

Abstract
This review article presents the fifth part (part E) in the series of stories on antiviral drug discovery. The ten stories belonging to this fifth part are dealing with (i) aurintricarboxylic acid; (ii) alkenyldiarylmethanes; (iii) human immunodeficiency virus (HIV) integrase inhibitors; (iv) lens epithelium‐derived growth factor as a potential target for HIV proviral DNA integration; (v) the status presens of neuraminidase inhibitors NAIs in the control of influenza virus infections; (vi) the status presens on respiratory syncytial virus inhibitors; (vii) tricyclic (1,N‐2‐ethenoguanine)‐based acyclovir and ganciclovir derivatives; (viii) glycopeptide antibiotics as antivirals targeted at viral entry; (ix) the potential (off‐label) use of cidofovir in the treatment of polyoma (JC and BK) virus infections; and (x) finally, thymidine phosphorylase as a target for both antiviral and anticancer agents. © 2009 Wiley Periodicals, Inc. Med Res Rev, 31, No. 1, 118–160, 2010
Keywords: ATA, ADAM, HIV integrase inhibitors, LEDGF, influenza, neuraminidase inhibitors, RSV (respiratory syncytial virus) inhibitors, tricyclic guanosine derivatives, glycopeptide antibiotics, polyoma virus infections, thymidine phosphorylase
1. INTRODUCTION
This review article corresponds to the fifth paper in the series of stories on antiviral drug discovery (part E), thus following up, in a similar, narrative style on the stories presented in part A (although, it was not labeled as such),1 part B,2 part C,3 and part D.4 The present article (part E) is entitled “Advents, Advances, and Adventures” because it heralds new opportunities for antiviral drug design, that have blossomed recently, and may represent important advances in our understanding of antiviral drug development, but considering the pitfalls involved they may also signal some adventures ahead.
The first story (story E1) concerns aurintricarboxylic acid (ATA), a compound accredited over the past 25 years with an ever‐increasing number of biological, including antiviral, activities.
From ATA originated the alkenyldiarylmethanes (ADAMs), which, as told in story E2, have been primarily pursued as anti‐human immunodeficiency virus (HIV)‐1 agents, alike the non‐nucleoside reverse transcriptase inhibitors (NNRTIs) HEPT (A8), TIBO (A9), rilpivirine (C9), and TBZ (D3).
Integrase inhibitors (INIs) have, with raltegravir, only recently made their appearance on the anti‐HIV drug scene (story E3), which is almost two decades after integrase was considered a potential target for anti‐HIV drug design.
Whether the host cell factor LEDGF (lens epithelium‐derived growth factor), which helps the HIV integrase in accomplishing the integration of the proviral DNA into the cellular genome could be considered as an attractive antiviral target remains an intriguing story (story E4).
For influenza (already covered in C3 and D5), the seasonal flu has continued to lead to its annual outbreaks (with, unexpectedly, an outbreak of influenza A (H1N1), tentatively called “Mexican”), but the so much feared bird flu (avian influenza A H5N1) has (fortunately) not (yet) arrived, while the stockpiled neuraminidase inhibitors (NAIs), particularly oseltamivir, are facing increasing emergence of drug‐resistant influenza H1N1 variants, which would be particularly worrisome should this extend to the “Mexican” H1N1 and to the avian H5N1 (story E5).
Although considered as only second in importance to influenza, respiratory syncytial virus (RSV) infections have received relatively little attention from a chemotherapeutic viewpoint. Available options are ribavirin and monoclonal antibody (palivizumab), and a number of fusion inhibitors, as well as one nucleocapsid binder (RSV‐604), are under scrutiny (story E6).
In the past, little, if any attention has been given to a class of tricyclic acyclovir or ganciclovir derivatives (containing 1,N‐2‐ethenoguanine) that besides being active as antiviral agents [for acyclovir, see C7] are also endowed with fluorescent properties (story E7).
Antibiotics have been rarely linked to antiviral activity, except for aminoquinolone derivatives that, being transcription inhibitors, deploy some activity against HIV. In story E8, I review the inhibitory effects of aglycosylated glycopeptide antibiotics (i.e. vancomycin, teicoplanin, etc.) on the entry of a number of viruses [particularly HIV, but also coronaviruses (including the severe acute respiratory syndrome coronavirus (SARS CoV)].
Story E9 follows up on D8, where I described the potential of cidofovir (off‐label) in the treatment of human papillomavirus (HPV)‐associated infections. In story E9 I now further address the potential of cidofovir for the treatment of polyoma (JC and BK) virus infections.
During our studies with (E)‐5‐(2‐bromovinyl)‐2′‐deoxyuridine (BVDU) (A3), we observed that the compound is readily cleaved through thymidine (dThd) phosphorylase to yield (E)‐5‐(2‐bromovinyl)uracil (BVU) and 2‐deoxyribose‐1‐phosphate. In story E10 I discuss dThd phosphorylase as a target for antiviral as well as antitumor activity.
A. ATA, a Salicylic Acid Derivative, With a Myriad of Antiviral Effects
ATA (Fig. 1) is a heterogeneous mixture of polymers formed when salicylic acid is treated with formaldehyde, sulfuric acid, and sodium nitrite.5, 6 ATA is often incorrectly represented as a triphenylmethane dye (Fig. 1) rather than as a polymer (Fig. 1).7 While ATA had been known for a long time to interact with a number of enzymes including DNA polymerases, RNA polymerases, reverse transcriptase (RNA‐dependent DNA polymerase), aminoacyl‐tRNA synthetase, ribonucleotide reductase, and ribonucleases (as reviewed by Cushman et al.7), it was considered as a nonspecific enzyme inhibitor,8 simply because ATA, being a polycarboxylate, would bind by electrostatic interactions to any protein that contains positively charged residues. [The use of ATA as an inhibitor of nucleases during nucleic isolation was advocated at a certain time.9
Figure 1.
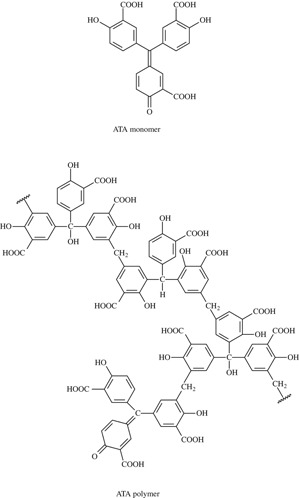
Aurintricarboxylic acid (ATA) is often incorrectly represented as a triphenylmethane dye rather than as a polymer.
Interest in ATA revived when it was shown to inhibit the cytopathogenicity of HIV in cell culture.10, 11 ATA was then shown to specifically interact with CD4, the cell's principal receptor for HIV, as it selectively prevented the binding of OKT4A/Leu‐3a monoclonal antibody to the CD4 cell receptor.12 In further studies, ATA also appeared to block the binding of monoclonal antibody to the viral envelope glycoprotein gp120.13 The lower molecular weight fractions of ATA bound to gp120, but not CD4, and, as they prevented the cytopathicity of HIV‐1 and HIV‐2, it could be concluded that the ATA fractions bound more avidly to gp120 than to CD4, and that the binding of ATA to gp120 in the absence of CD4 binding was sufficient for anti‐HIV activity.7 After a number of ATA monomer analogues (such as the triphenylcarbinol and triphenylmethane) had proven to be virtually inactive against HIV‐1,14 interest in ATA, as a potential lead compound for the treatment of HIV infections, largely waned.
Yet, new viruses emerged (or re‐emerged) and so did the interest in ATA. After ATA had been reported as a potent inhibitor of the SARS CoV,15 ATA was predicted, by using computer‐aided analysis, to bind to and inhibit the SARS CoV RNA‐dependent RNA polymerase (RdRp), thereby blocking viral transcription.16 However, whether ATA inhibits SARS CoV replication through interference with the RdRp function has not been directly demonstrated [considering the negative charges of ATA, one could indeed question how easily this compound enters the cells].
Then followed the report that ATA inhibits (the early stage of) vaccinia virus replication,17 as the result of two mechanisms of action: (i) blocking the extracellular signal‐regulated kinase (ERK) phosphorylation [activation of the ERK cascade is essential for vaccinia virus replication], and (ii) inhibiting the phosphatase activity of the viral enzyme H1L [which plays an essential role in initiating viral early gene transcription]. In fact, ATA would be able to interfere with various signaling pathways: viz. the ERK pathway,18 the JAK‐STAT signaling pathway,19 and the insuline‐like growth factor 1 signaling pathway.20
Given the current interest in new anti‐influenza therapeutics, a battery of approximately 2,000 structurally diverse compounds were screened, and ATA emerged as one of the most promising NAIs.21 Although ATA was originally assumed to inhibit influenza virus replication through the inhibition of the influenza viral polymerase (RdRp) activity,22, 23 Hung et al.21 found that ATA did not inhibit the influenza polymerase, but, instead, interfered with the viral neuraminidase by competing with sialic acid (although ATA is structurally distinct from sialic acid) at the enzyme's substrate binding site. At least, this was suggested by molecular modeling experiments.21
Another virus and accompanying viral target, that has received increasing attention is the S‐adenosylmethionine (SAM)‐dependent methyltransferase (MTase)3 associated with flaviviruses (i.e. dengue virus).4 Here, again, ATA has been identified, as the result of a combined computational and experimental screening approach, as a potent flaviviral MTase inhibitor, even the most potent known to date, according to Milani et al.24
Thus, there is no shortage of potential therapeutic applications for ATA, but separately from its eventual therapeutic usefulness, what has to be resolved in each case, is whether the proposed molecular target, despite all the molecular docking and computation, really accounts for the observed antiviral activity, or, given the myriad of possible interaction sites of ATA, only represent(s) one of these many sites with which the compound could theoretically interact.
B. ADAM: Non‐Nucleoside HIV‐1 Reverse Transcriptase Inhibitors that Should Go Beyond Just HIV‐1
Following up on the “ATA” lead (see supra in this series of stories), Mark Cushman and his collaborators then described cosalane (Fig. 2), which should conceptually be considered as an ATA derivative (missing one salicylic acid part) conjugated to cholestane: this compound acts primarily by the inhibition of viral (gp120‐cellular) CD4 binding,25 the cholestane part being responsible for anchoring the compound into the cell membrane. Why cholestane was, as such, not further developed for its anti‐HIV properties, has remained unclear (to me), but, instead, it opened the way to the ADAM derivatives, as potential NNRTIs, thus joining the wide array of NNRTIs (HEPT, TIBO, nevirapine, BHAP, TSAO, α‐APA, etc.) described at that time.26
Figure 2.

Structures of alkenyldiarylmethane (ADAM) non‐nucleoside reverse transcriptase inhibitors (NNRTIs).
The ADAM derivatives were from the very beginning recognized as genuine NNRTIs, as, for example, suggested for 3′,3′′‐dibromo‐4′,4′′‐dimethoxy‐5′,5′′‐bis(methoxycarbonyl)‐1,1‐diphenyl‐1‐heptene (compound 8 in Cushman et al.27; or compound 5 in Cushman et al.26) (Fig. 2), although structurally little resemblance could be observed between the “classical” NNRTIs (HEPT, TIBO, …) and the ADAMs, particularly with regard to the paradigmatic “butterfly” structure assigned to NNRTIs such as TBZs.4
Starting from the original ADAM derivatives,27 new “ADAMs” were derived with optimized potency,28 exhibiting an EC50 of 0.013 µM and a selectivity index of 2430 [i.e. methyl‐3′,3′′‐dichloro‐4′,4′′‐dimethoxy‐5,5′′‐bis(methoxycarbonyl)‐6,6‐diphenylhexenoate].29 This potency was even further increased to 0.0013 µM (EC50) and the selectivity increased to 10,000, if the chlorines were replaced by bromines, as in methyl‐3′,3′′‐dibromo‐4′,4′′‐dimethoxy‐5′,5′′‐bis(methoxycarbonyl)‐6,6‐diphenyl‐5‐hexenoate (Fig. 2)30 [solid‐phase synthesis of the ADAMs was described by Xu et al.31].
Further improvement of anti‐HIV‐1 potency, especially against NNRTI resistance mutations (i.e. A98G) were noted with ADAM derivatives based on the ethyl (instead of methyl) 5‐hexenoate, as in both the 3′,3′′‐dichloro‐ and 3′,3′′‐dibromo‐derivatives (Fig. 2), so that the latter compounds were at the time (year 2001) considered as the most promising ADAM derivatives.32 New methods were in the mean time devised for the preparation of new ADAM derivatives with nonidentical aromatic rings.33
The potential therapeutic usefulness of the ADAMs could be comprised by the metabolic instability that may be expected from their ester moieties that are likely to be hydrolyzed by nonspecific esterases present in human plasma.34 Therefore, structural modifications (i.e. thioesters) introduced in the parent compound 2 (Fig. 2), as in compound 23 (Fig. 2), resulted in enhanced stability (t1/2 = 55 min), diminished cytotoxicity (CC50>224 µM), while maintaining reasonable potency (EC50 = 1.8 µM).34
Similarly, modifications of compound 1 (Fig. 2) (including introduction of a benzo[d]isoxazole as in compound 17) (Fig. 2) enhanced stability (t1/2 = 22 min), diminished cytotoxicity (CC50 = 16 µM), while maintaining moderate antiviral potency (EC50 = 2.7 µM).35 For the benzoxazolyl derivative (E)‐5‐[1‐(3,7‐dimethyl‐2‐oxo‐2,3‐dihydro‐benzoxazol‐5‐yl)‐5‐methoxycarbonyl‐pent‐1‐enyl]‐2‐methoxy‐3‐methylbenzoic acid methyl ester (compound 7) (Fig. 2), EC50 values of 30 and 90 nM against HIV‐1 (RF and IIIB strains) were recorded.36 For (Z)‐5‐((E)‐1‐(3,7‐dimethyl‐2‐oxo‐2,3‐dihydrobenzo[d]oxazol‐5‐yl)‐5‐(3‐methyl‐1,2,4‐oxadiazol‐5‐yl)pent‐1‐enyl)‐N,2‐dimethoxy‐3‐methylbenzimidoyl fluoride (compound 6) this was accompanied by enhanced metabolic stability in rat plasma (t1/2 = 61 hr).37 At present, the most promising compound within the ADAM series is compound 3, (Z)‐2‐methoxy‐5‐[5‐methoxycarbonyl‐1‐(3‐methoxy‐7‐methylbenzo[d]isoxazol‐5‐yl)‐pent‐1‐enyl]‐3‐methylbenzoic acid methyl ester (Fig. 2) with EC50 values of 40 nM (HIV‐1 RF) and 20 nM (HIV‐1 IIIB).38
The primary objective has been to optimize the ADAMs by replacing the metabolically unstable methyl ester moieties with stable isosteres (i.e. benzo[d]isoxazole), while maintaining or enhancing the antiviral potency of the ADAMs as potential NNRTI‐type drugs. However, other potential applications of the ADAMs should not be ignored. They may not have much potential as PDE4 inhibitors [phosphodiesterase 4 (PDE4) belongs to the phosphodiesterase family of hydrolases responsible for regulating cellular cAMP levels, PDE4 playing a role in HIV infections, and PDE4B2 being specifically involved in inflammation], as they have only weak, if any, activity against PDE4B2.39 However, two distantly related ADAM derivatives, i.e. compounds 15 and 16,40 like colchicine, proved active as tubulin polymerization inhibitors (IC50 of 3.7 and 2.8 µM, respectively). This makes them worth pursuing as potential anticancer drug candidates.
C. HIV INIs, the Fifth Class of HIV Inhibitors, Following N(t)RTIs, NNRTIs, PIs, and CRIs
Exactly 25 years after the discovery of HIV as the causative agent of AIDS by Françoise Barré‐Sinoussi [Nobel Prize in Medicine or Physiology (2008)] and her co‐workers41 and by Mikulas Popovic and his co‐workers,42 25 compounds have been officially approved for the treatment of AIDS.43 The most recent compounds to complete the anti‐HIV drug armamentarium are the INIs,44, 45 presently only one (raltegravir)45 but soon to be expected two (raltegravir and elvitegravir).44 Raltegravir plus optimized background therapy provided better viral suppression than optimized background therapy alone for at least 48 weeks,46 and raltegravir has been considered a valuable addition to the current armamentarium for the treatment of patients infected with multi‐drug‐resistant HIV‐1.47
The era of the INIs started with Daria Hazuda's paper in Science48 on the inhibitory effects of “diketo acids”, i.e. L‐731988, on integration and replication of HIV in cell culture, providing the first proof of concept for HIV‐1 integrase (specifically, strand transfer) inhibitors to act as antiviral agents. While first demonstrated for L‐731988,48 this proof of principle has been extended to various related compounds, i.e. L‐870810 (a naphthyridine carboxamide)49 and L‐87081250 which proved efficacious in vivo against retroviral (simian‐human immunodeficiency virus) replication in vivo (rhesus macaques).
Hence, it did not come as a surprise that the compound MK‐0518 [4‐[N‐(4‐fluorobenzyl)carbamoyl]‐1‐methyl‐2‐[1‐methyl‐1‐(5‐methyl‐1,3,4‐oxadiazol‐2‐ylcarboxamido)ethyl]‐6‐oxo‐1,6‐dihydropyrimidin‐5‐olate (raltegravir)] (Fig. 3) selected as the clinical candidate for further development51 finally became licensed for clinical use as the first INI ever to be approved for the treatment of HIV infection. Meanwhile, L‐870812 has been advocated as a microbicide to prevent cell‐free and cell‐associated HIV infection.52 Should INIs ever be used topically as microbicides, they preferentially be combined, as is the rule for their systemic use as well, with other classes of HIV inhibitors, including reverse transcriptase inhibitors and viral entry inhibitors.
Figure 3.

HIV Integrase inhibitors (INIs) (in red: metal (Mg++ or Mn++) binding β‐diketo acid or the like portion of the molecules).
The runner‐up INI to be licensed for clinical use is elvitegravir (GS‐9137)53 (Fig. 3). While raltegravir is currently dosed at 400 mg twice daily, elvitegravir is dosed once daily at 150 mg [currently in combination with 100 mg of ritonavir as its booster; on the horizon, however, is the combination of elvitegravir with Gilead's own booster (pharmacoenhancer) GS 9350 and Truvada (tenofovir disoproxil fumarate and emtricitabine), which together will make a one pill once‐daily treatment (tentatively called “QUAD” as it will consist of four compounds)].
