

Abstract
Visible light photocatalysis and electrocatalysis are two powerful strategies for the promotion of chemical reactions. Here, these two modalities are combined in an electrophotocatalytic oxidation platform. This chemistry employs a trisaminocyclopropenium (TAC) ion catalyst, which is electrochemically oxidized to form a cyclopropenium radical dication intermediate. The radical dication undergoes photoexcitation with visible light to produce an excited state species with oxidizing power (3.33 V vs SCE) sufficient to oxidize benzene and halogenated benzenes via single electron transfer (SET), resulting in C–H/N–H coupling with azoles. A rationale for the strongly oxidizing behaviour of the photoexcited species is provided, while the stability of the catalyst is rationalized by a particular conformation of the cis-2,6-dimethylpiperidine moieties.
Keywords: Electrophotocatalysis, radical dication, trisaminocyclopropenium ion, oxidation, C–H functionalization
COMMUNICATION
A trisaminocyclopropenium (TAC) ion is used as a potent electrophotocatalyst for the selective oxidative coupling of benzene and other arenes. The potent oxidizing power derives from the open-shell character of the intermediate TAC radical dication.
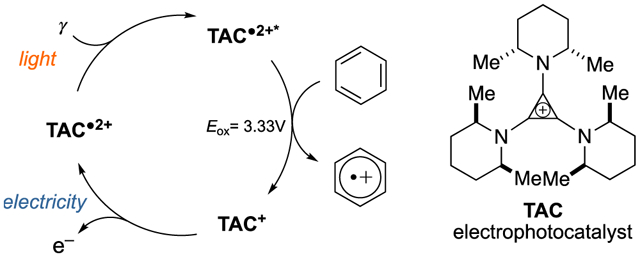
Due to the unique reactivity patterns of free radicals, single-electron oxidations offer a powerful means to induce chemical transformations.[1,2] In recent years, vigorous activity in the areas of visible light photocatalysis[3–4,5,6] and electrocatalysis[7–8,9,10] have enabled a range of powerful oxidative methods. With photocatalysis, the energy of visible light is leveraged to convert a ground-state photocatalyst (PC) into a strongly oxidizing photoexcited species, the potency of which can far exceed that of whatever terminal oxidant is employed (Fig. 1A). With electrocatalysis, an applied potential effects the oxidation of a substrate through the intermediacy of an electrocatalyst (EC) (Fig. 1B), which obviates the need for external oxidants and can improve the kinetics of electron transfer, prevent electrode passivation, and provide catalyst control for selective transformations. Given the substantial benefits of these two broad paradigms, processes that combine photocatalysis and electrocatalysis could be especially attractive. Nevertheless, the merger of electro- and photocatalysis in organic chemistry has rarely been achieved. Moutet and Reverdy demonstrated[11] the electrophotocatalytic oxidation of benzyl alcohol to benzaldehyde, albeit with only three turnovers and no reported yield. Meanwhile, Scheffold achieved the nucleophilic acylation of electron-deficient olefins using vitamin B12 as a reductive electrophotocatalyst.[12, 13] More recently, several elegant examples of the combination of electrochemical and photochemical processes were reported. For example, Xu demonstrated the alkylation of arenes with organotrifluoroborates using photoredox catalysis coupled with anodic re-oxidation,[14] and Stahl developed a Hofmann-Löffler-Freytag type C–H amination reaction using electrochemical iodination paired with photochemical homolysis.[15] Meanwhile, Grätzel and Hu have shown that the photoelectric effect can be utilized for the coupling of electron-rich arenes with azoles.[16]
Figure 1.
