

Abstract
Hepatitis A virus (HAV) is a faeco‐orally transmitted picornavirus and is one of the main causes of acute hepatitis worldwide. An overview of the molecular biology of HAV is presented with an emphasis on recent findings. Immune evasion strategies and a possible correlation between HAV and atopy are discussed as well. Despite the availability of efficient vaccines, antiviral drugs targeting HAV are required to treat severe cases of fulminant hepatitis, contain outbreaks, and halt the potential spread of vaccine‐escape variants. Additionally, such drugs could be used to shorten the period of illness and decrease associated economical costs. Several known inhibitors of HAV with various mechanisms of action will be discussed. Since none of these molecules is readily useable in the clinic and since the availability of an anti‐HAV drug would be of clinical importance, increased efforts should be targeted toward discovery and development of such antivirals.
Keywords: hepatitis A virus, virus replication, immune evasion, atopy, antiviral drugs
1. INTRODUCTION
Hepatitis A virus (HAV) is a major cause of enterically transmitted hepatitis worldwide, posing a global burden estimated in 2005 at 119 million infections of which 31 million resulted in symptomatic illness and 34,000 in death.1 The virus is transmitted faeco‐orally, mainly through close contact with infected individuals or by consumption of contaminated food and drinking water. The extreme environmental stability of the HAV particle contributes significantly to its transmission.2, 3 HAV epidemiology correlates with poor hygiene and living conditions. Consequently, virtually every adult in developing countries is seropositive due to childhood infection. On the contrary, in regions with improved hygiene standards, infection is often postponed to later age. In addition, disease severity is generally age‐dependent: infections are usually mild or asymptomatic in young children, whereas at older age, hepatitis A frequently presents with classic symptoms of acute hepatitis (e.g., jaundice, fatigue, general malaise, etc.) and a higher incidence of fulminant hepatitis, which may require liver transplantation.4 Fulminant hepatitis occurs especially in those aged over 50 for which mortality rates up to 5.4% have been reported.5 Additionally, HAV superinfections in chronic liver disease patients (e.g., hepatitis B or C) are believed to increase morbidity and mortality,6, 7, 8 although these findings are still subject to debate.9 Since highly efficient vaccines that provide long‐lasting immunity have become available, HAV mortality and morbidity has decreased dramatically.8, 10 However, occasional outbreaks of hepatitis A, sometimes resulting in fatal outcomes, still occur in industrialized countries. For instance, in the last decade, outbreaks have been linked to the consumption of contaminated green onions, semidried tomatoes, seafood, such as raw oysters and sushi, and other foodstuffs.11, 12, 13, 14
As posited earlier,15 research interest in the molecular biology and pathogenesis of HAV has decreased substantially since the availability of a safe and efficient vaccine. Nonetheless, HAV remains an intriguing and poorly understood virus and hepatitis A is still a public health problem in many countries. Here, we will review key aspects of the biology of HAV with an emphasis on recent findings and the unique characteristics of the virus. The potential clinical use of antivirals against HAV will be discussed as well.
2. HAV GENOME ORGANIZATION AND REPLICATION CYCLE
HAV is a nonenveloped, single‐stranded RNA virus with a positive‐sense genome. Despite the fact that HAV is classified within the family of the Picornaviridae, it exhibits quite some differences compared to other members of this family and is consequently the sole member of the genus Hepatovirus.16 Based on phylogenetic analysis of full length VP1 sequences, the single HAV serotype is divided into six genotypes (I–VI).17 Genotypes I to III are human and can be subdivided into subgenotypes A and B (and potentially subgenotype IC18), whereas genotypes IV to VI are of simian origin.17 The HAV genome (as shown in Fig. 1) is approximately 7.5 kb in length and consists of a 5′ untranslated region (UTR), a single open reading frame (ORF) and a 3′‐UTR with a polyadenosine tract. Like other picornaviruses, the HAV genome lacks a cap structure; instead a VPg protein (3B) is attached to the 5′ end. In the 5′‐UTR, six secondary structure domains can be found: domain I contains a hairpin structure, while the second domain comprises two stem loop structures followed by a polypyrimidine tract (pY1).19 The remaining domains form the type III internal ribosome entry site (IRES) allowing cap‐independent translation of the viral genome. The single ORF comprises the structural genes (VP1 to VP4 (= P1) and 2A) and the nonstructural genes (2B–2C and 3A–3D). The short 3′‐UTR contains two stem loops and/or a pseudoknot structure which are, together with domains I and II and the polypyrimidine tract of the 5′‐UTR, crucial for viral RNA synthesis.20
Figure 1.
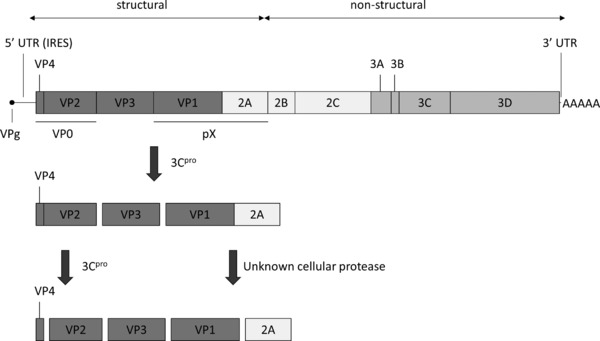
HAV genome organization and proteolytic processing of the structural proteins (IRES, internal ribosome entry site; UTR, untranslated region).
A. Receptor Binding and Cell Entry
In 1996, the HAV cellular receptor (HAVcr‐1) was identified on African green monkey kidney cells (AGMK) as an attachment and probably functional receptor.21 Consequently, the human homolog (HuHAVcr‐1) was identified and characterized as a human HAV receptor.22 HAVcr‐1 is also known to be a marker for acute ischemic kidney injury (in this context referred to as kidney injury molecule 1 or KIM‐1)23 and a regulator of T‐cell based immunity (in this context referred to as T‐cell immunoglobulin and mucin domain‐containing protein 1 or TIM‐1).24 Nevertheless, since HAVcr‐1 is also expressed on other organs,22 it is likely that additional receptors are required for HAV attachment and entry. For instance, it has been suggested that TIM‐3 promotes HAV entry without being a functional receptor.25 In addition, HAV‐specific IgA are reported to mediate infection of hepatocytes via the asialoglycoprotein receptor (ASGPR).26 This and the fact that IgA was also identified as a natural ligand for HAVcr‐127 may explain why (and how) IgA‐coated HAV can enter the hepatocytes through both HAVcr‐1 and ASGPR, thereby promoting enterohepatic circulation and continuous (endogenous) reinfections of the liver.28 This phenomenon is thought to play an essential role in prolonged and relapsing cases of hepatitis A. Only the emergence of avid IgG antibodies can break the cycle and eventually clear the infection.28
B. Uptake and Uncoating
Following receptor‐mediated binding to the cell surface, the HAV particle is internalized. This has been suggested to occur through receptor‐mediated endocytosis since HAV infection can be inhibited by blockers of endosomal acidification such as monensin, ammonium chloride, and chloroquine.29, 30, 31, 32 However, the precise mechanism remains unclear to date. Upon binding to its receptor the particle is destabilized, thus initiating the uncoating process and releasing the single‐stranded RNA into the host cell cytoplasm.33 Uncoating has been reported to be a slow and asynchronous process with a reported duration of 4–10 hr, in contrast to under an hour for poliovirus.34 It has been proposed that this asynchronicity may be due to the fact that the HAV inoculum contains a heterogeneous mixture of mature virions and provirions. These provirions still require a maturation cleavage (of VP0 into VP2 and VP4, as discussed below) following entry, prior to uncoating, and might therefore uncoat more slowly than mature virions.34, 35 During the uncoating process, dense, noninfectious HAV uncoating intermediates are formed. Intriguingly, these particles do not appear to have an altered sedimentation profile, thereby differing from the typical picornaviral A particle.36 Finally, both low pH and Ca2+ ions are reported to play an important role in HAV receptor binding, uncoating and during the maturation cleavage of provirions following virus entry (as described above).31, 32, 35, 37 Upon completion of uncoating, the single‐stranded RNA is released into the host cell by an unknown mechanism.
C. Translation
The viral RNA, being released into the cytoplasm, is then translated into a single polyprotein. Similar to other picornaviruses, HAV employs an IRES, located in the 5′ UTR, to direct cap‐independent translation of the viral genome using the host ribosomal machinery. Most picornaviruses affect the host cell protein synthesis to favor a more efficient translation of the viral mRNA. This process is mediated by a proteolytic cleavage of eukaryotic initiation factor 4G (eIF4G), thereby inducing a complete shutdown of capped mRNA translation.38 Intriguingly, unlike for other picornaviruses, it is thought that HAV IRES depends on eIF4G, as part of an intact eIF4F complex.39 The fact that HAV has to compete with an intact host cell machinery may also explain its poor replication in cell culture (see Section 5.). However, a recent publication by Redondo et al. suggested that HAV IRES‐driven translation can occur without intact eIF4G and that another factor may be crucial for this translation.38
Next, the translated polyprotein is processed co‐ and posttranslationally in a series of proteolytic cleavages into several functional precursor and mature proteins. The primary posttranslational cleavage occurs at the junction between P1–2A and 2B (instead of the junction between P1 and 2A as for other picornaviruses) (Fig. 1).40 All other processing steps (except the VP1–2A junction, as discussed below) are mediated by 3Cpro and its functional precursor 3ABCpro.41 This is different from other picornaviruses that may use Lpro, 2A, 3Cpro, and the 3CDpro precursor.42 Intriguingly, HAV protein 2A, unlike for other picornaviruses, has no proteolytic activity nor does it contain a ribosome‐skipping sequence,42 but seems to be implicated in morphogenesis (see Section 2.,F).