Structurally, elvitegravir (Fig. 3) is built upon the quinolone 3‐carboxylic acid scaffold [this pharmacophore has also served as the starting point for the design of a second generation of HIV‐1 INIs].54 Quinolones, such as the fluoroquinolone K‐1255 and the 6‐aminoquinolone (WM‐5),56 have been known as Tat‐dependent transcription inhibitors. It was, therefore, interesting to ascertain whether elvitegravir, being both a “diketo acid” and a “quinolone” should behave as an INI or transcription inhibitor. Unlike WM‐5, but like L‐870810, elvitegravir behaved as a genuine INI.57
Elvitegravir is a more potent INI than raltegravir,58 but its mechanism of action, i.e. inhibition of the strand transfer function of integrase must be similar, as both drugs exhibited a parallel resistance profile, the Q148 K and T66I mutations conferring the highest resistance to both drugs.58 In addition to the Q148 K and T66I mutations, a few other mutations, i.e. E92Q, L74 M and S230N have been shown to confer resistance to elvitegravir and, where examined, other INIs (i.e. L‐870810 and raltegravir) as well.59, 60, 61 While, on the one hand, the clinical relevance of these mutations remains to be demonstrated, i.e. with regard to possible reduced fitness of the viral mutants, they should, on the other hand, help in gaining further insight in the mode of action of INIs and, eventually facilitate the design of new INIs.
D. LEDGF as Cellular Cofactor for HIV Integrase: An Attractive Target for anti‐HIV Drug Design?
The LEDGF “story” started in 2000, when Peter Cherepanov and his coworkers62 demonstrated that using a synthetic gene, efficient expression of HIV‐1 integrase was achieved in human cells. Cherepanov et al. then found in the nuclei of human cells stably expressing the HIV integrase that it formed stable tetramers associated with the LEDGF/p75, a protein which had been implicated in the regulation of gene expression and cellular stress response.63 Moreover, LEDGF was found to robustly enhance the strand transfer activity of HIV integrase in vitro, and Cherepanov et al.63 concluded that LEDGF may constitute a novel target for anti‐HIV drug therapy.
In subsequent studies it was further shown that LEDGF/p75 accounts for the karyophilic, i.e. nuclear targeting, and chromosomal targeting of HIV‐1 integrase,64 that LEDG/p75 is essential for HIV DNA integration into chromosomal DNA,65 and LEDGF/p75 is the first example of a cellular protein controlling the location of HIV integration in human cells.66 In fact, LEDGF/p75 is able to interact with different lentiviral integrases, including those from bovine immunodeficiency virus, maedi‐visna virus, and equine infectious anemia virus, but nonlentiviral integrases such as those from beta‐, gamma‐, deltaretroviruses or spumaviruses do not possess detectable affinity for LEDGF.67
Poeschla thus concluded that LEDGF/p75 may act as a chromatin docking factor or receptor for lentiviral preintegration complexes; LEDGF/p75 tethers HIV integrase to chromatin, protects it from degradation, and strongly influences the genome‐wide pattern of HIV integration.68 Given the fact that LEDGF/p75 is a ubiquitous nuclear protein, tightly associated with chromatin throughout the cell cycle, and although an important but not strictly essential cofactor of lentiviral DNA integration,69 could such ubiquitous protein (that is not strictly essential for retroviral DNA integration) serve as a target for the development of a novel class of antiretroviral drugs?
Not the LEDGF per se, but its interaction with the integrase (thus the protein–protein interaction (PPI)), which is further illustrated in Figure 4A, 70 may serve as a potential target for drug discovery, but, therefore, several issues should be addressed71: (i) the target has to be validated as important for HIV‐1 replication; (ii) inhibition of the specific PPI should not be associated with toxicity; (iii) structural information on the PPI should be available; and (iv) identification of genuine inhibitors should provide ultimate proof‐of‐concept.71 The pursuit of LEDGF as a potential target for anti‐HIV drugs may seem as a Sisyphian task as we not only have to take into account the PPI interaction between the integrase's CCD and LEDGF's IBD (Fig. 4A), but also the role this PPI has to fulfill, that is the integration of the proviral DNA into the target chromosomal DNA, which makes it, stricto sensu, a “menage à quatre.”
Figure 4.

(A) Interface between the HIV‐1 integrase catalytic core domain (CCD) and the integrase‐binding domain (IBD) of LEDGF/p75 using Protein Data Bank crystal structure file 2BJ4 (panel A). Panel B shows a close‐up view of the interface. The integrase CCD monomers are colored purple and green, and the IBD subunit is orange. Integrase residues A128 and W131 are part of a hydrophobic patch, which accommodates the side chains of the LEDGF/p75 residues I365, F406 and V408. Data taken from Busschots et al.70 (B) Molecular docking of D77 into the HIV‐1 integrase catalytic core domain (CCD). Panel A. Chemical structure of D77, the benzoic acid derivative 4‐[(5‐bromo‐4‐ [2,4‐dioxo‐3‐(2‐oxo‐2‐phenylethyl)‐1,3‐thiazolidin‐5‐ylidene]methyl
[2,4‐dioxo‐3‐(2‐oxo‐2‐phenylethyl)‐1,3‐thiazolidin‐5‐ylidene]methyl ‐2‐ethoxyphenoxy)methyl]benzoic acid. Panel B. View on D77 as docked into CCD (amino acid residues W131, T125, Q95, and T174). Panel C. Closer view on D77 as docked into the CCD/IBD complex (showing amino acid residues W131, T125, Q95, and T174 of the CCD domain). Data taken from Du et al.73
‐2‐ethoxyphenoxy)methyl]benzoic acid. Panel B. View on D77 as docked into CCD (amino acid residues W131, T125, Q95, and T174). Panel C. Closer view on D77 as docked into the CCD/IBD complex (showing amino acid residues W131, T125, Q95, and T174 of the CCD domain). Data taken from Du et al.73
These tantalizing perspectives have not deterred several investigators to launch a search for small molecule PPI inhibitors (already nicknamed as SMPPIIs)70 or to screen for small‐molecule inhibitors of the HIV integrase‐LEDGF/p75 interaction by a luminescent proximity assay.72 A first (apparent) success has already been reported: that of D77, a benzoic acid derivative (Fig. 4B) that would function as a novel HIV‐1 inhibitor targeting the interaction between the integrase's CCD and the LEDGF's IBD73 [D77, as shown in Figure 4B, would (principally) interact with the CDD domain of the integrase]. This approach opens a wealth of possibilities, i.e. structure–activity relationship (SAR) studies, pharmacokinetics and toxicity, and, particularly, confirmatory studies to validate the presumed target of action and provide the ultimate proof‐of‐concept.
E. While Waiting for the Avian H5N1 Influenza Pandemic, What is the Position of NAIs (such as Oseltamivir)?
As put forward by Frederick Hayden,74 new antiviral agents for the treatment of influenza are urgently needed, one of the limitations of current drugs (i.e. oseltamivir) being the emergence of resistance among influenza A (H1N1) strains and the fear it may also emerge among avian influenza A (H5N1) (“bird flu”) strains. Highly pathogenic avian influenza A (H5N1) viruses are entrenched among poultry in parts of Asia, Africa, and the Middle East,75 and human infection with these viruses has been noted in many of countries including Vietnam,76 Indonesia (three clusters!),77 and Eastern Turkey78 [Indonesia has had the most human cases of highly pathogenic avian influenza A (H5N1) and one of the highest case‐fatality rates worldwide79].
It is re‐assuring that person‐to‐person transmission of avian influenza A (H5N1) is rather limited,80 and this is probably linked, as I explained before,81 to the differences in receptor specificity: avian receptors prefer a α(2–3) linkage, whereas humans prefer the α(2–6) linkage so that productive infections may have difficulty in jumping from one host (avian) to the other (human).
Oseltamivir has been considered as the golden grail and stockpiled for use should the need prevail: first, to treat infected individuals (to moderate disease severity); second, to protect family members of an index case (to interrupt transmission); and third, to all the people in an area surrounding an index case (ring prophylaxis).82 As it can be administered orally, oseltamivir (Fig. 5A) is the drug of choice for the therapy of both seasonal and avian influenza83 [with zanamivir (Fig. 5A) (that has to be inhaled via a rather sophisticated devide) ranking second].
Figure 5.

(A) Structural formulae of zanamivir, oseltamivir, peramivir, A‐322278, R‐125489 and CS‐8958. (B) Mechanism of development of resistance to oseltamivir. Binding of oseltamivir, but not zanamivir, to the neuraminidase active site requires a shape change that creates a pocket (Panel A), and mutations that prevent formation of this pocket may prevent binding of oseltamivir but permit binding of zanamivir (Panel B). Crystal structure analysis shows that binding of oseltamivir requires a rotation of the Glu276 residue away from the hydrophobic pentyloxy group of oseltamivir, which then makes hydrophobic contact with a methylene group on Glu276 (Panel C). However, binding of either sialic acid (the natural substrate) or zanamivir does not require a change in the position of Glu276. The His274Tyr substitution in the neuraminidase active site pushes the Glu276 further into the binding site and disrupts the hydrophobic pocket that is required only for oseltamivir binding. According to Moscona.117
While waiting for a still hypothetical avian influenza A H5N1 pandemic, the world was recently confronted with a new influenza (first called “pig,” then “Mexican”) H1N1 variant, which spread rapidly over the five Continents and thus, in epidemiological terms, gave rise to a pandemic. Although highly contagious, the “Mexican” H1N1 influenza variant has so far not proven more pathogenic than other seasonal influenza A H1N1 variants (although it started its journey in the off‐season). As all the other H1N1 variants, the “Mexican” H1N1 virus should normally be sensitive to the NAIs oseltamivir and zanamivir.
However, a worrying phenomenon that has been observed within the past few years is widespread oseltamivir resistance in influenza A (H1N1) in the United States,84, 85 Europe,86 Japan,87 Norway,88 and South Africa.89 It has been suggested that the H274Y mutation conferring resistance to oseltamivir leaves the influenza A (H5N1) virus severely compromised.90 However, this oseltamivir‐resistant virus can be pathogenic, and fatal, in an immunocompromised patient.91 On the contrary, oseltamivir allowed complete recovery of a bone‐marrow transplant recipient from an influenza A (H1N1) virus infection that did not respond to zanamivir.92
Influenza A virus strains resistant to NAIs (zanamivir, oseltamivir) would circulate at a stable and low level (1%) since these compounds were introduced in clinical practice.93 Influenza B viruses with reduced sensitivity to NAIs may also arise, but not as frequently as for the influenza A viruses.94 As originally pointed out by Peter Palese, influenza A may be effectively transmitted among guinea pigs (Personal communication, 8th International Symposium on NeuroVirology, San Diego, California, USA, 30 October‐2, November 2007), and this was effectively demonstrated by Bouvier et al. for oseltamivir‐resistant influenza A viruses.95 This opens new avenues not only for the study of the transmission of influenza A virus infections, but also for the chemotherapy and chemoprophylaxis of these infections.
Although resistance to oseltamivir has also been noted with influenza A (H3N2) virus [i.e. due to a deletion of four amino acids (Δ245–248) in the neuraminidase96], resistance to oseltamivir would emerge at a higher rate in influenza A (H1N1) virus than in influenza A (H3N2) virus or influenza B virus in children.97 For influenza A (H5N1) virus there might be a significant natural variation in sensitivity to oseltamivir,98 although reduced sensitivity of influenza A (H5N1) to oseltamivir has been noted at several occasions99, 100; and NAI‐resistant H5N1 influenza viruses (i.e. those carrying the H274Y mutation) may retain the high pathogenicity of the wild‐type virus both in vitro and in vivo (in mice).101 As learned from ferrets, it should be advisable to increase the dose of oseltamivir to treat the highly pathogenic avian influenza H5N1 infections in humans.102, 103
Novel neuraminidase mutations D197E104 and D198N105 have been described that show resistance to both oseltamivir and zanamivir, but the clinical relevance of these resistance mutations remains to be further elucidated. Taking together all the resistance mutations collected for oseltamivir, Skehel and colleagues considered it prudent for pandemic stockpiles of oseltamivir to be augmented by additional antiviral drugs including zanamivir.106
To stockpile oseltamivir (tamiflu) as a precautionary measure against an influenza pandemic is both risky and costly. It is recomforting, therefore, that intensive efforts have been made to improve the synthesis of oseltamivir [the starting material is shikimic acid normally obtained from the Chinese star anise107] and these efforts have indeed allowed a more practical and, at the same time, high‐yielding synthesis of oseltamivir.108, 109, 110, 111 An environmental issue, linked to its chemical stability, is that oseltamivir would not be easily removed or degraded in normal sewage water.112 This not only points to the stability of the compound, but also its propensity to contribute to far‐reaching anti‐influenza virus drug resistance.
Are new NAIs forthcoming that may supersede the classical NAIs oseltamivir and zanamivir? A‐322278 (Fig. 5A) certainly seems promising as it is active against the oseltamivir‐resistant H274Y A (H1N1) influenza virus mutant in mice.113 A‐322278, the 2‐methylpropanoyl prodrug of A‐315675, would have comparable activity to oseltamivir in immunocompetent and ‐compromised murine models of influenza virus infection.114 A‐315675 has potent inhibitory activity against oseltamivir‐resistant influenza viruses (N1 and N2 subtypes).115 In addition to zanamivir and oseltamivir, peramivir, A‐322278 and A‐315675, also R‐125489 and CS‐8958 (Fig. 5A) show high promise as anti‐influenza virus agents, the latter (CS‐8958) demonstrating long‐acting anti‐influenza virus activity.116
The mechanism of development of resistance to oseltamivir (Fig. 5B) as proposed by Moscona117 is paradigmatic, but is it correct? NAI inhibitors (whether zanamivir, oseltamivir or peramivir) are still trying to find their best way of application, prophylactic/therapeutic,118 or would injectable peramivir offer the ideal, if not final, solution?119 Drug‐resistant virus will remain an important factor for NAIs for many years to come,120 but the design, synthesis, and structure–activity relationship (SAR) studies will be continued to trying to cope with this challenge.121
Of primordial importance, if the avian H5N1 influenza virus, or any H1N1 influenza virus such as the “Mexican” variant, becomes a pandemic threat in humans, is that we are “prepared” [as we are already supposed to be for many years]; and the combination of oseltamivir with, for example, amantadine,122 or ribavirin,123 or double combinations of amantadine, oseltamivir and ribavirin124 may rank among the various measures that could be taken to curb influenza virus infections.
F. RSV: Still in Search of Specific Antiviral Drugs
In 1996, I reviewed the perspectives for the chemotherapy of RSV infections.125 At the time, ribavirin (given as an aerosol at 20 mg/mL) was the only antiviral agent approved for the treatment of RSV infection. Several new possible drug candidates were considered.125 What has happened with ribavirin and these drug candidates, and how does the chemotherapy (and/or–prophylaxis) of RSV infections look like nowadays?
Human RSV is the major cause of upper and lower respiratory tract infections in the pediatric population. These infections are particularly problematic in infants that are born prematurely or with congenital heart disease or chronic lung disease (CLD) or are otherwise immune compromised (ref.126; and references therein). Elderly and immunocompromised adults are also at increased risk for developing complications or even death associated with RSV infection (for a review, see Falsey & Walsh127).
Ribavirin still is the only antiviral agent approved for the treatment of RSV infection, but due to efficacy and toxicity issues, it has only limited utility.128 Inhaled (nebulized) ribavirin has remained the standard therapy for the treatment of RSV infections, despite it is costly and cumbersome. Pelaez et al.129 recently pointed to the efficacy of oral ribavirin in the treatment of lower respiratory tract infection after lung transplantation. They argued that further studies should be conducted to compare the long‐term efficacy of oral ribavirin vs. nebulized therapy for RSV.
After more than 40 years of research, there is no approved vaccine, and the only prophylactic therapies available are RSV‐IVIG, a polyclonal RSV immunoglobulin,130 and palivizumab (Synagis®), a humanized monoclonal antibody targeting the RSV fusion protein.131 While effective, this treatment is only administered to high‐risk pediatric patients. There is a clear need for new anti‐RSV therapeutics, with improved efficacy and safety for broader applications (for a review, see Meanwell & Krystal132).
RSV encodes three surface glycoproteins, the fusion protein F, the attachment glycoprotein G, and the small hydrophobic protein SH. Both the F and G glycoproteins are required for efficient infectivity in vivo, but the F protein alone is sufficient for virus binding and entry into cells in vitro.133
RSV is a major cause of hospitalization in preterm infants and infants with CLD.134 Prophylaxis with palivizumab significantly reduced the incidence of RSV‐related hospitalization relative to placebo and is generally well tolerated in high‐risk infants aged<2 years, including those with prematurity and bronchopulmonary dysplasia/CLD or hemodynamically significant congenital heart disease, which are risk factors for early or serious RSV infection. Palivizumab has been approved for use in these patients.135
Children who experience RSV lower respiratory tract infections early in life have high rates of subsequent recurrent wheezing. Palivizumab, by ameliorating or preventing this lower respiratory tract infection in preterm infants may reduce subsequent recurrent wheezing.136 Wu et al.137 described the selection of ultra‐potent anti‐RSV antibodies for preventing RSV infection. They applied an iterative mutagenesis approach, and were able to identify palivizumab Fab variants with up to 1500‐fold improvement and palivizumab IgG variants with up to 44‐fold improvement in the ability to neutralize RSV. Recently, a new, ultra‐potent antibody, motavizumab, has been developed for the prevention of RSV infections.138 Motavizumab binds to RSV F protein 70‐fold better than palivizumab, and it reduced pulmonary RSV titers to up to 100‐fold lower levels than did palivizumab. Motavizumab is currently being evaluated in pivotal clinical trials for RSV prophylaxis, and for the reduction of RSV‐related asthma.