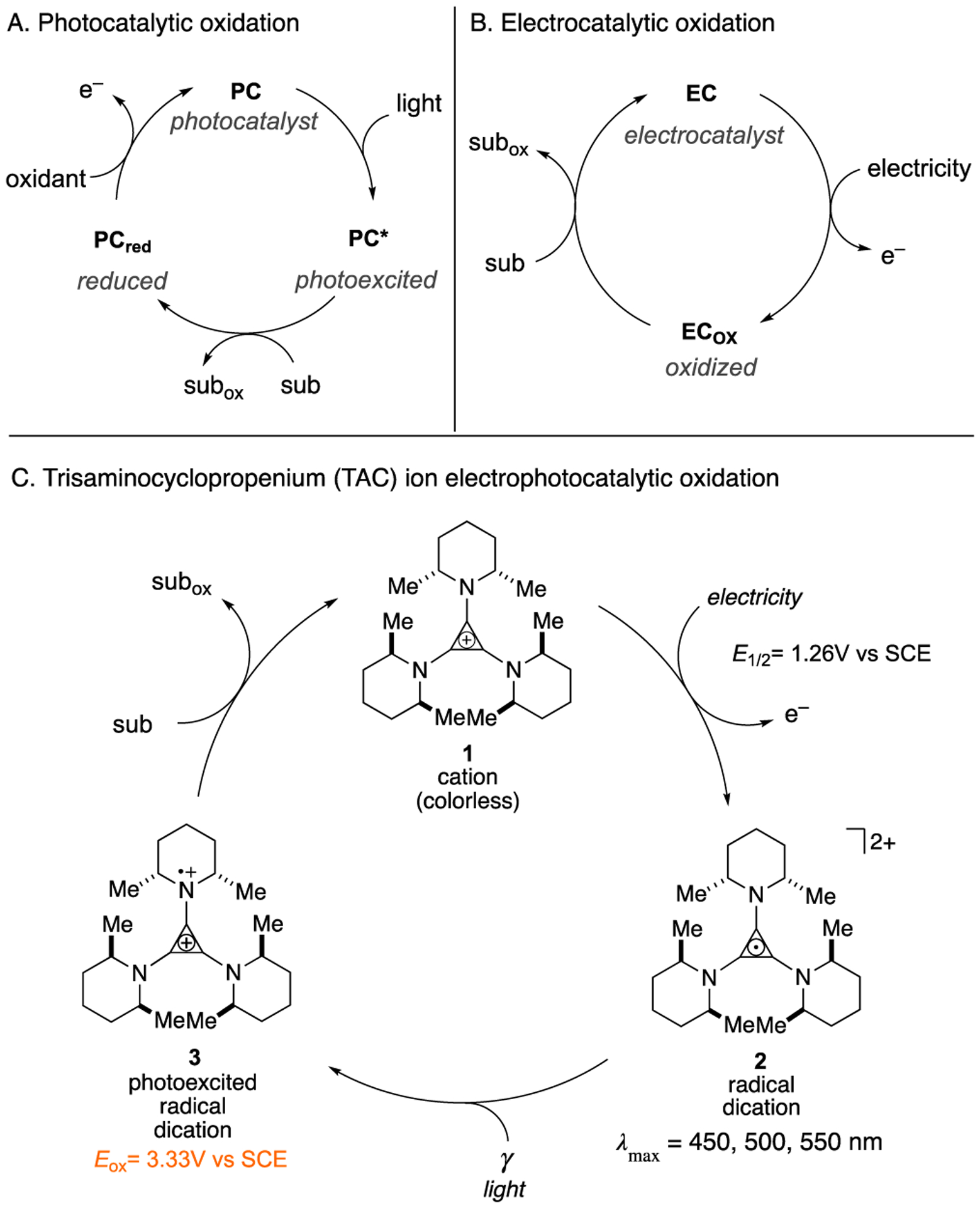
Generic paradigms for (A) photocatalytic and (B) electrocatalytic oxidations. (C) Electrophotocatalytic oxidation with TAC ion 1.
We envisioned the merger of light and electrical energy via the design of new single-component “electrophotocatalysts”. The major challenge in this endeavor resides in the identification of a species that (a) can undergo a facile one-electron redox event to yield a stable intermediate, (b) absorbs visible light once oxidized, (c) has a sufficiently long-lived excited state to interact with a substrate, and (d) is stable throughout the catalytic cycle. In this Communication, we show that trisaminocyclopropenium (TAC) ion 1[17] meets all of these criteria and can operate as a potent yet selective oxidative electrophotocatalyst (Figure 1C).
Trisaminocyclopropenium (TAC) ions[17] are unusual in that they can be reversibly oxidized to the corresponding stable radical dications. In 1971, Yoshida showed that these radical dications could be generated from the corresponding TAC monocations by electrochemical oxidation.[18] Johnson later measured the oxidation potential of a permethylated TAC to be only 1.12 V (vs. standard calomel electrode, SCE).[19] In 1975, Weiss reported that TAC radical dications generated by chemical oxidation formed deep red crystals that were stable enough for single crystal X-ray analysis.[20] Recently, Sanford demonstrated that TACs can serve as robust electrolytes, with possible applications in non-aqueous flow batteries.[21]
In our own work, we have found that trisaminocyclopropenium (TAC) ion 1 can be electrochemically oxidized (E1/2 = +1.26 V vs SCE) to the corresponding air-stable radical dication 2.[18–21] Given the deep red color of this species (λmax = 550, 500, 450 nm), we suspected it might function as a visible light photooxidant and therefore be a candidate for a potent new electrophotocatalyst. Indeed, based on the oxidation potential of 1 and the long wavelength absorption tail (600 nm) of 2,[22] the photoexcited TAC radical dication 3 has a calculated excited state reduction potential of E*1/2 = 3.33 V (vs SCE). For comparison, this value exceeds the excited state potentials of 9-mesityl-10-methylacridinium (E*1/2 = 2.06 V),[5] 3-cyano-1-methylquinolinium (E*1/2 = 2.72 V),[5] and even 2,6-dichloro-3,5-dicyanoquinone (DDQ) (E*1/2 = 3.18 V).[5] As such, we expected that 3 should be capable of oxidizing a variety of challenging substrates.
