

Abstract
This review highlights ten “hot topics” in current antiviral research: (i) new nucleoside derivatives (i.e., PSI‐352938) showing high potential as a direct antiviral against hepatitis C virus (HCV); (ii) cyclopropavir, which should be further pursued for treatment of human cytomegalovirus (HCMV) infections; (iii) North‐methanocarbathymidine (N‐MCT), with a N‐locked conformation, showing promising activity against both α‐ and γ‐herpesviruses; (iv) CMX001, an orally bioavailable prodrug of cidofovir with broad‐spectrum activity against DNA viruses, including polyoma, adeno, herpes, and pox; (v) favipiravir, which is primarily pursued for the treatment of influenza virus infections, but also inhibits the replication of other RNA viruses, particularly (‐)RNA viruses such as arena, bunya, and hanta; (vi) newly emerging antiarenaviral compounds which should be more effective (and less toxic) than the ubiquitously used ribavirin; (vii) antipicornavirus agents in clinical development (pleconaril, BTA‐798, and V‐073); (viii) natural products receiving increased attention as potential antiviral drugs; (ix) antivirals such as U0126 targeted at specific cellular kinase pathways [i.e., mitogen extracellular kinase (MEK)], showing activity against influenza and other viruses; and (x) two structurally unrelated compounds (i.e., LJ‐001 and dUY11) with broad‐spectrum activity against virtually all enveloped RNA and DNA viruses. © 2012 Wiley Periodicals, Inc. Med. Res. Rev., 00, No. 0, 1–34, 2012
Keywords: PSI‐352938, cyclopropavir, N‐MCT, CMX001, favipiravir, antiarenaviral, antipicornaviral, U0126, LJ‐001, dUY11, natural products
1. INTRODUCTION
The search for new antivirals has proceeded unabatedly. In previous reviews on “stories on antiviral drug discovery,” I have reviewed various subjects in which I was personally involved.1, 2, 3, 4, 5 In this review, hopefully the first of a new series, I will address “hot topics” in areas of antiviral research in which over the past few years significant progress has been made requiring due attention. Most of the compounds covered are still in the preclinical stage. Compounds that have successfully completed phase II and III clinical trials and progressing to approval (or have already been approved) are not subject of this review. Instead, this article is based on compounds in the pipeline that offer attractive perspectives as future antiviral drugs. In this sense, the present survey is a little arbitrary in the selection of the topics, which depends at least in part on my own prejudices and a number of other factors, not at least my own acquaintance with the subject.
An important impetus for initiating this review was based on the 24th ICAR Abstract Issue (Antiviral Research, vol. 90: A21–A78, 2011) covering the presentations at the 24th ICAR (International Conference on Antiviral Research), held in Sofia (Bulgaria) on May 8–11, 2011. Unfortunately, I could not attend the Conference, but guided by a certain devotion, I went through the Abstract Issue and spotted a number of interesting leads which I would like to reflect on in the present article: (i) nucleoside analogues, targeted at hepatitis C virus (HCV) NS5B RNA polymerase, such as PSI‐352938;6 (ii) cyclopropavir for the treatment of human cytomegalovirus (HCMV) infections;7 (iii) North‐methanocarbathymidine (N‐MCT) for the treatment of α‐herpesvirus [herpes simplex virus type 1 (HSV‐1) and type 2 (HSV‐2)] and γ‐herpesvirus [Epstein‐Barr virus (EBV) and Kaposi's sarcoma‐associated herpesvirus (KSHV) infection];8 (iv) CMX001 (1‐O‐hexadecyloxypropyl cidofovir) for the treatment of a broad variety of DNA virus infections;9 (v) favipiravir (T‐705) for the treatment of influenza virus infections and various other RNA virus infections;10 (vi) the increasing number of compounds found effective against arenaviruses;10, 11 (vii) new picornavirus (i.e., rhinovirus) inhibitors;12, 13 (viii) natural products with antiviral activity such as aglycoristocetin derivatives14 and tricin (4′,5,7‐trihydroxy‐3′,5′‐dimethoxyflavone);15 (ix) mitogen extracellular kinase (MEK) inhibitors, such as U0126, acting at a cellular target, and effective against influenza and other viruses;16 and (x) two unique sets of compounds which should be effective against virtually all enveloped DNA and RNA viruses.17
Taken together, compounds (i) through (x) cover all major viral pathogens: polyoma‐, adeno‐, herpes‐, pox‐, picorna‐, flavi‐, arena‐, myxo‐, bunya‐, and retroviruses, and particularly HSV, HCMV, hemorrhagic fever, influenza, and human immunodeficiency virus (HIV).
2. NUCLEOSIDE ANALOGUES TARGETED AT THE HCV NS5B POLYMERASE
2′‐Deoxy‐2′‐fluorocytidine (FdC) could be considered as the first nucleoside analogue (Fig. 1) found to inhibit the hepatitis C virus (HCV) replicon in cell culture, its 5′‐triphosphate inhibiting the NS5B polymerase.18 Introduction of a methyl group at the C‐2′ position, as in β‐d‐2′‐fluoro‐2′‐C‐methylcytidine (Fig. 1), conferred a similar potency as FdC in the HCV replicon assay.19 Although the 2′‐C‐methylcytidine (Fig. 1) demonstrated somewhat lesser activity against HCV than 2′‐deoxy‐2′‐fluoro‐2′‐C‐methylcytidine,19 the prodrug of 2′‐C‐methylcytidine, its 3′‐O‐valine ester, NM283 (valopicitabine) (Fig. 1) was developed (but later dropped) for phase II clinical trials.20
Figure 1.
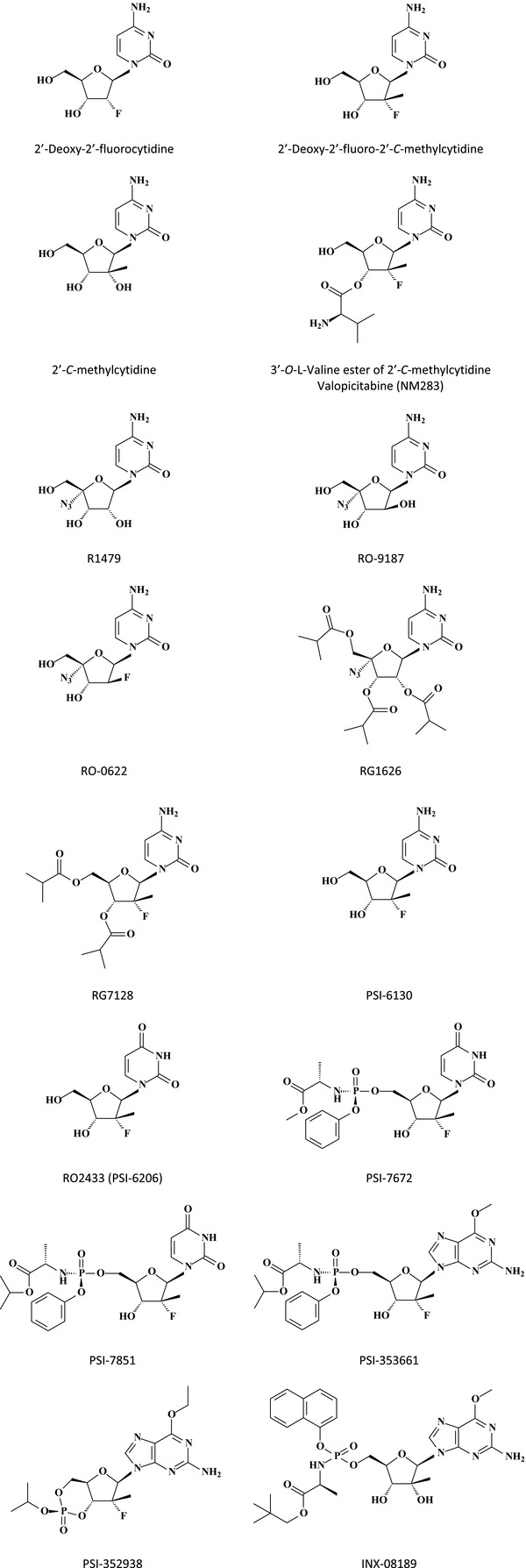
Structures of nucleoside analogues and their prodrugs active against HCV NS5B polymerase.
Introduction of a 4′‐azido group (Fig. 1), as in R1479,21, 22 RO‐9187, RO‐0622,23 4′‐azido‐2′‐deoxy‐2′,2′‐difluorocytidine, and 4′‐azido‐2′‐deoxy‐2′‐fluoroarabinocytidine significantly increased the potency in the HCV replicon system [i.e., with 50% effective concentration (EC50) values of 66 and 24 nM for the latter two compounds].24 The 2′,3′,5′‐triisobutyrate ester prodrug of 4′‐azido‐2′‐C‐methylcytidine (RG1626) (Fig. 1) has been (but is no longer) in development as inhibitor of HCV, according to Reddy et al.25
According to Reddy et al.,25 RG7128 (Fig. 1) is currently in phase IIb clinical trials. RG7128 is the 3′,5′‐diisobutyrate ester prodrug of PSI‐6130 (Fig. 1); in a 4‐week combination study with the current standard of care (soc) for HCV infection (i.e., combination of pegylated interferon‐α and ribavirin), RG7128 demonstrated efficacy in genotype 1, 2, and 3 patients, and, therefore, represents the first direct acting antiviral to show pan‐genotype HCV coverage in the clinic.
PSI‐6130 (β‐d‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methylcytidine) is a potent inhibitor of HCV replication in the HCV replicon system [for an efficient, diastereoselective synthesis of PSI‐6130, see Wang et al.26]. To be active at the NS5B RNA polymerase level, PSI‐6130 must be phosphorylated successively to its 5‐mono‐, di‐, and triphosphate, the final, active metabolite which acts as a chain terminator of the NS5B RNA polymerase.27 However, in addition to the 5′‐triphosphate of PSI‐6130, the 5′‐triphosphate of its uridine counterpart, β‐d‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methyluridine (RO2433) (Fig. 1), is also formed,28 and this second metabolite of PSI‐6130 is also a potent inhibitor of the HSV NS5B RNA polymerase.29 Deamination of PSI‐6130 occurs at the 5′‐monophosphate level.29 RO2433 (Fig. 1) itself is inactive in the HCV replicon system, but its phosphoramidate prodrug PSI‐7672 (Fig. 1) is active in this system, where it is released as its 5′‐monophosphate which can then be further phosphorylated intracellularly to the 5′‐di‐ and 5′‐triphosphate.29
From R02433 (a uridine homolog, which was renamed PSI‐6206), another phosphoramidate prodrug, PSI‐7851 (Fig. 1) was prepared, which proved to be pan‐genotype inhibitor of HCV replication, with, however, lesser activity against the S282T replicon mutant (while the S96T/N142T mutation remained fully susceptible to PSI‐7851).30 Inside the cell, PSI‐7851 is converted to the 5′‐monophosphate of PSI‐6206, and in this sense, PSI‐7851 can be considered as a prodrug of PSI‐7411.31 PSI‐7851 is, in fact, a mixture of two diastereoisomers, PSI‐7976 (Rp diastereomer) and PSI‐7977 (Sp diastereomer), the latter being the more active inhibitor of HCV RNA replication in the replicon system.31, 32 PSI‐7977 is currently being evaluated in phase II clinical trials.33
From 2′‐deoxy‐2′‐fluoro‐2′‐C‐methylguanosine‐5′‐monophosphate a phosphoramidate prodrug was prepared, PSI‐353661 (Fig. 1), which proved highly active against genotypes 1a, 1b, and 2a HCV RNA replication in the replicon system, genotype 1a and 2a infectious virus production, and HCV replicons harboring the NS5B S282T or S96T/N142T mutations.33 PSI‐353661 [(S)‐2‐{(S)‐[(1R,4R,5R)‐5‐(2‐amino‐6‐methoxy‐purin‐9‐yl)‐4‐(R)‐fluoro‐3‐hydroxy‐4‐methyl‐tetrahydro‐furan‐2‐yl‐methoxy]‐phenoxy‐phosphonylamino}‐propionic acid isopropyl ester] is the more active isomer of a mixture (PSI‐352879) of two diastereoisomers.