There is an experimental concern, originated from preclinical studies in cotton rats, which RSV escape mutants may emerge in immunosuppressed patients. Should palivizumab arise in humans, palivizumab may obviously be ineffective.139 Zhao and Sullender140 evaluated the potential for palivizumab‐resistant RSV mutants to arise in vivo. Cotton rats were immunosuppressed with cyclophosphamide. Three of the five animals had mixed populations of lung virus, and over 50% of the clones from the three animals revealed F gene mutations associated with resistance to palivizumab. Thus, prolonged pulmonary replication of RSV in the presence of palivizumab was followed by the appearance of viruses resistant to palivizumab.
Using an RNA interference (RNAi) approach, inhibition of both RSV and parainfluenza virus can be achieved in the mouse by intranasally administered short interfering RNAs (siRNAs),141 and a similar success has been obtained with intranasal siRNA nanoparticles targeting the viral NS1 gene (siNS1).142 Initial clinical studies on safety, tolerability, and pharmacokinetics favor the further pursuit of siRNAs (i.e. ALN‐RSV01) for RSV in humans.143 Similarly to siRNAs, RSV‐targeted deoxyribozymes (DNA zymes) potentially present a therapeutic (or prophylactic) approach for RSV diseases. Their activity is based on the ability to bind and cleave complementary RNA sequences, thus inhibiting protein expression. D71133 is an example of such deoxyribozyme that targets the conserved genomic RNA sequence of the RSV nucleocapsid protein: it has been shown to block RSV infection both in vitro144 and in vivo.145
Sidwell and Barnard146 recently reviewed the prospects of the principal drug candidates for the control of RSV infections. Prominent among the cited compounds were VP‐14637, JNJ‐2408068, BMS‐433771, and A‐60444 (the latter compound became later known as RSV604) (Fig. 6A). Two RSV fusion inhibitors, VP‐14637 (Fig. 6A) and JNJ‐2408068 (Fig. 6A), have been identified,147, 148 which are both targeted at the F1 glycoprotein [as could be deduced from the resistance mutations selected by VP‐14637 and JNJ‐2408068 (Fig. 6B)].
Figure 6.

(A) Structural formulae of RSV inhibitors. (B) RSV inhibitors interacting with the F protein. Location of the resistance mutations selected in the presence of VP‐14637 and JNJ‐2408068. Schematic view of the RSV F polyprotein indicating salient features: F1 and F2 subunits; FP, fusion peptide; HR1, heptad repeat 1; HR2, heptad repeat 2; TM, transmembrane domain. The numbers represent amino acid designations, the asterisks above the line indicate the resistance mutations selected by VP‐14637, and the arrows below the line indicate the resistance mutations selected by JNJ‐2408068. According to Douglas et al.148
VP‐14637 (Fig. 6A) was first reported by D.C. Pevear (ViroPharma) [as mentioned by Wyde et al.149] before it was further evaluated by Cihlar and his colleagues148, 150 and found to be targeted at the F protein of RSV. The efficacy of VP‐14637 against RSV was demonstrated in vitro, and in vivo in cotton rats following delivery by small droplet aerosol.151 According to Sidwell and Barnard,146 the compound was in phase I trials before a decision not to develop it further, in part due to developmental costs.
JNJ‐2408068, originally named R170591 and for the first time reported by Andries and his colleagues., has a structure (2‐[[2‐[[1‐(2‐aminoethyl)‐4‐piperidinyl]amino]‐4‐methyl‐1H‐benzimidazol‐1‐yl]‐6‐methyl‐3‐pyridinonol) (Fig. 6A), which is fundamentally different from that of VP‐14637, but nevertheless targeted at the F protein of HSV148 (Fig. 6B). JNJ‐2408068 is highly potent (EC50: 0.16 nM) but according to Douglas,150 it has limited oral bioavailability. Like VP‐14637, JNJ2408068 was found to protect cotton rats from experimental RSV infection.151
BMS‐433771 [1‐cyclopropyl‐1,3‐dihydro‐3‐[[1‐(4‐hydroxybutyl)‐1H‐benzi‐midazol‐2‐yl]methyl]‐2H‐imidazo[4,5‐c]pyridin‐2‐one] is another example of anti‐RSV agents targeted at the F protein.152 [To confirm that the mechanism of action was through inhibition of fusion, the K394R virus mutant was selected for resistance to BMS‐433771 in vitro, and found refractory to BMS‐433771 in vivo.] It proved orally efficacious against RSV infection in Balb/C mice and cotton rats.153, 154 Numerous structural variants of BMS‐433771 have been synthesized155, 156, 157, 158, 159, 160 but, most likely, BMS‐433771 still excelled in potency and oral bioavailability.
Powell and his colleagues have recently identified a new class of RSV inhibitors, namely that of 1,4‐benzodiazepines,161 which eventually led to the identification of RSV‐604 (Fig. 6A) as the clinical candidate.162 RSV‐604 can be considered as a truly novel inhibitor of RSV replication in that, being a benzodiazinylurea derivative, it is targeted at the nucleocapsid, and, unlike all the F protein inhibitors (see supra) active against RSV after it has infected the cells.163 In the conclusion of the latter paper, RSV‐604 was hailed as the “most promising candidate to date for the treatment of RSV disease in humans.” In the wake of data on in vivo efficacy and oral bioavailability, this sounds as a daring statement.
G. Fluorescent Tricyclic Analogues of Acyclovir and Ganciclovir Based on 1,N‐2‐ethenoguanine
Following up on the work of Beauchamp et al. on a series of acyclovir derivatives with modifications in the heterocyclic base (which did not yield significantly active compounds),164 we (Bozenna Golankiewicz, Jerzy Boryski, and I) started a new program around new acyclovir derivatives with one or more nitrogen centers blocked by methylation or incorporated into an additional ring structure. Marked activity against herpes simplex virus (HSV‐1 and HSV‐2), comparable to that of acyclovir, was obtained with 1‐methyl‐9‐[(2‐hydroxyethoxy)methyl]guanine and 9‐[(2‐hydroxyethoxy)methyl]‐1,N‐2‐isopropeno‐guanine (Fig. 7), compounds 2 and 5, respectively, in the original publication of Boryski et al.165 While N‐1 methylation allowed the antiviral activity of acyclovir to be preserved, it was virtually abolished following N‐3 methylation.166
Figure 7.
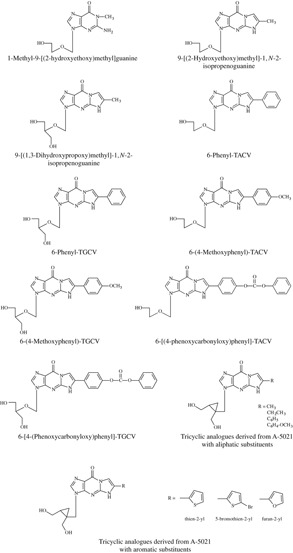
Tricyclic analogues of acyclovir, ganciclovir and A‐5021, based on 1,N‐2‐ethenoguanine.
The earliest report on 1,N‐2‐etheno derivatives of guanine has been that of Sattsangi et al.167 on 1,N‐2‐ethenoguanosine. 1,N‐2‐ethenoguanine was not only successfully implemented in acyclovir but also in ganciclovir. The resulting tricyclic analogue of ganciclovir (compound 2b in the original publication) (Fig. 7) proved markedly active not only against HSV‐1 and HSV‐2, but also varicella‐zoster virus (VZV) and cytomegalovirus 168 Further substitution of the methyl group in the 1,N‐2‐isopropeno moiety by a phenyl group as in 6‐phenyl‐TACV (tricyclic acyclovir) and 6‐phenyl‐TGCV (tricyclic ganciclovir) (Fig. 7), compounds 7 and 13 in the original publication169 give rise to fluorescent antiviral compounds which, on the one hand, showed an antiviral activity profile similar to that of their parent compounds (acyclovir and ganciclovir), and, on the other hand, showed relatively strong fluorescence, making them useful for the noninvasive diagnosis of herpesvirus infections.169 This line of research was then extended to 6‐(4‐methoxyphenyl)‐TACV and 6‐(4‐methoxyphenyl)‐TGCV (compounds 8 and 27 in the original publication) as fluorescent tricyclic analogues of acyclovir and ganciclovir.170
Yet further extension of the 6‐side chain in TACV and TGCV yielded the 6‐[4‐(phenoxycarbonyloxy)phenyl]substituted derivatives (compounds 11 and 19 in the original publication)171 (Fig. 7) with, again, combination of high antiherpetic activity and strong fluorescence. Similarly, the 6‐(4‐biphenylyl)substituted TACV and TGCV derivatives showed high selectivity against HSV‐1 together with fluorescent properties.172 Introduction of a fluorine i.e. in the 6‐phenyl part of the structure may make the tricyclic acyclovir and ganciclovir analogues amenable to 19F NMR studies.173
Meanwhile the fluorescent tricyclic acyclovir and ganciclovir derivatives had demonstrated a pronounced cytostatic activity (accompanied by a pronounced bystander effect) in HSV‐1 thymidine kinase (TK) gene‐transduced tumor cell lines.174 The importance of this particular observation should be viewed in the context of our earlier observation that tumor cells transformed with the HSV‐1 TK gene become highly sensitive to the cytostatic effects of anti‐herpetic drugs such as ganciclovir.175 In 1992 Culver et al. reported that rats bearing glioma tumors could be successfully treated with ganciclovir following in situ transfection by the HSV‐1 TK gene.176 This combined gene/chemotherapy approach for cancer has been further elaborated by Degrève et al.177 The fluorescent tricyclic acyclovir and ganciclovir may well fit in this approach as being fluorescent they could be readily monitored in biological fluids and tissues, and being more lipophilic than acyclovir or ganciclovir themselves they may better taken up from the blood into the central nervous system.174
Following acyclovir, ganciclovir, the relatively unknown 9‐ [cis‐1′,2′‐bis(hydroxymethyl)cycloprop‐1′‐yl]methyl
[cis‐1′,2′‐bis(hydroxymethyl)cycloprop‐1′‐yl]methyl guanine and especially its 1′S,2′R enantiomer A‐5021 served as the starting point for implanting the 1,N‐2‐etheno bridge. Although A‐5021, quite unfortunately, has not been further pursued as an antiviral drug (candidate), its credentials have been known since the late 1990s.178, 179, 180, 181 I have, personally, always emphasized the potential of A‐5021 as an antiviral drug,182 and, therefore, I favor the use of the scaffold of A‐5021 when building it up to tricyclic analogues, reminiscent of the tricyclic derivatives of acyclovir (TACV) and ganciclovir (TGCV) (see supra). The tricyclic analogues of A‐5021 reported by Ostrowski et al.183 confirm the potential of this approach.
guanine and especially its 1′S,2′R enantiomer A‐5021 served as the starting point for implanting the 1,N‐2‐etheno bridge. Although A‐5021, quite unfortunately, has not been further pursued as an antiviral drug (candidate), its credentials have been known since the late 1990s.178, 179, 180, 181 I have, personally, always emphasized the potential of A‐5021 as an antiviral drug,182 and, therefore, I favor the use of the scaffold of A‐5021 when building it up to tricyclic analogues, reminiscent of the tricyclic derivatives of acyclovir (TACV) and ganciclovir (TGCV) (see supra). The tricyclic analogues of A‐5021 reported by Ostrowski et al.183 confirm the potential of this approach.
Prevailing also for the concept of the tricyclic acyclovir and ganciclovir approach is that their activity against VZV could be easily extended to a number of A‐5021 derivatives, including its 6‐thien‐2‐yl, 6‐(5‐bromothien‐2‐yl), and 6‐furan‐2‐yl derivatives (Fig. 7).184
H. Glycopeptide (and Other) Antibiotic Derivatives: an as Yet to be Fully Explored Opportunity to Inhibit Cellular Entry of a Wide Variety of (Enveloped) Viruses
More than 20 ago, it must have been in the 1980's, I met with Dr. Maria Preobrazhenskaya [she went with me on a trip to our (remote) Campus of Kortrijk for a side visit]. She was (and still is) at the Gause Institute of New Antibiotics of the Russian Academy of Medical Sciences at Moscow (Russia). We decided to work on antibiotics, not as antibacterial agents, but as cytostatic agents, and from our original collaboration resulted several papers describing the cytostatic activity of a number of anthracycline (i.e. daunorubicin) derivatives185, 186, 187, 188 as well as streptonigrin derivatives.189, 190 [Incidentally, Ferenc Sztaricskai (see infra) was a co‐author on the latter paper.]
None of these compounds or any of their derivatives was further explored for their antiviral potential; and so was the antibiotic arcyriarubin.191 Yet, antibiotics belonging to this class of compounds (such as arcyriaflavin) have been reported to exhibit potent and selective inhibition of human cytomegalovirus (HCMV) replication,192 and bisindolylmaleimides such as arcyriaflavin A (Fig. 8) may even have potential usefulness as anti‐hepatitis C virus agents.193 In the mean time, novel derivatives of the antibiotic olivomycin194, 195 and novel anthracene‐9,10‐diones196 were reported, although they were considered only as cytostatic, and, regretfully, not as (potential) antiviral agents.
Figure 8.

Glycopeptide antibiotic (e.g. vancomycin, teicoplanin, eremomycin) aglycon derivatives.
Yet, the glycopeptide antibiotics vancomycin (Fig. 8), eremomycin, ristocetin and teicoplanin and their aglycons ( = nonglycosylated parts) have served as the starting points for the synthesis of (semisynthetic) derivatives with antiviral activity, i.e. compound 62 (a teicoplanin type aglycon) (Fig. 8) exhibiting an EC50 of 0.75 µM against HIV‐1.197 The mode of anti‐HIV action of these glycopeptide antibiotics could be ascribed to an inhibition of the viral entry process.197 From further work along these lines emanated several other glycopeptide derivatives, which, while inactive against bacteria, proved quite effective and selective as antiretroviral agents, i.e. compound 5a (Fig. 8)198 with an EC50 of 1.6 µM against HIV‐1. As these compounds had lost their antibacterial activity (i.e. ability to interact with the peptidoglycan glycopeptidyl transferase) they may be expected not to lead to bacterial resistance even after prolonged administration. Glycopeptide aglycon derivatives such as 5a 198 and 62 197 may therefore be considered promising candidate drugs for the chemotherapy and/or–prophylaxis of HIV infections.
Numerous glycopeptide antibiotics, primarily derived from eremomycin and teicoplanin, have been described for their antibacterial activity.199, 200, 201, 202, 203, 204, 205 As shown earlier with regard to their anti‐HIV activity,197, 198 many of these compounds also showed activity against the coronaviruses feline infectious peritonitis virus (FIPV) and human SARS CoV, i.e. compound 27 [EC50: 0.43 µM (HIV‐1), 5.4 µM (FPIV), and 14 µM (SARS CoV)] and compound 115 [EC50: 0.7 µM (HIV‐1), 1.6 µM (FIPV), and 8.0 µM (SARS CoV)] (Fig. 8).206 Although the mode of anti‐coronavirus action could, like the anti‐HIV‐1 mode of action, be attributed to an inhibition of the viral entry process, there was, in general, no close correlation between the EC50 values of the compounds for HIV‐1, FIPV, and SARS CoV.206
Whether any of the above mentioned glycopeptide aglycon derivatives would have any anti‐influenza virus activity is not known (and has probably never been assessed). It therefore came as a surprise that one particular glycopeptide aglycon derivative, namely aglycoristocetin with cyclobutene dione carrying an hydrophobic side chain (Fig. 8), compound 8e in the original publication, showed a specific, consistent and selective activity (EC50: 0.4 µM) against various influenza A and B viruses.207 Again the mode of action of compound 8e was assumed to be associated with the influenza virus entry process.207 This signals that (many of) the glycopeptide antibiotic derivatives reported by Maria Peobrazhenskaya and her co‐workers should be revisited for their potential anti‐influenza virus activity, and, vice versa, the compounds from Ferenc Sztaricskai and his coworkers should also be looked at for their potential activity against HIV and SARS CoV.
I. Is There a Role for Cidofovir in the Treatment of Polyomavirus (JC or BK) Infection: a Continuing Question?
Since we (Graciela Andrei, Robert Snoeck, Michel Vandeputte and I) described (in 1997) the inhibitory effects of acyclic nucleoside phosphonates, such as cidofovir, against the in vitro replication of polyomaviruses,208 cidofovir has been used with (anecdotal) success in the treatment of various diseases associated with the polyomaviruses JC [progressive multifocal leukoencephalopathy (PML)], BK (hemorrhagic cystitis (HC) and BK virus nephropathy (BKN)]. In 2003, I reviewed clinical data obtained with cidofovir in the treatment of polyomavirus (JC and BK)‐associated PML and HC, respectively.209 Here, I address further case reports that have appeared within the past 5‐year period on the potential usefulness of cidofovir in the treatment of polyomavirus‐associated infections.