To test this idea, we first targeted the Nicewicz-type oxidative coupling of benzene (Fig. 2).[23] With an oxidation potential of 2.48 V (vs SCE), the controlled functionalization of benzene presents a major challenge for both electrochemistry[24] and photochemistry. [25,26] Previous instances of this type of coupling reaction by Nicewicz,[23] Lei,[27] and Grätzel and Hu[16] have been conducted with more easily oxidized arenes using the 9-mesityl-10-methylacridinium photocatalyst; however, the oxidative functionalization of benzene remains a challenge. We found that, using a cell voltage of 1.5 V (Eanode = 1.4 V)[28] and irradiation with a 23 W compact fluorescent light (CFL) in acetonitrile with acetic acid,[29] 8 mol% TAC 1 catalyzed the oxidative coupling of benzene (4) and pyrazole (5) to furnish product 6 cleanly in 65% yield (Fig. 2A). The counterelectrode product, hydrogen gas, could be seen as bubbles formed at the cathode. Crucially, control experiments showed that no reaction occurred without the catalyst (1), light, or current. Furthermore, direct electrolysis at higher potentials up to 3.0 V resulted in very low yield and the visible formation of polymeric material. The stark difference between direct electrolysis and the electrophotocatalytic approach can be appreciated by comparison of the 1H NMR spectra of the electrophotocatalytic reaction versus that of direct electrolysis (Figure 2B, spectra 1 and 3 respectively), and especially by visual comparison of the two (Figure 2C).
Figure 2.
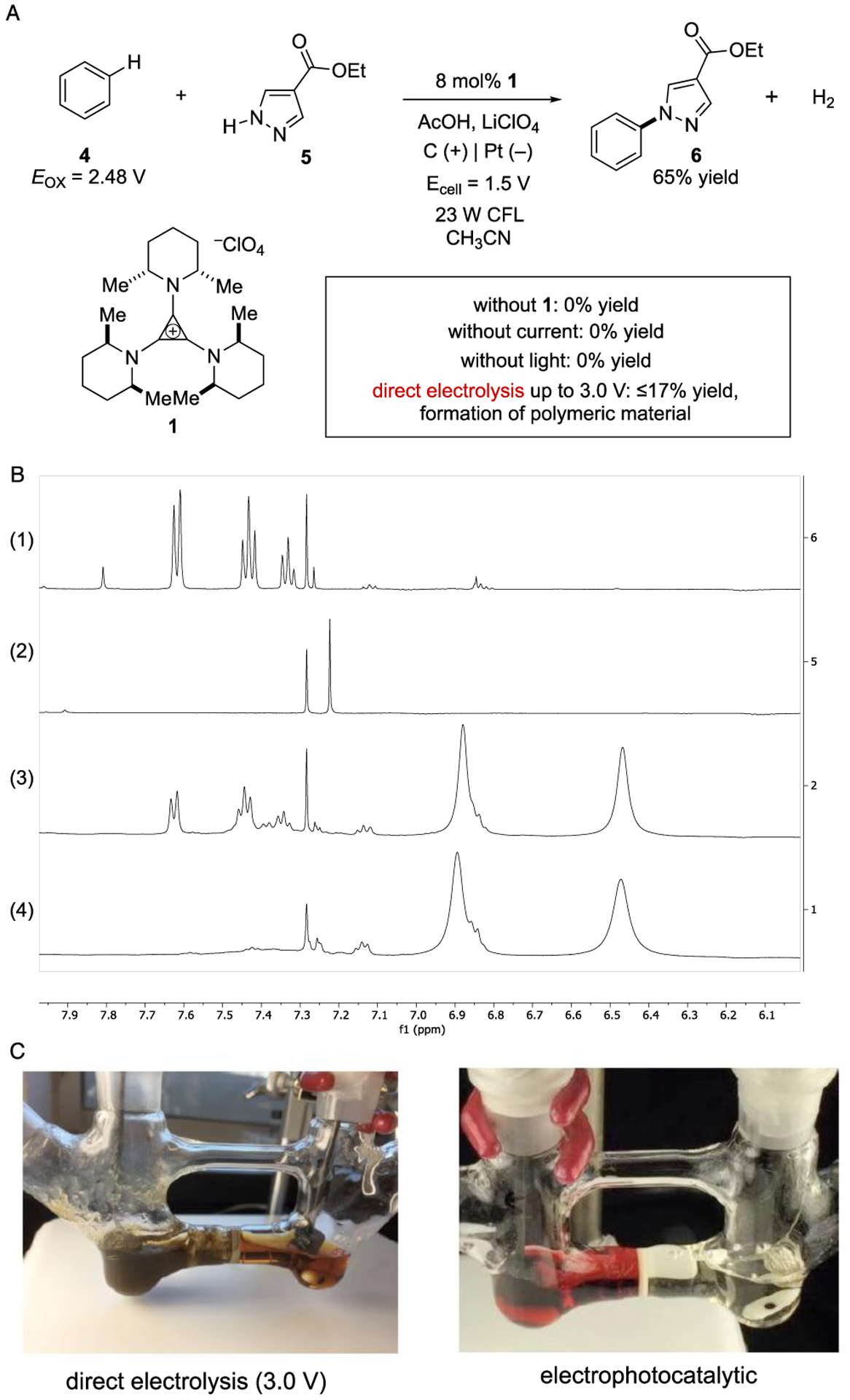
(A) Electrophotocatalytic C–H/N–H coupling of benzene and pyrazole 4. (B) 1H NMR spectra of (1) the reaction shown in Figure 2A; (2) the same conditions without catalyst 1 showing no reaction occurred; (3) the same reaction under direct electrolysis at 3.0 V; and (4) direct electrolysis of benzene at 3.0 V. (C) Visual comparison of attempted direct electrolysis and electrophotocatalytic reactions.