D. Regulating the Balance between Translation and Replication
Since translation of viral proteins and RNA replication are competing processes, they must be balanced properly to allow efficient viral replication.43 In order to tip the balance from translation to replication, the poly(A)‐binding protein (PABP), as part of the eIF4F complex, is cleaved by HAV 3Cpro. The N‐terminal cleavage product of PABP was shown to have an improved RNA‐binding capacity compared to uncleaved PABP and may act as a dominant negative for IRES‐mediated translation, thus favoring viral RNA synthesis.44 Additionally, proteolytic cleavage of the poly(rC)‐binding protein PCBP2, which interacts with the pyrimidine‐rich tract pY1 in the HAV 5′‐UTR, may also be implicated in regulating the balance between translation and viral RNA synthesis.19 Other enzymes that bind to the HAV IRES are polypyrimidine tract binding protein (PTB), glyceraldehyde 3‐phosphate dehydrogenase, and La autoantigen, which respectively enhance, suppress, and suppress translation.45, 46, 47
E. Replication
As for most RNA viruses, replication of the viral genome takes place in replication complexes that consist of rearranged cellular membranes containing both viral and host proteins. Studies revealed important roles for 2BC precursor and 2C proteins in the membrane rearrangements forming the replication complexes. These complexes are described as a tubular vesicular network and were thought to be of endoplasmatic reticulum (ER) origin.48, 49 However, a recent publication described the mitochondrial localization of the 3ABC precursor protein suggesting that HAV replication complexes may be derived from the outer mitochondrial membrane.50 In line with this hypothesis, exchanging the mitochondria‐targeting 3A transmembrane domain for the ER‐targeting poliovirus 3A transmembrane domain resulted in loss of replication competence.51
Within the replication complexes, the viral genome is transcribed into antisense RNA, which subsequently serves as a template for the production of new viral genomes. During this process, the 3D protein functions as an RNA‐dependent RNA polymerase. The 3B (VPg) protein serves as a starting point for primer‐independent transcription and is covalently linked to the 5′ genome end. A conserved replication element near the 5′ end of the 3Dpol‐coding sequence likely directs uridylylation of VPg by 3Dpol, in such way priming VPg for initiation of RNA replication.52 During replication, the 3A/3B junction remains uncleaved and the transmembrane protein 3A serves as an anchor tethering the growing HAV RNA strand and associated proteins to the membranes of the replication complex,42, 53 as is the case for other picornaviruses.54 It has also been reported that the 2C protein of HAV binds to the 3′ end of the antisense RNA. In this way, protein 2C may be implicated in anchoring the negative sense RNA template to the membranes of the replication complex.55
F. Capsid Assembly
Despite the fact that the multitiered capsid assembly process is only poorly understood, HAV differs at various steps from other picornaviruses. Following the initial cleavage at the 2A/2B junction by 3Cpro (as discussed above), the N‐terminal part of protein 2A coordinates proper folding of the P1–2A precursor protein. This precursor protein is then processed by 3Cpro and its stable precursor 3ABCpro into VP0 (consisting of VP4 and VP2), pX precursor (consisting of VP1 and 2A), and mature VP3 protein.42, 56 These building blocks assemble into pentamers and subsequently associate with viral RNA to form preprovirions. In a final processing step, VP0 will be cleaved into VP4 and VP2 and precursor pX into VP1 and 2A, yielding mature virus particles with only VP1, VP2, and VP3 proteins. The processing of pX is mediated by a yet unknown cellular protease.57 Although the cleavage can be performed by extracellular enzymes like factor Xa and trypsin,58, 59 maturation can also be executed by the lysosomal proteinase cathepsin L. This may indicate a role for lysosomal proteinases in maturation cleavage and potential targeting of HAV provirions to the early lysosomes for maturation cleavage.59 On the other hand, VP0 processing is thought to be a self‐catalytic process that is dependent on the presence of encapsidated RNA.60 The resulting VP4 is rather small compared to other picornaviruses and lacks an N‐terminal myristoylation signal.61 Note that the mature VP4 protein has never been identified in purified virus stocks and its role in capsid formation remains unclear.15, 60
G. Release
HAV is an enterically transmitted virus that replicates mostly in hepatocytes before it is excreted via the bile into the faeces. Despite the fact that the exact details of HAV release remain elusive, the mechanism seems to differ depending on the cell type infected and is thought to involve either a vesicle‐mediated cellular protein transport pathway or specialized hepatocellular transport proteins involved in bile secretion.62 Blank and colleagues demonstrated that following infection of polarized human intestinal epithelial Caco‐2 cells, release of progeny virus was largely restricted to the apical membrane.62 In this way, virus is secreted mainly into the intestinal lumen resulting in an amplification of the HAV inoculum in the intestines and thus an increased viral shedding and spreading of the virus. However, it is still unclear by which mechanism HAV reaches the blood stream. A role for transcytosis by M cells present in Peyer's patches in the ileum has been suggested, a mechanism that was found for poliovirus and reovirus as well.62 Another study described that infection of polarized human hepatocytes occurred most efficiently via the basolateral plasma membrane, after which more than 95% of progeny virus was exported through the basolateral membrane (into the bloodstream), rather than through the apical membrane (into the bile channels).63 This contrasts with the in vivo observation that bile and faecal titers are considerably higher than serum titers.15, 64 Reuptake and transcytosis of progeny virus by the hepatocytes into the bile channels is suggested as the mechanism for enteric secretion.
H. Particle Structure
HAV is a nonenveloped icosahedral particle of approximately 27 nm in diameter.65 The mature capsid is composed of 12 pentamers each consisting of five copies of VP1, VP2, and VP3. Unlike for other picornaviruses, VP4 appears not to be present in mature HAV particles (see Section 2.,F). Attempts to produce high‐resolution images of HAV particles remained unsuccessful so far. A 2006 review15 showed a medium‐resolution image obtained by cryo‐electron microscopy suggesting the absence of the well‐defined canyon surrounding the fivefold axis, in this way differing from other picornaviruses. However, these results have not been confirmed or published separately to date. HAV has a limited number of neutralization antigenic sites. The immunodominant antigenic site is composed of VP1 and VP3 residues and is conformation‐dependent.66 A second antigenic site is the glycophorin A binding site by which HAV can bind to erythrocytes, causing hemagglutination. This process is optimal at acidic pH, but is impaired at physiological pH. This suggests that HAV has evolved to escape erythrocyte binding and consequent clearance.67, 68 Indeed, a mutant of this binding site displayed increased clearance from the blood and lower overall fitness, suggesting evolutionary constraints and explaining the low level of antigenic variability of the glycophorin A binding site.68
3. IMMUNE EVASION MECHANISMS
Hepatitis A is clinically characterized by a prolonged asymptomatic phase before clinical illness becomes apparent and a very limited type I interferon response (only in week 1–2 after HAV challenge).69 HAV replication remains largely undetected by the immune system for several weeks after infection and intrahepatic RNA was shown to persist for over 48 weeks in experimentally infected chimpanzees.69 To this end, HAV employs several strategies to evade the host immune response that are rather different from those used by other picornaviruses.
A. Targeting MAVS and TRIF
One of these evasion tactics is to ablate the innate immunity's alarming system that induces IFN expression. HAV was shown to inhibit double‐stranded RNA (ds RNA)‐induced IFNβ gene expression70 by interfering with the retinoic acid‐inducible gene I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA‐5) signaling pathways (Fig. 2).71 RIG‐I recognizes single‐stranded 5′‐triphosphate RNA, while MDA‐5 detects single‐ and double‐stranded picornaviral RNA covalently linked to VPg.72 Both RIG‐I and MDA‐5 use an adaptor protein called mitochondrial antiviral signaling protein (MAVS, also known as IPS‐1, VISA, or Cardif) that is localized on the outer membrane of the mitochondria.50 Upon activation by RIG‐I or MDA‐5, MAVS recruits and activates TANK‐binding kinase 1 (TBK1) and inhibitor of NF‐κB kinase ε (IKKε). TBK1 and IKKε are both responsible for the phosphorylation of IFN regulatory factor 3 (IRF‐3), eventually leading to IRF‐3 dimerization, nuclear translocation, and induction of IFNβ transcription. HAV proteins 3ABCpro 50 and 2B72 have been described to interfere with MAVS, thereby disrupting the innate cellular antiviral defense mechanism. Precursor protein 3ABC is a stable polyprotein processing intermediate that requires both the 3A and the 3Cpro domain for the cleavage and inactivation of MAVS. The transmembrane domain of protein 3A ascertains mitochondrial localization while the 3Cpro catalytic site performs the actual proteolytic cleavage of MAVS. Mere 3Cpro, lacking the 3A domain, is incapable to perform this proteolysis.50 In addition, it has been demonstrated that protein 2B suppressed both MAVS functioning and the kinase activities of TBK1 and IKKε, thus synergistically suppressing RIG‐I/MDA‐5 signaling, although the exact mechanism still remains to be elucidated.72
Figure 2.
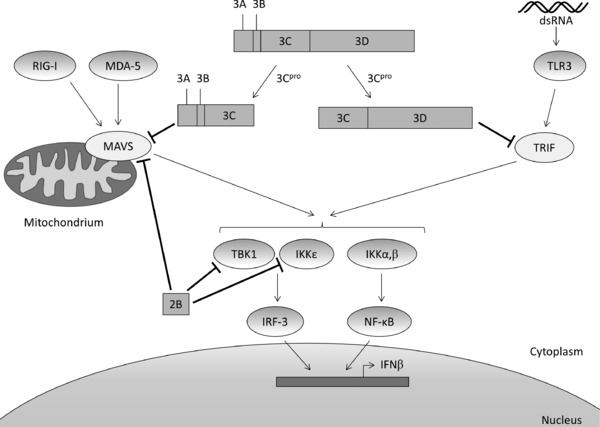
Inhibition of IFNβ transcription by HAV through proteolytic cleavage of MAVS (by 3ABC) and TRIF (by 3CD) and through direct inhibition of MAVS, IKKε, and TBK1 (by 2B). (dsRNA, double‐stranded RNA; IKK, inhibitor of NF‐κB kinase; IRF‐3, interferon regulatory factor 3; MAVS, mitochondrial antiviral signaling protein; MDA‐5, melanoma differentiation‐associated gene 5; RIG‐I, retinoic acid‐inducible gene I; TBK1, TANK‐binding kinase 1; TLR3, Toll‐like receptor 3; TRIF, Toll/IL‐1 receptor domain‐containing adaptor inducing IFNβ).