The most remarkable report, perhaps, is that of a dual infection with polyomavirus BK and acyclovir‐resistant herpes simplex virus (HSV) in a hematopoietic stem cell transplant (HSCT) recipient that was successfully treated with cidofovir (intravenously at a dose of 5 mg/kg weekly, three times, and then every other week for three additional times).210 That cidofovir may preferentially inhibit HSV, as well as HCMV infection could be rationalized on the basis of its specific inhibitory effect on the viral DNA polymerase,211 but polyomaviruses do not induce their own DNA polymerase, which makes it more difficult to explain the favorable results reported for cidofovir in the treatment of polymavirus (BK) virus infections,212, 213, 214 as reviewed by Rinaldo and Hirsch.215 [Incidentally, in the latter review, the structure of cidofovir was not correctly presented.] As to its mechanism of action, cidofovir would inhibit polyomavirus BK replication in human renal tubular cells at a transcription of late viral mRNA, downstream of viral early gene expression.216 Esterification of cidofovir with an ether lipid group, such as hexadecyloxopropyl, octadecyloxyethyl,or oleyloxyethyl, would further enhance the in vitro inhibitory effect of cidofovir on polyomavirus BK replication by 3 log's.217
BKN is an important cause of renal graft dysfunction in kidney transplant recipients [BK virus was first identified in 1971 in a kidney transplant patient with ureteric stenosis218] BK virus has been found in 50% of normal healthy kidneys but, when reactivated in a transplanted kidney, consequently to immunosuppression (i.e. with tacrolimus mycophenolate mofetil, and prednisolone) it may be causing interstitial nephritis nephropathy, which may lead to irreversible graft failure.219, 220 Polyomavirus has been considered a “hot problem” in renal transplantation.221
Cidofovir that is highly concentrated in tubuli and renal tissue, the primary sites of BK virus infection, has proven to have a beneficial effect on BKN, even if administered weekly at a low dose (0.5–1.0 mg/kg intravenously) for 4–10 weeks.212 This and other similarly favorable results obtained for cidofovir (reviewed by Bonvoisin et al.221) also including the first case in Japan of a kidney transplant recipient successfully treated with cidofovir for BKN222 should be followed up by randomized controlled studies to prospectively evaluate the effects of cidofovir in graft survival as well as BK viral load.221
Low‐dose cidofovir (weekly, 1 mg/kg intravenously) has also proven successful in the treatment of BK virus‐associated HC in recipients of HSCTs,223 and this observation, again, necessitates followup by a prospective trial. In the treatment of HC after allogeneic stem cell transplantation, cidofovir may even to be instilled directly into the bladder, as this regimen has been shown to decrease BK viral load and significantly improve the clinical outcome.214
Whether cidofovir, in addition to highly active anti‐retroviral therapy (HAART), would give an additional benefit, as compared with HAART alone in the treatment of PML in HIV‐infected patients could not be proven.224 The usefulness of cidofovir in the treatment of PML has continued to be controversial since my previous review in 2003.209 Cidofovir would be helpful in the treatment of PML according to some reports225, 226 but not according to others.227 The favorable response of PML, as visualized by magnetic resonance imaging of the brain, to treatment with cidofovir (5 mg/kg every 2 weeks) in combination with cytarabine (2 mg/kg/day for 5 days every 3 weeks) is illustrated in Figure 9. In this case an obvious regression of the lesions was noted.226
Figure 9.

Magnetic resonance imaging (MRI) of the brain before (3 panels on the left) and after (3 panels on the right) six regimens of cidofovir (5 mg/kg every 2 weeks, intravenously) in combination with cytarabine (2 mg/kg/day for 5 days every 3 weeks, intravenously) in a non‐AIDS patient with PML (progressive multifocal leukoencephalopathy). Data taken from Terrier et al.226
J. Thymidine Phosphorylase, a Target for Both Antiviral and Anticancer Agents
Thymidine phosphorylase (TPase)228 is one of the key enzymes involved in both the salvage and catabolism of pyrimidine 2′‐deoxynucleosides: TPase catalyzes the reversible phosphorolysis of 2′‐deoxythymidine, with formation of 2‐deoxy‐α‐D‐ribose‐1‐phosphate and thymine (Fig. 10A). TPase recognizes as substrate not only 2′‐deoxythymidine and 2′‐deoxyuridine but also a variety of 5‐substituted 2′‐deoxyuridine analogues, including BVDU (brivudin).229 [BVDU has in the mean time been licensed in several European countries for the treatment of herpes zoster, where it has proved efficacious as a single oral dose of 125 mg once daily for 7 days: see ref.1.] It is in fact the TPase, which converts BVDU into its free base BVU. BVU is an inhibitor of DPD (dihydropyrimidine dehydrogenase), the enzyme responsible for the hydrogenation of the antitumor agent 5‐fluorouracil (FU), and has been shown to enhance the toxicity of FU. Combination of BVDU with TPase inhibitors (see infra) should by preventing its degradation to BVU, in principle, enhance the antiviral activity of BVDU, and, concomitantly therewith, reduce the toxic side effects of FU, should the latter (or prodrugs thereof) be used (inadvertently) in combination with BVDU. Despite their attractive potential for increasing the antiviral activity of BVDU or other 5‐substituted 2′‐deoxyuridines (such as IDU or TFT: see ref.3). TPase inhibitors (see infra) have not been fully explored as (adjunct) antiviral agents.
Figure 10.

(A) Reversible phosphorolysis of 2′‐deoxythymidine by thymidine phosphorylase (TPase). (B) Inhibitors of thymidine phosphorylase (TPase).
Instead, after it had become clear that TPase is identical to platelet‐derived endothelial cell growth factor (PD‐ECGF, a factor that had been known since the mid 1980s,230, 231, 232, 233 the sole angiogenic (angiogenesis‐inducing) factor present in platelets,234 interest in TPase inhibitors shifted toward their potential as anticancer agents blocking angiogenesis. As reviewed previously,235 TPase is overexpressed in many solid tumors including breast, ovarian, colorectal, and pancreatic cancers, pointing to a role for this enzyme in tumor vascularization.
Which are the compounds that have been reported as TPase inhibitors? The first ones, and this dates from long before TPase was recognized to be an angiogenic factor, must have been 6‐amino‐5‐bromouracil and 6‐aminothymine (Fig. 10B), described in 1967 by Peter Langen and his coworkers.236 It took until 1998 before 7‐deazaxanthine (Fig. 10B), which could be viewed as an uracil derivative with a ring closure between C5 and C6, was described as a TPase inhibitor,237 and in 2000, a new approach, based on “multisubstrate” inhibitors of TPase, was announced.238, 239 This “multisubstrate” concept based upon 1‐(8‐phosphonooctyl)‐7‐deazaxanthine (TP‐65) was, however, not followed up (except for its anti‐angiogenic activity240) in further studies.
Novel 6‐substituted uracil derivatives that have been described as TPase inhibitors with anti‐angiogenic properties include 6‐(2‐aminoethyl)amino‐5‐chlorouracil (AEAC) (Fig. 10B).241 One of the most promising TPase inhibitors described for their inhibitory effects on angiogenesis has been the thymidine phosphorylase inhibitor (TPI) 5‐chloro‐6‐[1‐(2‐iminopyrrolidinyl)methyl]uracil hydrochloride (Fig. 10B).242 Characteristically, this compound potentiated the antitumor activity of IDU and TFT.243 In the light of what I explained above, it would not be too farfetched to postulate that TPI may potentiate the antiviral activity of IDU, TFT and… BVDU as well.
Acyclic nucleoside phosphonates, such as cidofovir, adefovir, and tenofovir,211 also referred by Holý as phosphonomethoxyalkyl analogues of nucleotides244 have gained such a wide notoriety, that it would almost seem “odd” if they (or at least some of them) would not be active against TPase (PD‐ECGF), and, indeed, substantial inhibition of PD‐ECGF (from Sprague–Dawley lymphoma) has been observed with a series of phosphonomethoxyalkyl thymines (in order of decreasing activity): (R)‐FPMPT>(S)‐FPMPT≥(R)‐HPMPT>(S)‐PMPT>(S)‐HPMPT≥(R)‐PMPT.245 [Inhibition of 2‐deoxy‐D‐ribose‐1‐phosphate release from the endothelial cells may represent the potential anti‐angiogenic target in this case.246] (R)‐ and (S)‐PMPT, ‐HPMPT and–FPMPT (Fig. 10B)247, 248 should be further followed up as TPase (PD‐ECGF) inhibitors for their anti‐angiogenic activity in particular, and anticancer activity in general.
As for the reverse transcriptase (RT), both nucleoside type (i.e. TPI) and nucleotide type (i.e. (R)‐FPMPT) of TPase inhibitors have been described. This analogy can be extended to non‐nucleoside type of TPase inhibitors such as KIN59 (5′‐O‐tritylinosine) (Fig. 10B), which like the non‐nucleoside RT inhibitors (NNRTIs), inhibit TPase via a non‐competitive mechanism of action249 and thus act as allosteric inhibitors of TPase.250 As recently demonstrated,251 5′‐O‐tritylinosine would fit into a cavity (with aspartic acid residue −203 being critical) located at a distance of about 11 Å from the substrate‐binding site of TPase.
2. CONCLUSIONS
The ten stories (E1–E10) that are subject of the present review offer a mix of premises and promises: they have done so over a period spanning one or two decades, and they are likely to continue to do so for the next 10 years or so.
E1. ATA analogues have continued to find new applications, one of the last being influenza virus, but given their lack of specificity, would they ever find their final “niche”?
E2. The ADAMs originated from the ATAs and are still looked upon as NNRTIs, but are they genuine NNRTIs (as are nevirapine, efavirenz, etravirine, rilpivirine, and others), or may they have, as yet uncovered, “side” effects worth pursuing?
E3. Among the HIV INIs, raltegravir was the first to cross the (finish) line, thereby fulfilling a long‐vested premise. Will elvitegravir be the second crossing the line? What are the prospects for a quadruple drug combination consisting of tenofovir, emtricitabine, elvitegravir, and a booster (pharmacoenhancer)?
E4. In the aftermath of the INIs, LEDGF may be considered as a fascinating but risky target, and the development of LEDGF inhibitors may be a roller coaster ride.
E5. There is, at present, more talking (and writing) about influenza A (H1N1) virus strains that are resistant to oseltamivir, than about an imminent avian influenza A (H5N1) pandemic. [Meanwhile, the influenza A H5N1 Mexican pandemic swept through the five Continents.] Oseltamivir should have been stockpiled by now, and its synthesis has been improved. It should still be the first choice drug to be used if needed to curb any influenza A pandemic (whether H1N1 or H5N1).
E6. In the meantime RSV has remained a real threat, at least in the very young and (not so very) old, and besides the monoclonal antibody palivizumab and the (questionable) use of ribavirin, we are still waiting for an effective and versatile anti‐RSV agent.
E7. From a purely chemical viewpoint it is surprising that tricyclic guanosine derivatives (based on 1H,‐2‐ethenoguanine) has received so little attention from the biomedical world, despite their unique combination: an anti‐herpesvirus activity combined with marked fluorescent properties.
E8. Antibiotics and antivirals, often envisaged as antipodes, have been reconciled with the demonstration that glycopeptide antibiotics (or their aglycons) are able to block the entry of enveloped viruses such as HIV. This unique property may well extend, and should be further explored for other enveloped viruses, such as coronaviruses (already done), myxoviruses (only barely touched upon), and herpesviruses (not even yet considered).
E9. Cidofovir (off‐label), and other acyclic nucleoside phosphonates have as yet unexplored potential for the treatment of what was once called papovaviruses. Their potential in the treatment of HPV infections has already been highlighted,4 but their equally promising potential for the treatment of polyomavirus infections should be further explored as well.
E10. TPase as target to increase the antiviral activity of compounds like BVDU against HSV‐1 and VZV that are readily degraded by dThd phosphorylase deserves (much) more attention than received so far and in its own right the dThd phosphorylase should be considered as a target for antitumor agents, given its angiogenic prowess thus offering the design of anti‐angiogenic compounds with antitumor potential.
Biographical Information
Erik De Clercq, MD, Ph.D. has been Chairman of the Department of Microbiology and Immunology of the Medical School at the Katholieke Universiteit Leuven (K.U.Leuven) as well as Chairman of the Board of the Rega Institute for Medical Research (until September 2006). He is currently President of the Rega Foundation and a director of the Belgian (Flemish) Royal Academy of Medicine, a member of the Academia Europaea and Fellow of the American Association for the Advancement of Science. He has also been the titular Prof. of the P. De Somer Chair for Microbiology. He has been teaching the courses of Cell Biology, Biochemistry and Microbiology at the K.U.Leuven (and Kortrijk) Medical School, until September 2006 and is still allowed to teach part of the Biochemistry course at the K.U.Leuven Campus Kortrijk. Professor De Clercq received in 1996 the Hoechst Marion Roussel (now called “Aventis”) award (American Society for Microbiology), and in 2000 the Maisin Prize for Biomedical Sciences (National Science Foundation, Belgium) for his pioneering efforts in the field of antiviral research. He is an honorary doctor of several Universities [i.e. Ghent (Belgium), Athens (Greece), Ferrara (Italy), Shandong (Jinan, China), Charles University (Prague, Czech Republic) and Budejovice (Czech Republic)]. In 2008 he was elected European Inventor of the Year (Life time achievement award from the European Union). His scientific interests are in the antiviral chemotherapy field, and, in particular, the development of new antiviral agents for various viral infections, including herpes simplex virus (HSV), varicella‐zoster virus (VZV), cytomegalovirus (CMV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), human papilloma virus (HPV) and hepatitis C virus (HCV). He has (co)‐discovered a number of antiviral drugs, currently used in the treatment of HSV infections (valaciclovir, Valtrex®, Zelitrex®), VZV infections (brivudin, Zostex®, Brivirac®, Zerpex®), CMV infections (cidofovir, Vistide®), HBV infections (adefovir dipivoxil, Hepsera®), and HIV infections (AIDS) (tenofovir disoproxil fumarate, marketed as Viread®, and, in combination with emtricitabine, as Truvada®, and, in combination with both emtricitabine and efavirenz, as Atripla®). Viread® has also been approved for the treatment of HBV infections (chronic hepatitis B).
Acknowledgements
Christiane Callebaut, who helped me in previous parts of this series 1–4 proved again of invaluable editorial assistance in this review article (part E). I owe her and the chemists referred to in E1 and E2 (Mark Cushman and his colleagues), E7 (Bozenna Golankiewicz and her colleagues) and E8 (Maria Preobrazhenskaya and her colleagues) my warmest thanks. A special acknowledgment is going to Peter Cherepanov now at Imperial College London (United Kingdom) as he laid the basis for the discovery of LEDGF (see E4). Dedication: This paper is dedicated to those (superb) friends who over the past decade introduced me to a honorary doctor's degree at their University: Prof. Dennis De Keukeleire (Ghent, Belgium); Prof. Nicolas Kolocouris (Athens, Greece), Prof. Stefano Manfredini and Prof. Pier Giovanni Baraldi (Ferrara, Italy), Prof. Xinyong Liu [Jinan (Shandong University), China], Prof. Anthonin Holý [Prague (Charles University), Czech Republic] and Prof. Libor Grubhoffer (Ceské Budějovice, Czech Republic). I owe them my sincere thanks for this esteemed recognition.