An investigation of reaction scope revealed that, in addition to the carboxyl group (Table 1, entry 1), aldehyde and ketone functional groups were well tolerated (entries 2 and 3). Oxidative phenylation of 4-chloropyrazole was also feasible (entry 4). Notably, we found that catalyst 1 was also capable of the oxidative functionalization of chlorobenzene (entries 5–9), and bromobenzene (entry 10). In these cases, the regioselectivity of the coupling was significantly influenced by the nature of the azole partner. In further probing the oxidizing capacity of this catalyst, we found that all three isomers of dichlorobenzene could also be made to engage in azole coupling (entries 11–13). Again, potentially sensitive carbonyl functionalities were found to be compatible with this process. The reaction of 1,2-dibromobenzene also gave rise to the coupled product (entry 14). In an attempt to find the limits of the oxidizing power of this catalyst, we examined the reaction of fluorobenzene and trifluorotoluene. Reaction of the former indeed led to the formation of the arylated adduct as a mixture of regioisomers (entry 15). On the other hand, trifluorotoluene did not undergo any observable reaction (entry 16), suggesting the trifluoromethyl group is too strongly deactivating for this catalyst. In addition to these challenging arenes, this method also works with more easily oxidized substrates such as mesitylene and other alkylated benzenes (entries 17–36).
Table 1.
Electrophotocatalytic coupling of arenes and nitrogen heteroaromatics.[a]
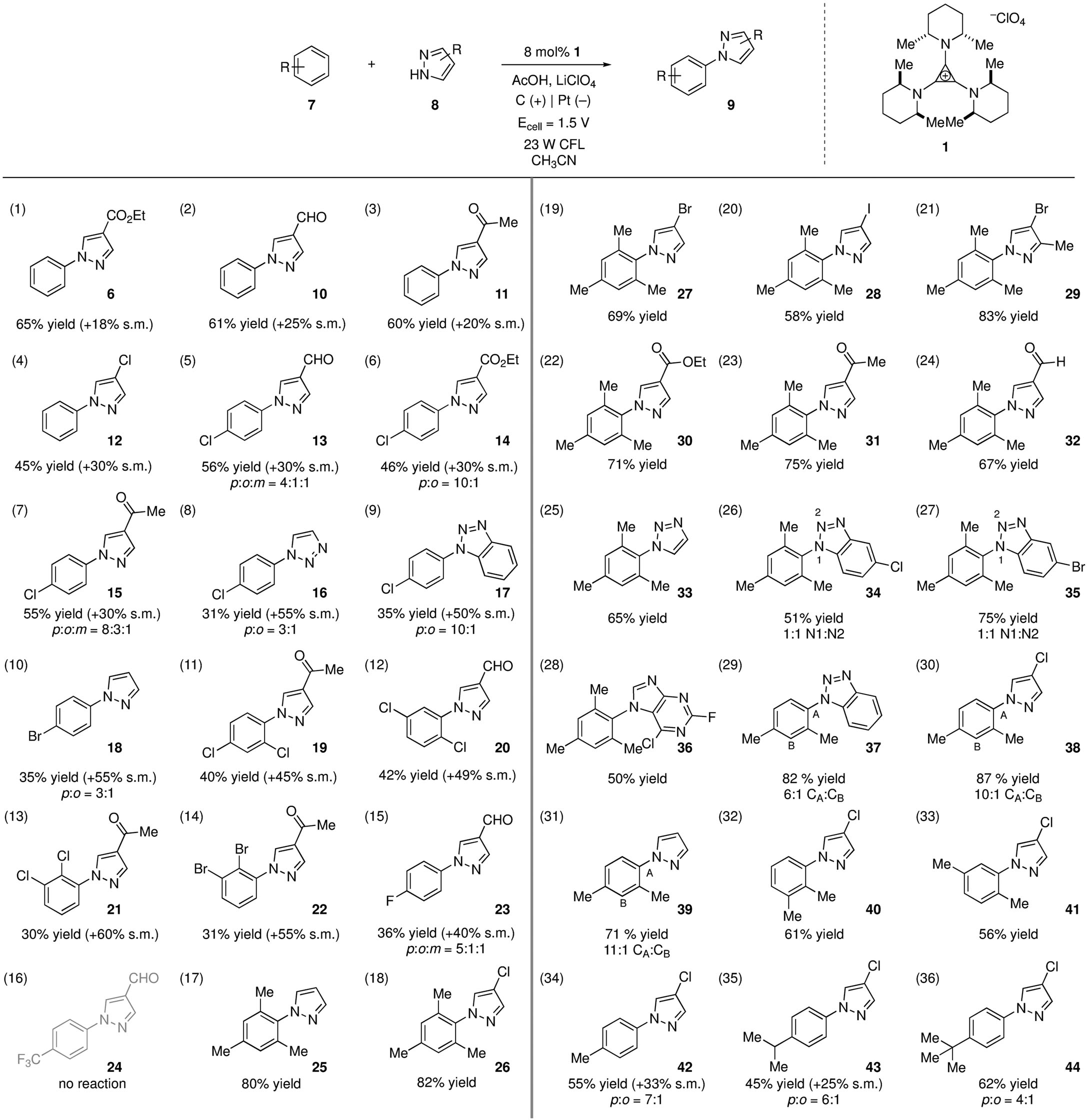
|
Entries 1–16 and 34–36 were conducted in a divided cell, while entries 17–33 were conducted in an undivided cell. Yields reflect isolated and purified material. Reactions were performed on a 0.4 mmol scale. Reaction times were generally 48–60 h. See supplementary materials for experimental details.