Recently, the 2′‐deoxy‐2′‐α‐fluoro‐2′‐β‐C‐methyl 3′,5′‐cyclic phosphate prodrug PSI‐352938 (Fig. 1) was described. Based on its antipan‐genotype HCV activity (EC50 for the replicons in the 0.1–0.2 μM range) also including activity against S282T or S96T/N142T mutations, ability to produce high intracellular 5′‐triphosphate levels both in vitro and in vivo, the synthetic accessibility of a single diastereomer,34 PSI‐352938 was selected for further development.25, 35 The compound is currently in phase I clinical trials.
Supportive of continued development as a clinical candidate for the treatment of HCV infection is INX‐08189 or (2S)‐neopentyl 2‐(2R,3R,4R)‐5‐(2‐amino‐6‐methoxy‐9H‐purin‐9‐yl)‐3,4‐dihydroxy‐4‐methyltetrahydrofuran‐2‐yl)(methoxy)(naphthalen‐1‐yloxy)(phosphorylamino) propanoate, an aryl‐phosphoramidate of 6‐O‐methyl‐2′‐C‐methyl guanosine; its EC50 for 1a, 1b, and 2a HCV replicons is around 0.01 μM.36 The separated diastereomers of INX‐08189 were shown to have similar activity in the replicon assay. INX‐08189 has completed investigational new drug (IND) enabling studies and has progressed to human clinical trials for the treatment of chronic HCV infection.37
3. CYCLOPROPAVIR
Cyclopropavir (Fig. 2) can be viewed as structurally related to acyclovir, ganciclovir, and penciclovir in that the acyclic side chain of the latter has been replaced by a methylenecyclopropane. Its antiviral properties have been known since a decade or so.38 Cyclopropavir has proven to be effective against herpesviruses.39, 40 It has proven particularly effective in animal models for cytomegalovirus (CMV) infection,41 including SCID‐hu (where SCID is severe combined immunodeficient) mouse models for HCMV.42 In addition, cyclopropavir has also been found effective in vitro against human herpesvirus type 6 (HHV‐6), like HCMV, a β‐herpesvirus.43
Figure 2.

Structures of cyclopropavir, valcyclopropavir, and the cyclic phosphonate of cyclopropavir.
Currently available drugs for the treatment of HCMV infections are ganciclovir, cidofovir, foscarnet, and valganciclovir (the valine ester of ganciclovir). Esterification of ganciclovir with l‐valine increased its oral bioavailability, as was previously also shown for acyclovir (valaciclovir) and would be later shown for the valine esters of 2′‐deoxy‐l‐cytidine (valtorcitabine) and 2′‐C‐methylcytidine (valopicitabine).20 Also, the l‐valine ester of cyclopropavir (valcyclopropavir) (Fig. 2) has been constructed.44 In mice, oral bioavailability of valcyclopropavir was 95%.
Against all those HCMV strains against which cyclopropavir was compared with ganciclovir, cyclopropavir displayed an EC50 that was five‐ to tenfold lower than that of ganciclovir.45 This was also the case for UL97 mutations that affected cyclopropavir and ganciclovir susceptibility. In fact, purified pUL97 phosphorylated cyclopropavir (to its monophosphate) 45‐fold more extensively than ganciclovir.46 This phosphorylation is stereoselective.
Cyclopropavir monophosphate is converted successively to its diphosphate and its triphosphate (the latter is the active metabolite interacting with the HCMV pUL54 DNA polymerase) by a single cellular enzyme, guanosine monophosphate kinase (GMPK), once the monophosphate is formed by a virally encoded kinase.47
While new and safe anti‐HCMV drugs are eagerly awaited, the fact that cyclopropavir has excellent activity against HCMV, combined with its specific phosphorylation by the viral enzyme pUL97 would seem to justify further development of the compound and its valine ester (valcyclopropavir), and eventually that of its phosphonate and cyclic phosphonate as well.48 It is curious, in this regard, that while both the phosphonate and cyclic phosphonate (Fig. 2) were equally active against HCMV, only the phosphonate, but not the cyclic phosphonate, was active against the γ‐herpesvirus, EBV.48
4. NORTH‐METHANOCARBATHYMIDINE (N‐MCT)
N‐methanocarbathymidine (N‐MCT) with a pseudosugar rigidly fixed in the Northern conformation (1R,2S,4S,5S)‐1‐(hydroxymethyl)‐2‐hydroxy‐4‐(5‐methyl‐2,4(1H,3H)‐dioxopyrimidin‐1‐yl)bicyclo[3.1.0]hexane (Fig. 3) was first synthesized by Marquez et al. in 1996.49 It was found to exhibit potent antiviral activity against HSV‐1 and HSV‐2. This was further corroborated in subsequent studies.50, 51, 52 N‐MCT appeared to be phosphorylated to the mono‐ and diphosphate by the HSV‐encoded thymidine kinase, and to inhibit the viral DNA polymerase through its triphosphate metabolite.50 Kinases would prefer substrates that adopt the S sugar conformation, whereas cellular DNA polymerases almost exclusively incorporated the triphosphate of the locked N conformer, notwithstanding the presence of higher triphosphate levels of the S‐conformer S‐MCT (Fig. 3).53
Figure 3.

Structures of carbocyclic thymidine, North‐methanocarbathymidine (N‐MCT), South‐methanocarbathymidine (S‐MCT), and D‐(+)‐iso‐methanocarbathymidine (D‐(+)‐iso‐MCT).
Antiviral activity against vaccinia virus was first shown with carbocyclic thymidine (Fig. 3).54 N‐MCT was found to be highly effective against orthopoxvirus infections in vivo (mice),55, 56 although the lung, nasal, brain virus reductions it achieved for vaccinia virus infection (1HD strain) were not nearly to the same extent as for cidofovir.57
Vaccinia virus lacking the F2L gene encoding functional deoxyuridine triphosphatase (dUTPase, that catalyzes the conversion of dUTP to dUMP) continued to replicate well in vitro and in vivo, but proved hypersensitive to the inhibitory effect of N‐MCT.58
As to its activity spectrum, N‐MCT is not only active against the α‐herpesviruses HSV‐1, HSV‐2, but also against the γ‐herpesviruses EBV55 and KSHV.59 Apparently, N‐MCT inhibits lytic KSHV DNA synthesis through its triphosphate metabolite produced in KSHV‐infected cells expressing a virally encoded thymidine kinase.59
Recently, a “greener” enantioselective synthesis of N‐MCT from 2‐deoxy‐d‐ribose has been reported60 and a new MCT distinct from N‐MCT, namely d‐(+)‐iso‐MCT (Fig. 3), has been described as a high‐affinity substrate for HSV‐1 thymidine kinase.61 N‐MCT shows potent anti‐HIV activity in human osteosarcoma (HOS) cells modified so as to contain, and express, the HSV‐1‐encoded thymidine kinase. Possible anti‐HIV activity of d‐(+)‐iso‐MCT may have been masked by cytotoxicity.61
N‐MCT represents an interesting conformational concept.62 Its therapeutic utility, however, remains to be demonstrated. As there are, at present, no therapeutic options for EBV and KSHV infections, these infections may well represent unique opportunities for the clinical potential of N‐MCT to be further explored.
5. CMX001 (HDP‐CDV)
CMX001 is the 1‐O‐hexadecyloxypropyl (HDP) prodrug of the acyclic nucleoside phosphonate cidofovir (CDV), representing an oral version of cidofovir with reduced (nephro)toxicity.63 The active form of HDP‐CDV (CMX001) is cidofovir (Fig. 4), which explains why, in principle, CMX001 should possess an activity spectrum similar to that of cidofovir, thus, encompassing DNA viruses, herpes‐, adeno‐, polyoma‐, and poxviruses. For all these indications, CMX001 could, given its oral bioavailability and safer (nephro)toxicity profile, replace cidofovir in future therapeutic regimens.
Figure 4.

Structures of cidofovir (CDV) and HDP‐CDV (CMX001).
Against disseminated or central nervous system (CNS) HSV infections, firm Chimerix (CMX) appears to be superior to acyclovir.64 Orally administered HDP‐CDV is four‐ to eightfold more active, on a molar basis, than intraperitoneally administered cidofovir against HCMV infection in SCID/hu mice.65 HDP‐cidofovir exhibits multiple‐log enhancement of antiviral activity against both HCMV and HSV replication in vitro,66 and oral treatment with HDP‐CDV is as effective as parenteral CDV for the treatment of murine CMV infections.67 As long awaited, oral HDP‐CDV improves the outcome of CMV infection in a congenital model for CMV infection in pregnant guinea pigs.68
Ether lipid esters of cidofovir, such as HDP‐CDV, are much more potent than cidofovir against adenovirus replication in vitro.69 HDP‐CDV (CMX001) suppressed adenovirus‐induced mortality in immunosuppressed hamsters, a powerful model to evaluate the efficacy of anti‐adenovirus agents.70 A first case of the successful eradication of disseminated adenovirus infection by CMX001 has been recently reported in a severely immunosuppressed pediatric stem cell transplant recipient.71
In line of earlier observations on the inhibitory effects of cidofovir against murine and primate polyomaviruses,72 cidofovir was shown to inhibit polyomavirus BK replication in human renal tubular cells.73 Either lipid esters of cidofor such as HDP‐CDV were then shown to inhibit polyomavirus BK replication in vitro at a 3 log10‐fold lower concentration than cidofovir.74 CMX001 proved highly effective in inhibiting polyomavirus BK replication in primary human renal tubular epithelial cells.75 This points to the potential of CMX001 in the treatment of BK virus nephropathy that is seen in 1–10% of kidney transplant recipients.
The JC polyomavirus infects human oligodendrocytes leading to the development of progressive multifocal leukoencephalopathy. CMX001 was shown to suppress polyomavirus JC in human fetal brain SVG cell cultures76 [the SVG cell line is derived from primary human brain cells transfected with Simian virus 40 (SV40) and expressing SV40 T antigen in these cells]. CMX001 was recently shown to inhibit polyomavirus JC replication in human brain progenitor‐derived astrocytes,77 and a case of progressive multifocal leukoencephalopathy (accompanied by idiopathic CD4+ lymphocytopenia) responded successfully to treatment with CMX001.78
Alkoxyalkyl esters of cidofovir, including HDP‐CDV, have been most intensively pursued for inhibition of orthopoxvirus replication.79 They were first proven active in vitro against vaccinia virus and cowpox,80 before their in vivo activity against the same viruses was demonstrated.81 Their efficacy was demonstrated in a lethal mousepox model (based on lethal, aerosol ectromelia virus infection in A/NCR mice).82 In an improved model for evaluating antipoxvirus therapies, based on the use of C57BL/6 mice infected with mousepox (ectromelia) virus, CMX001 proved more efficacious than in A/NCR mice.83
Ectromelia virus infection of mice serves as a model to support the licensure of antiorthopoxvirus therapeutics based on the “animal efficacy rule” because of the genetic similarity of ectromelia virus to variola and monkeypox viruses.84 In the lethal mousepox model, complete protection against mortality was achieved when administration of CMX001 was delayed until as late as 5 days postinfection.85 A single dose of 25 mg/kg of CMX001 administered 4 or 5 days postinfection sufficed to be effective in the mousepox model.86
CMX001 is also efficacious in the treatment of monkeypox virus infection in STATI‐deficient C57BL/6 mice.87 Against the highly virulent, interleukin‐4 expressing ectromelia virus recombinant, CMX001 may afford the highest efficacy when combined with another antiviral drug, ST‐246.88 The pre‐ and postexposure prophylactic efficacy of CMX001 has also been demonstrated in rabbits infected with rabbitpox virus.89, 90
Compared with CDV, CMX001 would have the advantages that it could be administered orally (whereas CDV needs to be administered intravenously) and, unlike CDV, CMX001 may not lead to nephrotoxicity.91 Furthermore, various additional alkoxyalkyl esters of CDV have been described, that is, 1‐O‐octadecyl‐2‐O‐benzyl‐sn‐glycero‐3‐CDV (ODBG‐CDV),92, 93 which may be worth further exploring for potential advantages over HDP‐CDV (CMX001).