Toll‐like receptor 3 (TLR3) provides an additional recognition mechanism for dsRNA. Activation also results in IRF‐3 phosphorylation and induction of IFNβ transcription, but unlike RIG‐I and MDA‐5, the downstream action of TLR3 is mediated through TRIF (Toll/IL‐1 receptor domain‐containing adaptor inducing IFNβ), which activates the TBK1 and IKKε kinases. A recent study reported the proteolytic cleavage of TRIF by the 3CD precursor.73 In analogy with the specificity of 3ABCpro‐induced cleavage of MAVS, 3Cpro without the 3D domain is not capable of performing this cleavage. This suggests that the 3Dpol sequence is required to modify the substrate specificity without requiring the catalytic polymerase activity. In addition, proteolytic cleavage of TRIF cannot be performed by the 3ABC intermediate.73
Cleavage of TRIF and MAVS has been reported for other picornaviruses as well, for example, coxsackievirus B3 (MAVS and TRIF)74 and enterovirus 71 (TRIF).75 Unlike for HAV, 3Cpro of both viruses appeared to be sufficient for proteolysis. Interestingly, both MAVS and TRIF are also cleaved by the HCV NS3/4A protease. Despite the fact that the HAV and HCV proteases differ in cleavage specificity and are phylogenetically unrelated, they target the same pair of adaptor proteins, which may be considered a remarkable example of convergent evolution.73
B. Coding Biases
A second immune evasion strategy employed by HAV can be found in its codon bias or nonuniform usage of codons during translation.76 Due to the lack of mechanisms to induce cellular shut off, HAV has to compete with host mRNA for translation (as described above). Within this context, HAV has strategically adopted a codon bias toward rare codons, resulting in a highly deoptimized coding usage.77, 78 This phenomenon contributes considerably to the low replication efficiency of HAV in several ways and seems to serve multiple purposes: first, the deoptimized codon use may reflect a strategy by which rare tRNAs are preferred over abundant tRNAs thereby avoiding competition with cellular tRNAs in the absence of an adequate host cell translation shut off.77 Second, due to a decreased translation and replication rate, the virus is capable of keeping the cellular amount of dsRNA to a minimum, thus escaping the host cell antiviral response.78 In addition, CpG‐containing codons are particularly suppressed in the HAV coding sequence.79, 80 This particular codon deoptimization is proposed to be associated with the recognition of unmethylated CpG by the innate immunity as a pathogen marker, contrary to the methylated CpG pairs of the host. Also in this way, HAV can evade the attention of the immune system. Third, the use of rare codons, usually grouped in clusters, is important for translation kinetics as such cluster may lead to a transient translation stop, a phenomenon known as ribosome stalling.81 It has been suggested that this transient stop may be an important prerequisite for proper folding of HAV structural proteins thereby contributing to the low antigenic variability and extreme environmental stability of HAV.77, 78, 82 Consequently, when HAV is adapted to replicate in vitro under circumstances of chemically induced cellular shut off, the resulting strains (with a “re‐deoptimized” coding sequence) display reduced environmental stability.83 Given the preference of HAV for reduced RNA replication and translation levels, it is now comprehensible why HAV would employ such inefficient type III IRES84, 85: a very efficient IRES would not be compatible with escaping innate immunity and with the many ribosome stalls during translation.83
In conclusion, the coding biases found in the HAV genome may result from evolutionary pressures toward (i) environmental stability (and thus controlled translation kinetics to obtain a resilient viral particle) and (ii) evading the host immune system.79 Additional insights into the mechanisms at action would prove very valuable with regards to our understanding of virus evolution and virus–host interplay.
C. Modulation of Regulatory T‐Cell Activity
In addition to manipulation of the innate immunity, HAV also modifies the adaptive immunity through its receptor binding. HAVcr‐1 is a phosphatidylserine (PtdSer) receptor and functions as a potent T‐cell costimulatory molecule that regulates activation of and tolerance induction (thus suppressing autoimmune responses) in T cells.24 A recent study reported that HAVcr‐1 is constitutively expressed on regulatory T (Treg) cells.86 The function of these specialized T cells is to limit the magnitude of the immune response to diverse pathogens in such way avoiding a hyperactive immune response and subsequent collateral (tissue) damage.87 Several microorganisms activate Treg in order to limit inflammation and tissue injury, as is the case for chronic HBV and HCV infections.87 Conversely, binding of HAV to its receptor inhibits Treg cell functioning.86 In this way, the host is overwhelmed by anti‐self‐responses since Treg cells normally suppress autoimmune reactions, allowing HAV to escape the attention of the immune system. In addition, HAV inhibits the production of transforming growth factor β by Treg cells, thereby blocking T‐effector‐mediated anti‐HAV responses. Complementary, HAV stimulates IL‐22 production that limits immune‐mediated liver damage.86
Taken together, by inhibiting Treg function, HAV creates an environment that favors viral replication and establishment of infection. These findings may also provide an explanation why HAV may elicit autoimmune hepatitis and extrahepatic manifestations including autoimmune hemolytic anemia, even though these complications are rare.88
4. A LINK BETWEEN HEPATITIS A AND ATOPY
The hygiene hypothesis states that the uprising of atopic disease, including asthma, allergic rhinitis, and atopic dermatitis in industrialized countries is due to the increased hygiene.89 As HAV serostatus can be regarded as a hygiene marker, its correlation with atopy was studied. It has been reported by several groups that HAV infection is negatively correlated with hay fever, asthma, and other atopic diseases.89, 90 However, other studies were unable to replicate such effect.91, 92 In addition, McIntire et al. described that previous HAV infection may protect against atopy in individuals carrying a six amino acid insertion (designated 157insMTTTVP) in the gene encoding HAVcr‐1 (which is present in 46–64% of the population).93 However, more recent studies failed to fully replicate these results.94, 95 In addition, 157insMTTTVP appears to increase the susceptibility to severe HAV infection.96 A double mechanistic basis was suggested to (partially) explain this observation: (i) HAV binds more efficiently to the 157insMTTTVP‐carrying HAVcr‐1, resulting in a more effective receptor and (ii) natural killer T cells expressing this long form of HAVcr‐1 were more cytolytic against HAV‐infected hepatocytes.96 Thus, the 157insMTTTVP may protect from atopy, but predispose to severe HAV infections.
In conclusion, a possible relationship between a previous HAV infection, protection from atopic disease and HAVcr‐1 polymorphisms is still subject of debate, but represents an interesting area of research. If HAV truly has a protecting effect, several questions should be addressed: does infection need to occur during childhood? Has vaccination a similar effect? Can this relationship be used in finding a therapeutic target for atopy? Insights into this relationship may prove crucial in combating the rise of atopic disease in industrialized countries.
5. CELL CULTURE
Unlike for most picornaviruses, culturing wild‐type (wt) HAV in cell culture has proven to be quite a challenge as these strains replicate only very marginally in vitro (e.g., reference 97). HAV can be adapted to growth in cell culture through serial passaging; a process that introduces cell culture adapting mutations stepwise into the viral genome. Such mutations have been found in the P2 region (especially in 2B and 2C)98, 99 and in the 5′‐UTR,100, 101 but also in other parts of the genome.98 Intriguingly, these cell culture adapting mutations result in HAV strains that are highly attenuated in vivo (e.g., references 102, 103). A major advance in culturing HAV was the selection of a Huh7 cell line (designated Huh7‐A‐I) that allows genetically stable growth of wt HAV without the accumulation of cell culture adapting mutations.104
Although wt HAV does not induce a cytopathic effect (CPE) in cell culture, cytopathic strains may arise during prolonged serial passaging.105, 106 CPE appears to be cell type specific and is mostly found in monkey kidney cell lines like FRhK‐4 and BS‐C‐1. These selected virus strains induce apoptosis through ribosomal RNA degradation by RNase L107, 108 and consequent caspase activation.109 Interestingly, it seems possible to induce CPE with cell culture adapted noncytopathic strains under specific conditions of lower temperature (<34°C) and decreased cell density, both in FRhK‐4 and human A549 cells.110 Similarly, we observed CPE for the PA21 strain (genotype IIIa) in FRhK‐4 cells under these conditions (unpublished results).
6. THE NEED FOR ANTIVIRALS?
Despite the availability of an efficient vaccine, an antiviral drug against HAV would be of great use. First, since HAV growth in vitro is rather limited, the production of vaccines remains a painstaking process, which in part explains the high cost.15 This cost is particularly a problem in relatively poor regions with improved hygiene where infections occur at later age and consequently are more severe.4 Second, antivirals could shorten the period of illness and decrease symptoms and associated economic costs in infected unvaccinated patients. Early treatment of these infected unvaccinated persons may also prevent severe cases of fulminant hepatitis. Third, an antiviral could be a useful tool in rapidly containing epidemics. Lastly, the potential emergence of vaccine‐escape variants has been reported recently;18 an anti‐HAV drug could therefore be instrumental in halting the spread of such virus strains. In conclusion, there is certainly a need for antiviral drugs for hepatitis A, given that they are safe, efficacious, and preferably cheap.
Research into HAV antiviral drug discovery was mostly performed during the late eighties and early nineties. Afterwards, research efforts and funding waned due to the introduction of vaccines,15 although some interesting work has been done on amantadine and 3Cpro inhibitors in the last decade. An overview of the most relevant inhibitors is presented here (also see Table I).
Table I.
Overview of Reported Inhibitors of HAV with (Putative) Targets and Calculated Values for 50% Effective Concentrations (EC50), 50% Cytotoxic Concentrations (CC50), and Selectivity Indices (SI) When Provided
| Antiviral activity | Toxicity | SI | (Putative) target | Reference | |
|---|---|---|---|---|---|
| IFNα‐2a | EC50: 90 IU/mL | CC50: >10,000 IU/mL | >100 | Induction of antiviral state | 112 |
| Amantadine | EC50: 58 μM | CC50: 310 μM | 5.3 | HAV IRES‐mediated translation | 117 |
| Guanidine HCl | Variable inhibition between 0.1 and 3 mM | ND | ND | Protein 2C | 99, 107, 119, 120, 121 |
| Iota‐carrageenan | EC50: 2.5 μg/mL | CC50: >1000 μg/mL | >400 | Attachment | 122 |
| Atropine | 84% reduction in viral titer at 1.7 mM | CC50: >3.5 mM | ND | Attachment and/or uncoating | 123 |
| Glycyrrhizin | EC50: 325 μM | CC50: 5 mM | 15 | Membrane penetration | 117 |
| 4,6′‐dichloroflavan | EC50: 6 nM | CC50: 45 μM | 7500 | Entry and/or uncoating | 127 |
| 4,6′‐dichloroisoflavan | EC50: 6 nM | CC50: 90 μM | 15,000 | Entry and/or uncoating | 127 |
| Ribavirin | EC50: 94 μM | CC50: 430 μM | 4.6 | Inosine 5′‐monophosphate dehydrogenase | 117 |
| Pyrazofurin | EC50: 0.62 μM | CC50: 28 μM | 45 | Orotidine 5′‐monophosphate decarboxylase | 117 |
| Protamine | 97% reduction in viral titer at 100 μg/mL | CC50: 200 μg/mL | ND | RNA polymerase | 123 |
| Ac‐LAAQ′‐fluoromethylketone | 96% reduction in viral titer at 5 μM | ND | ND | Protein 3Cpro | 143 |
| Halopyridinyl ester | EC50: 53 nM | ND | ND | Protein 3Cpro | 151 |
ND, not determined.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
A. Interferon
In 1984, IFNβ was already demonstrated to efficiently inhibit the replication of HAV in human embryo fibroblasts.111 HAV‐infected cells were completely cleared following five passages with 1000 IU/mL of IFNβ. Later, another study reported an effective concentration 50 (EC50, minimal concentration required to reduce virus replication by 50%) of 90 IU/mL and a selectivity index (SI = 50% cytotoxic concentration (CC50)/EC50) of more than 100 for IFNα‐2a.112 In addition to the in vitro activity, a case study describes the in vivo efficacy of IFNβ in three patients with fulminant hepatitis A and one patient with severe hepatic failure due to HAV. Following a treatment with 3 million units per day, liver functioning ameliorated and all four patients survived.113 However, in addition to its high price and parenteral administration, the clinical use of IFN is associated with severe side effects hampering usage in less developed regions.