REFERENCES
- 1. De Clercq E. The discovery of antiviral agents: Ten different compounds, ten different stories. Med Res Rev 2008;28:929–953. [DOI] [PubMed] [Google Scholar]
- 2. De Clercq E. Antiviral drug discovery: Ten more compounds, and ten more stories (part B). Med Res Rev 2009;29:571–610. [DOI] [PubMed] [Google Scholar]
- 3. De Clercq E. Another ten stories in antiviral drug discovery (part C): “old” and “new” antivirals, strategies, and perspectives. Med Res Rev 2009;29:611–645. [DOI] [PubMed] [Google Scholar]
- 4. De Clercq E. Ten stories on antiviral drug discovery (part D): Paradigms, paradoxes and paraductions. Med Res Rev 2009, in press. [DOI] [PubMed] [Google Scholar]
- 5. González RG, Blackburn BJ, Schleich T. Fractionation and structural elucidation of the active components of aurintricarboxylic acid, a potent inhibitor of protein nucleic acid interactions. Biochim Biophys Acta 1979;562:534–545. [DOI] [PubMed] [Google Scholar]
- 6. Cushman M, Kanamathareddy S. Synthesis of the covalent hydrate of the incorrectly assumed structure of aurintricarboxylic acid (ATA). Tetrahedron 1990;46:1491–1498. [Google Scholar]
- 7. Cushman M, Wang PL, Chang SH, Wild C, De Clercq E, Schols D, Goldman ME, Bowen JA. Preparation and anti‐HIV activities of aurintricarboxylic acid fractions and analogues: Direct correlation of antiviral potency with molecular weight. J Med Chem 1991;34:329–337. [DOI] [PubMed] [Google Scholar]
- 8. Bina‐Stein M, Tritton TR. Auritricarboxylic acid is a nonspecific enzyme inhibitor. Mol Pharmacol 1976;12:191–193. [PubMed] [Google Scholar]
- 9. Hallick RB, Chelm BK, Gray PW, Orozco EM, Jr. Use of aurintricarboxylic acid as an inhibitor of nucleases during nucleic acid isolation. Nucleic Acids Res 1977;4:3055–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balzarini J, Mitsuya H, De Clercq E, Broder S. Aurintricarboxylic acid and Evans Blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T‐cell lymphotropic virus type III/lymphadenopathy‐associated virus. Biochem Biophys Res Commun 1986;136:64–71. [DOI] [PubMed] [Google Scholar]
- 11. Baba M, Schols D, Pauwels R, Balzarini J, De Clercq E. Fuchsin acid selectively inhibits human immunodeficiency virus (HIV) replication in vitro. Biochem Biophys Res Commun 1988;155:1404–1411. [DOI] [PubMed] [Google Scholar]
- 12. Schols D, Baba M, Pauwels R, Desmyter J, De Clercq E. Specific interaction of aurintricarboxylic acid with the human immunodeficiency virus/CD4 cell receptor. Proc Natl Acad Sci USA 1989;86:3322–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schols D, Pauwels R, Desmyter J, De Clercq E. Dextran sulfate and other polyanionic anti‐HIV compounds specifically interact with the viral gp120 glycoprotein expressed by T‐cells persistently infected with HIV‐1. Virology 1990;175:556–561. [DOI] [PubMed] [Google Scholar]
- 14. Cushman M, Kanamathareddy S, De Clercq E, Schols D, Goldman ME, Bowen JA. Synthesis and anti‐HIV activities of low molecular weight aurintricarboxylic acid fragments and related compounds. J Med Chem 1991;34:337–342. [DOI] [PubMed] [Google Scholar]
- 15. He R, Adonov A, Traykova‐Adonova M, Cao J, Cutts T, Grudesky E, Deschambaul Y, Berry J, Drebot M, Li X. Potent and selective inhibition of SARS coronavirus replication by aurintricarboxylic acid. Biochem Biophys Res Commun 2004;320:1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yap Y, Zhang X, Andonov A, He R. Structural analysis of inhibition mechanisms of aurintricarboxylic acid on SARS‐CoV polymerase and other proteins. Comput Biol Chem 2005;3:212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Myskiw C, Deschambault Y, Jefferies K, He R, Cao J. Aurintricarboxylic acid inhibits the early stage of vaccinia virus replication by targeting both cellular and viral factors. J Virol 2007;81:3027–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsi CJ, Chao Y, Chen CW, Lin WW. Aurintricarboxylic acid protects against cell death caused by lipopolysaccharide in macrophages by decreasing inducible nitric‐oxide synthase induction via IkappaB kinase, extracellular signal‐regulated kinase, and p38 mitogen‐activated protein kinase inhibition. Mol Pharmacol 2002;62:90–101. [DOI] [PubMed] [Google Scholar]
- 19. Chen CW, Chao Y, Chang YH, Hsu MJ, Lin WW. Inhibition of cytokine‐induced JAK‐STAT signalling pathways by an endonuclease inhibitor aurintricarboxylic acid. Br J Pharmacol 2002;137:1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beery R, Haimsohn M, Wertheim N, Hemi R, Nir U, Karasik A, Kanety H, Geier A. Activation of the insulin‐like growth factor 1 signaling pathway by the antiapoptotic agents aurintricarboxylic acid and evans blue. Endocrinology 2001;142:3098–3107. [DOI] [PubMed] [Google Scholar]
- 21. Hung HC, Tseng CP, Yang JM, Ju YW, Tseng SN, Chen YF, Chao YS, Hsieh HP, Shih SR, Hsu JT. Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antiviral Res 2009;81:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steward DL, Martin J, Grollman AP. Inhibition of influenza virus by triphenylmethane compounds. Ann NY Acad Sci 1977;284:638–649. [DOI] [PubMed] [Google Scholar]
- 23. Liao LL, Horwitz SB, Huang MT, Grollman AP, Steward D, Martin J. Triphenylmethane dyes as inhibitors of reverse transcriptase, ribonucleic acid polymerase, and protein synthesis. Structure–activity relationships. J Med Chem 1975;18:117–120. [DOI] [PubMed] [Google Scholar]
- 24. Milani M, Mastrangelo E, Bollati M, Selisko B, Decroly E, Bouvet M, Canard B, Bolognesi M. Flaviviral methyltransferase/RNA interaction: Structural basis for enzyme inhibition. Antiviral Res 2009;83:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cushman M, Golebiewski WM, McMahon JB, Buckheit RW, Jr , Clanton DJ, Weislow O, Haugwitz RD, Bader JP, Graham L, Rice WG. Design, synthesis, and biological evaluation of cosalane, a novel anti‐HIV agent which inhibits multiple features of virus reproduction. J Med Chem 1994;37:3040–3050. [DOI] [PubMed] [Google Scholar]
- 26. Cushman M, Golebiewski WM, Buckheit RW, Jr , Graham L, Rice WG. Synthesis and biological evaluation of an alkenyldiarylmethane (ADAM) which acts as a novel non‐nucleoside HIV‐1 reverse transcriptase inhibitor. Bioorg Med Chem Lett 1995;5:2713–2716. [Google Scholar]
- 27. Cushman M, Golebiewski WM, Graham L, Turpin JA, Rice WG, Fliakas‐Boltz V, Buckheit RW, Jr. Synthesis and biological evaluation of certain alkenyldiarylmethanes as anti‐HIV‐1 agents which act as non‐nucleoside reverse transcriptase inhibitors. J Med Chem 1996;39:3217–3227. [DOI] [PubMed] [Google Scholar]
- 28. Cushman M, Casimiro‐Garcia A, Williamson K, Rice WG. Synthesis of a non‐nucleoside reverse transcriptase inhibitor in the alkenyldiarylmethane (ADAM) series with optimized potency and therapeutic index. Bioorg Med Chem Lett 1998;8:195–198. [DOI] [PubMed] [Google Scholar]
- 29. Cushman M, Casimiro‐Garcia A, Hejchman E, Ruell JA, Huang M, Schaeffer CA, Williamson K, Rice WG, Buckheit RW, Jr. New alkenyldiarylmethanes with enhanced potencies as anti‐HIV agents which act as non‐nucleoside reverse transcriptase inhibitors. J Med Chem 1998;41:2076–2089. [DOI] [PubMed] [Google Scholar]
- 30. Casimiro‐Garcia A, Micklatcher M, Turpin JA, Stup TL, Watson K, Buckheit RW, Cushman M. Novel modifications in the alkenyldiarylmethane (ADAM) series of non‐nucleoside reverse transcriptase inhibitors. J Med Chem 1999;42:4861–4874. [DOI] [PubMed] [Google Scholar]
- 31. Xu G, Loftus TL, Wargo H, Turpin JA, Buckheit RW, Jr , Cushman M. Solid‐phase synthesis of the alkenyldiarylmethane (ADAM) series of non‐nucleoside HIV‐1 reverse transcriptase inhibitors. J Org Chem 2001;66:5958–5964. [DOI] [PubMed] [Google Scholar]
- 32. Xu G, Micklatcher M, Silvestri MA, Hartman TL, Burrier J, Osterling MC, Wargo H, Turpin JA, Buckheit RW, Jr , Cushman M. The biological effects of structural variation at the meta position of the aromatic rings and at the end of the alkenyl chain in the alkenyldiarylmethane series of non‐nucleoside reverse transcriptase inhibitors. J Med Chem 2001;44:4092–4113. [DOI] [PubMed] [Google Scholar]
- 33. Xu G, Hartman TL, Wargo H, Turpin JA, Buckheit RW, Cushman M. Synthesis of alkenyldiarylmethane (ADAM) non‐nucleoside HIV‐1 reverse transcriptase inhibitors with non‐identical aromatic rings. Bioorg Med Chem 2002;10:283–290. [DOI] [PubMed] [Google Scholar]
- 34. Silvestri MA, Nagarajan M, De Clercq E, Pannecouque C, Cushman M. Design, synthesis, anti‐HIV activities, and metabolic stabilities of alkenyldiarylmethane (ADAM) non‐nucleoside reverse transcriptase inhibitors. J Med Chem 2004;47:3149–3162. [DOI] [PubMed] [Google Scholar]
- 35. Deng BL, Hartman TL, Buckheit RW, Jr , Pannecouque C, De Clercq E, Cushman M. Replacement of the metabolically labile methyl esters in the alkenyldiarylmethane series of non‐nucleoside reverse transcriptase inhibitors with isoxazolone, isoxazole, oxazolone, or cyano substituents. J Med Chem 2006;49:5316–5323. [DOI] [PubMed] [Google Scholar]
- 36. Deng BL, Cullen MD, Zhou Z, Hartman TL, Buckheit RW, Jr , Pannecouque C, De Clercq E, Fanwick PE, Cushman M. Synthesis and anti‐HIV activity of new alkenyldiarylmethane (ADAM) non‐nucleoside reverse transcriptase inhibitors (NNRTIs) incorporating benzoxazolone and benzisoxazole rings. Bioorg Med Chem 2006;14:2366–2374. [DOI] [PubMed] [Google Scholar]
- 37. Sakamoto T, Cullen MD, Hartman TL, Watson KM, Buckheit RW, Pannecouque C, De Clercq E, Cushman M. Synthesis and anti‐HIV activity of new metabolically stable alkenyldiarylmethane non‐nucleoside reverse transcriptase inhibitors incorporating N‐methoxy imidoyl halide and 1,2,4‐oxadiazole systems. J Med Chem 2007;50:3314–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deng BL, Zhao Y, Hartman TL, Watson K, Buckheit RW, Jr , Pannecouque C, De Clercq E, Cushman M. Synthesis of alkenyldiarylmethanes (ADAMs) containing benzo[d]isoxazole and oxazolidin‐2‐one rings, a new series of potent non‐nucleoside HIV‐1 reverse transcriptase inhibitors. Eur J Med Chem 2009;44:1210–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cullen MD, Cheung YF, Houslay MD, Hartman TL, Watson KM, Buckheit RW, Jr , Pannecouque C, De Clercq E, Cushman M. Investigation of the alkenyldiarylmethane non‐nucleoside reverse transcriptase inhibitors as potential cAMP phosphodiesterase‐4B2 inhibitors. Bioorg Med Chem Lett 2008;18:1530–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cullen MD, Sarkar T, Hamel E, Hartman TL, Watson KM, Buckheit RW, Jr , Pannecouque C, De Clercq E, Cushman M. Inhibition of tubulin polymerization by select alkenyldiarylmethanes. Bioorg Med Chem Lett 2008;18:469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barré‐Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler‐Blin C, Vézinet‐Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Isolation of a T‐lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983;220:868–871. [DOI] [PubMed] [Google Scholar]
- 42. Popovic M, Sarin PS, Robert‐Gurroff M, Kalyanaraman VS, Mann D, Minowada J, Gallo RC. Isolation and transmission of human retrovirus (human t‐cell leukemia virus). Science 1983;219:856–859. [DOI] [PubMed] [Google Scholar]
- 43. De Clercq E. Anti‐HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents 2009;33:307–320. [DOI] [PubMed] [Google Scholar]
- 44. Berger DS. Comparing two integrase inhibitors. Posit Aware 2008;19:49–51. [PubMed] [Google Scholar]
- 45. Havlir DV. HIV integrase inhibitors—out of the pipeline and into the clinic. N Engl J Med 2008;359:416–418. [DOI] [PubMed] [Google Scholar]
- 46. Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Gatell JM, Rockstroh JK, Katlama C, Yeni P, Lazzarin A, Clotet B, Zhao J, Chen J, Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Nguyen BY, Teppler H. Raltegravir with optimized background therapy for resistant HIV‐1 infection. N Engl J Med 2008;359:339–354. [DOI] [PubMed] [Google Scholar]
- 47. Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, Lazzarin A, Clotet B, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Zhao J, Chen J, Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Teppler H, Nguyen BY. Subgroup and resistance analyses of raltegravir for resistant HIV‐1 infection. N Engl J Med 2008;359:355–365. [DOI] [PubMed] [Google Scholar]
- 48. Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV‐1 replication in cells. Science 2000;287:646–650. [DOI] [PubMed] [Google Scholar]
- 49. Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Jr , Egbertson MS, Vacca JP, Huff JR, Felock PJ, Witmer MV, Stillmock KA, Danovich R, Grobler J, Miller MD, Espeseth AS, Jin L, Chen IW, Lin JH, Kassahun K, Ellis JD, Wong BK, Xu W, Pearson PG, Schleif WA, Cortese R, Emini E, Summa V, Holloway MK, Young SD. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV‐1 integrase. Proc Natl Acad Sci USA 2004;101:11233–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, Dornadula G, Danovich RM, Witmer MV, Wilson KA, Tussey L, Schleif WA, Gabryelski LS, Jin L, Miller MD, Casimiro DR, Emini EA, Shiver JW. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 2004;305:528–532. [DOI] [PubMed] [Google Scholar]
- 51. Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV‐integrase inhibitor for the treatment of HIV‐AIDS infection. J Med Chem 2008;51:5843–5855. [DOI] [PubMed] [Google Scholar]
- 52. Terrazas‐Aranda K, Van Herrewege Y, Hazuda D, Lewi P, Costi R, Di Santo R, Cara A, Vanham G. Human immunodeficiency virus type 1 (HIV‐1) integration: A potential target for microbicides to prevent cell‐free or cell‐associated HIV‐1 infection. Antimicrob Agents Chemother 2008;52:2544–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, Elion R, Farthing C, Zhong L, Cheng AK, McColl D, Kearney BP. Antiviral activity, pharmacokinetics, and dose response of the HIV‐1 integrase inhibitor GS‐9137 (JTK‐303) in treatment‐naive and treatment‐experienced patients. J Acquir Immune Defic Syndr 2006;43:1–5. [DOI] [PubMed] [Google Scholar]
- 54. Dayam R, Al‐Mawsawi LQ, Zawahir Z, Witvrouw M, Debyser Z, Neamati N. Quinolone 3‐carboxylic acid pharmacophore: Design of second generation HIV‐1 integrase inhibitors. J Med Chem 2008;51:1136–1144. [DOI] [PubMed] [Google Scholar]
- 55. Witvrouw M, Daelemans D, Pannecouque C, Neyts J, Andrei G, Snoeck R, Vandamme AM, Balzarini J, Desmyter J, Baba M, De Clercq E. Broad‐spectrum antiviral activity and mechanism of antiviral action of the fluoroquinolone derivative K‐12. Antivir Chem Chemother 1998;9:403–411. [DOI] [PubMed] [Google Scholar]
- 56. Parolin C, Gatto B, Del Vecchio C, Pecere T, Tramontano E, Cecchetti V, Fravolini A, Masiero S, Palumbo M, Palù G. New anti‐human immunodeficiency virus type 1 6‐aminoquinolones: Mechanism of action. Antimicrob Agents Chemother 2003;47:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Daelemans D, Lu R, De Clercq E, Engelman A. Characterization of a replication‐competent, integrase‐defective human immunodeficiency virus (HIV)/simian virus 40 chimera as a powerful tool for the discovery and validation of HIV integrase inhibitors. J Virol 2007;81:4381–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. Comparison of raltegravir and elvitegravir on HIV‐1 integrase catalytic reactions and on a series of drug‐resistant integrase mutants. Biochemistry 2008;47:9345–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK‐303/GS‐9137). J Virol 2008;82:764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hombrouck A, Voet A, Van Remoortel B, Desadeleer C, De Maeyer M, Debyser Z, Witvrouw M. Mutations in human immunodeficiency virus type 1 integrase confer resistance to the naphthyridine L‐870,810 and cross‐resistance to the clinical trial drug GS‐9137. Antimicrob Agents Chemother 2008;56:2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, Dockx K, Strijbos R, Smits V, Vos A, Meersseman G, Jochmans D, Vermeire K, Schols D, Hallenberger S, Hertogs K. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J Virol 2008;82:10366–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cherepanov P, Pluymers W, Claeys A, Proost P, De Clercq E, Debyser Z. High‐level expression of active HIV‐1 integrase from a synthetic gene in human cells. FASEB J 2000;14:1389–1399. [DOI] [PubMed] [Google Scholar]
- 63. Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV‐1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem 2003;278:372–381. [DOI] [PubMed] [Google Scholar]