6. FAVIPIRAVIR (T‐705)
Favipiravir (T‐705) (Fig. 5) is currently in clinical trials in Japan (phase III) and the United States (phase II) for the treatment of influenza virus infections (as mentioned by Buys et al.94). The in vitro and in vivo activities of T‐705 (6‐fluoro‐3‐hydroxy‐2‐pyrazinecarboxamide) were first reported in 2002 by Furuta et al.95 The compound showed potent inhibitory activity against influenza A, B, and C viruses, some activity against picorna‐ and paramyxoviruses, but no activity whatsoever against DNA viruses. From its structure, it was immediately clear that the mode of action of T‐705 had to be different from that of the M2 ion channel inhibitors amantadine and rimantadine, as well as that of the neuraminidase inhibitors zanamivir and oseltamivir.
Figure 5.

Structures of T‐705, T‐1105, and T‐1106, and of T‐705 metabolites.
This mechanism of action was further addressed by Furuta et al.96 Within the cells, T‐705 would be converted to T‐705 ribofuranosyl monophosphate by a purine (adenine, hypoxanthine/guanine) phosphoribosyl transferase. Two phosphorylations would then generate T‐705–4‐ribofuranosyl‐5′‐triphosphate (T‐705 RTP), the active metabolite of T‐705.96 That the latter is indeed the active metabolite of T‐705 was also shown in cells infected with the highly pathogenic influenza A (H5N1) virus.97, 98
Meanwhile, Sidwell et al.99 had shown that T‐705, when orally administered at dosages from 30 to 300 mg/kg (once or twice daily), prevented death due to a lethal avian influenza A (H5N1) virus infection in mice. In vitro, favipiravir proved antivirally active against influenza A (H1N1) virus strains that were resistant to antiviral drugs such as oseltamivir,100 and, in vivo, synergistic effects were obtained with favipiravir combined with oseltamivir against influenza A (H5N1) infection.101
The carboxamide group present in favipiravir (T‐705) is reminiscent of the carboxamide present in ribavirin. This carboxamide group is also present in T‐1105 and T‐1106, two compounds that are structurally related to T‐705 (Fig. 5). All the compounds share, with ribavirin, a broad‐spectrum activity against various RNA viruses. Thus, T‐1105 shows activity against foot‐and‐mouth disease virus (FMDV) and, as it can be administered through food, it could be a powerful tool to control foot‐and‐mouth disease in pigs.102 T‐1106 has proven efficacious against Yellow fever virus (YFV) in a hamster model of YFV infection103 and is also active against bovine viral diarrhea virus (BVDV),102 another flavivirus that could be considered as a surrogate virus for hepatitis C virus (HCV).
T‐705 is also efficacious in the YFV hamster model, although the dose of T‐705 required for efficacy in hamsters is higher than that of T‐1106 required for efficacy. Yet, T‐705 improved the disease parameters in YFV‐infected hamsters, which may indicate its potential utility in the treatment of YFV infection in humans.104
In rodents (mice or hamsters), orally administered T‐705 is also effective against West Nile virus (WNV),105 another flavivirus related to YFV. Whether T‐705 or any of its analogues T‐1105 or T‐1106 would be effective against two other mosquito borne flaviviruses, dengue virus and Japanese encephalitis virus, remains an intriguing possibility worth exploring. Western equine encephalitis virus (WEEV), an alphavirus belonging to the broad family of the flaviviridae, would seem to respond to T‐705 treatment.106
The antiviral activity spectrum of T‐705 extends to arenaviruses: that is, Junin virus (JUNV), Pichinde virus, Tacaribe virus, Machupovirus (MACV), and Guanarito virus (GTOV) replication in cell culture could be inhibited by T‐705, and as previously noted for its inhibitory effect on influenza virus, the antiarenavirus activity of T‐705 could be reversed by the addition of purine, but not pyrimidine nucleosides.107, 108 T‐705 also proved efficacious against Pichinde virus infection in hamsters, even when treatment was begun after the animals fell ill, the day before the animals began to succumb to the disease.109 Thus, for the treatment of late stage arenaviral hemorrhagic fever, T‐705 could be considered as an alternative option to ribavirin. This would seem very important for severe arenavirus infections, such as Lassa fever, in humans.
Besides arenavirus infections, bunyaviruses, that is, Punta Toro, La Crosse, Rift Valley fever, and sandfly fever virus, have also proven sensitive to inhibition by T‐705 in vitro, and, for Punta Toro virus, also in vivo (mice and hamsters).107 In hamsters infected with Punta Toro virus, T‐1106 was more efficacious than T‐705, but in mice, T‐705 was the more effective.110
Of the bunyaviridae, the hantaviruses Dobrava and Maporal were found to be sensitive to the inhibitory effects of T‐705.94 Maporal virus is phylogenetically similar to Andes virus, the principal cause of hantavirus cardiopulmonary syndrome (HCPS) in Argentina. It would now seem mandatory to further explore the efficacy of T‐705 in the suitable hantavirus models in hamsters and/or mice.
In conclusion, T‐705 is an intriguing new antiviral compound that should be further explored not only for its clinical potential in the prevention/therapy of influenza virus infections, but also for its broad activity spectrum against (−) and (+) RNA strand viruses, its target of action (RNA‐dependent RNA polymerase for all these RNA viruses ?), and its structure‐activity relationship and pharmacodynamics relative to that of related structural analogues such as T‐1105 and T‐1106.
7. ARENAVIRUS INHIBITORS
Since McCormick's pioneering paper in 1986111 on the effective therapy of Lassa fever with ribavirin, ribavirin has remained the only antiviral drug available for the treatment of arenavirus infections. The inhibitory effect of ribavirin on arenaviruses might, at least partially, be attributed to lethal mutagenesis.112 In recent years, antiarenaviral drug development has received increasing attention,113, 114 one of the new antiarenaviral drug candidates being favipiravir (T‐705) (see preceding section).
The Tacaribe arenavirus infection model has been employed to explore the antiviral potential of novel aristeromycin analogues,115 and new imidazo[2,1‐b]thiazole carbohydrate derivatives have been found to inhibit the replication of the Argentine hemorrhagic fever virus Junin.116 Small interfering (si)RNAs targeting the conserved RNA termini of Lassa fever virus117 offer therapeutic potential and so do interferon‐α and interferon‐γ,118 whereas antimicrobial cationic peptides were found active against JUNV as well as HSV‐1 and HSV‐2.119
Lassa fever virus is restricted by the bone marrow stromal antigen 2 (BST‐2), also called tetherin, which besides inhibiting the release of HIV‐1, also inhibits the egress of arenaviruses.120 Tetherin could thus be considered an innate immunity strategy to suppress arenavirus replication.
The Lassa fever viral nucleoprotein (NP) is endowed with several functions (i.e., a 3′‐5′ exoribonuclease a the C‐domain, involved in suppressing interferon induction, and a m7GpppN cap‐binding site at the N‐domain, protecting the cap against cap snatching), which may serve as potential targets for chemotherapeutic intervention.121
One of the most fascinating targets for novel antiarenavirus strategies is the arenavirus envelope glycoprotein complex (GPC) processing by the cellular site 1 protease (S1P), which is strictly required for the production of infectious progeny and cell‐to‐cell virus propagation.122 The small molecule PF‐429242 (Fig. 6) was recently reported to be a potent S1P inhibitor in vitro (cell‐based assay).123, 124 This correlated with the compound's potent antiviral activity against Lassa fever virus in cell culture.125
Figure 6.

Arenavirus inhibitors targeted at viral entry.
Of the small molecular weight inhibitors targeted at arenaviral entry, in particular the viral glycoprotein GP2, the first to be announced chosen for drug development was ST‐294 (Fig. 6).126 The arenavirus GP is synthesized as a single polypeptide that undergoes posttranslational processing to yield the mature virion glycoproteins GP1 and GP2. GP1 is involved in receptor binding, whereas GP2 is similar to the fusion proteins of other enveloped viruses such as retroviruses, paramyxoviruses, and filoviruses. A series of small molecules, including 17C8 (Fig. 6) have been identified to be targeted at the arenavirus GP.127
These small molecule entry inhibitors (including ST‐294 and ST‐193) interact with the envelope GPC of arenaviruses so as to stabilize the complex against pH‐induced activation of membrane fusion in the endosome.128 Both ST‐294 and ST‐193 (Fig. 6) inhibit the pH‐induced dissociation of the receptor‐binding GP1 subunit from GPC. ST‐294 and ST‐193 thus stabilize the GPC against pH‐acidification which would otherwise initiate the fusion process.129
The antiviral potency of ST‐193 (Fig. 6) against Lassa virus and other arenavirus pseudotypes is within the range of 0.2–12 nM. The sensitivity to ST‐193 is dictated by a segment of about 30 amino acids within the GP2 subunit. This region includes the carboxy‐terminal region of the ectodomain of the transmembrane domain of the envelope protein.130
The small molecule arenavirus inhibitor ST‐193 was compared with ribavirin in a guinea pig model for Lassa virus infection, and found to increase the survival rate from 0% (control, ribavirin) to 62.5% (ST‐193).
What now remains to be established is how ST‐193 compares with other arenavirus inhibitors such as favipiravir (T‐705) and PF‐429242, both in terms of efficacy and safety, and whether its efficacy can be extrapolated from Lassa to other arenavirus infections such as Junin, Machupo, Sabia, and Guanarito.
8. PLECONARIL AND ANTIPICORNAVIRUS AGENTS REVISITED
Although picornaviruses encompass a number of important human pathogens, including the enteroviruses polio, Coxsackie A and B, and echo, and rhinoviruses, there is still no single antipicornavirus agent approved for clinical use. Yet, a wealth of compounds has been shown to inhibit picornaviruses, including, especially for Coxsackie B virus, a number of natural products (see Section 8.). Prominent among the currently envisaged antipicornavirus therapies131 are the original Winthrop compounds (disoxaril derivatives), which engage in a specific binding to the viral capsid.
Pleconaril (Fig. 7), the prototype of this class of compounds,132 had been shown to inhibit the replication of various entero‐ and rhinoviruses133 and had demonstrated tentative efficacy against potentially life‐threatening enterovirus infections,134 before, in 2002, it was rejected by the US FDA for the treatment of common cold.135 Several double‐blind placebo‐controlled trials with pleconaril in infants with enterovirus meningitis,136 and adults with common cold137 were conducted, and Pevear et al.138 showed that the efficacy of pleconaril in reducing the duration and severity of common cold symptoms (when it was administered within 24 hr of symptom onset) was related to the virus susceptibility to pleconaril. De Palma et al.139 in their review mentioned that a phase II double‐blind, placebo‐controlled trial to evaluate the effects of pleconaril nasal spray on common cold symptoms and asthma exacerbations following rhinovirus exposure was completed in 2007, but that results of this trial have not yet been divulged.
Figure 7.

Structures of pleconaril, BTA‐798, and V‐073.