B. Amantadine
Amantadine was initially thought to inhibit HAV by increasing the intravesicular pH. However, its antiviral effect required an extended incubation when compared to NH4Cl and other lipophilic amines (e.g., methylamine, dansylcadaverine).29 In line with these findings, inhibition of HAV IRES‐dependent translation was later reported as a mechanism of action (MOA).114 A follow‐up study confirmed the moderate activity of amantadine in cell‐based assays.115 In addition, an increased effect was observed when amantadine was combined with IFNα or IL‐29 (IFNλ1).115, 116 However, in addition to a fairly limited effect in vitro (EC50 of 58 μM and a SI around 5117), pharmacokinetic studies demonstrated maximum plasma concentrations (C max) following a multiple dosage scheme well below the reported EC50,118 limiting its use in a clinical context.
C. The Curious Case of Guanidine HCl
For guanidine HCl, a protein 2C‐targeting enterovirus inhibitor, mixed results were reported. The earliest publication detected no inhibition at 1 and 2 mM,119 but other groups found moderate inhibition (around 50%) following treatment with 3 mM120 or 2.5 mM.107 Cho and colleagues reported reversible inhibition at 2 mM but also reported enhancement of replication at 0.1–1 mM.121 Conversely to these results, Yi et al. reported strong inhibition of viral replication at concentrations as low as 0.1 mM.99 Several factors may explain the inconsistency of these data: (i) mutations in the different viral genomes studied, specifically in the 2C region; (ii) the cell type used in the antiviral assay; (iii) the multiplicity of infection (MOI), which was found to be an important determinant for the antiviral activity107; and (iv) the detection method employed.
D. Miscellaneous Small Molecule Inhibitors of the HAV Replication Cycle
Only limited information is available on other compounds with anti‐HAV activity. Early steps of the replication cycle can be blocked by iota‐carrageenan, which probably functions as an inhibitor of virus attachment and receptor binding.122 Atropine, at 1.7 mM, proved to moderately reduce HAV replication by targeting the early steps of the HAV replication cycle as well (attachment and possibly uncoating).123 Due to toxicity and limited antiviral activity, the clinical potential of both compounds is low. For glycyrrhizin, an aqueous extract of the licorice root, an EC50 of 325μM and a SI of 15 were reported.117, 124 Subsequent studies suggested inhibition of membrane penetration as the putative MOA.124 In addition to its anti‐HAV activity, glycyrrhizin proved also to be active against a relatively large panel of other viruses, including HBV, HCV, HSV, and HIV, with several proposed mechanisms of action, as reviewed by Fiore et al.125 However, glycyrrhizin may cause pseudohyperaldosteronism due to its aldosterone‐like effects. In addition, the compound is readily metabolized following oral and intravenous administration and the attained plasma concentrations are considerably lower than the reported EC50,126 hampering clinical use for treatment of HAV‐infected patients.
Probably the most potent small molecules reported thus far are 4′,6‐dichloroflavan and 4′,6‐dichloroisoflavan (EC50 values of 6 nM).127 Inhibition of cell entry or viral uncoating were posited as possible mechanisms of action for the antiviral activity against HAV. These drugs were also found to be effective against rhinoviruses and poliovirus in vitro.127 However, 4′,6‐dichloroflavan failed to protect volunteers from experimental rhinovirus infection when administered both orally or intranasally,128, 129 raising doubts concerning its in vivo efficacy against HAV as well.
Other early stage inhibitors include chlorpromazine and chloroquine.31 The former is thought to block endocytosis of nonclathrin‐coated vesicles, while the latter inhibits endosome acidification.31 Monensin, another endosome acidification blocker, yielded minor to extensive inhibition of HAV replication in different studies.30, 31, 62
In addition to early stage inhibitors, several other HAV inhibitors were identified to target different steps of the intracellular replication process including RNA synthesis, translation, and capsid assembly. However, the information on their precise MOA is very limited. For instance, ribavirin was demonstrated to have a moderate effect at 100 μM,130 showing an EC50 of 94 μM and a selectivity index of around 4.117 Improved anti‐HAV activity was reported for pyrazofurin, an orotidine 5′‐monophosphate decarboxylase inhibitor, but further development was halted due to toxicity (interference with nucleotide metabolism).117 Protamine reduced HAV replication with 97% at 100 μg/mL and was suggested to function as a RNA polymerase inhibitor.123 In addition, inhibitors of oxidative phosphorylation and 2‐deoxy‐d‐glucose, a glycolysis inhibitor, were identified as inhibitors of HAV replication by unknown mechanisms.30 Other inhibitors include brefeldin A62, 107 and cycloheximide,107 inhibitors of ER‐to‐golgi transport and protein synthesis, respectively. A moderate anti‐HAV activity was also reported for amphotericin B131 and methisoprinol,132 drugs used against fungal infections and as an immunomodulator in the treatment of subacute sclerosing panencephalitis, respectively. However, no data are available on the MOA.
In recent years, several new anti‐HAV compounds were synthesized, but the antiviral activity remained rather limited.133, 134, 135 Other papers reported highly potent inhibitors of HAV replication, but did not report toxicity data, making it impossible to judge whether or not these are selective inhibitors.136, 137, 138
E. Rational Design of 3Cpro Inhibitors
Considerable efforts have been undertaken to rationally design and develop inhibitors for HAV and human enterovirus 3Cpro and the related coronavirus 3C‐like protease (3CLpro).139 These enzymes have a topology comparable to that of the chymotrypsin‐like serine proteases, but are in fact cysteine proteases.139 Consequently, the general research strategy is to introduce thiol‐reactive groups into the catalytic site. Several of such protease inhibitors have been developed over the years, both modified peptides and nonpeptidic analogs. Despite the fact that the modified peptides are easier to design (by merely mimicking the natural protease substrate), nonpeptidic and peptidomimetic inactivators are preferred, since they can be modified in order to increase stability and cellular and gastrointestinal uptake, properties that are difficult to attain for peptides. A similar approach has already been applied to rhino‐ and enterovirus 3Cpro and resulted in the molecule rupintrivir that was halted after unsuccessful phase II clinical trials.140
The first HAV 3Cpro inhibitors developed were modified peptides based on the acetyl‐Leu‐Ala‐Ala sequence, serving as competitors for the natural peptide substrate Leu‐Arg‐Thr.141 Based on these findings, peptidyl aldehyde and monofluoromethylketone (FMK) inhibitors were developed.142, 143 Resolving the crystal structure of HAV 3Cpro in complex with a peptidyl FMK inhibitor indicated that the inhibitor covalently binds the catalytic cysteine.144 One of these peptidyl FMK inhibitors was also tested ex vivo and was shown to reduce HAV replication by 96%.143 However, cellular uptake appeared to be a major problem. In addition, several azaglutamine derivatives were found to be irreversible peptidic inhibitors.145
On the other hand, several nonpeptidic antagonists were reported as well. Asymetric azodicarboxamides were found to be irreversible inhibitors of HAV and HRV 3Cpro.146 Serine and threonine β‐lactones also inhibited HAV 3Cpro.147, 148 Other inhibitors include monophenyl pseudoxazolones of glycine (IC50 of 4–6 μM)149 and cathepsin K inhibitor‐based keto‐glutamines.150 Lastly, halopyridinyl esters were reported as the most potent nonpeptide HAV 3Cpro inhibitors thus far with IC50's as low as 53 nM.151
Although recent publications in this field seem to focus increasingly on HRV 3Cpro and coronavirus 3CLpro,152 it would be wise not to neglect possible anti‐HAV activity for potential future (off‐label) use of a marketed HRV or coronavirus protease inhibitor for severe HAV infections.
F. RNA Interference
RNA interference (RNAi) has been suggested as a future treatment of severe HAV infections.153 Small interfering RNA (siRNA) targeting nonstructural genes can effectively block replication.153, 154 Inhibition was also reported when targeting domains IIIC and V of the HAV IRES.155 Three consecutive applications of siRNA over 14 days resulted in a 3 log10 infectivity titer reduction in persistently infected cells. However, resistance emerged fairly quickly through mutation of the target site, necessitating combination of different siRNAs.154 Once clinical RNAi therapy would be an established treatment for other (infectious) diseases, it would be interesting to further explore this for hepatitis A. However, such clinical applications of RNAi are currently still at the experimental stage.
G. Novel Antiviral Assays
We recently reported the development of three assays to screen for and identify small molecule inhibitors of HAV replication.156 A CPE reduction assay based on the cell culture adapted HM175/18f strain (genotype IB) is amendable to high‐throughput screening. RT‐qPCR‐based virus yield assays for HM175/18f and genotype IIIA strain PA21 were developed as well, allowing for confirmation and further characterization of the antiviral activity of selected molecules. The known inhibitors IFNα and amantadine were used to validate these assays. Using these assays, three enterovirus inhibitors with different targets were evaluated for anti‐HAV activity. Pleconaril, a known capsid binder,140 yielded (not unexpectedly) a very limited activity; inhibition by rupintrivir, a 3Cpro inhibitor, proved to be strain‐dependent156; and enviroxime, a direct inhibitor of phosphatidylinositol‐4‐kinase IIIβ (PI4KIIIβ) that induces resistance mutations in enterovirus protein 3A,157 was inactive in all three systems. Recently, PI4KIIIβ has been identified as an essential host factor in the replication strategy of enteroviruses. Following recruitment by enterovirus protein 3A to ER‐derived organelle membranes, a phosphatidylinositol‐4‐phosphate‐rich environment is created, promoting viral RNA replication. The observation that HAV protein 3A is targeted toward the outer mitochondrial membrane instead of the ER,50 together with the observed lack of activity for enviroxime in two different cell lines,156 suggests that PI4KIIIβ kinase activity does not play a role in HAV replication.
H. Small Animal Models
A small animal model would be an additional prerequisite for the development and validation of anti‐HAV compounds. The host range of HAV is restricted to humans and several nonhuman primates, for example, tamarins and chimpanzees.158 Some efforts have been undertaken toward the development of small animal models, for example, virus adaptation to mouse cell lines or infection of guinea pigs.159, 160 However, so far no robust model for clinical hepatitis A is available. Very recently, it has been found that the chimeric SCID/Alb‐uPA mouse model, which supports the engraftment and proliferation of transplanted human hepatocytes, is susceptible to HAV infection (Pang D., personal communication). Despite the fact that these results still need to be published, they could represent a major leap forward for anti‐HAV research.
