACE2 is known to be an important protector of lung damage in experimental ARDS, we and other groups found no solid association between alleles of the two ACE genes (ACE and ACE2) and the severity of ARDS after SARS infection 52, 53, 98.
Several immunogenetic studies have been reported in association with SARS infection. Among 37 Taiwanese SARS patients, HLA‐B*4601 was associated with both predisposition to infection and severity of infection 99. However, the association of this allele was replicated in another Chinese community of Hong Kong involving 90 SARS patients 100. HLA‐B*0703 was found to be a predisposition allele in the latter study. It should be noted that this latter allele is rare and is found in ∼3% of the general population. Hence, this allele cannot be considered a major predisposition factor for SARS infection 100. Immunogenotype may play a role in determining the severity of host responses. There is considerable variability in the prevalence of immunogenotypes among different populations and the significance of detecting so‐called ‘predisposing’ alleles in clinical practice is questionable. More studies are needed to uncover fully the real genetic determinants for both predisposition to infection and the host–pathogen interaction after infection with the virus.
Conclusion
A considerable amount of knowledge of SARS infection has accumulated as a result of almost 3 years of research since the emergence of SARS. Some key issues about the pathogen, SARS‐CoV, have been addressed. These include the rapid discovery of SARS‐CoV receptors and the actions of some of the specific viral proteins in different host cells. Understanding the molecular basis of differences in host cell responses to SARS‐CoV infection will be crucial in delineating its pathogenesis. It is also clear from clinical and experimental data that host immune responses may be the key determinant in disease progression after initial SARS‐CoV infection. Future studies aimed at characterization of the variability of host immune and inflammatory responses will be important in understanding this new emerging infectious disease.
Acknowledgements
SARS research projects are supported by Research Grant Council CUHK4507/03M to NLST, and the Health, Welfare and Food Bureau of the Hong Kong Special Administration Region Government (NLST and KFT; Ref 01030702).
AWIL and KFT are enlisted inventors of Hong Kong Patent No 1072161 and US patent application 60/579,333 (filed on 14 June 2004). Ethical approvals were granted for research in the authors' individual laboratories involving SARS patients, following the ethical standards of the local committee on human experimentation and in accordance with the Helsinki Declaration of 1975, as revised in 1983.
References
- 1. Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt GM, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med 2003; 348: 1986–1994. [DOI] [PubMed] [Google Scholar]
- 2. Stadler K, Masignani V, Eickmann M, Becker S, Abrignani S, Klenk HD, et al. SARS—beginning to understand a new virus. Nature Rev Microbiol 2003; 1: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hui DS, Sung JJ. Treatment of severe acute respiratory syndrome. Chest 2004; 126: 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong RS, Wu A, To KF, Lee N, Lam CW, Wong CK, et al. Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. Br Med J 2003; 326: 1358–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leung WK, To KF, Chan PK, Chan HL, Wu AK, Lee N, et al. Enteric involvement of severe acute respiratory syndrome‐associated coronavirus infection. Gastroenterology 2003; 125: 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan HL, Leung WK, To KF, Chan PK, Lee N, Wu A, et al. Retrospective analysis of liver function derangement in severe acute respiratory syndrome. Am J Med 2004; 116: 566–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wong WM, Ho JC, Ooi GC, Mok T, Chan J, Hung IF, et al. Temporal patterns of hepatic dysfunction and disease severity in patients with SARS. J Am Med Assoc 2003; 290: 2663–2665. [DOI] [PubMed] [Google Scholar]