Pleconaril has in the meantime been found to shorten the course of illness, compared to placebo, in patients with enteroviral meningitis, but the benefit appeared to be modest after adjusting for confounding variables.140 Pleconaril did not have any effect on viral replication in a common variable immunodeficiency (CVID) patient with parechovirus‐associated enteropathy.141 New pleconaril derivatives have been reported to be active against pleconaril‐resistant Coxsackie B virus.142
Meanwhile, new benzimidazole derivatives have been synthesized and found active against Coxsackie B3 virus,143 and a small interfering (si)RNA has been shown to block Coxsackie B virus replication.144 Synergistic activity against Coxsackie B3 virus was obtained if the siRNA was combined with the soluble Coxsackievirus‐adenovirus receptor.145 A new class of compounds, which be structurally described as 9‐arylpurines, was recently described to inhibit a variety of enteroviruses, that is, Coxsackie A16, A21, A24, Coxsackie B3, and echovirus 9, at low micromolar concentrations.146
In earlier papers dating from 2003 to 2004, we have demonstrated that Coxsackie B3 virus‐induced myocarditis in mice can be inhibited by mycophenolate mofetil,147 2‐(3,4‐dichlorophenoxy)‐5‐nitrobenzonitrile,148 and the interferon inducer ampligen [poly(I).poly(C12,U)].149
Three antipicornavirus agents are currently in clinical development: pleconaril, BTA‐798, and V‐073150 (Fig. 7). Pleconaril is under development as an oral formulation for the treatment of rhinovirus infections in high‐risk patients with chronic lung diseases.139, 151, 152 Oral BTA‐798 is in phase II trial for symptomatic human rhinovirus infection in asthmatic adults.139 V‐073 is under further scrutiny for the treatment of poliovirus infections. Their EC50 values are, respectively, for BTA‐798 0.02 μM against human rhinovirus type 14 and 0.2 μM against enterovirus 71, 0.06 μM for pleconaril against human rhinovirus type 14, and 0.026 μM for V‐073 against poliovirus type 1.150 Pleconaril, BTA‐798 and V‐073 behave as capsid binders. They could, if required, be combined to provide an additive to slightly synergistic antiviral effect.150
As reviewed recently,153 antivirals directed against human rhinoviruses could be used to treat the common cold, but also be employed therapeutically or prophylactically to prevent asthma and chronic obstructive pulmonary disease (COPD) exacerbations in high‐risk patients.
9. NATURAL PRODUCTS WITH ANTIVIRAL ACTIVITY
Natural compounds, primarily those originating from plants, have received increasing attention for their antiviral potential. Typical examples are constituents of Ardisia chimensis 154 and caffeoylquinic acids from Schefflera heptaphylla,155 both originating from Southern China and found active particularly against Coxsackie B3 virus.
Norsesquiterpenoids isolated from the roots of Phyllanthus emblica showed, again, some activity against Coxsackie B3 virus156 and triterpenoids isolated from S. heptaphylla (Fig. 8) were accredited with broader activity against Coxsackie B3, influenza A, respiratory syncytial virus (RSV), and HSV‐1.157
Figure 8.

Structures of natural products.
Calycosin‐7‐O‐beta‐d‐glucopyranoside (Fig. 8), the main isoflavonoid isolated from Astragalus membranaceus, also showed activity against Coxsackie B3 virus in vitro, and would improve the survival rate of mice infected with Coxsackie B3 virus.158
Flavans 7‐O‐galloyltricetifavan and 7,4′‐di‐O‐galloyltricetifavan isolated from the leaves of Pithecellobium clypearia would show activity against the same array of viruses (Coxsackie B3, influenza A, RSV, and HSV‐1) as the triterpenoids mentioned above.159
Homoisoflavonoids 3‐benzyl‐4‐chromones (Fig. 8) showed activity against Coxsackie virus B1, B3, B4, A9, and echovirus 30, but not against poliovirus.160
The flavone 4′,5,7‐trihydroxy‐3′,5′‐dimethoxyflavone (tricin), isolated from the bamboo Sasa albo‐marginata was found effective against HCMV at an EC50 of 0.17 μg/mL which means a stronger antiviral activity than ganciclovir.161 Tricin has also been accredited with antiinfluenza virus activity.162 To increase the oral bioavailability of tricin, it has been conjugated with alanine‐glutamic acid. The prodrug of tricin (tricin‐alanine‐glutamic acid) showed excellent oral bioavailability upon oral administration in rats.163
The isoflavone genistein (Fig. 8) was originally isolated from fermentation broth of Pseudomonas sp.164 and initially described as a tyrosine‐specific protein kinase inhibitor.165 Only recently, genistein has been shown to inhibit arenavirus infection,166 putatively by inhibiting arenavirus entry which occurs through a cholesterol‐dependent clathrin‐mediated endocytic mechanism.167 Genistein has been found to increase the survival rate of hamsters infected with the arenavirus Pirital virus, a surrogate model for hemorrhagic fever causing arenaviruses.168
Raoulic acid (Fig. 8) is the principal ingredient of Raoulia australis. It was shown to inhibit picornaviruses, that is, rhinovirus 2 (HRV2), rhinovirus 3 (HRV3), Coxsackie B3, Coxsackie B4, and enterovirus 71 at EC50 values in the range of 0.1–0.4 μg/mL, that is, at lower concentrations than those at which the aforementioned triterpenoids and flavans showed antiviral activity.169 However, raoulic acid did not show activity against influenza A or B.169
Terameprocol is a methylated derivative of nordihydroguaiaretic acid, a phenolic antioxidant extracted from the creosote bush Larrea tridentate.170 Terameprocol (Fig. 8) has been found to inhibit the growth of poxviruses, that is, cowpox and vaccinia, by preventing the spread of virus particles from cell to cell.170 Nigericin (also known as antibiotic K‐178, helexin C, azalomycin M, antibiotic X‐464, and polyetherin A) has also been shown to inhibit poxvirus replication.171 It had been previously reported to inhibit poliovirus and influenza virus replication.172, 173
Aglycoristocetin derivatives with a cyclobutenedione carrying hydrophobic chains such as methylene bis(phenylene) (Fig. 8) inhibit influenza A and B virus infections, probably by interference with the viral entry process.174 These types of compounds (derivatives of glycopeptide antibiotics) have been previously shown to inhibit the replication of retro‐ and coronaviruses and this inhibitory effect was also attributed to interference with virus entry.175, 176
Brassinosteroids, that is (22S,23S)‐3β‐bromo‐5α,22,23‐trihydroxy stigmastan‐6‐one (Fig. 8), represent naturally occurring polyhydroxy steroidal plant hormones modulating the growth and differentiation of plant cells. Their antiviral activity has been well documented.177, 178 Brassinosteroids are particularly active against arenaviruses such as Tacaribe, Pichinde, and Junin.179
Biyouyanagins A and B were originally obtained from Hypericum chinense and shown to be active against HIV.180 These compounds possessing antiarenavirus and anti‐HIV properties have been recently obtained by total synthesis, and their originally assigned structures were revised (Fig. 8).181
In conclusion, a wealth of natural products has been reported to possess antiviral properties. In the majority of these cases the chemical structure was well identified but the full antiviral activity spectrum of the compounds still needs to be evaluated, their mode of action elucidated, and, most importantly, their therapeutic value delineated.
10. MEK INHIBITORS
U0126 (Fig. 9) is the prototype of the MEK (mitogen‐activated protein/extracellular signal‐regulated kinase) inhibitors acting at the tiered serine/threonine kinase Raf/MEK/extra‐cellular regulated kinase (ERK) signaling pathway, able to suppress the propagation of the pandemic H1N1 influenza virus and highly pathogenic avian influenza virus in vitro and in vivo.182 Among the MEK inhibitors, PD 0325901 and PD 184352 (Fig. 9) have been used in clinical trials against cancer.183, 184 They are also inhibitory to influenza virus infection in vitro.182 MEK inhibitors such as U0126 not only reduce virus titers in vitro and in vivo, but also reduce proinflammatory cytokine expression.185
Figure 9.

Structures of MEK inhibitors U0126, PD 184352, PD 0325901, and PD 098059.
MEK inhibitors, such as U0126, have also been shown to suppress influenza B virus propagation.186 Most importantly, to date this happened without the emergence of any resistant virus variants, demonstrating that influenza viruses cannot easily adapt to interference with cellular functions.
Influenza virus infections require the induction of a variety of cytokines including those that are regulated by transcription factors of the activating protein‐1 (AP‐1) family and the NK (Jun‐N‐terminal kinase) pathway.187 These different protein kinase pathways may ultimately lead to RANTES production in influenza virus‐infected human bronchial epithelial cells.188
The Raf/MEK/ERK cascade is the prototype of mitogen‐activated protein (MAP) kinase cascades: inhibition of Raf‐signaling results in nuclear retention of viral ribonucleoprotein complexes (RNPs), and concomitant inhibition of virus production. Signaling through the mitogenic cascade seems to be essential for influenza virus production.189
The Raf, MEK, and ERK pathway not only plays an important role in the replication of influenza A and B virus, but also in the replication of HIV,190, 191 Coxsackie virus B3,192 coronavirus,193 and HSV.194 MEK inhibitors such as U0126 should therefore impair the propagation of these viruses, as has been specifically shown for U0126 against HSV‐2194 and X4 HIV‐1.191
The Raf/MEK/ERK signaling cascade is activated upon infection with Borna disease virus (BDV), a noncytolytic highly neurotropic single‐stranded RNA virus, the only known member of the Bornaviridae (Mononegavirales) and, again, the MEK inhibitor U0126 was found to block spread of BDV in cultures cells.195
The pathogenesis of hemorrhages in dengue hemorrhagic fever (DHF)/dengue shock syndrome (DSS) is poorly understood. The hemorrhages may be related to the induction of the plasminogen activator inhibitor type 1 (PAI‐1) via activation of the MEK/ERK pathway, the MEK inhibitor U0126 almost completely suppressed PAI‐1 expression,196 and may therefore be assumed to suppress hemorrhages in DHF/DSS.
The MEK/ERK pathway is also associated with the MAP kinases (MAPKs), which may contribute to the visna virus‐induced processes leading to neurodegenerative pathology. Treatment of visna virus‐infected cells with PD 98059 (Fig. 9), which had since long been recognized as a specific inhibitor of MAPK,197, 198 abolished visna virus replication,199 attesting as to the potential of PD 98059 to prevent the neuropathology of visna virus.
The replication of HCMV depends on a number of protein kinase pathways. Sorafenib is a multitargeted tyrosine kinase inhibitor registered for anticancer treatment (Nexavar®, Bayer and Onyx Pharmaceuticals). Through the MAPK signaling pathway, sorafenib may also interfere with the replication of HCMV.200 Imatinib may suppress HCMV replication through inactivation of the platelet‐derived growth factor‐α receptor (PDGFR), which is a critical receptor required for HCMV infection.201
Various protein kinase inhibitors containing a quinazoline moiety such as gefitinib (Iressa®, AstraZeneca and Teva) exert anti‐HCMV activity in vitro and in vivo (gefitinib also inhibits the HCMV kinase UL97).202 Furthermore, HCMV replication depends on the MEK/ERK pathway, and could therefore be suppressed by MEK inhibitors such as PD 98059.203
In conclusion, MEK inhibitors may exhibit a broad‐spectrum activity against a multitude of viruses, including influenza, herpes simplex, HIV and other retroviruses, dengue, corona, and HCMV. Although the antiviral effects could not be considered as highly specific, the advantage of MEK inhibitors, in view of their action targeted at a cellular process, is that they are unlikely to lead to the (rapid) emergence of drug resistance.