7. CONCLUSION
Despite the fact that hepatitis A is a vaccine‐preventable disease and infections are usually mild, continued attention for this pathogen is warranted. Outbreaks and occasional cases of fatal fulminant hepatitis A still occur and as the age of infection shifts upwards, the severity of the infection increases as well. Morbidity and mortality may also increase in patients with chronic liver disease, for example, due to HBV or HCV. Additionally, a recent report warned for the potential emergence of vaccine‐escape variants.18 Taken together, these reasons warrant continuation and expansion of the current vaccination programs, but also the development of antiviral drugs against HAV, even though this may not be particularly interesting from a commercial point of view. So far no potent or selective inhibitors have been reported. We therefore propose to focus antiviral efforts on marketed drugs and antivirals that are currently in clinical development. Off‐label use of such drug would be a much appreciated therapy. In fact, we are currently employing our CPE reduction assay for screening of a library of FDA‐approved/marketed drugs and with promising results so far. Also on the fundamental biology level, HAV remains a largely understudied virus, despite many interesting features. Further investigation could provide unique insights into pathogenesis, tissue tropism determinants, and replication strategies for HAV and picornaviruses in general.
ACKNOWLEDGMENT
Yannick Debing thanks the Research Foundation—Flanders (FWO) of which he is a fellow. This work is supported by the European Union Seventh Framework Program (FP7/2007–2013) under SILVER grant agreement no. 260644 and by KU Leuven, geconcerteerde onderzoeksactie (GOA/10/014).
Biographies
Yannick Debing was born in 1988 and graduated in 2011 as a pharmacist and master in drug development and is currently a Ph.D. student at the Rega Institute, University of Leuven. His research project addresses the development of novel antiviral strategies against hepatitis A virus and hepatitis E virus.
Johan Neyts was born in 1966 and is full professor of Virology at the University of Leuven, Belgium. His research is focused on the development of novel antiviral strategies against a number of viruses including picornaviruses (such as rhinoviruses that cause exacerbations of asthma and COPD), flaviviruses (mainly dengue), and the hepatitis C virus. His work has been published in several book chapters, in more than 280 peer‐reviewed papers and in several patents. Three molecules discovered in his lab as HCV inhibitors made it to advanced clinical studies in man for the treatment of chronic HCV infections. He is on the editorial board of a number of journals, member of several national and international scientific committees and advisory boards as well as on the board of the “International Society for Antiviral Research.” He is co‐founder and Chief Scientific Officer of Okapi Sciences NV, a company developing antivirals for veterinary use. He teaches medical virology at the school of dentistry and the school of medicine at the University of Leuven. He has been honored with a number of awards including from the International Society for Antiviral Research, the Royal Belgian Academy of Medicine, and the Belgian Fund for Scientific Research.
Hendrik Jan Thibaut was born in 1984 and obtained his Ph.D. degree in virology in 2012 at the University of Leuven. He is currently involved in several projects on picornaviruses in the research group of Prof. J. Neyts at the Rega Institute, University of Leuven. His research focuses on the identification and characterization of novel antiviral strategies for the treatment of infections with enteroviruses, with a special interest in protein 2C and the role of the host cell in virus morphogenesis.
REFERENCES
- 1. Rein D. Modeling the global burden of hepatitis A virus infections in 1990 and 2005. Paper presented at The Liver Meeting 2011. The 62nd Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA; 2011 November 3–8. [Google Scholar]
- 2. Siegl G, Weitz M, Kronauer G. Stability of hepatitis A virus. Intervirology 1984;22:218–226. [DOI] [PubMed] [Google Scholar]
- 3. Abad FX, Pintó RM, Bosch A. Survival of enteric viruses on environmental fomites. Appl Environ Microbiol 1994;60:3704–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. FitzSimons D, Hendrickx G, Vorsters A, Van Damme P. Hepatitis A and E: Update on prevention and epidemiology. Vaccine 2010;28:583–588. [DOI] [PubMed] [Google Scholar]
- 5. WHO . The Immunological Basis for Immunization Series—Module 18: Hepatitis A. Geneva, Switzerland: WHO Press; 2011. 39 p. [Google Scholar]
- 6. Keeffe E. Hepatitis A in patients with chronic liver disease—Severity of illness and prevention with vaccination. J Viral Hepat 2000;7(Suppl 1):15–17. [DOI] [PubMed] [Google Scholar]
- 7. Vento S, Garofano T, Renzini C, Cainelli F, Casali F, Ghironzi G, Ferraro T, Concia E. Fulminant hepatitis associated with hepatitis A virus superinfection in patients with chronic hepatitis C. N Engl J Med 1998;338:286–290. [DOI] [PubMed] [Google Scholar]
- 8. Vogt TM, Wise ME, Bell BP, Finelli L. Declining hepatitis A mortality in the United States during the era of hepatitis A vaccination. J Infect Dis 2008;197:1282–1288. [DOI] [PubMed] [Google Scholar]
- 9. Shouval D. Hepatitis: New doubts about preventing HAV superinfection in chronic HCV. Nat Rev Gastroenterol Hepatol 2012;9:367–368. [DOI] [PubMed] [Google Scholar]
- 10. Ott JJ, Irving G, Wiersma ST. Long‐term protective effects of hepatitis A vaccines. A systematic review. Vaccine 2012;31:3–11. [DOI] [PubMed] [Google Scholar]
- 11. Wheeler C, Vogt TM, Armstrong GL, Vaughan G, Weltman A, Nainan OV, Dato V, Xia G, Waller K, Amon J, Lee TM, Highbaugh‐Battle A, Hembree C, Evenson S, Ruta MA, Williams IT, Fiore AE, Bell BP. An outbreak of hepatitis A associated with green onions. N Engl J Med 2005;353:890–897. [DOI] [PubMed] [Google Scholar]
- 12. Donnan EJ, Fielding JE, Gregory JE, Lalor K, Rowe S, Goldsmith P, Antoniou M, Fullerton KE, Knope K, Copland JG, Bowden DS, Tracy SL, Hogg GG, Tan A, Adamopoulos J, Gaston J, Vally H. A multistate outbreak of hepatitis A associated with semidried tomatoes in Australia, 2009. Clin Infect Dis 2012;54:775–781. [DOI] [PubMed] [Google Scholar]
- 13. Guillois‐Bécel Y, Couturier E, Le Saux JC, Roque‐Afonso AM, Le Guyader FS, Le Goas A, Pernès J, Le Bechec S, Briand A, Robert C, Dussaix E, Pommepuy M, Vaillant V. An oyster‐associated hepatitis A outbreak in France in 2007. Euro Surveill 2009;14:pii:19144. [PubMed] [Google Scholar]
- 14. Tominaga A, Kanda T, Akiike T, Komoda H, Ito K, Abe A, Aruga A, Kaneda S, Saito M, Kiyohara T, Wakita T, Ishii K, Yokosuka O, Sugiura N. Hepatitis A outbreak associated with a revolving sushi bar in Chiba, Japan: Application of molecular epidemiology. Hepatol Res 2012;42:828–834. [DOI] [PubMed] [Google Scholar]
- 15. Martin A, Lemon SM. Hepatitis A virus: From discovery to vaccines. Hepatology 2006;43:S164–S172. [DOI] [PubMed] [Google Scholar]
- 16. International Committee on Taxonomy of Viruses . Virus Taxonomy: 2011 Release (current) [Internet] [updated 2012; cited 2013 Feb 6]. http://ictvonline.org/virusTaxonomy.asp?version=2011.
- 17. Cristina J, Costa‐Mattioli M. Genetic variability and molecular evolution of hepatitis A virus. Virus Res 2007;127:151–157. [DOI] [PubMed] [Google Scholar]
- 18. Pérez‐Sautu U, Costafreda MI, Caylà J, Tortajada C, Lite J, Bosch A, Pintó RM. Hepatitis a virus vaccine escape variants and potential new serotype emergence. Emerg Infect Dis 2011;17:734–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang B, Seitz S, Kusov Y, Zell R, Gauss‐Müller V. RNA interaction and cleavage of poly(C)‐binding protein 2 by hepatitis A virus protease. Biochem Biophys Res Commun 2007;364:725–730. [DOI] [PubMed] [Google Scholar]
- 20. Dollenmaier G, Weitz M. Interaction of glyceraldehyde‐3‐phosphate dehydrogenase with secondary and tertiary RNA structural elements of the hepatitis A virus 3′ translated and non‐translated regions. J Gen Virol 2003;84:403–414. [DOI] [PubMed] [Google Scholar]
- 21. Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone SM. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J 1996;15:4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 22. Feigelstock D, Thompson P, Mattoo P, Zhang Y, Kaplan GG. The human homolog of HAVcr‐1 codes for a hepatitis A virus cellular receptor. J Virol. 1998;72:6621–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule‐1 (KIM‐1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up‐regulated in renal cells after injury. J Biol Chem 1998;273:4135–4142. [DOI] [PubMed] [Google Scholar]
- 24. Rodriguez‐Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev 2009;229:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sui L, Zhang W, Chen Y, Zheng Y, Wan T, Zhang W, Yang Y, Fang G, Mao J, Cao X. Human membrane protein Tim‐3 facilitates hepatitis A virus entry into target cells. Int J Mol Med 2006;17:1093–1099. [PubMed] [Google Scholar]
- 26. Dotzauer A, Gebhardt U, Bieback K, Göttke U, Kracke A, Mages J, Lemon SM, Vallbracht A. Hepatitis A virus‐specific immunoglobulin A mediates infection of hepatocytes with hepatitis A virus via the asialoglycoprotein receptor. J Virol 2000;74:10950–10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tami C, Silberstein E, Manangeeswaran M, Freeman GJ, Umetsu SE, DeKruyff RH, Umetsu DT, Kaplan GG. Immunoglobulin A (IgA) is a natural ligand of hepatitis A virus cellular receptor 1 (HAVCR1), and the association of IgA with HAVCR1 enhances virus‐receptor interactions. J Virol 2007;81:3437–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dotzauer A, Heitmann A, Laue T, Kraemer L, Schwabe K, Paulmann D, Flehmig B, Vallbracht A. The role of immunoglobulin A in prolonged and relapsing hepatitis A virus infections. J Gen Virol 2012;93:754–760. [DOI] [PubMed] [Google Scholar]