- 8. Chu KH, Tsang WK, Tang CS, Lam MF, Lai FM, To KF, et al. Acute renal impairment in coronavirus‐associated severe acute respiratory syndrome. Kidney Int 2005; 67: 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Farcas GA, Poutanen SM, Mazzulli T, Willey BM, Butany J, Asa SL, et al. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis 2005; 191: 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J, et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS‐CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol 2004; 203: 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu W, Tang F, Fontanet A, Zhan L, Zhao QM, Zhang PH, et al. Long‐term SARS coronavirus excretion from patient cohort, China. Emerg Infect Dis 2004; 10: 1841–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leung GM, Chung PH, Tsang T, Lim W, Chan SK, Chau P, et al. SARS‐CoV antibody prevalence in all Hong Kong patient contacts. Emerg Infect Dis 2004; 10: 1653–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiu WK, Cheung PC, Ng KL, Ip PL, Sugunan VK, Luk DC, et al. Severe acute respiratory syndrome in children: experience in a regional hospital in Hong Kong. Pediatr Crit Care Med 2003; 4: 279–283. [DOI] [PubMed] [Google Scholar]
- 14. Cheung OY, Chan JW, Ng CK, Koo CK. The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology 2004; 45: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 2003; 200: 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, Reid AH, et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol 2003; 34: 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsiao CH, Wu MZ, Hsieh SW, Chien LC, Hwang KC, Su IJ. Clinicopathology of severe acute respiratory syndrome: an autopsy case report. J Formos Med Assoc 2004; 103: 787–792. [PubMed] [Google Scholar]
- 18. Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 2005; 18: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lang ZW, Zhang LJ, Zhang SJ, Meng X, Li JQ, Song CZ, et al. A clinicopathological study of three cases of severe acute respiratory syndrome (SARS). Pathology 2003; 35: 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicholls JM, Poon LL, Lee KC, Ng WF, Lai ST, Leung CY, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003; 361: 1773–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tse GM, To KF, Chan PK, Lo AW, Ng KC, Wu A, et al. Pulmonary pathological features in coronavirus associated severe acute respiratory syndrome (SARS). J Clin Pathol 2004; 57: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. To KF, Tong JH, Chan PK, Au FW, Chim SS, Chan KC, et al. Tissue and cellular tropism of the coronavirus associated with severe acute respiratory syndrome: an in‐situ hybridization study of fatal cases. J Pathol 2004; 202: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chan WS, Wu C, Chow SC, Cheung T, To KF, Leung WK, et al. Coronaviral hypothetical and structural proteins were found in the intestinal surface enterocytes and pneumocytes of severe acute respiratory syndrome (SARS). Mod Pathol 2005; 18: 1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chong PY, Chui P, Ling AE, Franks TJ, Tai DY, Leo YS, et al. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: challenges in determining a SARS diagnosis. Arch Pathol Lab Med 2004; 128: 195–204. [DOI] [PubMed] [Google Scholar]
- 25. Chow KC, Hsiao CH, Lin TY, Chen CL, Chiou SH. Detection of severe acute respiratory syndrome‐associated coronavirus in pneumocytes of the lung. Am J Clin Pathol 2004; 121: 574–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakajima N, Asahi‐Ozaki Y, Nagata N, Sato Y, Dizon F, Paladin FJ, et al. SARS coronavirus‐infected cells in lung detected by new in situ hybridization technique. Jpn J Infect Dis 2003; 56: 139–141. [PubMed] [Google Scholar]
- 27. Shieh WJ, Hsiao CH, Paddock CD, Guarner J, Goldsmith CS, Tatti K, et al. Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS‐associated coronavirus in lung of a fatal case of severe acute respiratory syndrome in Taiwan. Hum Pathol 2005; 36: 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leung TW, Wong KS, Hui AC, To KF, Lai ST, Ng WF, et al. Myopathic changes associated with severe acute respiratory syndrome: a postmortem case series. Arch Neurol 2005; 62: 1113–1117. [DOI] [PubMed] [Google Scholar]
- 29. Chau TN, Lee KC, Yao H, Tsang TY, Chow TC, Yeung YC, et al. SARS‐associated viral hepatitis caused by a novel coronavirus: report of three cases. Hepatology 2004; 39: 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fouchier RA, Kuiken T, Schutten M, van Amerongen G, van Doornum GJ, van den Hoogen BG, et al. Aetiology: Koch's postulates fulfilled for SARS virus. Nature 2003; 423: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003; 362: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rowe T, Gao G, Hogan RJ, Crystal RG, Voss TG, Grant RL, et al. Macaque model for severe acute respiratory syndrome. J Virol 2004; 78: 11401–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qin C, Wang J, Wei Q, She M, Marasco WA, Jiang H, et al. An animal model of SARS produced by infection of Macaca mulatta with SARS coronavirus. J Pathol 2005; 206: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. WHO . In Severe Acute Respiratory Syndrome (SARS). World Health Organization; 2004; http://www.who.int/csr/en/snrs/en. [Google Scholar]