- 64. Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV‐1 integrase in human cells. J Biol Chem 2003;278:33528–33539. [DOI] [PubMed] [Google Scholar]
- 65. Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. An essential role for LEDGF/p75 in HIV integration. Science 2006;314:461–464. [DOI] [PubMed] [Google Scholar]
- 66. Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 2005;11:1287–1289. [DOI] [PubMed] [Google Scholar]
- 67. Cherepanov P. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res 2007;35:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Poeschla EM. Integrase, LEDGF/p75 and HIV replication. Cell Mol Life Sci 2008;65:1403–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Engelman A, Cherepanov P. The lentiviral integrase binding protein LEDGF/p75 and HIV‐1 replication. PLoS Pathogens 2008;4:e1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Busschots K, De Rijck J, Christ F, Debyser Z. In search of small molecules blocking interactions between HIV proteins and intracellular cofactors. Mol Biosyst 2009;5:21–31. [DOI] [PubMed] [Google Scholar]
- 71. Greene WC, Debyser Z, Ikeda Y, Freed EO, Stephens E, Yonemoto W, Buckheit RW, Esté JA, Cihlar T. Novel targets for HIV therapy. Antiviral Res 2008;80:251–265. [DOI] [PubMed] [Google Scholar]
- 72. Hou Y, McGuinness DE, Prongay AJ, Feld B, Ingravallo P, Ogert RA, Lunn CA, Howe JA. Screening for antiviral inhibitors of the HIV integrase‐LEDGF/p75 interaction using the AlphaScreen luminescent proximity assay. J Biomol Screen 2008;13:406–414. [DOI] [PubMed] [Google Scholar]
- 73. Du L, Zhao Y, Chen J, Yang L, Zheng Y, Tang Y, Shen X, Jiang H. D77, one benzoic acid derivative, functions as a novel anti‐HIV‐1 inhibitor targeting the interaction between integrase and cellular LEDGF/p75. Biochem Biophys Res Commun 2008;375:139–144. [DOI] [PubMed] [Google Scholar]
- 74. Hayden F. Developing new antiviral agents for influenza treatment: What does the future hold? Clin Infect Dis 2009;48:S3–S13. [DOI] [PubMed] [Google Scholar]
- 75. Writing Committee of the Second World Health Organization Consultation on Clinical Aspects of Human Infection with Avian Influenza A (H5N1) virus . Update on avian influenza A (H5N1) virus infection in humans. N Engl J Med 2008;358:261–273. [DOI] [PubMed] [Google Scholar]
- 76. Hien ND, Ha NH, Van NT, Ha NT, Lien TT, Thai NQ, Trang VD, Shimbo T, Takahashi Y, Kato Y, Kawana A, Akita S, Kudo K. Human infection with highly pathogenic avian influenza virus (H5N1) in northern Vietnam, 2004–2005. Emerg Infect Dis 2009;15:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kandun IN, Wibisono H, Sedyaningsih ER, Yusharmen, Hadisoedarsuno W, Purba W, Santoso H, Septiawati C, Tresnaningsih E, Heriyanto B, Yuwono D, Harun S, Soeroso S, Giriputra S, Blair PJ, Jeremijenko A, Kosasih H, Putnam SD, Samaan G, Silitonga M, Chan KH, Poon LL, Lim W, Klimov A, Lindstrom S, Guan Y, Donis R, Katz J, Cox N, Peiris M, Uyeki TM. Three Indonesian clusters of H5N1 virus infection in 2005. N Engl J Med 2006;355:2186–2194. [DOI] [PubMed] [Google Scholar]
- 78. Oner AF, Bay A, Arslan S, Akdeniz H, Sahin HA, Cesur Y, Epcacan S, Yilmaz N, Deger I, Kizilyildiz B, Karsen H, Ceyhan M. Avian influenza A (H5N1) infection in Eastern Turkey in 2006. N Engl J Med 2006;355:2179–2185. [DOI] [PubMed] [Google Scholar]
- 79. Kandun IN, Tresnaningsih E, Purba WH, Lee V, Samaan G, Harun S, Soni E, Septiawati C, Setiawati T, Sariwati E, Wandra T. Factors associated with case fatality of human H5N1 virus infections in Indonesia: A case series. Lancet 2008;372:744–749. [DOI] [PubMed] [Google Scholar]
- 80. Wang H, Feng Z, Shu Y, Yu H, Zhou L, Zu R, Huai Y, Dong J, Bao C, Wen L, Wang H, Yang P, Zhao W, Dong L, Zhou M, Liao Q, Yang H, Wang M, Lu X, Shi Z, Wang W, Gu L, Zhu F, Li Q, Yin W, Yang W, Li D, Uyeki TM, Wang Y. Probable limited person‐to‐person transmission of highly pathogenic avian influenza A (H5N1) virus in China. Lancet 2008;371:1427–1434. [DOI] [PubMed] [Google Scholar]
- 81. De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov 2006;5:1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Griffiths PD. Whatever happened to bird flu? Rev Med Virol 2008;18:1–3. [DOI] [PubMed] [Google Scholar]
- 83. Beigel J, Bray M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res 2008;78:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dharan NJ, Gubareva LV, Meyer JJ, Okomo‐Adhiambo M, McClinton RC, Marshall SA, St George K, Epperson S, Brammer L, Klimov AI, Bresee JS, Fry AM. Infections with oseltamivir‐resistant influenza A(H1N1) virus in the United States. J Am Med Assoc 2009;301:1034–1041. [DOI] [PubMed] [Google Scholar]
- 85. Gooskens J, Jonges M, Claas EC, Meijer A, van den Broek PJ, Kroes AM. Morbidity and mortality associated with nosocomial transmission of oseltamivir‐resistant influenza A(H1N1) virus. J Am Med Assoc 2009;301:1042–1046. [DOI] [PubMed] [Google Scholar]
- 86. Lackenby A, Hungnes O, Dudman SG, Meijer A, Paget WJ, Hay AJ, Zambon MC. Emergence of resistance to oseltamivir among influenza A (H1N1) viruses in Europe. Euro Surveill 2008;13:1–2. [DOI] [PubMed] [Google Scholar]
- 87. Tamura D, Mitamura K, Yamazaki M, Fujino M, Nirasawa M, Kimura K, Kiso M, Shimizu H, Kawakami C, Hiroi S, Takahashi K, Hata M, Minagawa H, Kimura Y, Kaneda S, Sugita S, Horimoto T, Sugaya N, Kawaoka Y. Oseltamivir‐resistant influenza A viruses circulating in Japan. J Clin Microbiol 2009;47:1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hauge SH, Dudman S, Borgen K, Lackenby A, Hungnes O. Oseltamivir‐resistant influenza viruses A (H1N1), Norway, 2007–2008. Emerg Infect Dis 2009;15:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Besselaar TG, Naidoo D, Buys A, Gregory V, McAnerney J, Manamela JM, Blumberg L, Schoub BD. Widespread oseltamivir resistance in influenza A viruses (H1N1), South Africa. Emerg Infect Dis 2008;14:1809–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ives JA, Carr JA, Mendel DB, Tai CY, Lambkin R, Kelly L, Oxford JS, Hayden FG, Roberts NA. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Res 2002;55:307–317. [DOI] [PubMed] [Google Scholar]
- 91. van der Vries E, van den Berg B, Schutten M. Fatal oseltamivir‐resistant influenza virus infection. N Engl J Med 2008;359:1074–1076. [DOI] [PubMed] [Google Scholar]
- 92. Medeiros R, Rameix‐Welti MA, Lorin V, Ribaud P, Manuguerra JC, Socie G, Scieux C, Naffakh N, van der Werf S. Failure of zanamivir therapy for pneumonia in a bone‐marrow transplant recipient infected by a zanamivir‐sensitive influenza A (H1N1) virus. Antivir Ther 2007;12:571–576. [PubMed] [Google Scholar]
- 93. Escuret V, Frobert E, Bouscambert‐Duchamp M, Sabatier M, Grog I, Valette M, Lina B, Morfin F, Ferraris O. Detection of human influenza A (H1N1) and B strains with reduced sensitivity to neuraminidase inhibitors. J Clin Virol 2008;41:25–28. [DOI] [PubMed] [Google Scholar]
- 94. Hatakeyama S, Sugaya N, Ito M, Yamazaki M, Ichikawa M, Kimura K, Kiso M, Shimizu H, Kawakami C, Koike K, Mitamura K, Kawaoka Y. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. J Am Med Assoc 2007;13:1435–1442. [DOI] [PubMed] [Google Scholar]
- 95. Bouvier NM, Lowen AC, Palese P. Oseltamivir‐resistant influenza A viruses are transmitted efficiently among guinea pigs by direct contact but not by aerosol. J Virol 2008;82:10052–10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Abed Y, Baz M, Boivin G. A novel neuraminidase deletion mutation conferring resistance to oseltamivir in clinical influenza A/H3N2 virus. J Infect Dis 2009;199:180–183. [DOI] [PubMed] [Google Scholar]
- 97. Stephenson I, Democratis J, Lackenby A, McNally T, Smith J, Pareek M, Ellis J, Bermingham A, Nicholson K, Zambon M. Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clin Infect Dis 2009;48:389–396. [DOI] [PubMed] [Google Scholar]
- 98. Rameix‐Welti MA, Agou F, Buchy P, Mardy S, Aubin JT, Véron M, van der Werf S, Naffakh N. Natural variation can significantly alter the sensitivity of influenza A (H5N1) viruses to oseltamivir. Antimicrob Agents Chemother 2006;50:3809–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. McKimm‐Breschkin JL, Selleck PW, Usman TB, Johnson MA. Reduced sensitivity of influenza A (H5N1) to oseltamivir. Emerg Infect Dis 2007;13:1354–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Le MT, Wertheim HF, Nguyen HD, Taylor W, Hoang PV, Vuong CD, Nguyen HL, Nguyen HH, Nguyen TQ, Nguyen TV, Van TD, Ngoc BT, Bui TN, Nguyen BG, Nguyen LT, Luong ST, Phan PH, Pham HV, Nguyen T, Fox A, Nguyen CV, Do HQ, Crusat M, Farrar J, Nguyen HT, de Jong MD, Horby P. Influenza A H5N1 clade 2.3.4 virus with a different antiviral susceptibility profile replaced clade 1 virus in humans in northern Vietnam. PloS ONE 2008;3:e3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yen HL, Ilyushina NA, Salomon R, Hoffmann E, Webster RG, Govorkova EA. Neuraminidase inhibitor‐resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J Virol 2007;81:12418–12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Govorkova EA, Ilyushina NA, Boltz DA, Douglas A, Yilmaz N, Webster RG. Efficacy of oseltamivir therapy in ferrets inoculated with different clades of H5N1 influenza virus. Antimicrob Agents Chemother 2007;51:1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Boltz DA, Rehg JE, McClaren J, Webster RG, Govorkova EA. Oseltamivir prophylactic regimens prevent H5N1 influenza morbidity and mortality in a ferret model. J Infect Dis 2008;197:1315–1323. [DOI] [PubMed] [Google Scholar]
- 104. Hurt AC, Iannello P, Jachno K, Komadina N, Hampson AW, Barr IG, McKimm‐Breschkin JL. Neuraminidase inhibitor‐resistant and ‐sensitive influenza B viruses isolated from an untreated human patient. Antimicrob Agents Chemother 2006;50:1872–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ison MG, Gubareva LV, Atmar RL, Treanor J, Hayden FG. Recovery of drug‐resistant influenza virus from immunocompromised patients: A case series. J Infect Dis 2006;193:760–764. [DOI] [PubMed] [Google Scholar]
- 106. Collins PJ, Haire LF, Lin YP, Liu J, Russell RJ, Walker PA, Skehel JJ, Martin SR, Hay AJ, Gamblin SJ. Crystal structures of oseltamivir‐resistant influenza virus neuraminidase mutants. Nature 2008;453:1258–1262. [DOI] [PubMed] [Google Scholar]
- 107. Lagoja IM, De Clercq E. Anti‐influenza virus agents: Synthesis and mode of action. Med Res Rev 2008;28:1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Farina V, Brown JD. Tamiflu: The supply problem. Angew Chem Int Ed 2006;45:7330–7334. [DOI] [PubMed] [Google Scholar]
- 109. Satoh N, Akiba T, Yokoshima S, Fukuyama T. A practical synthesis of (‐)‐oseltamivir. Angew Chem Int Ed 2007;46:5734–5736. [DOI] [PubMed] [Google Scholar]
- 110. Ishikawa H, Suzuki T, Hayashi Y. High‐yielding synthesis of the anti‐influenza neuraminidase inhibitor (‐)‐oseltamivir by three “one‐pot” operations. Angew Chem Int Ed 2009;48:1304–1307. [DOI] [PubMed] [Google Scholar]
- 111. Carbain B, Collins PJ, Callum L, Martin SR, Hay AJ, McCauley J, Streicher H. Efficient synthesis of highly active phospha‐isosteres of the influenza neuraminidase inhibitor oseltamivir. Chem Med Chem 2009;4:335–337. [DOI] [PubMed] [Google Scholar]
- 112. Fick J, Lindberg RH, Tysklind M, Haemig PD, Waldenström J, Wallensten A, Olsen B. Antiviral oseltamivir is not removed or degraded in normal sewage water treatment: Implications for development of resistance by influenza A virus. PLoS ONE 2007;2:e986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Baz M, Abed Y, Nehmé B, Boivin G. Activity of the oral neuraminidase inhibitor A‐322278 against the oseltamivir‐resistant H274Y (A/H1N1) influenza virus mutant in mice. Antimicrob Agents Chemother 2009;53:791–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ison MG, Mishin VP, Braciale TJ, Hayden FG, Gubareva LV. Comparative activities of oseltamivir and A‐322278 in immunocompetent and immunocompromised murine models of influenza virus infection. J Infect Dis 2006;193:765–772. [DOI] [PubMed] [Google Scholar]
- 115. Abed Y, Nehmé B, Baz M, Boivin G. Activity of the neuraminidase inhibitor A‐315675 against oseltamivir‐resistant influenza neuraminidases of N1 and N2 subtypes. Antiviral Res 2008;77:163–166. [DOI] [PubMed] [Google Scholar]
- 116. Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S. CS‐8958, a prodrug of the new neuraminidase inhibitor R‐125489, shows long‐acting anti‐influenza virus activity. Antimicrob Agents Chemother 2009;53:186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Moscona A. Global transmission of oseltamivir‐resistant influenza. N Engl J Med 2009;360:953–955. [DOI] [PubMed] [Google Scholar]
- 118. Barroso L, Treanor J, Gubareva L, Hayden FG. Efficacy and tolerability of the oral neuraminidase inhibitor peramivir in experimental human influenza: Randomized, controlled trials for prophylaxis and treatment. Antivir Ther 2005;10:901–910. [PubMed] [Google Scholar]
- 119. Yun NE, Linde NS, Zacks MA, Barr IG, Hurt AC, Smith JN, Dziuba N, Holbrook MR, Zhang L, Kilpatrick JM, Arnold CS, Paessler S. Injectable peramivir mitigates disease and promotes survival in ferrets and mice infected with the highly virulent influenza virus, A/Vietnam/1203/04 (H5N1). Virology 2008;374:198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Baz M, Abed Y, Boivin G. Characterization of drug‐resistant recombinant influenza A/H1N1 viruses selected in vitro with peramivir and zanamivir. Antiviral Res 2007;74:159–162. [DOI] [PubMed] [Google Scholar]
- 121. Zhang J, Wang Q, Fang H, Xu W, Liu A, Du G. Design, synthesis, inhibitory activity, and SAR studies of pyrrolidine derivatives as neuraminidase inhibitors. Bioorg Med Chem 2007;15:2749–2758. [DOI] [PubMed] [Google Scholar]
- 122. Ilyushina NA, Hoffmann E, Salomon R, Webster RG, Govorkova EA. Amantadine‐oseltamivir combination therapy for H5N1 influenza virus infection in mice. Antivir Ther 2007;12:363–370. [PubMed] [Google Scholar]
- 123. Ilyushina NA, Hay A, Yilmaz N, Boon AC, Webster RG, Govorkova EA. Oseltamivir‐ribavirin combination therapy for highly pathogenic H5N1 influenza virus infection in mice. Antimicrob Agents Chemother 2008;52:3889–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Smee DF, Hurst BL, Wong MH, Bailey KW, Morrey JD. Effects of double combinations of amantadine, oseltamivir, and ribavirin on influenza A (H5N1) virus infections in cell culture and in mice. Antimicrob Agents Chemother 2009;53:2120–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. De Clercq E. Perspectives for the chemotherapy of respiratory syncytial virus (RSV) infections. Int J Antimicrob Agents 1996;7:193–202. [DOI] [PubMed] [Google Scholar]
- 126. Collins PL, Chanock RM, Murphy BR. Respiratory syncytial virus. In: Knipe DM, Howley PM, editors. Fields virology. 4th ed., Vol. 1. Philadelphia: Lippincott Williams & Wilkins; 2001. pp 1443–1485. [Google Scholar]
- 127. Falsey AR, Walsh EE. Respiratory syncytial virus infection in adults. Clin Microbiol Rev 2000;13:371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Anderson LJ, Parker RA, Strikas RL. Association between respiratory syncytial virus outbreaks and lower respiratory tract deaths of infants and young children. J Infect Dis 1990;161:640–646. [DOI] [PubMed] [Google Scholar]
- 129. Pelaez A, Lyon GM, Force SD, Ramirez AM, Neujahr DC, Foster M, Naik PM, Gal AA, Mitchell PO, Lawrence EC. Efficacy of oral ribavirin in lung transplant patients with respiratory syncytial virus lower respiratory tract infection. J Heart Lung Transplant 2009;28:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Groothuis JR, Simoes E, Levin MJ, Hall CB, Long CE, Rodriguez WJ, Arrobio J, Meissner HC, Fulton DR, Welliver RC, Tristram DA, Siber GR, Prince GA, Van Raden M, Hemming VG. Prophylactic administration of respiratory syncytial virus immune globulin to high‐risk infants and young children. N Engl J Med 1993;329:1524–1530. [DOI] [PubMed] [Google Scholar]
- 131. Johnson S, Oliver C, Prince GA, Hemming VG, Pfarr DS, Wang SC, Dormitzer M, O'Grady J, Koenig S, Tamura JK, Woods R, Bansal G, Couchenour D, Tsao E, Hall WC, Young JF. Development of a humanized monoclonal antibody (MEDI‐493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis 1997;176:1215–1224. [DOI] [PubMed] [Google Scholar]