11. BROAD‐SPECTRUM ANTIVIRAL AGENTS TARGETING ENTRY OF ENVELOPED VIRUSES
In the February 16, 2010, issue of the Proc Natl Acad Sci (USA) appeared a paper on a broad‐spectrum antiviral agent LJ‐001 targeting entry of envelope viruses,204 followed in the October 5, 2010, issue of the same Journal by a remarkably similar antiviral activity of a structurally unrelated inhibitor dUY11.205 LJ‐001 is a rhodanine derivative and dUY11 is a rigid amphipathic fusion inhibitor (RAFI) derived from 2′‐deoxyuridine (Fig. 10). They both possess a rigid and planar hydrophobic moiety. With their hydrophobic moiety, LJ‐001 and dUY11 would intercalate into the lipid bilayer of the viral envelope, thereby affecting the virus‐cell fusion process. Both LJ‐001 and dUY11 should, in principle, be active against all enveloped viruses. For two important human pathogens, HCV and HSV (‐1 and ‐2), this activity was demonstrated, but for several others (i.e., yellow fever, dengue, Japanese encephalitis), this was not. In fact, the compounds should be compared side by side for their spectrum of activity. Admittedly, they should not easily lead to drug resistance development, but other issues should be further addressed, that is, in vivo activity and selectivity, biodistribution, drug formulation, and pharmacodynamics, before their therapeutic value could be assessed. LJ‐001 and dUY11 herald a new approach or strategy to combat enveloped virus infections, but the question can be raised whether they are druggable as well.
Figure 10.

Structures of dUY11 and LJ‐001.
ACKNOWLEDGMENT
The author thanks Mrs. Christiane Callebaut for her proficient editorial assistance.
Biography
Erik De Clercq, M.D., Ph.D., has been Chairman of the Department of Microbiology and Immunology of the Medical School at the Katholieke Universiteit Leuven (K.U. Leuven) as well as Chairman of the Board of the Rega Institute for Medical Research, until September 2006 when he was forced to retire. He is a member the Belgian (Flemish) Royal Academy of Medicine, a member of the Academia Europaea, and Fellow of the American Association for the Advancement of Science. He has also been the titular of the Prof. P. De Somer Chair for Microbiology. He has been teaching the courses of Cell Biology, Biochemistry, and Microbiology at the K.U. Leuven (and Kortrijk) Medical School until September 2006, and continued to teach part of the Biochemistry course at the K.U. Leuven Campus Kortrijk until 2009. Since 2008, he has been teaching the course of “Biochemistry at the Service of Medicine” at the Faculty of Sciences at the University of South Bohemia (Česke Budĕjovice, Czech Republic). Professor De Clercq received in 1996 the Hoechst Marion Roussel (now called “Aventis”) award (American Society for Microbiology), in 2000 the Maisin Prize for Biomedical Sciences (National Science Foundation, Belgium), and in 2010 the IS3NA John A. Montgomery Award and the Dr. Paul Janssen Award for Biomedical Research for his pioneering efforts in the field of antiviral research. He is a honorary doctor of several Universities [i.e., Ghent (Belgium), Athens (Greece), Ferrara (Italy), Shandong (Jinan, China), Charles University (Prague, Czech Republic), Česke Budĕjovice (Czech Republic), Tours (France), and Hull (United Kingdom)]. In 2008, he was elected European Inventor of the Year (Life time achievement award) and in 2011 Alumnus of the Year 2011 (Faculty of Medicine, K.U. Leuven, Belgium). His scientific interests are in the antiviral chemotherapy field, and, in particular, the development of new antiviral agents for various viral infections, including herpes simplex virus (HSV), varicella‐zoster virus (VZV), cytomegalovirus (CMV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), human papilloma virus (HPV), and hepatitis C virus (HCV). He has (co)‐discovered a number of antiviral drugs, currently used in the treatment of HSV infections (valaciclovir, Valtrex®, Zelitrex®), VZV infections (brivudin, Zostex®, Brivirac®, Zerpex®), CMV infections (cidofovir, Vistide®), HBV infections (adefovir dipivoxil, Hepsera®), and HIV infections (AIDS) (tenofovir disoproxil fumarate, marketed as Viread®, and, in combination with emtricitabine, as Truvada®; and in combination with both emtricitabine and efavirenz, as Atripla®). The combination of Truvada® with rilpivirine (Edurant®) has been approved in 2011 (Complera®, Eviplera®) for the treatment of HIV infections (AIDS). Viread® has since 2008 been approved for the treatment of HBV infections (chronic hepatitis B).
REFERENCES
- 1. De Clercq E. The discovery of antiviral agents: Ten different compounds, ten different stories. Med Res Rev 2008;28:929–953. [DOI] [PubMed] [Google Scholar]
- 2. De Clercq E. Antiviral drug discovery: Ten more compounds, and ten more stories (part B). Med Res Rev 2009;29:571–610. [DOI] [PubMed] [Google Scholar]
- 3. De Clercq E. Another ten stories in antiviral drug discovery (part C): “Old” and “new” antivirals, strategies and perspectives. Med Res Rev 2009;29:611–645. [DOI] [PubMed] [Google Scholar]
- 4. De Clercq E. Yet another ten stories on antiviral drug discovery (part D): Paradigms, paradoxes and paraductions. Med Res Rev 2010;30:667–707. [DOI] [PubMed] [Google Scholar]
- 5. De Clercq E. The next ten stories on antiviral drug discovery (part E): Advents, advances and adventures. Med Res Rev 2011;31:118–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lam AM, Espiritu C, Bansal S, Steuer HM, Zennou V, Otto MJ, Furman PA. In vitro selection of HCV replicons with reduced sensitivity to PSI‐352938, a cyclicphosphate prodrug of β‐d‐2′‐α‐F‐2′‐β‐C‐methylguanosine. Antiviral Res 2011;90:A22, abstract no 10. [Google Scholar]
- 7. Prichard M, Hartline C, Gill R, Bowlin T, James S. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antiviral Res 2011;90:A24, abstract no 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cardin R, Bravo F, Clark J, Earwood J, Glazer R, Rahman A, Bernstein D. In vivo efficacy of N‐methanocarbathymidine (N‐MCT) against herpes simplex virus type 2 in neonatal guinea pigs. Antiviral Res 2011;90:A24–25, abstract no 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lanier ER, Painter W, Tippin TK, Anderson M, Lampert BM, Mommeja‐Marin H, Trost LC. CMX001 (hexadecyloxypropyl cidofovir) antiviral activity against adenovirus in patients correlates with drug levels and viral sensitivity. Antiviral Res 2011;90:A27–28, abstract no 33. [Google Scholar]
- 10. Gowen BB, Mendenhall M, Russell A, Smee DF, Furuta Y. Favipiravir (T‐705) treatment of experimental arenaviral infection initiated after the onset of clinical disease. Antiviral Res 2011;90:A52, abstract no 107. [Google Scholar]
- 11. Pasquato A, Rochat C, de la Torre JC, Kunz S. Combinatorial anti‐arenaviral therapy with the small molecule SKI‐1/S1P inhibitor PF‐429242 and ribavirin. Antiviral Res 2011;90:A62, abstract no 136. [Google Scholar]
- 12. Braun H, Makarov VA, Riabova OB, Komarova ES, Richter M, Wutzler P, Schmidtke M. OBR‐5–340—A novel pyrazolo‐pyrimidine derivative with strong antiviral activity against Coxsackievirus B3 in vitro and in vivo. Antiviral Res 2011;90:A29; abstract no 39. [Google Scholar]
- 13. Richter M, Makarov VA, Riabova OB, Wutzler P, Schmidtke M. Identification and characterization of OBR‐5–340 – a novel broad‐spectrum anti‐human rhinovirus (HRV) inhibitor. Antiviral Res 2011;90:A40, abstract no 73. [Google Scholar]
- 14. Vanderlinden E, Vanstreels E, Vermeire K, Daelemans D, Herczegh P, Sztaricskai F, Naesens L. Mechanistic studies on a novel hydrophobic derivative of aglycoristocetin with potent and broad activity against influenza viruses. Antiviral Res 2011;90:A47, abstract 91. [Google Scholar]
- 15. Murayama T, Li Y, Yamada R, Sadanari H, Matsubara K, Tuchida Y, Koketsu M, Watanabe K. Inhibition of human cytomegalovirus replication by tricin (4′,5,7‐trihydroxy‐3′,5′‐dimethoxyflavone). Antiviral Res 2011;90:A60, abstract no 131. [Google Scholar]
- 16. Droebner K, Ludwig S, Pleschka S, Planz O. Antiviral activity of the MEK‐inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res 2011;90:A32, abstract no 47. [DOI] [PubMed] [Google Scholar]
- 17. Colpitts CC, Ustinov AV, Korshun VA, Schang LM. Rigid amphipathic fusion inhibitors (RAFIs) inhibit infectivity of enveloped viruses by targeting envelope lipids to prevent fusion with cellular membranes. Antiviral Res 2011; A30, abstract no 43. [Google Scholar]
- 18. Stuyver LJ, McBrayer TR, Whitaker T, Tharnish PM, Ramesh M, Lostia S, Cartee L, Shi J, Hobbs A, Schinazi RF, Watanabe KA, Otto MJ. Inhibition of the subgenomic hepatitis C virus replicon in huh‐7 cells by 2′‐deoxy‐2′‐fluorocytidine. Antimicrob Agents Chemother 2004;48:651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. Design, synthesis, and antiviral activity of 2′‐deoxy‐2′‐fluoro‐2′‐C‐methylcytidine, a potent inhibitor of hepatitis C virus replication. J Med Chem 2005;48:5504–5508. [DOI] [PubMed] [Google Scholar]
- 20. Pierra C, Amador A, Benzaria S, Cretton‐Scott E, D′Amours M, Mao J, Mathieu S, Moussa A, Bridges EG, Standring DN, Sommadossi JP, Storer R, Gosselin G. Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti‐HCV agent 2′‐C‐methylcytidine. J Med Chem 2006;49:6614–6620. [DOI] [PubMed] [Google Scholar]
- 21. Klumpp K, Lévêque V, Le Pogam S, Ma H, Jiang WR, Kang H, Granycome C, Singer M, Laxton C, Hang JQ, Sarma K, Smith DB, Heindl D, Hobbs CJ, Merrett JH, Symons J, Cammack N, Martin JA, Devos R, Nájera I. The novel nucleoside analog R1479 (4′‐azidocytidine) is a potent inhibitor of NS5B‐dependent RNA synthesis and hepatitis C virus replication in cell culture. J Biol Chem 2006;281:3793–3799. [DOI] [PubMed] [Google Scholar]
- 22. Smith DB, Martin JA, Klumpp K, Baker SJ, Blomgren PA, Devos R, Granycome C, Hang J, Hobbs CJ, Jiang WR, Laxton C, Le Pogam S, Leveque V, Ma H, Maile G, Merrett JH, Pichota A, Sarma K, Smith M, Swallow S, Symons J, Vesey D, Najera I, Cammack N. Design, synthesis, and antiviral properties of 4′‐substituted ribonucleosides as inhibitors of hepatitis C virus replication: The discovery of R1479. Bioorg Med Chem Lett 2007;17:2570–2576. [DOI] [PubMed] [Google Scholar]
- 23. Klumpp K, Kalayanov G, Ma H, Le Pogam S, Leveque V, Jiang WR, Inocencio N, De Witte A, Rajyaguru S, Tai E, Chanda S, Irwin MR, Sund C, Winqist A, Maltseva T, Eriksson S, Usova E, Smith M, Alker A, Najera I, Cammack N, Martin JA, Johansson NG, Smith DB. 2′‐deoxy‐4′‐azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2′‐alpha‐hydroxyl groups. J Biol Chem 2008;283:2167–2175. [DOI] [PubMed] [Google Scholar]
- 24. Smith DB, Kalayanov G, Sund C, Winqvist A, Maltseva T, Leveque VJ, Rajyaguru S, Le Pogam S, Najera I, Benkestock K, Zhou XX, Kaiser AC, Maag H, Cammack N, Martin JA, Swallow S, Johansson NG, Klumpp K, Smith M. The design, synthesis, and antiviral activity of monofluoro and difluoro analogues of 4′‐azidocytidine against hepatitis C virus replication: The discovery of 4′‐azido‐2′‐deoxy‐2′‐fluorocytidine and 4′‐azido‐2′‐dideoxy‐2′,2′‐difluorocytidine. J Med Chem 2009;52:2971–2978. [DOI] [PubMed] [Google Scholar]