- 35. Martina BE, Haagmans BL, Kuiken T, Fouchier RA, Rimmelzwaan GF, Van Amerongen G, et al. Virology: SARS virus infection of cats and ferrets. Nature 2003; 425: 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, et al. Severe acute respiratory syndrome coronavirus‐like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A 2005; 102: 14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li W, Greenough TC, Moore MJ, Vasilieva N, Somasundaran M, Sullivan JL, et al. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin‐converting enzyme 2. J Virol 2004; 78: 11429–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426: 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193‐amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin‐converting enzyme 2. J Biol Chem 2004; 279: 3197–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS‐CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun 2003; 312: 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prabakaran P, Xiao X, Dimitrov DS. A model of the ACE2 structure and function as a SARS‐CoV receptor. Biochem Biophys Res Commun 2004; 314: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, Fey GH, et al. Susceptibility to SARS coronavirus S protein‐driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res Commun 2004; 319: 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moore MJ, Dorfman T, Li W, Wong SK, Li Y, Kuhn JH, et al. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin‐converting enzyme 2. J Virol 2004; 78: 10628–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nie Y, Wang P, Shi X, Wang G, Chen J, Zheng A, et al. Highly infectious SARS‐CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem Biophys Res Commun 2004; 321: 994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004; 203: 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chan PK, To KF, Lo AW, Cheung JL, Chu I, Au FW, et al. Persistent infection of SARS coronavirus in colonic cells in vitro . J Med Virol 2004; 74: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin‐converting enzyme 2 protects from severe acute lung failure. Nature 2005; 436: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burrell LM, Johnston CI, Tikellis C, Cooper ME. ACE2, a new regulator of the renin–angiotensin system. Trends Endocrinol Metabol 2004; 15: 167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus‐induced lung injury. Nature Med 2005; 11: 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pohlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A 2005; 102: 7988–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Itoyama S, Keicho N, Quy T, Phi NC, Long HT, Ha le D, et al. ACE1 polymorphism and progression of SARS. Biochem Biophys Res Commun 2004; 323: 1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chan KC, Tang NL, Hui DS, Chung GT, Wu AK, Chim SS, et al. Absence of association between angiotensin converting enzyme polymorphism and development of adult respiratory distress syndrome in patients with severe acute respiratory syndrome: a case control study. BMC Infect Dis 2005; 5: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chiu RW, Tang NL, Hui DS, Chung GT, Chim SS, Chan KC, et al. ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome. Clin Chem 2004; 50: 1683–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, Schwartz O, et al. pH‐dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC‐SIGN. J Virol 2004; 78: 5642–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jeffers SA, Tusell SM, Gillim‐Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L‐SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A 2004; 101: 15748–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gramberg T, Hofmann H, Moller P, Lalor PF, Marzi A, Geier M, et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005; 340: 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marzi A, Gramberg T, Simmons G, Moller P, Rennekamp AJ, Krumbiegel M, et al. DC‐SIGN and DC‐SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J Virol 2004; 78: 12090–12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine upregulation in SARS coronavirus infected human monocyte derived dendritic cells. Blood 2005; 106: 2366–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Kooyk Y, Geijtenbeek TB. DC‐SIGN: escape mechanism for pathogens. Nature Rev Immunol 2003; 3: 697–709. [DOI] [PubMed] [Google Scholar]