Notably, whereas reactions of benzene and the halogenated benzenes were optimally conducted in a divided cell, reactions with the alkylated benzenes could be performed in an undivided cell. We measured the consumed current for a reaction in the divided cell as 2.05 F/mol, which accords well with the theoretical value. On the other hand, an undivided cell reaction was found to consume 3.44 F/mol. We believe the excess charge consumption is due to the competing, unproductive reduction of the TAC radical dication 2 back to TAC 1. Additionally, we note that the measured current typically begins at 4–5 mA, but quickly drops to and remains at ≤1 mA for the rest of the reaction. Such a current profile is expected, assuming that the photooxidation or some subsequent step is rate-limiting, because once all of the catalyst is oxidized only a very low steady state amount of TAC 1 would remain to be oxidized.
A mechanistic rationale for these electrophotocatalytic reactions is shown in Figure 3. Electrochemical oxidation of the colorless TAC cation 1 generates the radical dication 2. Photoexcitation then leads to the reactive intermediate 3, which can oxidize benzene (4) via SET to produce the radical cation 45 with concomitant direct regeneration of 1. Nucleophilic trapping of 45 by pyrazole 5 followed by deprotonation leads to the radical 46. Further oxidation of this radical by 2 or directly at the anode, followed by rearomatization via loss of proton then reveals the coupled product 6. Although this rationale is in line with previous reports of this type of arene-azole coupling,[16, 17, 27] it is plausible that in some cases the pyrazole substrates, rather than the arenes, undergo initial oxidation given the higher oxidation potentials of many of the arenes in our study. We view this as a less likely possibility because the arene is used in vast excess, the chemistry works even with strongly deactivated pyrazoles (e.g. 5), we have observed small amounts of oligomeric products arising from benzene oxidation, and the attempted reaction of pyrazole by itself resulted in no conversion. However, the mechanistic possibilities for this reaction are clearly complex, and further study is warranted.
Figure 3.