- 25. Reddy PG, Bao D, Chang W, Chun BK, Du J, Nagarathnam D, Rachakonda S, Ross BS, Zhang HR, Bansal S, Espiritu CL, Keilman M, Lam AM, Niu C, Steuer HM, Furman PA, Otto MJ, Sofia MJ. 2′‐deoxy‐2′‐α‐fluoro‐2′‐β‐C‐methyl 3′,5′‐cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: Discovery of PSI‐352938. Bioorg Med Chem Lett 2010;20:7376–7380. [DOI] [PubMed] [Google Scholar]
- 26. Wang P, Chun BK, Rachakonda S, Du J, Khan N, Shi J, Stec W, Cleary D, Ross BS, Sofia MJ. An efficient and diastereoselective synthesis of PSI‐6130: A clinically efficacious inhibitor of HCV NS5B polymerase. J Org Chem 2009;74:6819–6824. [DOI] [PubMed] [Google Scholar]
- 27. Murakami E, Bao H, Ramesh M, McBrayer TR, Whitaker T, Micolochick Steuer HM, Schinazi RF, Stuyver LJ, Obikhod A, Otto MJ, Furman PA. Mechanism of activation of beta‐D‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob Agents Chemother 2007;51:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma H, Jiang WR, Robledo N, Leveque V, Ali S, Lara‐Jaime T, Masjedizadeh M, Smith DB, Cammack N, Klumpp K, Symons J. Characterization of the metabolic activation of hepatitis C virus nucleoside inhibitor beta‐D‐2′‐Deoxy‐2′‐fluoro‐2′‐C‐methylcytidine (PSI‐6130) and identification of a novel active 5′‐triphosphate species. J Biol Chem 2007;282:29812–29820. [DOI] [PubMed] [Google Scholar]
- 29. Murakami E, Niu C, Bao H, Micolochick Steuer HM, Whitaker T, Nachman T, Sofia MA, Wang P, Otto MJ, Furman PA. The mechanism of action of beta‐D‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methylcytidine involves a second metabolic pathway leading to beta‐D‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methyluridine 5′‐triphosphate, a potent inhibitor of the hepatitis C virus RNA‐dependent RNA polymerase. Antimicrob Agents Chemother 2008;52:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lam AM, Murakami E, Espiritu C, Steuer HM, Niu C, Keilman M, Bao H, Zennou V, Bourne N, Julander JG, Morrey JD, Smee DF, Frick DN, Heck JA, Wang P, Nagarathnam D, Ross BS, Sofia MJ, Otto MJ, Furman PA. PSI‐7851, a pronucleotide of beta‐D‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methyluridine monophosphate, is a potent and pan‐genotype inhibitor of hepatitis C virus replication. Antimicrob Agents Chemother 2010;54:3187–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA. Mechanism of activation of PSI‐7851 and its diastereoisomer PSI‐7977. J Biol Chem 2010;285:34337–34347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. Discovery of a β‐d‐2′‐deoxy‐2′‐α‐fluoro‐2′‐β‐C‐methyluridine nucleotide prodrug (PSI‐7977) for the treatment of hepatitis C virus. J Med Chem 2010;53:7202–7218. [DOI] [PubMed] [Google Scholar]
- 33. Furman PA, Murakami E, Niu C, Lam AM, Espiritu C, Bansal S, Bao H, Tolstykh T, Micolochick Steuer H, Keilman M, Zennou V, Bourne N, Veselenak RL, Chang W, Ross BS, Du J, Otto MJ, Sofia MJ. Activity and the metabolic activation pathway of the potent and selective hepatitis C virus pronucleotide inhibitor PSI‐353661. Antiviral Res 2011;91:120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reddy PG, Chun BK, Zhang HR, Rachakonda S, Ross BS, Sofia MJ. Stereoselective synthesis of PSI‐352938: A β‐D‐2′‐deoxy‐2′‐α‐fluoro‐2′‐β‐C‐methyl‐3′,5′‐cyclic phosphate nucleotide prodrug for the treatment of HCV . J Org Chem 2011;76:3782–3790. [DOI] [PubMed] [Google Scholar]
- 35. Lam AM, Espiritu C, Murakami E, Zennou V, Bansal S, Micolochick Steuer HM, Niu C, Keilman M, Bao H, Bourne N, Veselenak RL, Reddy PG, Chang W, Du J, Nagarathnam D, Sofia MJ, Otto MJ, Furman PA. Inhibition of hepatitis C virus replicon RNA synthesis by PSI‐352938, a cyclic phosphate prodrug of β‐D‐2′‐deoxy‐2′‐α‐fluoro‐2′‐β‐C‐methylguanosine. Antimicrob Agents Chemother 2011;55:2566–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vernachio JH, Bleiman B, Bryant KD, Chamberlain S, Hunley D, Hutchins J, Ames B, Gorovits E, Ganguly B, Hall A, Kolykhalov A, Liu Y, Muhammad J, Raja N, Walters CR, Wang J, Williams K, Patti JM, Henson G, Madela K, Aljarah M, Gilles A, McGuigan C. INX‐08189, a phosphoramidate prodrug of 6‐O‐methyl‐2′‐C‐methyl guanosine, is a potent inhibitor of hepatitis C virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob Agents Chemother 2011;55:1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McGuigan C, Madela K, Aljarah M, Gilles A, Brancale A, Zonta N, Chamberlain S, Vernachio J, Hutchins J, Hall A, Ames B, Gorovits E, Ganguly B, Kolykhalov A, Wang J, Muhammad J, Patti JM, Henson G. Design, synthesis and evaluation of a novel double pro‐drug: INX‐08189. A new clinical candidate for hepatitis C virus. Bioorg Med Chem Lett 2010;20:4850–4854. [DOI] [PubMed] [Google Scholar]
- 38. Zemlicka J. Methylenecyclopropane analogues of nucleosides as anti‐herpes agents In: De Clercq E, Ed. Advances in Antiviral Drug Design, Vol. 5. Elsevier Science; 2007. p 113–165. [Google Scholar]
- 39. Zhou S, Breitenbach JM, Borysko KZ, Drach JC, Kern ER, Gullen E, Cheng YC, Zemlicka J. Synthesis and antiviral activity of (Z)‐ and (E)‐2,2‐[bis(hydroxymethyl)cyclopropylidene]methylpurines and ‐pyrimidines: Second‐generation methylenecyclopropane analogues of nucleosides. J Med Chem 2004;47:566–575. [DOI] [PubMed] [Google Scholar]
- 40. Kern ER, Kushner NL, Hartline CB, Williams‐Aziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob Agents Chemother 2005;49:1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kern ER, Bidanset DJ, Hartline CB, Yan Z, Zemlicka J, Quenelle DC. Oral activity of a methylenecyclopropane analog, cyclopropavir, in animal models for cytomegalovirus infections. Antimicrob Agents Chemother 2004;48:4745–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kern ER. Pivotal role of animal models in the development of new therapies for cytomegalovirus infections. Antiviral Res 2006;71:164–171. [DOI] [PubMed] [Google Scholar]
- 43. De Clercq E, Naesens L. In search of effective anti‐HHV‐6 agents. J Clin Virol 2006;37(Suppl 1):S82–S86. [DOI] [PubMed] [Google Scholar]
- 44. Wu Z, Drach JC, Prichard MN, Yanachkova M, Yanachkov I, Bowlin TL, Zemlicka J. L‐valine ester of cyclopropavir: A new antiviral prodrug. Antivir Chem Chemother 2009;20:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chou S, Bowlin TL. Cytomegalovirus UL97 mutations affecting cyclopropavir and ganciclovir susceptibility. Antimicrob Agents Chemother 2011;55:382–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gentry BG, Kamil JP, Coen DM, Zemlicka J, Drach JC. Stereoselective phosphorylation of cyclopropavir by pUL97 and competitive inhibition by maribavir. Antimicrob Agents Chemother 2010;54:3093–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gentry BG, Gentry SN, Jackson TL, Zemlicka J, Drach JC. Phosphorylation of antiviral and endogenous nucleotides to di‐ and triphosphates by guanosine monophosphate kinase. Biochem Pharmacol 2011;81:43–49. [DOI] [PubMed] [Google Scholar]
- 48. Mhaske SB, Ksebati B, Prichard MN, Drach JC, Zemlicka J. Phosphonate analogues of cyclopropavir phosphates and their E‐isomers. Synthesis and antiviral activity. Bioorg Med Chem 2009;17:3892–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem 1996;39:3739–3747. [DOI] [PubMed] [Google Scholar]
- 50. Zalah L, Huleihel M, Manor E, Konson A, Ford H, Jr , Marquez VE, Johns DG, Agbaria R. Metabolic pathways of N‐methanocarbathymidine, a novel antiviral agent, in native and herpes simplex virus type 1 infected Vero cells. Antiviral Res 2002;55:63–75. [DOI] [PubMed] [Google Scholar]
- 51. Russ P, Schelling P, Scapozza L, Folkers G, De Clercq E, Marquez VE. Synthesis and biological evaluation of 5‐substituted derivatives of the potent antiherpes agent (north)‐methanocarbathymine. J Med Chem 2003;46:5045–5054. [DOI] [PubMed] [Google Scholar]
- 52. Huleihel M, Talishanisky M, Ford H, Jr , Marquez VE, Kelley JA, Johns DG, Agbaria R. Dynamics of the antiviral activity of N‐methanocarbathymidine against herpes simplex virus type 1 in cell culture. Int J Antimicrob Agents 2005;25:427–432. [DOI] [PubMed] [Google Scholar]
- 53. Marquez VE, Ben‐Kasus T, Barchi JJ, Jr , Green KM, Nicklaus MC, Agbaria R. Experimental and structural evidence that herpes 1 kinase and cellular DNA polymerase(s) discriminate on the basis of sugar pucker. J Am Chem Soc 2004;126:543–549. [DOI] [PubMed] [Google Scholar]
- 54. Béres J, Sági G, Tömösközi I, Gruber L, Baitz‐Gács E, Otvös L, De Clercq E. Stereospecific synthesis and antiviral properties of different enantiomerically pure carbocyclic 2′‐deoxyribonucleoside analogues derived from common chiral pools: (+)‐(1R,5S)‐ and (‐)‐(1S,5R)‐2‐oxabicyclo[3.3.0]oct‐6‐en‐3‐one. J Med Chem 1990;33:1353–1360. [DOI] [PubMed] [Google Scholar]
- 55. Prichard MN, Keith KA, Quenelle DC, Kern ER. Activity and mechanism of action of N‐methanocarbathymidine against herpesvirus and orthopoxvirus infections. Antimicrob Agents Chemother 2006;50:1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smee DF, Wandersee MK, Bailey KW, Wong MH, Chu CK, Gadthula S, Sidwell RW. Cell line dependency for antiviral activity and in vivo efficacy of N‐methanocarbathymidine against orthopoxvirus infections in mice. Antiviral Res 2007;73:69–77. [DOI] [PubMed] [Google Scholar]
- 57. Smee DF, Hurst BL, Wong MH, Glazer RI, Rahman A, Sidwell RW. Efficacy of N‐methanocarbathymidine in treating mice infected intranasally with the IHD and WR strains of vaccinia virus. Antiviral Res 2007;76:124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Prichard MN, Kern ER, Quenelle DC, Keith KA, Moyer RW, Turner PC. Vaccinia virus lacking the deoxyuridine triphosphatase gene (F2L) replicates well in vitro and in vivo, but is hypersensitive to the antiviral drug (N)‐methanocarbathymidine. Virol J 2008;5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu W, Burnette A, Dorjsuren D, Roberts PE, Huleihel M, Shoemaker RH, Marquez VE, Agbaria R, Sei S. Potent antiviral activity of north‐methanocarbathymidine against Kaposi's sarcoma‐associated herpesvirus. Antimicrob Agents Chemother 2005;49:4965–4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ludek OR, Marquez VE. A greener enantioselective synthesis of the antiviral agent North‐methanocarbathymidine (N‐MCT) from 2‐deoxy‐d‐ribose. Tetrahedron 2009;65:8461–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Comin MJ, Vu BC, Boyer PL, Liao C, Hughes SH, Marquez VE. D‐(+)‐iso‐methanocarbathymidine: A high‐affinity substrate for herpes simplex virus 1 thymidine kinase. ChemMedChem 2008;3:1129–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marquez VE, Hughes SH, Sei S, Agbaria R. The history of N‐methanocarbathymidine: The investigation of a conformational concept leads to the discovery of a potent and selective nucleoside antiviral agent. Antiviral Res 2006;71:268–275. [DOI] [PubMed] [Google Scholar]