- 60. Chiu RW, Chim SS, Lo YM. Molecular epidemiology of SARS—from Amoy Gardens to Taiwan. N Engl J Med 2003; 349: 1875–1876. [DOI] [PubMed] [Google Scholar]
- 61. Chinese SMEC . Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 2004; 303: 1666–1669. [DOI] [PubMed] [Google Scholar]
- 62. Marra MA, Jones SJ, Astell CR, Holt RA, Brooks‐Wilson A, Butterfield YS, et al. The Genome sequence of the SARS‐associated coronavirus. Science 2003; 300: 1399–1404. [DOI] [PubMed] [Google Scholar]
- 63. Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003; 300: 1394–1399. [DOI] [PubMed] [Google Scholar]
- 64. Tan YJ, Teng E, Shen S, Tan TH, Goh PY, Fielding BC, et al. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J Virol 2004; 78: 6723–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fielding BC, Tan YJ, Shuo S, Tan TH, Ooi EE, Lim SG, et al. Characterization of a unique group‐specific protein (U122) of the severe acute respiratory syndrome coronavirus. J Virol 2004; 78: 7311–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chow SC, Ho CY, Tam TT, Wu C, Cheung T, Chan PK, et al. Specific epitopes of the structural and hypothetical proteins elicit variable humoral responses in SARS patients. J Clin Pathol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Law PT, Wong CH, Au TC, Chuck CP, Kong SK, Chan PK, et al. The 3a protein of severe acute respiratory syndrome‐associated coronavirus induces apoptosis in Vero E6 cells. J Gen Virol 2005; 86: 1921–1930. [DOI] [PubMed] [Google Scholar]
- 68. Shen S, Lin PS, Chao YC, Zhang A, Yang X, Lim SG, et al. The severe acute respiratory syndrome coronavirus 3a is a novel structural protein. Biochem Biophys Res Commun 2005; 330: 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ito N, Mossel EC, Narayanan K, Popov VL, Huang C, Inoue T, et al. Severe acute respiratory syndrome coronavirus 3a protein is a viral structural protein. J Virol 2005; 79: 3182–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu CJ, Chen YC, Hsiao CH, Kuo TC, Chang SC, Lu CY, et al. Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus. FEBS Lett 2004; 565: 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tan YJ, Tham PY, Chan DZ, Chou CF, Shen S, Fielding BC, et al. The severe acute respiratory syndrome coronavirus 3a protein up‐regulates expression of fibrinogen in lung epithelial cells. J Virol 2005; 79: 10083–10087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tan YJ, Fielding BC, Goh PY, Shen S, Tan TH, Lim SG, et al. Overexpression of 7a, a protein specifically encoded by the severe acute respiratory syndrome coronavirus, induces apoptosis via a caspase‐dependent pathway. J Virol 2004; 78: 14043–14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bellingan GJ. The pulmonary physician in critical care * 6: the pathogenesis of ALI/ARDS. Thorax 2002; 57: 540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chollet‐Martin S. Polymorphonuclear neutrophil activation during the acute respiratory distress syndrome. Intensive Care Med 2000; 26: 1575–1577. [DOI] [PubMed] [Google Scholar]
- 75. To KF, Chan PK, Chan KF, Lee WK, Lam WY, Wong KF, et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol 2001; 63: 242–246. [DOI] [PubMed] [Google Scholar]
- 76. Headley AS, Tolley E, Meduri GU. Infections and the inflammatory response in acute respiratory distress syndrome. Chest 1997; 111: 1306–1321. [DOI] [PubMed] [Google Scholar]
- 77. Lee N, Chan PK, Ip M, Wong E, Ho J, Ho C, et al. Anti‐SARS‐CoV IgG response in relation to disease severity of severe acute respiratory syndrome. J Clin Virol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yang YH, Huang YH, Chuang YH, Peng CM, Wang LC, Lin YT, et al. Autoantibodies against human epithelial cells and endothelial cells after severe acute respiratory syndrome (SARS)‐associated coronavirus infection. J Med Virol 2005; 77: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 2005; 288: L3–L15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Salazar‐Mather TP, Hamilton TA, Biron CA. A chemokine‐to‐cytokine‐to‐chemokine cascade critical in antiviral defense. J Clin Invest 2000; 105: 985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Glass WG, Rosenberg HF, Murphy PM. Chemokine regulation of inflammation during acute viral infection. Curr Opin Allergy Clin Immunol 2003; 3: 467–473. [DOI] [PubMed] [Google Scholar]