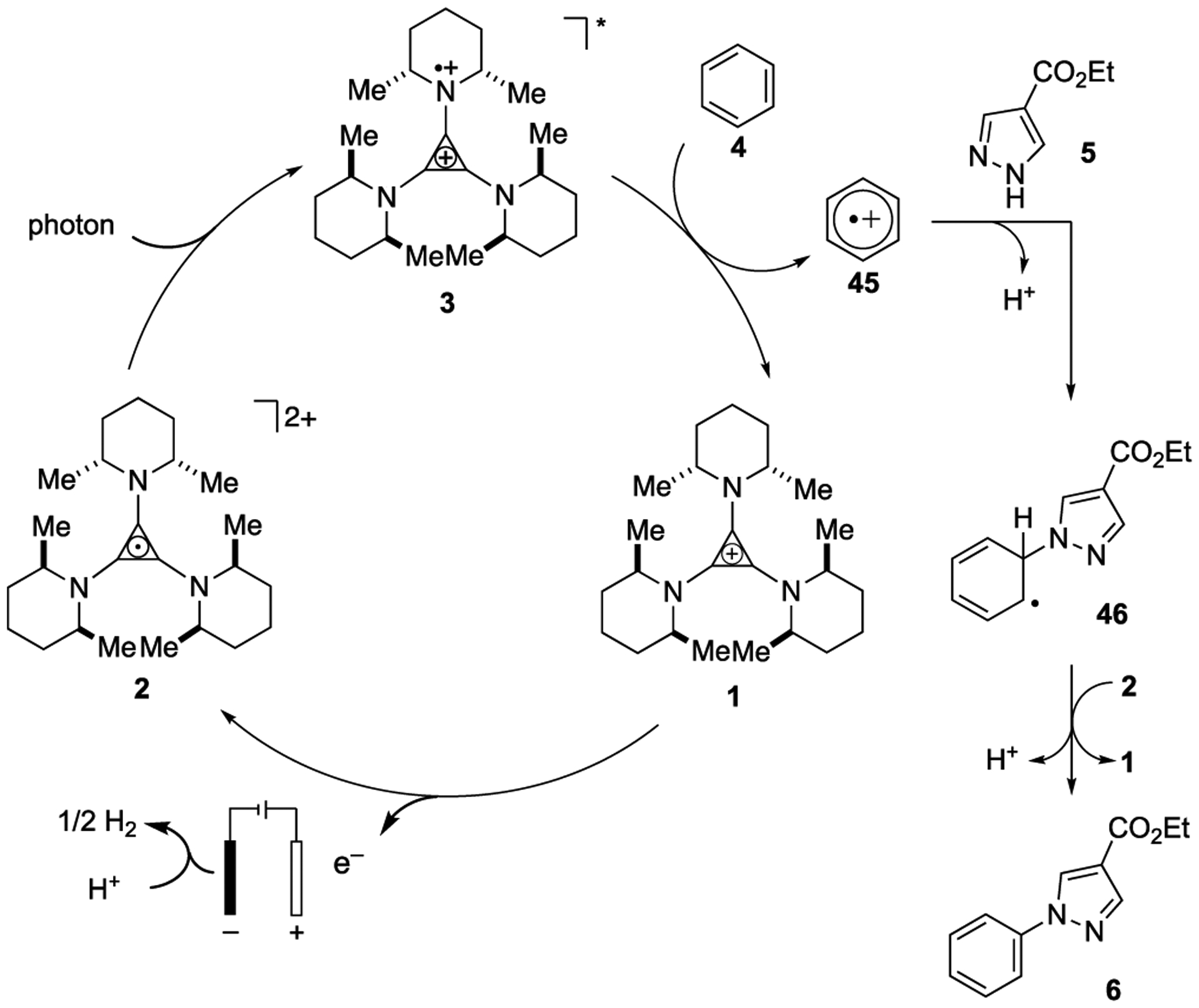
Mechanistic rationale.
Notably, radical dication 2 represents an unusual example of an open-shell, doublet photocatalyst (Figure 4A).[30] We used time-dependent density functional theory (TD-DFT) to characterize the low-lying excited electronic states of the TAC radical dication 2. As predicted,[20] the two lowest energy absorptions result from transitions from the nearly degenerate HOMO-1 and HOMO-2 orbitals to the SOMO (Figure 4B). These transitions result in an excited state SOMO-HOMO level inversion,[31] with the resulting hole residing in a sub-frontier orbital. The energy gained from repopulation of this low-lying orbital helps to explain the exceptionally potent oxidizing power of this photoexcited species.
Figure 4.