- 63. Hostetler KY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antiviral Res 2009;82:A84‐A98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Quenelle DC, Lampert B, Collins DJ, Rice TL, Painter GR, Kern ER. Efficacy of CMX001 against herpes simplex virus infections in mice and correlations with drug distribution studies. J Infect Dis 2010;202:1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bidanset DJ, Beadle JR, Wan WB, Hostetler KY, Kern ER. Oral activity of ether lipid ester prodrugs of cidofovir against experimental human cytomegalovirus infection. J Infect Dis 2004;190:499–503. [DOI] [PubMed] [Google Scholar]
- 66. Beadle JR, Hartline C, Aldern KA, Rodriguez N, Harden E, Kern ER, Hostetler KY. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple‐log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob Agents Chemother 2002;46:2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kern ER, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Quenelle DC. Oral treatment of murine cytomegalovirus infections with ether lipid esters of cidofovir. Antimicrob Agents Chemother 2004;48:3516–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bravo FJ, Bernstein DI, Beadle JR, Hostetler KY, Cardin RD. Oral hexadecyloxypropyl‐cidofovir therapy in pregnant guinea pigs improves outcome in the congenital model of cytomegalovirus infection. Antimicrob Agents Chemother 2011;55:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hartline CB, Gustin KM, Wan WB, Ciesla SL, Beadle JR, Hostetler KY, Kern ER. Ether lipid‐ester prodrugs of acyclic nucleoside phosphonates: Activity against adenovirus replication in vitro. J Infect Dis 2005;191:396–399. [DOI] [PubMed] [Google Scholar]
- 70. Toth K, Spencer JF, Dhar D, Sagartz JE, Buller RM, Painter GR, Wold WS. Hexadecyloxypropyl‐cidofovir, CMX001, prevents adenovirus‐induced mortality in a permissive, immunosuppressed animal model. Proc Natl Acad Sci USA 2008;105:7293–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Paolino K, Sande J, Perez E, Loechelt B, Jantausch B, Painter W, Anderson M, Tippin T, Lanier ER, Fry T, DeBiasi RL. Eradication of disseminated adenovirus infection in a pediatric hematopoietic stem cell transplantation recipient using the novel antiviral agent CMX001. J Clin Virol 2011;50:167–170. [DOI] [PubMed] [Google Scholar]
- 72. Andrei G, Snoeck R, Vandeputte M, De Clercq E. Activities of various compounds against murine and primate polyomaviruses. Antimicrob Agents Chemother 1997;41:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bernhoff E, Gutteberg TJ, Sandvik K, Hirsch HH, Rinaldo CH. Cidofovir inhibits polyomavirus BK replication in human renal tubular cells downstream of viral early gene expression. Am J Transplant 2008;8:1413–1422. [DOI] [PubMed] [Google Scholar]
- 74. Randhawa P, Farasati NA, Shapiro R, Hostetler KY. Ether lipid ester derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob Agents Chemother 2006;50:1564–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rinaldo CH, Gosert R, Bernhoff E, Finstad S, Hirsch HH. 1‐O‐hexadecyloxypropyl cidofovir (CMX001) effectively inhibits polyomavirus BK replication in primary human renal tubular epithelial cells. Antimicrob Agents Chemother 2010;54:4714–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jiang ZG, Cohen J, Marshall LJ, Major EO. Hexadecyloxypropyl‐cidofovir (CMX001) suppresses JC virus replication in human fetal brain SVG cell cultures. Antimicrob Agents Chemother 2010;54:4723–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gosert R, Rinaldo CH, Wernli M, Major EO, Hirsch HH. CMX001 (1‐O‐hexadecyloxypropyl‐cidofovir) inhibits polyomavirus JC replication in human brain progenitor‐derived astrocytes. Antimicrob Agents Chemother 2011;55:2129–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Patel A, Patel J, Ikwuagwu J. Treatment of progressive multifocal leukoencephalopathy and idiopathic CD4+ lymphocytopenia. J Antimicrob Chemother 2010;65:2489–2492. [DOI] [PubMed] [Google Scholar]
- 79. Painter GR, Hostetler KY. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection. Trends Biotechnol 2004;22:423–427. [DOI] [PubMed] [Google Scholar]
- 80. Kern ER, Hartline C, Harden E, Keith K, Rodriguez N, Beadle JR, Hostetler KY. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents Chemother 2002;46:991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Quenelle DC, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Kern ER. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob Agents Chemother 2004;48:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 2004;318:474–481. [DOI] [PubMed] [Google Scholar]
- 83. Parker S, Siddiqui AM, Oberle C, Hembrador E, Lanier R, Painter G, Robertson A, Buller RM. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti‐poxvirus therapies. Virology 2009;385:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Parker S, Siddiqui AM, Painter G, Schriewer J, Buller RM. Ectromelia virus infections of mice as a model to support the licensure of anti‐orthopoxvirus therapeutics. Viruses 2010;2:1918–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Parker S, Touchette E, Oberle C, Almond M, Robertson A, Trost LC, Lampert B, Painter G, Buller RM. Efficacy of therapeutic intervention with an oral ether‐lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res 2008;77:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Parker S, Schriewer J, Oberle C, Robertson A, Lanier R, Painter G, Buller RM. Using biomarkers to stage disease progression in a lethal mousepox model treated with CMX001. Antivir Ther 2008;13:863–873. [PMC free article] [PubMed] [Google Scholar]
- 87. Stabenow J, Buller RM, Schriewer J, West C, Sagartz JE, Parker S. A mouse model of lethal infection for evaluating prophylactics and therapeutics against Monkeypox virus. J Virol 2010;84:3909–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen N, Bellone CJ, Schriewer J, Owens G, Fredrickson T, Parker S, Buller RM. Poxvirus interleukin‐4 expression overcomes inherent resistance and vaccine‐induced immunity: Pathogenesis, prophylaxis, and antiviral therapy. Virology 2011;409:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rice AD, Adams MM, Wallace G, Burrage AM, Lindsey SF, Smith AJ, Swetnam D, Manning BR, Gray SA, Lampert B, Foster S, Lanier R, Robertson A, Painter G, Moyer RW. Efficacy of CMX001 as a post exposure antiviral in New Zealand white rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 2011;3:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rice AD, Adams MM, Lampert B, Foster S, Lanier R, Robertson A, Painter G, Moyer RW. Efficacy of CMX001 as a prophylactic and presymptomatic antiviral agent in New Zealand white rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 2001;3:63–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lanier R, Trost L, Tippin T, Lampert B, Robertson A, Foster S, Rose M, Painter W, O′Mahony R, Almond M, Painter G. Development of CMX001 for the treatment of poxvirus infections. Viruses 2010;2:2740–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wan WB, Beadle JR, Hartline C, Kern ER, Ciesla SL, Valiaeva N, Hostetler KY. Comparison of the antiviral activities of alkoxyalkyl and alkyl esters of cidofovir against human and murine cytomegalovirus replication in vitro. Antimicrob Agents Chemother 2005;49:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ruiz J, Beadle JR, Buller RM, Schreiwer J, Prichard MN, Keith KA, Lewis KC, Hostetler KY. Synthesis, metabolic stability and antiviral evaluation of various alkoxyalkyl esters of cidofovir and 9‐(S)‐[3‐hydroxy‐2‐(phosphonomethoxy)propyl]adenine. Bioorg Med Chem 2011;19:2950–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Buys KK, Jung KH, Smee DF, Furuta Y, Gowen BB. Maporal virus as a surrogate for pathogenic New World hantaviruses and its inhibition by favipiravir. Antivir Chem Chemother 2011;21:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Watanabe Y, Narita H, Shiraki K. In vitro and in vivo activities of anti‐influenza virus compound T‐705. Antimicrob Agents Chemother 2002;46:977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Furuta Y, Takahashi K, Kuno‐Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. Mechanism of action of T‐705 against influenza virus. Antimicrob Agents Chemother 2005;49:981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Smee DF, Hurst BL, Egawa H, Takahashi K, Kadota T, Furuta Y. Intracellular metabolism of favipiravir (T‐705) in uninfected and influenza A (H5N1) virus‐infected cells. J Antimicrob Chemother 2009;64:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kiso M, Takahashi K, Sakai‐Tagawa Y, Shinya K, Sakabe S, Le QM, Ozawa M, Furuta Y, Kawaoka Y. T‐705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc Natl Acad Sci USA 2010;107:882–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sidwell RW, Barnard DL, Day CW, Smee DF, Bailey KW, Wong MH, Morrey JD, Furuta Y. Efficacy of orally administered T‐705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob Agents Chemother 2007;51:845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sleeman K, Mishin VP, Deyde VM, Furuta Y, Klimov AI, Gubareva LV. In vitro antiviral activity of favipiravir (T‐705) against drug‐resistant influenza and 2009 A(H1N1) viruses. Antimicrob Agents Chemother 2010;54:2517–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Smee DF, Hurst BL, Wong MH, Bailey KW, Tarbet EB, Morrey JD, Furuta Y. Effects of the combination of favipiravir (T‐705) and oseltamivir on influenza A virus infections in mice. Antimicrob Agents Chemother 2010;54:126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. T‐705 (favipiravir) and related compounds: Novel broad‐spectrum inhibitors of RNA viral infections. Antiviral Res 2009;82:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Julander JG, Furuta Y, Shafer K, Sidwell RW. Activity of T‐1106 in a hamster model of yellow fever virus infection. Antimicrob Agents Chemother 2007;51:1962–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Julander JG, Shafer K, Smee DF, Morrey JD, Furuta Y. Activity of T‐705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T‐1106. Antimicrob Agents Chemother 2009;53:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Morrey JD, Taro BS, Siddharthan V, Wang H, Smee DF, Christensen AJ, Furuta Y. Efficacy of orally administered T‐705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 2008;80:377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Julander JG, Smee DF, Morrey JD, Furuta Y. Effect of T‐705 treatment on western equine encephalitis in a mouse model. Antiviral Res 2009;82:169–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gowen BB, Wong MH, Jung KH, Sanders AB, Mendenhall M, Bailey KW, Furuta Y, Sidwell RW. In vitro and in vivo activities of T‐705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother 2007;51:3168–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mendenhall M, Russell A, Juelich T, Messina EL, Smee DF, Freiberg AN, Holbrook MR, Furuta Y, de la Torre JC, Nunberg JH, Gowen BB. T‐705 (favipiravir) inhibition of arenavirus replication in cell culture. Antimicrob Agents Chemother 2011;55:782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gowen BB, Smee DF, Wong MH, Hall JO, Jung KH, Bailey KW, Stevens JR, Furuta Y, Morrey JD. Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T‐705 efficacy and reduced toxicity suggests an alternative to ribavirin. PLoS One 2008;3:e3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gowen BB, Wong MH, Jung KH, Smee DF, Morrey JD, Furuta Y. Efficacy of favipiravir (T‐705) and T‐1106 pyrazine derivatives in phlebovirus disease models. Antiviral Res 2010;86:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, Johnson KM, Elliott LH, Belmont‐Williams R. Lassa fever. Effective therapy with ribavirin. N Engl J Med 1986;314:20–26. [DOI] [PubMed] [Google Scholar]