- 29. Superti F, Seganti L, Orsi N, Divizia M, Gabrieli R, Panà A. The effect of lipophilic amines on the growth of hepatitis A virus in Frp/3 cells. Arch Virol 1987;96:289–296. [DOI] [PubMed] [Google Scholar]
- 30. Superti F, Seganti L, Orsi N, Divizia M, Gabrieli R, Panà A. Effect of cellular function inhibitors on the infection of Frp/3 cells by hepatitis A virus. Med Microbiol Immunol 1989;178:29–36. [DOI] [PubMed] [Google Scholar]
- 31. Bishop NE. Examination of potential inhibitors of hepatitis A virus uncoating. Intervirology 1998;41:261–271. [DOI] [PubMed] [Google Scholar]
- 32. Bishop NE. Conformational changes in the hepatitis A virus capsid in response to acidic conditions. J Med Microbiol 1999;48:443–450. [DOI] [PubMed] [Google Scholar]
- 33. Silberstein E, Xing L, Van de Beek W, Lu J, Cheng H, Kaplan GG. Alteration of hepatitis A virus (HAV) particles by a soluble form of HAV cellular receptor 1 containing the immunoglobin‐and mucin‐like regions. J Virol 2003;77:8765–8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bishop NE, Anderson DA. Uncoating kinetics of hepatitis A virus virions and provirions. J Virol 2000;74:3423–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bishop NE. Effect of low pH on the hepatitis A virus maturation cleavage. Acta Virol 1999;43:291–296. [PubMed] [Google Scholar]
- 36. Bishop NE. Hepatitis A virus replication: An intermediate in the uncoating process. Intervirology 2000;43:36–47. [DOI] [PubMed] [Google Scholar]
- 37. Bishop NE, Anderson DA. Early interactions of hepatitis A virus with cultured cells: Viral elution and the effect of pH and calcium ions. Arch Virol 1997;142:2161–2178. [DOI] [PubMed] [Google Scholar]
- 38. Redondo N, Sanz MA, Steinberger J, Skern T, Kusov Y, Carrasco L. Translation directed by hepatitis A virus IRES in the absence of active eIF4F complex and eIF2. PLoS One 2012;7:e52065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borman AM, Kean KM. Intact eukaryotic initiation factor 4G is required for hepatitis A virus internal initiation of translation. Virology 1997;237:129–136. [DOI] [PubMed] [Google Scholar]
- 40. Schultheiss T, Kusov YY, Gauss‐Müller V. Proteinase 3C of hepatitis A virus (HAV) cleaves the HAV polyprotein P2‐P3 at all sites including VP1/2A and 2A/2B. Virology 1994;198:275–281. [DOI] [PubMed] [Google Scholar]
- 41. Kusov Y, Gauss‐Müller V. Improving proteolytic cleavage at the 3A/3B site of the hepatitis A virus polyprotein impairs processing and particle formation, and the impairment can be complemented in trans by 3AB and 3ABC. J Virol 1999;73:9867–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Palma AM, Vliegen I, De Clercq E, Neyts J. Selective inhibitors of picornavirus replication. Med Res Rev 2008;28:823–884. [DOI] [PubMed] [Google Scholar]
- 43. Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive‐stranded RNA virus. Genes Dev 1998;12:2293–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang B, Morace G, Gauss‐Müller V, Kusov Y. Poly(A) binding protein, C‐terminally truncated by the hepatitis A virus proteinase 3C, inhibits viral translation. Nucleic Acids Res 2007;35:5975–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schultz DE, Hardin CC, Lemon SM. Specific interaction of glyceraldehyde 3‐phosphate dehydrogenase with the 5’‐nontranslated RNA of hepatitis A virus. J Biol Chem 1996;271:14134–14142. [DOI] [PubMed] [Google Scholar]
- 46. Yi M, Schultz DE, Lemon SM. Functional significance of the interaction of hepatitis A virus RNA with glyceraldehyde 3‐phosphate dehydrogenase (GAPDH): Opposing effects of GAPDH and polypyrimidine tract binding protein on internal ribosome entry site function. J Virol 2000;74:6459–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cordes S, Kusov Y, Heise T, Gauss‐Müller V. La autoantigen suppresses IRES‐dependent translation of the hepatitis A virus. Biochem Biophys Res Commun 2008;368:1014–1019. [DOI] [PubMed] [Google Scholar]
- 48. Teterina NL, Bienz K, Egger D, Gorbalenya AE, Ehrenfeld E. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology 1997;237:66–77. [DOI] [PubMed] [Google Scholar]
- 49. Gosert R, Egger D, Bienz K. A cytopathic and a cell culture adapted hepatitis A virus strain differ in cell killing but not in intracellular membrane rearrangements. Virology 2000;266:157–169. [DOI] [PubMed] [Google Scholar]
- 50. Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci USA 2007;104:7253–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feng Z, Lemon SM. Hepatitis A virus In: Ehrenfeld E, Domingo E, Roos RP, Eds. The Picornaviruses. Washington, DC: ASM Press; 2010. p 383–410. [Google Scholar]
- 52. Yang Y, Yi M, Evans DJ, Simmonds P, Lemon SM. Identification of a conserved RNA replication element (cre) within the 3Dpol‐coding sequence of hepatoviruses. J Virol 2008;82:10118–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ciervo A, Beneduce F, Morace G. Polypeptide 3AB of hepatitis A virus is a transmembrane protein. Biochem Biophys Res Commun 1998;249:266–274. [DOI] [PubMed] [Google Scholar]
- 54. Xiang W, Cuconati A, Paul AV, Cao X, Wimmer E. Molecular dissection of the multifunctional poliovirus RNA‐binding protein 3AB. RNA 1995;1:892–904. [PMC free article] [PubMed] [Google Scholar]
- 55. Banerjee R, Dasgupta A. Interaction of picornavirus 2C polypeptide with the viral negative‐strand RNA. J Gen Virol 2001;82:2621–2627. [DOI] [PubMed] [Google Scholar]
- 56. Cohen L, Bénichou D, Martin A. Analysis of deletion mutants indicates that the 2A polypeptide of hepatitis A virus participates in virion morphogenesis. J Virol 2002;76:7495–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martin A, Bénichou D, Chao SF, Cohen LM, Lemon SM. Maturation of the hepatitis A virus capsid protein VP1 is not dependent on processing by the 3Cpro proteinase. J Virol 1999;73:6220–6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rachow A, Gauss‐Müller V, Probst C. Homogeneous hepatitis A virus particles. Proteolytic release of the assembly signal 2A from procapsids by factor Xa. J Biol Chem 2003;278:29744–29751. [DOI] [PubMed] [Google Scholar]
- 59. Morace G, Kusov Y, Dzagurov G, Beneduce F, Gauss‐Muller V. The unique role of domain 2A of the hepatitis A virus precursor polypeptide P1–2A in viral morphogenesis. BMB Rep 2008;41:678–683. [DOI] [PubMed] [Google Scholar]
- 60. Bishop NE, Anderson DA. RNA‐dependent cleavage of VP0 capsid protein in provirions of hepatitis A virus. Virology 1993;197:616–623. [DOI] [PubMed] [Google Scholar]
- 61. Tesar M, Jia XY, Summers DF, Ehrenfeld E. Analysis of a potential myristoylation site in hepatitis A virus capsid protein VP4. Virology 1993;194:616–626. [DOI] [PubMed] [Google Scholar]
- 62. Blank CA, Anderson DA, Beard M, Lemon SM. Infection of polarized cultures of human intestinal epithelial cells with hepatitis A virus: Vectorial release of progeny virions through apical cellular membranes. J Virol 2000;74:6476–6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Snooks MJ, Bhat P, Mackenzie J, Counihan NA, Vaughan N, Anderson DA. Vectorial entry and release of hepatitis A virus in polarized human hepatocytes. J Virol 2008;82:8733–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lemon SM. Type A viral hepatitis. New developments in an old disease. N Engl J Med 1985;313:1059–1067. [DOI] [PubMed] [Google Scholar]
- 65. Feinstone SM, Kapikian AZ, Purcell RH. Hepatitis A: Detection by immune electron microscopy of a viruslike antigen associated with acute illness. Science 1973;182:1026–1028. [DOI] [PubMed] [Google Scholar]
- 66. Mattioli S, Imberti L, Stellini R, Primi D. Mimicry of the immunodominant conformation‐dependent antigenic site of hepatitis A virus by motifs selected from synthetic peptide libraries. J Virol 1995;69:5294–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sánchez G, Aragonès L, Costafreda MI, Ribes E, Bosch A, Pintó RM. Capsid region involved in hepatitis A virus binding to glycophorin A of the erythrocyte membrane. J Virol 2004;78:9807–9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Costafreda MI, Ribes E, Franch A, Bosch A, Pintó RM. A single mutation in the glycophorin A binding site of hepatitis A virus enhances virus clearance from the blood and results in a lower fitness variant. J Virol 2012;86:7887–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lanford RE, Feng Z, Chavez D, Guerra B, Brasky KM, Zhou Y, Yamane D, Perelson AS, Walker CM, Lemon SM. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc Natl Acad Sci USA 2011;108:11223–11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brack K, Berk I, Magulski T, Lederer J, Dotzauer A, Vallbracht A. Hepatitis A virus inhibits cellular antiviral defense mechanisms induced by double‐stranded RNA. J Virol 2002;76:11920–11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fensterl V, Grotheer D, Berk I, Schlemminger S, Vallbracht A, Dotzauer A. Hepatitis A virus suppresses RIG‐I‐mediated IRF‐3 activation to block induction of beta interferon. J Virol 2005;79:10968–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Paulmann D, Magulski T, Schwarz R, Heitmann L, Flehmig B, Vallbracht A, Dotzauer A. Hepatitis A virus protein 2B suppresses beta interferon (IFN) gene transcription by interfering with IFN regulatory factor 3 activation. J Gen Virol 2008;89:1593–1604. [DOI] [PubMed] [Google Scholar]
- 73. Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K, Lemon SM. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease‐polymerase processing intermediate, 3CD. PLoS Pathog 2011;7:e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mukherjee A, Morosky SA, Delorme‐Axford E, Dybdahl‐Sissoko N, Oberste MS, Wang T, Coyne CB. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 2011;7:e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll‐like receptor 3. J Virol 2011;85:8811–8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Novoa EM, Ribas de Pouplana L. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet 2012;28:574–581. [DOI] [PubMed] [Google Scholar]