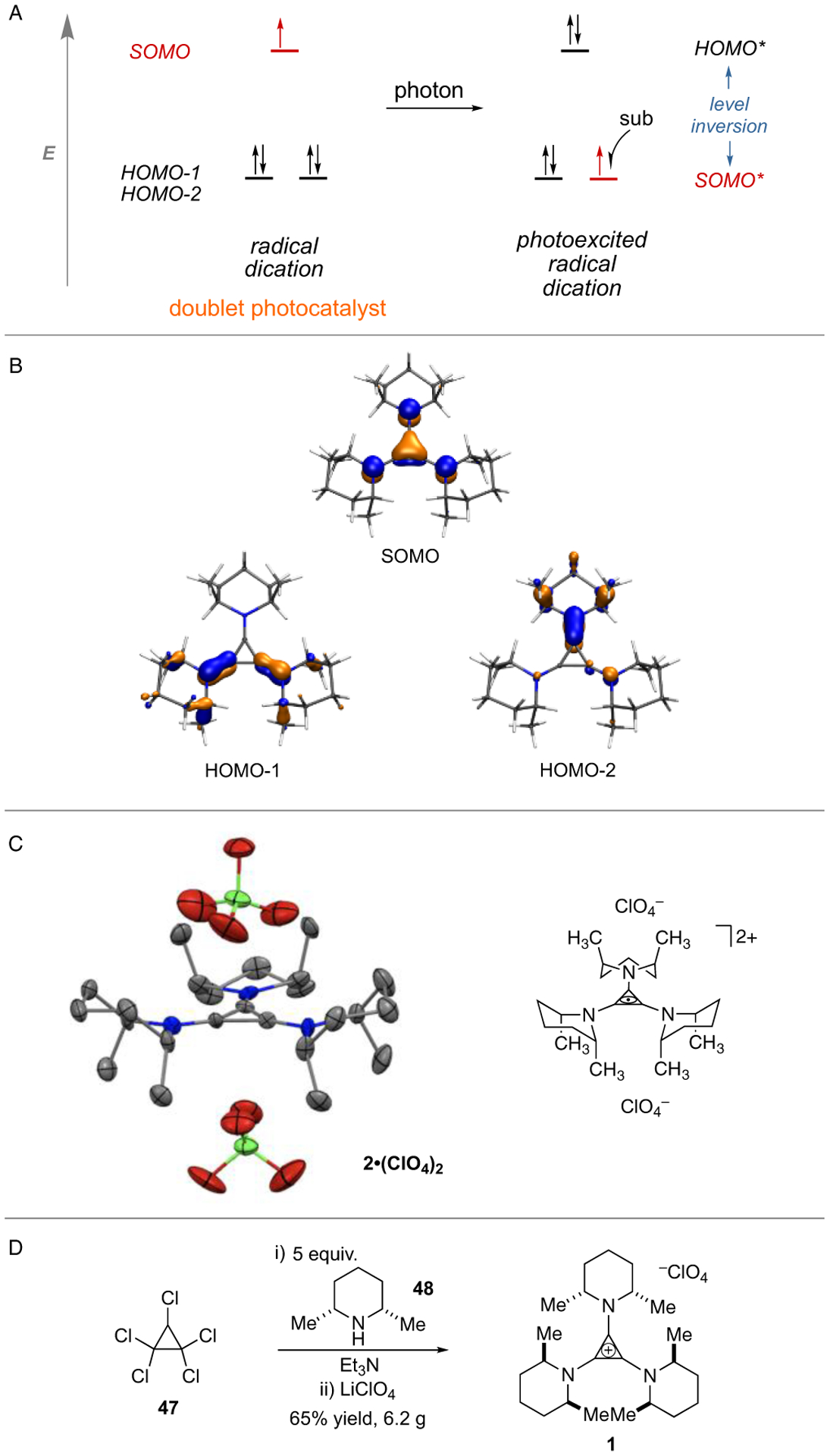
(A) Level inversion of a photoexcited TAC radical dication. (B) TD-DFT calculated orbitals of TAC radical dication 2. (C) X-ray structure of TAC radical dication 2. (D) Synthesis of TAC catalyst 1.
Despite such a strongly oxidizing character, the TAC catalyst is remarkably stable, remaining >95% intact during a typical electrophotocatalytic reaction. To explain this stability, we were able to obtain X-ray quality crystals of the TAC radical dication 2 as the bis-perchlorate salt (Figure 4C).[32] The most conspicuous aspect of this structure is the conformational bias of the three piperidine units, which places all six methyl substituents in the axial position to avoid what would otherwise be a severe allylic-type strain with the cyclopropenium ring. This conformation thus places the piperidine α-hydrogens orthogonal to the TAC π-system, rendering them unavailable for deprotonation or hydrogen atom abstraction, the common destructive process of aminyl radical cations. In support of this claim, we have calculated that the conformation in which one of the piperdine rings has the methyl substituents equatorial is 13.4 kcal higher in energy. We have also found that the TAC radical dications derived from 2-methylpiperidine or piperidine rapidly decompose upon irradiation.
Finally, we note that the TAC catalyst 1 is a bench stable solid that can be synthesized by the reaction of pentachlorocyclopropane (47) and cis-2,6-dimethylpiperidine (48) (both commercially available) in a one-pot procedure on a multi-gram scale and with no column purification (Figure 4D). The TAC catalyst 1 should thus be readily available for further investigations of electrophotocatalysis.