- 82. Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 2004; 136: 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ward SE, Loutfy MR, Blatt LM, Siminovitch KA, Chen J, Hinek A, et al. Dynamic changes in clinical features and cytokine/chemokine responses in SARS patients treated with interferon alfacon‐1 plus corticosteroids. Antivir Ther 2005; 10: 263–275. [PubMed] [Google Scholar]
- 84. Jiang Y, Xu J, Zhou C, Wu Z, Zhong S, Liu J, et al. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med 2005; 171: 850–857. [DOI] [PubMed] [Google Scholar]
- 85. Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, et al. An interferon‐gamma‐related cytokine storm in SARS patients. J Med Virol 2005; 75: 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tang NL, Chan PK, Wong CK, To KF, Wu A, Sung YM, et al. Early enhanced expression of IP‐10 (CXCL‐10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome (SARS). Clin Chem 2005; 51: 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Glass WG, Subbarao K, Murphy B, Murphy PM. Mechanisms of host defense following severe acute respiratory syndrome‐coronavirus (SARS‐CoV) pulmonary infection of mice. J Immunol 2004; 173: 4030–4039. [DOI] [PubMed] [Google Scholar]
- 88. Agostini C, Facco M, Siviero M, Carollo D, Galvan S, Cattelan AM, et al. CXC chemokines IP‐10 and mig expression and direct migration of pulmonary CD8+/CXCR3+ T cells in the lungs of patients with HIV infection and T‐cell alveolitis. Am J Respir Crit Care Med 2000; 162: 1466–1473. [DOI] [PubMed] [Google Scholar]
- 89. Belperio JA, Keane MP, Burdick MD, Lynch JP 3rd, Zisman DA, Xue YY, et al. Role of CXCL9/CXCR3 chemokine biology during pathogenesis of acute lung allograft rejection. J Immunol 2003; 171: 4844–4852. [DOI] [PubMed] [Google Scholar]
- 90. Ng PC, Lam CW, Li AM, Wong CK, Leung TF, Cheng FW, et al. Chemokine response in children with SARS. Arch Dis Child 2005; 90: 422–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Marshall RP, Webb S, Hill MR, Humphries SE, Laurent GJ. Genetic polymorphisms associated with susceptibility and outcome in ARDS. Chest 2002; 121: 68S–69S. [DOI] [PubMed] [Google Scholar]
- 92. Villar J, Flores C, Mendez‐Alvarez S. Genetic susceptibility to acute lung injury. Crit Care Med 2003; 31: S272–S275. [DOI] [PubMed] [Google Scholar]
- 93. Menges T, Hermans PW, Little SG, Langefeld T, Boning O, Engel J, et al. Plasminogen‐activator‐inhibitor‐1 4G/5G promoter polymorphism and prognosis of severely injured patients. Lancet 2001; 357: 1096–1097. [DOI] [PubMed] [Google Scholar]
- 94. Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med 2002; 166: 646–650. [DOI] [PubMed] [Google Scholar]
- 95. Gibot S, Cariou A, Drouet L, Rossignol M, Ripoll L. Association between a genomic polymorphism within the CD14 locus and septic shock susceptibility and mortality rate. Crit Care Med 2002; 30: 969–973. [DOI] [PubMed] [Google Scholar]
- 96. Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, et al. Polymorphisms of human SP‐A, SP‐B, and SP‐D genes: association of SP‐B Thr131Ile with ARDS. Clin Genet 2000; 58: 181–191. [DOI] [PubMed] [Google Scholar]
- 97. Takatsuka H, Takemoto Y, Mori A, Okamoto T, Kanamaru A, Kakishita E. Common features in the onset of ARDS after administration of granulocyte colony‐stimulating factor. Chest 2002; 121: 1716–1720. [DOI] [PubMed] [Google Scholar]
- 98. Itoyama S, Keicho N, Hijikata M, Quy T, Phi NC, Long HT, et al. Identification of an alternative 5′‐untranslated exon and new polymorphisms of angiotensin‐converting enzyme 2 gene: lack of association with SARS in the Vietnamese population. Am J Med Genet A 2005; 136: 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lin M, Tseng HK, Trejaut JA, Lee HL, Loo JH, Chu CC, et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med Genet 2003; 4: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ng MH, Lau KM, Li L, Cheng SH, Chan WY, Hui PK, et al. Association of human‐leukocyte‐antigen class I (B*0703) and class II (DRB1*0301) genotypes with susceptibility and resistance to the development of severe acute respiratory syndrome. J Infect Dis 2004; 190: 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]