- 77. Sánchez G, Bosch A, Pintó RM. Genome variability and capsid structural constraints of hepatitis a virus. J Virol 2003;77:452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pintó RM, Aragonès L, Costafreda MI, Ribes E, Bosch A. Codon usage and replicative strategies of hepatitis A virus. Virus Res 2007;127:158–163. [DOI] [PubMed] [Google Scholar]
- 79. D’ Andrea L, Pintó RM, Bosch A, Musto H, Cristina J. A detailed comparative analysis on the overall codon usage patterns in hepatitis A virus. Virus Res 2011;157:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bosch A, Mueller S, Pintó RM. Codon biases and viral fitness In: Ehrenfeld E, Domingo E, Roos RP, Eds. The Picornaviruses. Washington, DC: ASM Press; 2010. p 271–283. [Google Scholar]
- 81. Marin M. Folding at the rhythm of the rare codon beat. Biotechnol J 2008;3:1047–1057. [DOI] [PubMed] [Google Scholar]
- 82. Aragonès L, Guix S, Ribes E, Bosch A, Pintó RM. Fine‐tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathog 2010;6:e1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pintó RM, D'Andrea L, Pérez‐Rodriguez FJ, Costafreda MI, Ribes E, Guix S, Bosch A. Hepatitis A virus evolution and the potential emergence of new variants escaping the presently available vaccines. Future Microbiol 2012;7:331–346. [DOI] [PubMed] [Google Scholar]
- 84. Brown EA, Zajac AJ, Lemon SM. In vitro characterization of an internal ribosomal entry site (IRES) present within the 5′ nontranslated region of hepatitis A virus RNA: Comparison with the IRES of encephalomyocarditis virus. J Virol 1994;68:1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Borman AM, Le Mercier P, Girard M, Kean KM. Comparison of picornaviral IRES‐driven internal initiation of translation in cultured cells of different origins. Nucleic Acids Res 1997;25:925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Manangeeswaran M, Jacques J, Tami C, Konduru K, Amharref N, Perrella O, Casasnovas JM, Umetsu DT, DeKruyff RH, Freeman GJ, Perella A, Kaplan GG. Binding of hepatitis A virus to its cellular receptor 1 inhibits T‐regulatory cell functions in humans. Gastroenterology. 2012;142:1516–1525.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Belkaid Y, Tarbell K. Regulatory T cells in the control of host‐microorganism interactions (*). Annu Rev Immunol 2009;27:551–589. [DOI] [PubMed] [Google Scholar]
- 88. Jeong S‐H, Lee H‐S. Hepatitis A: Clinical manifestations and management. Intervirology 2010;53:15–19. [DOI] [PubMed] [Google Scholar]
- 89. Matricardi PM, Rosmini F, Panetta V, Ferrigno L, Bonini S. Hay fever and asthma in relation to markers of infection in the United States. J Allergy Clin Immunol 2002;110:381–387. [DOI] [PubMed] [Google Scholar]
- 90. Linneberg A, Ostergaard C, Tvede M, Andersen LP, Nielsen NH, Madsen F, Frølund L, Dirksen A, Jørgensen T. IgG antibodies against microorganisms and atopic disease in Danish adults: The Copenhagen Allergy Study. J Allergy Clin Immunol 2003;111:847–853. [DOI] [PubMed] [Google Scholar]
- 91. Jarvis D, Luczynska C, Chinn S, Burney P. The association of hepatitis A and Helicobacter pylori with sensitization to common allergens, asthma and hay fever in a population of young British adults. Allergy 2004;59:1063–1067. [DOI] [PubMed] [Google Scholar]
- 92. Gonzalez‐Quintela A, Gude F, Boquete O, Aguilera A, Rey J, Meijide LM, Fernandez‐Merino MC, Vidal C. Association of hepatitis A virus infection with allergic sensitization in a population with high prevalence of hepatitis A virus exposure. Allergy 2005;60:98–103. [DOI] [PubMed] [Google Scholar]
- 93. McIntire JJ, Umetsu SE, Macaubas C, Hoyte EG, Cinnioglu C, Cavalli‐Sforza LL, Barsh GS, Hallmayer JF, Underhill PA, Risch NJ, Freeman GJ, DeKruyff RH, Umetsu DT. Immunology: Hepatitis A virus link to atopic disease. Nature 2003;425:576. [DOI] [PubMed] [Google Scholar]
- 94. Gao P‐S, Mathias RA, Plunkett B, Togias A, Barnes KC, Beaty TH, Huang SK. Genetic variants of the T‐cell immunoglobulin mucin 1 but not the T‐cell immunoglobulin mucin 3 gene are associated with asthma in an African American population. J. Allergy Clin Immunol 2005;115:982–988. [DOI] [PubMed] [Google Scholar]
- 95. Chen J‐P, Zhao W‐L, He N‐H, Gui Q, Xiong J‐P, Zhou H‐M, Wang Y, Chen S, Zhou P. Association of hepatitis A exposure and TIM‐1 with childhood allergic asthma. J Asthma 2012;49:697–702. [DOI] [PubMed] [Google Scholar]
- 96. Kim HY, Eyheramonho MB, Pichavant M, Gonzalez Cambaceres C, Matangkasombut P, Cervio G, Kuperman S, Moreiro R, Konduru K, Manangeeswaran M, Freeman GJ, Kaplan GG, DeKruyff RH, Umetsu DT, Rosenzweig SD. A polymorphism in TIM1 is associated with susceptibility to severe hepatitis A virus infection in humans. J Clin Invest 2011;121:1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Binn LN, Lemon SM, Marchwicki RH, Redfield RR, Gates NL, Bancroft WH. Primary isolation and serial passage of hepatitis A virus strains in primate cell cultures. J Clin Microbiol 1984;20:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Emerson SU, Huang YK, Purcell RH. 2B and 2C mutations are essential but mutations throughout the genome of HAV contribute to adaptation to cell culture. Virology 1993;194:475–480. [DOI] [PubMed] [Google Scholar]
- 99. Yi M, Lemon SM. Replication of subgenomic hepatitis A virus RNAs expressing firefly luciferase is enhanced by mutations associated with adaptation of virus to growth in cultured cells. J Virol 2002;76:1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Day SP, Murphy P, Brown EA, Lemon SM. Mutations within the 5′ nontranslated region of hepatitis A virus RNA which enhance replication in BS‐C‐1 cells. J Virol 1992;66:6533–6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schultz DE, Honda M, Whetter LE, McKnight KL, Lemon SM. Mutations within the 5’ nontranslated RNA of cell culture‐adapted hepatitis A virus which enhance cap‐independent translation in cultured African green monkey kidney cells. J Virol 1996;70:1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cohen JI, Rosenblum B, Ticehurst JR, Daemer RJ, Feinstone SM, Purcell RH. Complete nucleotide sequence of an attenuated hepatitis A virus: Comparison with wild‐type virus. Proc Natl Acad Sci USA 1987;84:2497–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Taylor KL, Murphy PC, Asher LV, LeDuc JW, Lemon SM. Attenuation phenotype of a cell culture‐adapted variant of hepatitis A virus (HM175/p16) in susceptible New World owl monkeys. J Infect Dis 1993;168:592–601. [DOI] [PubMed] [Google Scholar]
- 104. Konduru K, Kaplan GG. Stable growth of wild‐type hepatitis A virus in cell culture. J Virol 2006;80:1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lemon SM, Murphy PC, Shields PA, Ping LH, Feinstone SM, Cromeans T, Jansen RW. Antigenic and genetic variation in cytopathic hepatitis A virus variants arising during persistent infection: Evidence for genetic recombination. J Virol 1991;65:2056–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Brack K, Frings W, Dotzauer A, Vallbracht A. A cytopathogenic, apoptosis‐inducing variant of hepatitis A virus. J Virol 1998;72:3370–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kulka M, Chen A, Ngo D, Bhattacharya SS, Cebula TA, Goswami BB. The cytopathic 18f strain of hepatitis A virus induces RNA degradation in FrhK4 cells. Arch Virol 2003;148:1275–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kulka M, Calvo MS, Ngo DT, Wales SQ, Goswami BB. Activation of the 2–5OAS/RNase L pathway in CVB1 or HAV/18f infected FRhK‐4 cells does not require induction of OAS1 or OAS2 expression. Virology 2009;388:169–184. [DOI] [PubMed] [Google Scholar]
- 109. Goswami BB, Kulka M, Ngo D, Cebula TA. Apoptosis induced by a cytopathic hepatitis A virus is dependent on caspase activation following ribosomal RNA degradation but occurs in the absence of 2’‐5’ oligoadenylate synthetase. Antiviral Res 2004;63:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wales SQ, Ngo D, Hida K, Kulka M. Temperature and density dependent induction of a cytopathic effect following infection with non‐cytopathic HAV strains. Virology 2012;430:30–42. [DOI] [PubMed] [Google Scholar]
- 111. Vallbracht A, Hofmann L, Wurster KG, Flehmig B. Persistent infection of human fibroblasts by hepatitis A virus. J Gen Virol 1984;65:609–615. [DOI] [PubMed] [Google Scholar]
- 112. Crance JM, Lévêque F, Chousterman S, Jouan A, Trépo C, Deloince R. Antiviral activity of recombinant interferon‐alpha on hepatitis A virus replication in human liver cells. Antiviral Res 1995;28:69–80. [DOI] [PubMed] [Google Scholar]
- 113. Yoshiba M, Inoue K, Sekiyama K. Interferon for hepatitis A. Lancet 1994;343:288–289. [DOI] [PubMed] [Google Scholar]
- 114. Kanda T, Yokosuka O, Imazeki F, Fujiwara K, Nagao K, Saisho H. Amantadine inhibits hepatitis A virus internal ribosomal entry site‐mediated translation in human hepatoma cells. Biochem Biophys Res Commun 2005;331:621–629. [DOI] [PubMed] [Google Scholar]
- 115. Yang L, Kiyohara T, Kanda T, Imazeki F, Fujiwara K, Gauss‐Müller V, Ishii K, Wakita T, Yokosuka O. Inhibitory effects on HAV IRES‐mediated translation and replication by a combination of amantadine and interferon‐alpha. Virol J 2010;7:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kanda T, Wu S, Kiyohara T, Nakamoto S, Jiang X, Miyamura T, Imazeki F, Ishii K, Wakita T, Yokosuka O. Interleukin‐29 suppresses hepatitis A and C viral internal ribosomal entry site‐mediated translation. Viral Immunol 2012;25:379–386. [DOI] [PubMed] [Google Scholar]
- 117. Crance JM, Biziagos E, Passagot J, Van Cuyck‐Gandré H, Deloince R. Inhibition of hepatitis A virus replication in vitro by antiviral compounds. J Med Virol 1990;31:155–160. [DOI] [PubMed] [Google Scholar]
- 118. PDR Network . Amantadine—Concise Monograph [Internet]. Montvale (NJ): PDR Network, LLC [updated 2013; cited 2013 Feb 6]. http://www.pdr.net/drugpages/concisemonograph.aspx?concise=411.