Supplementary Material
Acknowledgements
We thank Prof. Ged Parkin (Columbia University) for use of the X-ray diffractometer, Profs. Richard Friesner and David Reichman (Columbia University) for providing computational resources, and Prof. Song Lin (Cornell University) for helpful discussions.
Funding: Research reported in this publication was supported by the National Institutes of Health under R35 GM127135. The electrochemical studies of T.J.S. and C.N. were based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC001944. M.R. acknowledges the National Science Foundation for a Graduate Research Fellowship (DGE-16-44869). We thank the National Science Foundation (CHE-0619638) for acquisition of an X-ray diffractometer.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Studer A, Curran DP, Angew. Chem. Int. Ed 2016, 55, 58–102; Angew. Chem. 2016, 128, 58–106. [DOI] [PubMed] [Google Scholar]
- [2].Cho DW, Yoon UC, Mariano PS, Acc. Chem. Res 2011, 44, 204–215. [DOI] [PubMed] [Google Scholar]
- [3].Shaw MH, Twilton J, MacMillan DWC, J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schultz DM, Yoon TP Science 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Romero NA, Nicewicz DA, Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- [6].Narayanam JM, Stephenson CR, Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- [7].Moeller KD, Tetrahedron 2000, 56, 9527–9554. [Google Scholar]
- [8].Yoshida J, Kataoka K, Horcajada R, Nagaki A, Chem. Rev 2008, 108, 2265–2299. [DOI] [PubMed] [Google Scholar]
- [9].Francke R, Little RD, Chem. Soc. Rev 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]
- [10].Yan M, Kawamata Y, Baran PS, Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moutet J-C, Reverdy G, J. Chem. Soc., Chem. Commun 1982, 654–655. [Google Scholar]
- [12].Scheffold R, Orlinski R, J. Am. Chem. Soc 1983, 105, 7200–7202. [Google Scholar]
- [13].Steckhan E, Top. Curr. Chem 1987, 142, 1–69. [Google Scholar]
- [14].Yan H, Hou Z-W, Xu H-C, Angew. Chem. Int. Ed 2019, 58, 4592–4595; Angew. Chem. 2019, 131, 4640–4643. [DOI] [PubMed] [Google Scholar]
- [15].Wang F, Stahl SS, Angew. Chem. Int. Ed 2019, 58, 6385–6390; Angew. Chem. 2019, 131, 6451–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang L, Liardet L, Luo J, Ren D, Grätzel M, Hu X, Nature Catal. 2, 2019, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bandar JS, Lambert TH, Synthesis 2013, 45, 2485–2498. [Google Scholar]
- [18].Gerson F, Plattner G, Yoshida Z, Mol. Phys 1971, 21, 1027–1032. [Google Scholar]
- [19].Johnson RW, Tetrahedron Lett. 1976, 17, 589–592. [Google Scholar]
- [20].Weiss R, Schloter K, Tetrahedron Lett. 1975, 16, 3491–3494. [Google Scholar]
- [21].Sevov CS, Samaroo SK, Sanford MS, Adv. Energy Mater 2017, 7, 1602027. [Google Scholar]
- [22].Silvi M, Verrier C, Rey YP, Buzzetti L, Melchiorre P, Nat. Chem 2017, 9, 868–873. [DOI] [PubMed] [Google Scholar]
- [23].Romero NA, Margrey KA, Tay NE, Nicewicz DA, Science 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- [24].Merkel PB, Luo P, Dinnocenzo JP, Farid S, J. Org. Chem 2009, 74, 5163–5173. [DOI] [PubMed] [Google Scholar]
- [25].Ohkubo K, Kobayashi T, Fukuzumi S, Angew. Chem. Int. Ed 2011, 50, 8652–8655; Angew. Chem. 2011, 123, 8811–8814. [DOI] [PubMed] [Google Scholar]
- [26].Ohkubo K, Fujimoto A, Fukuzumi S, J. Am. Chem. Soc 2013, 135, 5368–5371. [DOI] [PubMed] [Google Scholar]
- [27].Niu L, Yi H, Wang S, Liu T, Liu J, Lei A, Nat. Commun 2017, 8, 14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].The working electrode potential was measured by inserting a reference electrode at the beginning of the reaction.
- [29].Because the electrochemical reaction is balanced by reduction of protons to hydrogen gas, a certain concentration of acid is necessary for reaction to occur.
- [30].Christensen JA, Phelan BT, Chaudhuri S, Acharya A, Batista VS, Wasielewski MR, J. Am. Chem. Soc 2018, 140, 5290–5299. [DOI] [PubMed] [Google Scholar]
- [31].Kumar A, Sevilla MD, J. Phys. Chem. B 2018, 122, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Crystallographic data for compound 2 are available free of charge from the Cambridge Crystallographic Data Centre under CCDC 1892753.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.