- 132. Meanwell NA, Krystal M. Respiratory syncytial virus: Recent progress towards the discovery of effective prophylactic and therapeutic agents. Drug Discov Today 2000;5:241–252. [DOI] [PubMed] [Google Scholar]
- 133. Karron RA, Buonagurio DA, Georgiu AF, Whitehead SS, Adamus JE, Clements‐Mann ML, Harris DO, Randolph VB, Udem SA, Murphy BR, Sidhu MS. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: Clinical evaluation and molecular characterization of a cold‐passaged, attenuated RSV subgroup B mutant. Proc Natl Acad Sci USA 1997;94:13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pedraz C, Carbonell‐Estrany X, Figueras‐Aloy J, Quero J, the Iris Study Group . Effect of palivizumab prophylaxis in decreasing respiratory syncytial virus hospitalizations in premature infants. Pediatr Infect Dis J 2003;22:823–827. [DOI] [PubMed] [Google Scholar]
- 135. Fenton C, Scott LJ, Plosker GL. Palivizumab. A review of its use as prophylaxis for serious respiratory syncytial virus infection. Pediatr Drugs 2004;6:171–197. [DOI] [PubMed] [Google Scholar]
- 136. Simoes EA, Groothuis JR, Carbonell‐Estrany X, Rieger CH, Mitchell I, Fredrick LM, Kimpen JL, Palivizumab. Long‐Term Respiratory Outcomes Study Group . Palivizumab prophylaxis, respiratory syncytial virus, and subsequent recurrent wheezing. J Pediatr 2007;151:34–42. [DOI] [PubMed] [Google Scholar]
- 137. Wu H, Pfarr DS, Tang Y, An L‐L, Patel NK, Watkins JD, Huse WD, Kiener PA, Young JF. Ultra‐potent antibodies against respiratory syncytial virus: Effects of binding kinetics and binding valence on viral neutralization. J Mol Biol 2005;350:126–144. [DOI] [PubMed] [Google Scholar]
- 138. Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of motavizumab, an ultra‐potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol 2007;368:652–665. [DOI] [PubMed] [Google Scholar]
- 139. Zhao X, Chen FP, Sullender WM. Respiratory syncytial virus escape mutant derived in vitro resists palivizumab prophylaxis in cotton rats. Virology 2004;318:608–612. [DOI] [PubMed] [Google Scholar]
- 140. Zhao X, Sullender WM. In vivo selection of respiratory syncytial viruses resistant to palivizumab. J Virol 2005;79:3962–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bitko V, Musiyenko A, Shulyayeva O, Barik S. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med 2005;11:50–55. [DOI] [PubMed] [Google Scholar]
- 142. Zhang W, Yang H, Kong X, Mohapatra S, San Juan‐Vergara H, Hellermann G, Behera S, Singam R, Lockey RF, Mohapatra SS. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat Med 2005;11:56–62. [DOI] [PubMed] [Google Scholar]
- 143. DeVincenzo J, Cehelsky JE, Alvarez R, Elbashir S, Harborth J, Toudjarska I, Nechev L, Murugaiah V, Van Vliet A, Vaishnaw AK, Meyers R. Evaluation of the safety, tolerability and pharmacokinetics of ALN‐RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral Res 2008;77:225–231. [DOI] [PubMed] [Google Scholar]
- 144. Zhou J, Yang XQ, Xie YY, Zhao XD, Jiang LP, Wang LJ, Cui YX. Inhibition of respiratory syncytial virus of subgroups A and B using deoxyribozyme DZ1133 in mice. Virus Res 2007;130:241–248. [DOI] [PubMed] [Google Scholar]
- 145. Xie YY, Zhao XD, Jiang LP, Liu HL, Wang LJ, Fang P, Shen KL, Xie ZD, Wu YP, Yang XQ. Inhibition of respiratory syncytial virus in cultured cells by nucleocapsid gene targeted deoxyribozyme (DNAzyme). Antiviral Res 2006;71:31–41. [DOI] [PubMed] [Google Scholar]
- 146. Sidwell RW, Barnard DL. Respiratory syncytial virus infections: Recent prospects for control. Antiviral Res 2006;71:379–390. [DOI] [PubMed] [Google Scholar]
- 147. Douglas JL, Panis ML, Ho E, Lin K‐Y, Krawczyk SH, Grant DM, Cai R, Swaminathan S, Cihlar T. Inhibition of respiratory syncytial virus fusion by the small molecule VP‐14637 via specific interactions with F protein. J Virol 2003;77:5054–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Douglas JL, Panis ML, Ho E, Lin KY, Krawczyk SH, Grant DM, Cai R, Swaminathan S, Chen X, Cihlar T. Small molecules VP‐14637 and JNJ‐2408068 inhibit respiratory syncytial virus fusion by similar mechanisms. Antimicrob Agents Chemother 2005;49:2460–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wyde PR, Laquerre S, Chetty SN, Gilbert BE, Nitz TJ, Pevear DC. Antiviral efficacy of VP14637 against respiratory syncytial virus in vitro and in cotton rats following delivery by small droplet aerosol. Antiviral Res 2005;68:18–26. [DOI] [PubMed] [Google Scholar]
- 150. Douglas JL. In search of a small‐molecule inhibitor for respiratory syncytial virus. Expert Rev Anti‐Infect Ther 2004;2:625–639. [DOI] [PubMed] [Google Scholar]
- 151. Wyde PR, Chetty SN, Timmerman P, Gilbert BE, Andries K. Short duration aerosols of JNJ 2408068 (R170591) administered prophylactically or therapeutically protect cotton rats from experimental respiratory syncytial virus infection. Antiviral Res 2003;60:221–231. [DOI] [PubMed] [Google Scholar]
- 152. Cianci C, Yu KL, Combrink K, Sin N, Pearce B, Wang A, Civiello R, Voss S, Luo G, Kadow K, Genovesi EV, Venables B, Gulgeze H, Trehan A, James J, Lamb L, Medina I, Roach J, Yang Z, Zadjura L, Colonno R, Clark J, Meanwell N, Krystal M. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob Agents Chemother 2004;48:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Cianci C, Genovesi EV, Lamb L, Medina I, Yang Z, Zadjura L, Yang H, D'Arienzo C, Sin N, Yu KL, Combrink K, Li Z, Colonno R, Meanwell N, Clark J, Krystal M. Oral efficacy of a respiratory syncytial virus inhibitor in rodent models of infection. Antimicrob Agents Chemother 2004;48:2448–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cianci C, Meanwell N, Krystal M. Antiviral activity and molecular mechanism of an orally active respiratory syncytial virus fusion inhibitor. J Antimicrob Chemother 2005;55:289–292. [DOI] [PubMed] [Google Scholar]
- 155. Yu KL, Zhang Y, Civiello RL, Kadow KF, Cianci C, Krystal M, Meanwell NA. Fundamental structure–activity relationships associated with a new structural class of respiratory syncytial virus inhibitor. Bioorg Med Chem Lett 2003;13:2141–2144. [DOI] [PubMed] [Google Scholar]
- 156. Yu KL, Zhang Y, Civiello RL, Trehan AK, Pearce BC, Yin Z, Combrink KD, Gulgeze HB, Wang XA, Kadow KF, Cianci CW, Krystal M, Meanwell NA. Respiratory syncytial virus inhibitors. Part 2: Benzimidazol‐2‐one derivatives. Bioorg Med Chem Lett 2004;14:1133–1137. [DOI] [PubMed] [Google Scholar]
- 157. Yu KL, Wang XA, Civiello RL, Trehan AK, Pearce BC, Yin Z, Combrink KD, Gulgeze HB, Zhang Y, Kadow KF, Cianci CW, Clarke J, Genovesi EV, Medina I, Lamb L, Wyde PR, Krystal M, Meanwell NA. Respiratory syncytial virus fusion inhibitors. Part 3: Water‐soluble benzimidazol‐2‐one derivatives with antiviral activity in vivo. Bioorg Med Chem Lett 2005;16:1115–1122. [DOI] [PubMed] [Google Scholar]
- 158. Yu KL, Sin N, Civiello RL, Wang XA, Combrink KD, Gulgeze HB, Venables BL, Wright JJ, Dalterio RA, Zadjura L, Marino A, Dando S, D'Arienzo C, Kadow KF, Cianci CW, Li Z, Clarke J, Genovesi EV, Medina I, Lamb L, Colonno RJ, Yang Z, Krystal M, Meanwell NA. Respiratory syncytial virus fusion inhibitors. Part 4: Optimization for oral bioavailability. Bioorg Med Chem Lett 2007;17:895–901. [DOI] [PubMed] [Google Scholar]
- 159. Wang XA, Cianci CW, Yu KL, Combrink KD, Thuring JW, Zhang Y, Civiello RL, Kadow KF, Roach J, Li Z, Langley DR, Krystal M, Meanwell NA. Respiratory syncytial virus fusion inhibitors. Part 5: Optimization of benzimidazole substitution patterns towards derivatives with improved activity. Bioorg Med Chem Lett 2007;17:4592–4598. [DOI] [PubMed] [Google Scholar]
- 160. Combrink KD, Gulgeze HB, Thuring JW, Yu KL, Civiello RL, Zhang Y, Pearce BC, Yin Z, Langley DR, Kadow KF, Cianci CW, Li Z, Clarke J, Genovesi EV, Medina I, Lamb L, Yang Z, Zadjura L, Krystal M, Meanwell NA. Respiratory syncytial virus fusion inhibitors. Part 6: An examination of the effect of structural variation of the benzimidazol‐2‐one heterocycle moiety. Bioorg Med Chem Lett 2007;17:4784–4790. [DOI] [PubMed] [Google Scholar]
- 161. Carter MC, Alber DG, Baxter RC, Bithell SK, Budworth J, Chubb A, Cockerill GS, Dowdell VC, Henderson EA, Keegan SJ, Kelsey RD, Lockyer MJ, Stables JN, Wilson LJ, Powell KL. 1,4‐Benzodiazepines as inhibitors of respiratory syncytial virus. J Med Chem 2006;49:2311–2319. [DOI] [PubMed] [Google Scholar]
- 162. Henderson EA, Alber DG, Baxter RC, Bithell SK, Budworth J, Carter MC, Chubb A, Cockerill GS, Dowdell VC, Fraser IJ, Harris RA, Keegan SJ, Kelsey RD, Lumley JA, Stables JN, Weerasekera N, Wilson LJ, Powell KL. 1,4‐Benzodiazepines as inhibitors of respiratory syncytial virus. The identification of a clinical candidate. J Med Chem 2007;50:1685–1692. [DOI] [PubMed] [Google Scholar]
- 163. Chapman J, Abbott E, Alber DG, Baxter RC, Bithell SK, Henderson EA, Carter MC, Chambers P, Chubb A, Cockerill GS, Collins PL, Dowdell VC, Keegan SJ, Kelsey RD, Lockyer MJ, Luongo C, Najarro P, Pickles RJ, Simmonds M, Taylor D, Tyms S, Wilson LJ, Powell KL. RSV604, a novel inhibitor of respiratory syncytial virus replication. Antimicrob Agents Chemother 2007;51:3346–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Beauchamp LM, Dolmatch BL, Schaeffer HJ, Collins P, Bauer DJ, Keller PM, Fyfe JA. Modifications on the heterocyclic base of acyclovir: Syntheses and antiviral properties. J Med Chem 1985;28:982–987. [DOI] [PubMed] [Google Scholar]
- 165. Boryski J, Golankiewicz B, De Clercq E. Synthesis and antiviral activity of novel N‐substituted derivatives of acyclovir. J Med Chem 1988;31:1351–1355. [DOI] [PubMed] [Google Scholar]
- 166. Golankiewicz B, Ostrowski T, Boryski J, De Clercq E. Synthesis of acyclowyosine and acyclo‐3‐methylguanosine, as probes for some chemical and biological properties resulting from the N‐3 substitution of guanosine and its analogues. J Chem Soc Perkin Trans 1. 1991;589–593. [Google Scholar]
- 167. Sattsangi PD, Leonard NJ, Frihart CR. 1,N2‐ethenoguanine and N2,3‐ethenoguanine. Synthesis and comparison of the electronic spectral properties of these linear and angular triheterocycles related to the Y bases. J Org Chem 1977;42:3292–3296. [DOI] [PubMed] [Google Scholar]
- 168. Boryski J, Golankiewicz B, De Clercq E. Synthesis and antiviral activity of 3‐substituted derivatives of 3,9‐dihydro‐9‐oxo‐5H‐imidazo[1,2‐a]purines, tricyclic analogues of acyclovir and ganciclovir. J Med Chem 1991;34:2380–2383. [DOI] [PubMed] [Google Scholar]
- 169. Golankiewicz B, Ostrowski T, Andrei G, Snoeck R, De Clercq E. Tricyclic analogues of acyclovir and ganciclovir. Influence of substituents in the heterocyclic moiety on the antiviral activity. J Med Chem 1994;37:3187–3190. [DOI] [PubMed] [Google Scholar]
- 170. Golankiewicz B, Ostrowski T, Goslinski T, Januszczyk P, Zeidler J, Baranowski D, de Clercq E. Fluorescent tricyclic analogues of acyclovir and ganciclovir. A structure–antiviral activity study. J Med Chem 2001;44:4284–4287. [DOI] [PubMed] [Google Scholar]
- 171. Goslinski T, Golankiewicz B, De Clercq E, Balzarini J. Synthesis and biological activity of strongly fluorescent tricyclic analogues of acyclovir and ganciclovir. J Med Chem 2002;45:5052–5057. [DOI] [PubMed] [Google Scholar]
- 172. Goslinski T, Wenska G, Golankiewicz B, Balzarini J, De Clercq E. Synthesis and fluorescent properties of 6‐(4‐biphenylyl)‐3,9‐dihydro‐9‐oxo‐5H‐imidazo[1,2‐a]purine analogues of acyclovir and ganciclovir. Nucleosides Nucleotides Nucleic Acids 2003;22:911–914. [DOI] [PubMed] [Google Scholar]
- 173. Ostrowski T, Golankiewicz B, De Clercq E, Balzarini J. Fluorosubstitution and 7‐alkylation as prospective modifications of biologically active 6‐aryl derivatives of tricyclic acyclovir and ganciclovir analogues. Bioorg Med Chem 2005;13:2089–2096. [DOI] [PubMed] [Google Scholar]
- 174. Balzarini J, Ostrowski T, Goslinski T, De Clercq E, Golankiewicz B. Pronounced cytostatic activity and bystander effect of a novel series of fluorescent tricyclic acyclovir and ganciclovir derivatives in herpes simplex virus thymidine kinase gene‐transduced tumor cell lines. Gene Ther 2002;9:1173–1182. [DOI] [PubMed] [Google Scholar]
- 175. Balzarini J, De Clercq E, Ayusawa D, Seno T. Murine mammary FM3A carcinoma cells transformed with the herpes simplex virus type 1 thymidine kinase gene are highly sensitive to the growth‐inhibitory properties of (E)‐5‐(2‐bromovinyl)‐2′‐deoxyuridine and related compounds. FEBS Lett 1985;185:95–100. [DOI] [PubMed] [Google Scholar]
- 176. Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector‐producer cells for treatment of experimental brain tumors. Science 1992;256:1550–1552. [DOI] [PubMed] [Google Scholar]
- 177. Degrève B, Johansson M, De Clercq E, Karlsson A, Balzarini J. Differential intracellular compartmentalization of herpetic thymidine kinases (TKs) in TK gene‐transfected tumor cells: Molecular characterization of the nuclear localization signal of herpes simplex virus type 1 TK. J Virol 1998;72:9535–9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Sekiyama T, Hatsuya S, Tanaka Y, Uchiyama M, Ono N, Iwayama S, Oikawa M, Suzuki K, Okunishi M, Tsuji T. Synthesis and antiviral activity of novel acyclic nucleosides: Discovery of a cyclopropyl nucleoside with potent inhibitory activity against herpesviruses. J Med Chem 1998;41:1284–1298. [DOI] [PubMed] [Google Scholar]
- 179. Iwayama S, Ono N, Ohmura Y, Suzuki K, Aoki M, Nakazawa H, Oikawa M, Kato T, Okunishi M, Nishiyama Y, Yamanishi K. Antiherpesvirus activities of (1′S,2′R)‐9‐[[1′,2′‐bis(hydroxymethyl)cycloprop‐1′‐yl]methyl]guanine (A‐5021) in cell culture. Antimicrob Agents Chemother 1998;42:1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Ono N, Iwayama S, Suzuki K, Sekiyama T, Nakazawa H, Tsuji T, Okunishi M, Daikoku T, Nishiyama Y. Mode of action of (1′S,2′R)‐9‐[[1′,2′‐bis(hydroxymethyl) cycloprop‐1′‐yl]methyl]guanine (A‐5021) against herpes simplex virus type 1 and type 2 and varicella‐zoster virus. Antimicrob Agents Chemother 1998;42:2095–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Onishi T, Mukai C, Nakagawa R, Sekiyama T, Aoki M, Suzuki K, Nakazawa H, Ono N, Ohmura Y, Iwayama S, Okunishi M, Tsuji T. Synthesis and antiviral activity of novel anti‐VZV 5‐substituted uracil nucleosides with a cyclopropane sugar moiety. J Med Chem 2000;43:278–282. [DOI] [PubMed] [Google Scholar]
- 182. De Clercq E, Andrei G, Snoeck R, De Bolle L, Naesens L, Degrève B, Balzarini J, Zhang Y, Schols D, Leyssen P, Ying C, Neyts J. Acyclic/carbocyclic guanosine analogues as anti‐herpesvirus agents. Nucleosides Nucleotides Nucleic Acids 2001;20:271–285. [DOI] [PubMed] [Google Scholar]
- 183. Ostrowski T, Golankiewicz B, De Clercq E, Balzarini J. Synthesis and biological activity of tricyclic analogues of 9‐[[cis‐1′,2′‐bis(hydroxymethyl)cycloprop‐1′‐yl]methyl]guanine. Bioorg Med Chem 2006;14:3535–3542. [DOI] [PubMed] [Google Scholar]
- 184. Ostrowski T, Golankiewicz B, De Clercq E, Andrei G, Snoeck R. Synthesis and anti‐VZV activity of 6‐heteroaryl derivatives of tricyclic acyclovir and 9‐{[cis‐1′,2′‐bis(hydroxymethyl)cycloprop‐1′‐yl]methyl}guanine analogues. Eur J Med Chem 2009;44:3313–3317. [DOI] [PubMed] [Google Scholar]
- 185. Korolev AM, Lazko EI, Preobrazhenskaya MN, Balzarini J, De Clercq E. Amides of anthracycline antibiotics and N‐carboxymethylascorbigen. Khim Pharm Zhurnal 1991;25:42–45. [Google Scholar]
- 186. Olsufyeva EN, Backinowsky LV, Preobrazhenskaya MN, Balzarini J, De Clercq E. New analogues of anthracycline antibiotics containing 2,3,6‐trideoxy‐3‐amino‐3‐C‐methyl‐L‐arabino‐hexose (L‐eremosamine). Sov J of Bioorg Chem 1991;17:316–322. [Google Scholar]
- 187. Preobrazhenskaya MN, Bakina EV, Povarov LS, Lazhko EI, Aleksandrova LG, Balzarini J, De Clercq E. Synthesis and cytostatic properties of daunorubicin derivatives, containing N‐phenylthiourea or N‐ethylthiourea moieties in the 3′‐position. J Antibiotics 1991;44:192–199. [DOI] [PubMed] [Google Scholar]
- 188. Olsufyeva EN, Brusentsov NA, Todorova N, Balzarini J, De Clercq E, Preobrazhenskaya MN. Daunorubicin derivatives obtained from daunorubicin and nucleoside dialdehydes. Nucleosides Nucleotides 1997;16:87–95. [Google Scholar]
- 189. Tolstikov VV, Preobrazhenskaya MN, Balzarini J, De Clercq E. Chemical modification of antibiotic streptonigrin: Synthesis and properties of 2′‐decarboxy‐2′‐aminostreptonigrin (streptonigrone‐2′‐imine). J Antibiotics 1992;45:1002–1004. [DOI] [PubMed] [Google Scholar]