- 119. Siegl G, Eggers HJ. Failure of guanidine and 2‐(alpha‐hydroxybenzyl)benzimidazole to inhibit replication of hepatitis A virus in vitro. J Gen Virol 1982;61:111–114. [DOI] [PubMed] [Google Scholar]
- 120. Cromeans T, Fields HA, Sobsey MD. Replication kinetics and cytopathic effect of hepatitis A virus. J Gen Virol 1989;70:2051–2062. [DOI] [PubMed] [Google Scholar]
- 121. Cho MW, Ehrenfeld E. Rapid completion of the replication cycle of hepatitis A virus subsequent to reversal of guanidine inhibition. Virology 1991;180:770–780. [DOI] [PubMed] [Google Scholar]
- 122. Girond S, Crance JM, Van Cuyck‐Gandre H, Renaudet J, Deloince R. Antiviral activity of carrageenan on hepatitis A virus replication in cell culture. Res Virol 1991;142:261–270. [DOI] [PubMed] [Google Scholar]
- 123. Biziagos E, Crance JM, Passagot J, Deloince R. Inhibitory effects of atropine, protamine, and their combination on hepatitis A virus replication in PLC/PRF/5 cells. Antimicrob Agents Chemother 1990;34:1112–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Crance JM, Lévêque F, Biziagos E, Van Cuyck‐Gandré H, Jouan A, Deloince R. Studies on mechanism of action of glycyrrhizin against hepatitis A virus replication in vitro. Antiviral Res 1994;23:63–76. [DOI] [PubMed] [Google Scholar]
- 125. Fiore C, Eisenhut M, Krausse R, Ragazzi E, Pellati D, Armanini D, Bielenberg J. Antiviral effects of Glycyrrhiza species. Phytother Res 2008;22:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yamamura Y, Kawakami J, Santa T, Kotaki H, Uchino K, Sawada Y, Tanaka N, Iga T. Pharmacokinetic profile of glycyrrhizin in healthy volunteers by a new high‐performance liquid chromatographic method. J Pharm Sci 1992;81:1042–1046. [DOI] [PubMed] [Google Scholar]
- 127. Superti F, Seganti L, Orsi N, Divizia M, Gabrieli R, Panà A, Stein ML. Effect of isoflavans and isoflavenes on the infection of Frp/3 cells by hepatitis A virus. Antiviral Res 1989;11:247–254. [DOI] [PubMed] [Google Scholar]
- 128. Phillpotts RJ, Wallace J, Tyrrell DA, Freestone DS, Shepherd WM. Failure of oral 4′,6‐dichloroflavan to protect against rhinovirus infection in man. Arch Virol 1983;75:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Al‐Nakib W, Willman J, Higgins PG, Tyrrell DA, Shepherd WM, Freestone DS. Failure of intranasally administered 4′, 6‐dichloroflavan to protect against rhinovirus infection in man. Arch Virol 1987;92:255–260. [DOI] [PubMed] [Google Scholar]
- 130. Widell A, Hansson BG, Oberg B, Nordenfelt E. Influence of twenty potentially antiviral substances on in vitro multiplication of hepatitis A virus. Antiviral Res 1986;6:103–112. [DOI] [PubMed] [Google Scholar]
- 131. Cuyck‐Gandré HV, Job A, Burckhart MF, Girond S, Crance JM. Use of digoxigenin‐labeled RNA probe to test hepatitis A virus antiviral drugs. Pathol Biol 1995;43:411–415. [PubMed] [Google Scholar]
- 132. Divizia M, Venuti A, Degener AM, Perez‐Bercoff R, Panà A. Methisoprinol‐effect on the replication cycle of human hepatitis A virus. Microbiologica 1992;15:323–328. [PubMed] [Google Scholar]
- 133. Rashad AA, El‐Sabbagh OI, Baraka MM, Ibrahim SM, Pannecouque C, Andrei G, Snoeck R, Balzarini J, Mostafa A. Design, synthesis and preliminary antiviral screening of new N‐phenylpyrazole and dihydroisoxazole derivatives. Med Chem Res 2010;19:1025–1035. [Google Scholar]
- 134. el‐Sabbagh OI, Rady HM. Synthesis of new acridines and hydrazones derived from cyclic beta‐diketone for cytotoxic and antiviral evaluation. Eur J Med Chem 2009;44:3680–3686. [DOI] [PubMed] [Google Scholar]
- 135. Rashad AE, Hegab MI, Abdel‐Megeid RE, Micky JA, Abdel‐Megeid FME. Synthesis and antiviral evaluation of some new pyrazole and fused pyrazolopyrimidine derivatives. Bioorg Med Chem 2008;16:7102–7106. [DOI] [PubMed] [Google Scholar]
- 136. Shamroukh AH, Ali MA. Anti‐HAV activity of some newly synthesized triazolo[4,3‐b]pyridazines. Arch Pharm (Weinheim) 2008;341:223–230. [DOI] [PubMed] [Google Scholar]
- 137. Sayed HH, Hashem AI, Yousif NM, El‐Sayed WA. Conversion of 3‐arylazo‐5‐phenyl‐2(3H)‐furanones into other heterocycles of anticipated biological activity. Arch Pharm (Weinheim) 2007;340:315–319. [DOI] [PubMed] [Google Scholar]
- 138. Rashad AE, Mohamed MS, Zaki MEA, Fatahala SS. Synthesis and biological evaluation of some pyrrolo[2,3‐d]pyrimidines. Arch Pharm (Weinheim) 2006;339:664–669. [DOI] [PubMed] [Google Scholar]
- 139. Lall MS, Jain RP, Vederas JC. Inhibitors of 3C cysteine proteinases from Picornaviridae . Curr Top Med Chem 2004;4:1239–1253. [DOI] [PubMed] [Google Scholar]
- 140. Thibaut HJ, De Palma AM, Neyts J. Combating enterovirus replication: State‐of‐the‐art on antiviral research. Biochem Pharmacol 2012;83:185–192. [DOI] [PubMed] [Google Scholar]
- 141. Jewell DA, Swietnicki W, Dunn BM, Malcolm BA. Hepatitis A virus 3C proteinase substrate specificity. Biochemistry 1992;31:7862–7869. [DOI] [PubMed] [Google Scholar]
- 142. Malcolm BA, Lowe C, Shechosky S, McKay RT, Yang CC, Shah VJ, Simon RJ, Vederas JC, Santi DV. Peptide aldehyde inhibitors of hepatitis A virus 3C proteinase. Biochemistry 1995;34:8172–8179. [DOI] [PubMed] [Google Scholar]
- 143. Morris TS, Frormann S, Shechosky S, Lowe C, Lall MS, Gauss‐Müller V, Purcell RH, Emerson SU, Vederas JC, Malcolm BA. In vitro and ex vivo inhibition of hepatitis A virus 3C proteinase by a peptidyl monofluoromethyl ketone. Bioorg Med Chem 1997;5:797–807. [DOI] [PubMed] [Google Scholar]
- 144. Yin J, Cherney MM, Bergmann EM, Zhang J, Huitema C, Pettersson H, Eltis LD, Vederas JC, James MN. An episulfide cation (thiiranium ring) trapped in the active site of HAV 3C proteinase inactivated by peptide‐based ketone inhibitors. J Mol Biol 2006;361:673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Huang Y, Malcolm BA, Vederas JC. Synthesis and testing of azaglutamine derivatives as inhibitors of hepatitis A virus (HAV) 3C proteinase. Bioorg Med Chem 1999;7:607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hill RD, Vederas JC. Azodicarboxamides: A new class of cysteine proteinase inhibitor for hepatitis A virus and human rhinovirus 3C enzymes. J Org Chem 1999;64:9538–9546. [Google Scholar]
- 147. Lall MS, Karvellas C, Vederas JC. Beta‐lactones as a new class of cysteine proteinase inhibitors: Inhibition of hepatitis A virus 3C proteinase by N‐Cbz‐serine beta‐lactone. Org Lett 1999;1:803–806. [DOI] [PubMed] [Google Scholar]
- 148. Lall MS, Ramtohul YK, James MNG, Vederas JC. Serine and threonine beta‐lactones: A new class of hepatitis A virus 3C cysteine proteinase inhibitors. J Org Chem 2002;67:1536–1547. [DOI] [PubMed] [Google Scholar]
- 149. Ramtohul YK, Martin NI, Silkin L, James MNG, Vederas JC. Synthesis of pseudoxazolones and their inhibition of the 3C cysteine proteinases from hepatitis A virus and human rhinovirus‐14. J Chem Soc Perkin Trans 1 2002;1351–1359. [Google Scholar]
- 150. Ramtohul YK, James MNG, Vederas JC. Synthesis and evaluation of keto‐glutamine analogues as inhibitors of hepatitis A virus 3C proteinase. J Org Chem 2002;67:3169–3178. [DOI] [PubMed] [Google Scholar]
- 151. Huitema C, Zhang J, Yin J, James MNG, Vederas JC, Eltis LD. Heteroaromatic ester inhibitors of hepatitis A virus 3C proteinase: Evaluation of mode of action. Bioorg Med Chem 2008;16:5761–5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wang H‐M, Liang P‐H. Picornaviral 3C protease inhibitors and the dual 3C protease/coronaviral 3C‐like protease inhibitors. Expert Opin Ther Pat 2010;20:59–71. [DOI] [PubMed] [Google Scholar]
- 153. Kanda T, Kusov Y, Yokosuka O, Gauss‐Müller V. Interference of hepatitis A virus replication by small interfering RNAs. Biochem Biophys Res Commun 2004;318:341–345. [DOI] [PubMed] [Google Scholar]
- 154. Kusov Y, Kanda T, Palmenberg A, Sgro J‐Y, Gauss‐Müller V. Silencing of hepatitis A virus infection by small interfering RNAs. J Virol 2006;80:5599–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kanda T, Zhang B, Kusov Y, Yokosuka O, Gauss‐Müller V. Suppression of hepatitis A virus genome translation and replication by siRNAs targeting the internal ribosomal entry site. Biochem Biophys Res Commun 2005;330:1217–1223. [DOI] [PubMed] [Google Scholar]
- 156. Debing, Y , Kaplan, GG , Neyts, J , Jochmans, D . Rapid and convenient assays to assess potential inhibitory activity on in vitro hepatitis A replication. Antiviral Res 2013;98:325–331. [DOI] [PubMed] [Google Scholar]
- 157. Van der Schaar HM, Van der Linden L, Lanke KHW, Strating JRPM, Pürstinger G, De Vries E, de Haan CAM, Neyts J, van Kuppeveld FJM. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res 2012;22:1576–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Purcell RH, Emerson SU. Animal models of hepatitis A and E. ILAR J 2001;42:161–177. [DOI] [PubMed] [Google Scholar]
- 159. Feigelstock DA, Thompson P, Kaplan GG. Growth of hepatitis A virus in a mouse liver cell line. J Virol 2005;79:2950–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hornei B, Kämmerer R, Moubayed P, Frings W, Gauss‐Müller V, Dotzauer A. Experimental hepatitis A virus infection in guinea pigs. J Med Virol 2001;64:402–409. [DOI] [PubMed] [Google Scholar]