- 190. Tolstikov VV, Holpne Kozlova NV, Oreshkina TD, Osipova TV, Preobrazhenskaya MN, Sztaricskai F, Balzarini J, De Clercq E. Amides of antibiotic streptonigrin and amino dicarboxylic acids or aminosugars: Synthesis and biological evaluation. J Antibiotics 1992;45:1020–1025. [DOI] [PubMed] [Google Scholar]
- 191. Lakatosh SA, Balzarini J, Andrei G, Snoeck R, De Clercq E, Preobrazhenskaya MN. Synthesis and cytotoxic activity of Nind‐alkoxy derivatives of antibiotic arcyriarubin and dechloro‐rebeccamycin aglycon. J Antibiotics 2002;55:768–773. [DOI] [PubMed] [Google Scholar]
- 192. Slater MJ, Baxter R, Bonser RW, Cockerill S, Gohil K, Parry N, Robinson E, Randall R, Yeates C, Snowden W, Walters A. Synthesis of N‐alkyl substituted indolocarbazoles as potent inhibitors of human cytomegalovirus replication. Bioorg Med Chem Lett 2001;11:1993–1995. [DOI] [PubMed] [Google Scholar]
- 193. Murakami Y, Noguchi K, Yamagoe S, Suzuki T, Wakita T, Fukazawa H. Identification of bisindolylmaleimides and indolocarbazoles as inhibitors of HCV replication by tube‐capture‐RT‐PCR. Antiviral Res 2009; in press. [DOI] [PubMed] [Google Scholar]
- 194. Tevyashova AN, Zbarsky EN, Balzarini J, Shtil AA, Dezhenkova LG, Bukhman VM, Zbarsky VB, Preobrazhenskaya MN. Modification of the antibiotic olivomycin I at the 2′‐keto group of the side chain. Novel derivatives, antitumor and topoisomerase I‐poisoning activity. J Antibiotics 2009;62:37–41. [DOI] [PubMed] [Google Scholar]
- 195. Tevyashova AN, Zbarsky EN, Balzarini J, Shtil AA, Dezhenkova LG, Bukhman VM, Zbarsky VB, Preobrazhenskaya MN. Modification of the antibiotic olivomycin I at the 2′‐keto group of the side chain. Novel derivatives, antitumor and topoisomerase I‐poisoning activity. J Antibiotics 2009;62:117. [DOI] [PubMed] [Google Scholar]
- 196. Shchekotikhin AE, Glazunova VA, Dezhenkova LG, Luzikov YN, Sinkevich YB, Kovalenko LV, Buyanov VN, Balzarini J, Huang FC, Lin JJ, Huang HS, Shtil AA, Preobrazhenskaya MN. Synthesis and cytotoxic properties of 4,11‐bis[(aminoethyl)amino]anthra[2,3‐b]thiophene‐5,10‐diones, novel analogues of antitumor anthracene‐9,10‐diones. Bioorg Med Chem 2009;17:1861–1869. [DOI] [PubMed] [Google Scholar]
- 197. Balzarini J, Pannecouque C, De Clercq E, Pavlov AY, Printsevskaya SS, Miroshnikova OV, Reznikova MI, Preobrazhenskaya MN. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J Med Chem 2003;46:2755–2764. [DOI] [PubMed] [Google Scholar]
- 198. Printsevskaya SS, Solovieva SE, Olsufyeva EN, Mirchink EP, Isakova EB, De Clercq E, Balzarini J, Preobrazhenskaya MN. Structure–activity relationship studies of a series of antiviral and antibacterial aglycon derivatives of the glycopeptide antibiotics vancomycin, eremomycin, and dechloroeremomycin. J Med Chem 2005;48:3885–3890. [DOI] [PubMed] [Google Scholar]
- 199. Pavlov AY, Berdnikova TF, Olsufyeva EN, Mazhko EI, Malkova IV, Preobrazhenskaya MN, Testa RT, Petersen PJ. Synthesis and biological activity of derivatives of glycopeptide antibiotics eremomycin and vancomycin nitrosated, acylated or carbamoylated at the N‐terminal. J Antibiotics 1993;46:1731–1739. [DOI] [PubMed] [Google Scholar]
- 200. Pavlov AY, Berdnikova TF, Olsufyeva EN, Malkova IV, Preobrazhenskaya MN, Risbridger GD. Modification of glycopeptide antibiotic eremomycin by the action of alkylhalides and study on antibacterial activity of the compounds obtained. J Antibiotics 1994;47:225–232. [DOI] [PubMed] [Google Scholar]
- 201. Pavlov AY, Berdnikova TF, Olsufyeva EN, Miroshnikova OV, Filiposyanz ST, Preobrazhenskaya MN, Scottani C, Colombo L, Goldstein BP. Carboxamides and hydrazide of glycopeptide antibiotic eremomycin synthesis and antibacterial activity. J Antibiotics 1996;49:194–198. [DOI] [PubMed] [Google Scholar]
- 202. Pavlov AY, Lazhko EI, Preobrazhenskaya MN. A new type of chemical modification of glycopeptides antibiotics;aminomethylated derivatives of eremomycin and their antibacterial activity. J Antibiotics 1997;50:509–513. [DOI] [PubMed] [Google Scholar]
- 203. Pavlov AY, Preobrazhenskaya MN, Malabarba A, Ciabatti R, Colombo L. Mono and double modified teicoplanin aglycon derivatives on the amino acid no. 7; structure–activity relationship. J Antibiotics 1998;51:73–81. [DOI] [PubMed] [Google Scholar]
- 204. Pavlov A, Miroshnikova OV, Printsevskaya SS, Olsufyeva EN, Preobrazhenskaya MN, Goldman RG, Bransrom AA, Baizman ER, Longley CB. Synthesis of hydrophobic N′‐mono and N′,N″‐double alkylated eremomycins inhibiting the transglycosylation stage of bacterial cell wall biosynthesis. J Antibiotics 2001;54:455–459. [DOI] [PubMed] [Google Scholar]
- 205. Printsevskaya SS, Pavlov AY, Olsufyeva EN, Mirchink EP, Isakova EB, Reznikova MI, Goldman RC, Branstrom AA, Baizman ER, Longley CB, Sztaricskai F, Batta G, Preobrazhenskaya MN. Synthesis and mode of action of hydrophobic derivatives of the glycopeptide antibiotic eremomycin and des‐(N‐methyl‐D‐leucyl)eremomycin against glycopeptide‐sensitive and resistant bacteria. J Med Chem 2002;45:1340–1347. [DOI] [PubMed] [Google Scholar]
- 206. Balzarini J, Keyaerts E, Vijgen L, Egberink H, De Clercq E, Van Ranst M, Printsevskaya SS, Olsufyeva EN, Solovieva SE, Preobrazhenskaya MN. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Res 2006;72:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Naesens L, Vanderlinden E, Roth E, Jeko J, Andrei G, Snoeck R, Pannecouque C, Illyés E, Batta G, Herczegh P, Sztaricskai F. Anti‐influenza virus activity and structure–activity relationship of aglycoristocetin derivatives with cyclobutenedione carrying hydrophobic chains. Antiviral Res 2009;82:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Andrei G, Snoeck R, Vandeputte M, De Clercq E. Activities of various compounds against murine and primate polyomaviruses. Antimicrob Agents Chemother 1997;41:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. De Clercq E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin Microbiol Rev 2003;16:569–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Andrei G, Fiten P, Goubau P, van Landuyt H, Gordts B, Selleslag D, De Clercq E, Opdenakker G, Snoeck R. Dual infection with polyomavirus BK and acyclovir‐resistant herpes simplex virus successfully treated with cidofovir in a bone marrow transplant recipient. Transplant Infect Dis 2007;9:126–131. [DOI] [PubMed] [Google Scholar]
- 211. De Clercq E, Holý A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat Rev Drug Discov 2005;4:928–940. [DOI] [PubMed] [Google Scholar]
- 212. Kuypers DR, Vandooren AK, Lerut E, Evenepoel P, Claes K, Snoeck R, Naesens L, Vanrenterghem Y. Adjuvant low‐dose cidofovir therapy for BK polyomavirus interstitial nephritis in renal transplant recipients. Am J Transplant 2005;5:1997–2004. [DOI] [PubMed] [Google Scholar]
- 213. Fanourgiakis P, Georgala A, Vekemans M, Triffet A, De Bruyn JM, Duchateau V, Martiat P, De Clercq E, Snoeck R, Wollants E, Rector A, Van Ranst M, Aoun M. Intravesical instillation of cidofovir in the treatment of hemorrhagic cystitis caused by adenovirus type 11 in a bone marrow transplant recipient. Clin Infect Dis 2005;40:199–201. [DOI] [PubMed] [Google Scholar]
- 214. Bridges B, Donegan S, Badros A. Cidofovir bladder instillation for the treatment of BK hemorrhagic cystitis after allogeneic stem cell transplantation. Am J Hematol 2006;81:535–537. [DOI] [PubMed] [Google Scholar]
- 215. Rinaldo CH, Hirsch HH. Antivirals for the treatment of polyomavirus BK replication. Expert Rev Anti‐Infect Ther 2007;5:105–115. [DOI] [PubMed] [Google Scholar]
- 216. Bernhoff E, Gutteberg TJ, Sandvik K, Hirsch HH, Rinaldo CH. Cidofovir inhibits polyomavirus BK replication in human renal tubular cells downstream of viral early gene expression. Am J Transplant 2008;18:1413–1422. [DOI] [PubMed] [Google Scholar]
- 217. Randhawa P, Farasati NA, Shapiro R, Hostetler KY. Ether lipid ester derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob Agents Chemother 2006;50:1564–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971;1:1253–1257. [DOI] [PubMed] [Google Scholar]
- 219. Hirsch HH, Brennan DC, Drachenberg CB, Ginevri F, Gordon J, Limaye AP, Mihatsch MJ, Nickeleit V, Ramos E, Randhawa P, Shapiro R, Steiger J, Suthanthiran M, Trofe J. Polyomavirus‐associated nephropathy in renal transplantation: Interdisciplinary analyses and recommendations. Transplantation 2005;79:1277–1286. [DOI] [PubMed] [Google Scholar]
- 220. White LH, Casian A, Hilton R, Macphee IA, Marsh J, Sweny P, Trevitt R, Frankel AH, Warrens AN, Pan‐Thames Renal Audit Group . BK virus nephropathy in renal transplant patients in London. Transplantation 2008;85:1008–1015. [DOI] [PubMed] [Google Scholar]
- 221. Bonvoisin C, Weekers L, Xhignesse P, Grosch S, Milicevic M, Krzesinski JM. Polyomavirus in renal transplantation: A hot problem. Transplantation 2008;85:S42–S48. [DOI] [PubMed] [Google Scholar]
- 222. Akioka K, Okamoto M, Ushigome H, Nobori S, Kaihara S, Yoshimura N. BK virus‐associated nephropathy in a kidney transplant recipient successfully treated with cidofovir, the first case in Japan. Int J Urol 2008;15:369–371. [DOI] [PubMed] [Google Scholar]
- 223. Savona MR, Newton D, Frame D, Levine JE, Mineishi S, Kaul DR. Low‐dose cidofovir treatment of BK virus‐associated hemorrhagic cystitis in recipients of hematopoietic stem cell transplant. Bone Marrow Transplant 2007;39:783–787. [DOI] [PubMed] [Google Scholar]
- 224. Kraemer C, Evers S, Nolting T, Arendt G, Husstedt IW. Cidofovir in combination with HAART and survival in AIDS‐associated progressive multifocal leukoencephalopathy. J Neurol 2008;255:526–531. [DOI] [PubMed] [Google Scholar]
- 225. Viallard JF, Lazaro E, Ellie E, Eimer S, Camou F, Caubet O, Lafon ME, Fleury H, Pellegrin JL. Improvement of progressive multifocal leukoencephalopathy after cidofovir therapy in a patient with a destructive polyarthritis. Infection 2007;35:33–36. [DOI] [PubMed] [Google Scholar]
- 226. Terrier B, Hummel A, Fakhouri F, Jablonski M, Hügle T, Gasnault J, Sanson M, Martinez F. Progressive multifocal leukoencephalopathy in a non‐AIDS patient: High efficiency of combined cytarabine and cidofovir. Rev Med Interne 2007;28:488–491. [DOI] [PubMed] [Google Scholar]
- 227. Pöhlmann C, Hochauf K, Röllig C, Schetelig J, Wunderlich O, Bandt D, Ehninger G, Jacobs E, Rohayem J. Chlorpromazine combined with cidofovir for treatment of a patient suffering from progressive multifocal leukoencephalopathy. Intervirology 2007;50:412–417. [DOI] [PubMed] [Google Scholar]
- 228. Friedkin M, Robert D. The enzymatic synthesis of nucleosides. I. Thymidine phosphorylase in mammalian tissue. J Biol Chem 1954;207:245–256. [PubMed] [Google Scholar]
- 229. Desgranges C, Razaka G, Rabaud M, Bricaud H, Balzarini J, De Clercq E. Phosphorolysis of (E)‐5‐(2‐bromovinyl)‐2′‐deoxyuridine (BVDU) and other 5‐substituted‐2′‐deoxyuridines by purified human thymidine phosphorylase and intact blood platelets. Biochem Pharmacol 1983;32:3583–3590. [DOI] [PubMed] [Google Scholar]
- 230. Miyazono K, Okabe T, Urabe A, Takaku F, Heldin CH. Purification and properties of an endothelial cell growth factor from human platelets. J Biol Chem 1987;262:4098–4103. [PubMed] [Google Scholar]
- 231. Usuki K, Saras J, Waltenberger J, Miyazono K, Pierce G, Thomason A, Heldin CH. Platelet‐derived endothelial cell growth factor has thymidine phosphorylase activity. Biochem Biophys Res Commun 1992;184:1311–1316. [DOI] [PubMed] [Google Scholar]
- 232. Sumizawa T, Furukawa T, Haraguchi M, Yoshimura A, Takeyasu A, Ishizawa M, Yamada Y, Akiyama S. Thymidine phosphorylase activity associated with platelet‐derived endothelial cell growth factor. J Biochem 1993;114:9–14. [DOI] [PubMed] [Google Scholar]
- 233. Haraguchi M, Miyadera K, Uemura K, Sumizawa T, Furukawa T, Yamada K, Akiyama S, Yamada Y. Angiogenic activity of enzymes. Nature 1994;368:198. [DOI] [PubMed] [Google Scholar]
- 234. Moghaddam A, Zhang HT, Fan TP, Hu DE, Lees VC, Turley H, Fox SB, Gatter KC, Harris AL, Bicknell R. Thymidine phosphorylase is angiogenic and promotes tumor growth. Proc Natl Acad Sci USA 1995;92:998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Liekens S, De Clercq E, Neyts J. Angiogenesis: Regulators and clinical applications. Biochem Pharmacol 2001;61:253–270. [DOI] [PubMed] [Google Scholar]
- 236. Langen P, Etzold G, Bärwolff D, Preussel B. Inhibition of thymidine phosphorylase by 6‐aminothymine and derivatives of 6‐aminouracil. Biochem Pharmacol 1967;16:1833–1837. [DOI] [PubMed] [Google Scholar]
- 237. Balzarini J, Gamboa AE, Esnouf R, Liekens S, Neyts J, De Clercq E, Camarasa MJ, Pérez‐Pérez MJ. 7‐Deazaxanthine, a novel prototype inhibitor of thymidine phosphorylase. FEBS Lett 1998;438:91–95. [DOI] [PubMed] [Google Scholar]
- 238. Esteban‐Gamboa A, Balzarini J, Esnouf R, De Clercq E, Camarasa MJ, Pérez‐Pérez MJ. Design, synthesis, and enzymatic evaluation of multisubstrate analogue inhibitors of Escherichia coli thymidine phosphorylase. J Med Chem 2000;43:971–983. [DOI] [PubMed] [Google Scholar]
- 239. Balzarini J, Degrève B, Esteban‐Gamboa A, Esnouf R, De Clercq E, Engelborghs Y, Camarasa MJ, Pérez‐Pérez MJ. Kinetic analysis of novel multisubstrate analogue inhibitors of thymidine phosphorylase. FEBS Lett 2000;483:181–185. [DOI] [PubMed] [Google Scholar]
- 240. Liekens S, Bilsen F, De Clercq E, Priego EM, Camarasa MJ, Pérez‐Pérez MJ, Balzarini J. Anti‐angiogenic activity of a novel multi‐substrate analogue inhibitor of thymidine phosphorylase. FEBS Lett 2002;510:83–88. [DOI] [PubMed] [Google Scholar]
- 241. Klein RS, Lenzi M, Lim TH, Hotchkiss KA, Wilson P, Schwartz EL. Novel 6‐substituted uracil analogs as inhibitors of the angiogenic actions of thymidine phosphorylase. Biochem Pharmacol 2001;62:1257–1263. [DOI] [PubMed] [Google Scholar]
- 242. Matsushita S, Nitanda T, Furukawa T, Sumizawa T, Tani A, Nishimoto K, Akiba S, Miyadera K, Fukushima M, Yamada Y, Yoshida H, Kanzaki T, Akiyama S. The effect of a thymidine phosphorylase inhibitor on angiogenesis and apoptosis in tumors. Cancer Res 1999;59:1911–1916. [PubMed] [Google Scholar]
- 243. Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, Yamada Y, Asao T. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′‐deoxyribonucleosides. Biochem Pharmacol 2000;59:1227–1236. [DOI] [PubMed] [Google Scholar]
- 244. Holý A. Phosphonomethoxyalkyl analogs of nucleotides. Curr Pharm Des 2003;9:2567–2592. [DOI] [PubMed] [Google Scholar]
- 245. Votruba I, Pomeisl K, Tloust'ová E, Holý A, Otová B. Inhibition of thymidine phosphorylase (PD‐ECGF) from SD‐lymphoma by phosphonomethoxyalkyl thymines. Biochem Pharmacol 2005;69:1517–1521. [DOI] [PubMed] [Google Scholar]
- 246. Brown NS, Bicknell R. Thymidine phosphorylase, 2‐deoxy‐D‐ribose and angiogenesis. Biochem J 1998;334:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Pomeisl K, Votruba I, Holý A, Pohl R. Syntheses of pyrimidine acyclic nucleoside phosphonates as potent inhibitors of thymidine phosphorylase (PD‐ECGF) from SD‐lymphoma. Nucleosides Nucleotides Nucleic Acids 2007;26:1025–1028. [DOI] [PubMed] [Google Scholar]
- 248. Pomeisl K, Votruba I, Holý A, Pohl R. Syntheses of base and side‐chain modified pyrimidine 1‐[2‐(phosphonomethoxy)propyl] derivatives as potent inhibitors of thymidine phosphorylase (PD‐ECGF) from SD‐lymphoma. Collect Czech Chem Commun 2006;71:595–624. [Google Scholar]
- 249. Liekens S, Hernández AI, Ribatti D, De Clercq E, Camarasa MJ, Pérez‐Pérez MJ, Balzarini J. The nucleoside derivative 5′‐O‐trityl‐inosine (KIN59) suppresses thymidine phosphorylase‐triggered angiogenesis via a noncompetitive mechanism of action. J Biol Chem 2004;279:29598–29605. [DOI] [PubMed] [Google Scholar]
- 250. Casanova E, Hernandez AI, Priego EM, Liekens S, Camarasa MJ, Balzarini J, Pérez‐Pérez MJ. 5′‐O‐tritylinosine and analogues as allosteric inhibitors of human thymidine phosphorylase. J Med Chem 2006;49:5562–5570. [DOI] [PubMed] [Google Scholar]
- 251. Bronckaers A, Aguado L, Negri A, Camarasa MJ, Balzarini J, Pérez‐Pérez MJ, Gago F, Liekens S. Identification of aspartic acid‐203 in human thymidine phosphorylase as an important residue for both catalysis and non‐competitive inhibition by the small molecule “crystallization chaperone” 5′‐O‐tritylinosine (KIN59). Biochem Pharmacol 2009;78:231–240. [DOI] [PubMed] [Google Scholar]