- 112. Moreno H, Gallego I, Sevilla N, de la Torre JC, Domingo E, Martín V. Ribavirin can be mutagenic for arenaviruses. J Virol 2011;85:7246–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lee AM, Pasquato A, Kunz S. Novel approaches in anti‐arenaviral drug development. Virology 2011;411:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Charrel RN, Coutard B, Baronti C, Canard B, Nougairede A, Frangeul A, Morin B, Jamal S, Schmidt CL, Hilgenfeld R, Klempa B, de Lamballerie X. Arenaviruses and hantaviruses: From epidemiology and genomics to antivirals. Antiviral Res 2011;90:102–114. [DOI] [PubMed] [Google Scholar]
- 115. Gowen BB, Wong MH, Larson D, Ye W, Jung KH, Sefing EJ, Skirpstunas R, Smee DF, Morrey JD, Schneller SW. Development of a new tacaribe arenavirus infection model and its use to explore antiviral activity of a novel aristeromycin analog. PLoS One 2010;5:e12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Barradas JS, Errea MI, D′Accorso NB, Sepúlveda CS, Damonte EB. Imidazo[2,1‐b]thiazole carbohydrate derivatives: Synthesis and antiviral activity against Junin virus, agent of Argentine hemorrhagic fever. Eur J Med Chem 2011;46:259–264. [DOI] [PubMed] [Google Scholar]
- 117. Müller S, Günther S. Broad‐spectrum antiviral activity of small interfering RNA targeting the conserved RNA termini of Lassa virus. Antimicrob Agents Chemother 2007;51:2215–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Asper M, Sternsdorf T, Hass M, Drosten C, Rhode A, Schmitz H, Günther S. Inhibition of different Lassa virus strains by alpha and gamma interferons and comparison with a less pathogenic arenavirus. J Virol 2004;78:3162–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Albiol Matanic VC, Castilla V. Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int J Antimicrob Agents 2004;23:382–389. [DOI] [PubMed] [Google Scholar]
- 120. Radoshitzky SR, Dong L, Chi X, Clester JC, Retterer C, Spurgers K, Kuhn JH, Sandwick S, Ruthel G, Kota K, Boltz D, Warren T, Kranzusch PJ, Whelan SP, Bavari S. Infectious Lassa virus, but not filoviruses, is restricted by BST‐2/tetherin. J Virol 2010;84:10569–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010;468:779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Emonet SE, Urata S, de la Torre JC. Arenavirus reverse genetics: New approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 2011;411:416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hawkins JL, Robbins MD, Warren LC, Xia D, Petras SF, Valentine JJ, Varghese AH, Wang IK, Subashi TA, Shelly LD, Hay BA, Landschulz KT, Geoghegan KF, Harwood HJ, Jr . Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element‐binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther 2008;326:801–808. [DOI] [PubMed] [Google Scholar]
- 124. Hay BA, Abrams B, Zumbrunn AY, Valentine JJ, Warren LC, Petras SF, Shelly LD, Xia A, Varghese AH, Hawkins JL, Van Camp JA, Robbins MD, Landschulz K, Harwood HJ, Jr . Aminopyrrolidineamide inhibitors of site‐1 protease. Bioorg Med Chem Lett 2007;17:4411–4414. [DOI] [PubMed] [Google Scholar]
- 125. Urata S, Yun N, Pasquato A, Paessler S, Kunz S, de la Torre JC. Antiviral activity of a small‐molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J Virol 2011;85:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bolken TC, Laquerre S, Zhang Y, Bailey TR, Pevear DC, Kickner SS, Sperzel LE, Jones KF, Warren TK, Amanda Lund S, Kirkwood‐Watts DL, King DS, Shurtleff AC, Guttieri MC, Deng Y, Bleam M, Hruby DE. Identification and characterization of potent small molecule inhibitor of hemorrhagic fever New World arenaviruses. Antiviral Res 2006;69:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lee AM, Rojek JM, Spiropoulou CF, Gundersen AT, Jin W, Shaginian A, York J, Nunberg JH, Boger DL, Oldstone MB, Kunz S. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J Biol Chem 2008;283:18734–18742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Thomas CJ, Casquilho‐Gray HE, York J, DeCamp DL, Dai D, Petrilli EB, Boger DL, Slayden RA, Amberg SM, Sprang SR, Nunberg JH. A specific interaction of small molecule entry inhibitors with the envelope glycoprotein complex of the Junín hemorrhagic fever arenavirus. J Biol Chem 2011;286:6192–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. York J, Dai D, Amberg SM, Nunberg JH. pH‐induced activation of arenavirus membrane fusion is antagonized by small‐molecule inhibitors. J Virol 2008;82:10932–10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Larson RA, Dai D, Hosack VT, Tan Y, Bolken TC, Hruby DE, Amberg SM. Identification of a broad‐spectrum arenavirus entry inhibitor. J Virol 2008;82:10768–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Barnard DL. Current status of anti‐picornavirus therapies. Curr Pharm Des 2006;12:1379–1390. [DOI] [PubMed] [Google Scholar]
- 132. Diana GD, Rudewicz P, Pevear DC, Nitz TJ, Aldous SC, Aldous DJ, Robinson DT, Draper T, Dutko FJ, Aldi C. Picornavirus inhibitors: Trifluoromethyl substitution provides a global protective effect against hepatic metabolism. J Med Chem 1995;38:1355–1371. [DOI] [PubMed] [Google Scholar]
- 133. Pevear DC, Tull TM, Seipel ME, Groarke JM. Activity of pleconaril against enteroviruses. Antimicrob Agents Chemother 1999;43:2109–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rotbart HA, Webster AD. Treatment of potentially life‐threatening enterovirus infections with pleconaril. Clin. Infect. Dis. 2001;32:228–235. [DOI] [PubMed] [Google Scholar]
- 135. Senior K. FDA panel rejects common cold treatment. Lancet Infect Dis 2002;2:264. [DOI] [PubMed] [Google Scholar]
- 136. Abzug MJ, Cloud G, Bradley J, Sánchez PJ, Romero J, Powell D, Lepow M, Mani C, Capparelli EV, Blount S, Lakeman F, Whitley RJ, Kimberlin DW; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Double blind placebo‐controlled trial of pleconaril in infants with enterovirus meningitis. Pediatr Infect Dis J 2003;22:335–341. [DOI] [PubMed] [Google Scholar]
- 137. Hayden FG, Herrington DT, Coats TL, Kim K, Cooper EC, Villano SA, Liu S, Hudson S, Pevear DC, Collett M, McKinlay M; Pleconaril Respiratory Infection Study Group. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: Results of 2 double‐blind, randomized, placebo‐controlled trials. Clin Infect Dis 2003;36:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pevear DC, Hayden FG, Demenczuk TM, Barone LR, McKinlay MA, Collett MS. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob Agents Chemother 2005;49:4492–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. De Palma AM, Vliegen I, De Clercq E, Neyts J. Selective inhibitors of picornavirus replication. Med Res Rev 2008;28:823–884. [DOI] [PubMed] [Google Scholar]
- 140. Desmond RA, Accortt NA, Talley L, Villano SA, Soong SJ, Whitley RJ. Enteroviral meningitis: Natural history and outcome of pleconaril therapy. Antimicrob Agents Chemother 2006;50:2409–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. van de Ven AA, Douma JW, Rademaker C, van Loon AM, Wensing AM, Boelens JJ, Sanders EA, van Montfrans JM. Pleconaril‐resistant chronic parechovirus‐associated enteropathy in agammaglobulinaemia. Antivir Ther 2011;16:611–614. [DOI] [PubMed] [Google Scholar]
- 142. Schmidtke M, Wutzler P, Zieger R, Riabova OB, Makarov VA. New pleconaril and [(biphenyloxy)propyl]isoxazole derivatives with substitutions in the central ring exhibit antiviral activity against pleconaril‐resistant coxsackievirus B3. Antiviral Res 2009;81:56–63. [DOI] [PubMed] [Google Scholar]
- 143. Cheng J, Xie J, Luo X. Synthesis and antiviral activity against Coxsackie virus B3 of some novel benzimidazole derivatives. Bioorg Med Chem Lett 2005;15:267–269. [DOI] [PubMed] [Google Scholar]
- 144. Ahn J, Jun ES, Lee HS, Yoon SY, Kim D, Joo CH, Kim YK, Lee H. A small interfering RNA targeting coxsackievirus B3 protects permissive HeLa cells from viral challenge. J Virol 2005;79:8620–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Werk D, Pinkert S, Heim A, Zeichhardt H, Grunert HP, Poller W, Erdmann VA, Fechner H, Kurreck J. Combination of soluble coxsackievirus‐adenovirus receptor and anti‐coxsackievirus siRNAs exerts synergistic antiviral activity against coxsackievirus B3. Antiviral Res 2009;83:298–306. [DOI] [PubMed] [Google Scholar]
- 146. Aguado L, Thibaut HJ, Priego EM, Jimeno ML, Camarasa MJ, Neyts J, Pérez‐Pérez MJ. 9‐Arylpurines as a novel class of enterovirus inhibitors. J Med Chem 2010;53:316–324. [DOI] [PubMed] [Google Scholar]
- 147. Padalko E, Verbeken E, Matthys P, Aerts JL, De Clercq E, Neyts J. Mycophenolate mofetil inhibits the development of Coxsackie B3‐virus‐induced myocarditis in mice. BMC Microbiol 2003;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Padalko E, Verbeken E, De Clercq E, Neyts J. Inhibition of coxsackie B3 virus induced myocarditis in mice by 2‐(3,4‐dichlorophenoxy)‐5‐nitrobenzonitrile. J Med Virol 2004;72:263–267. [DOI] [PubMed] [Google Scholar]
- 149. Padalko E, Nuyens D, De Palma A, Verbeken E, Aerts JL, De Clercq E, Carmeliet P, Neyts J. The interferon inducer ampligen [poly(I)‐poly(C12U)] markedly protects mice against coxsackie B3 virus‐induced myocarditis. Antimicrob Agents Chemother 2004;48:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Thibaut HJ, Leyssen P, Puerstinger G, Muigg A, Neyts J, De Palma AM. Towards the design of combination therapy for the treatment of enterovirus infections. Antiviral Res 2011;90:213–217. [DOI] [PubMed] [Google Scholar]
- 151. De Palma AM, Heggermont W, Lanke K, Coutard B, Bergmann M, Monforte AM, Canard B, De Clercq E, Chimirri A, Pürstinger G, Rohayem J, van Kuppeveld F, Neyts J. The thiazolobenzimidazole TBZE‐029 inhibits enterovirus replication by targeting a short region immediately downstream from motif C in the nonstructural protein 2C . J Virol 2008;82:4720–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. De Palma AM, Pürstinger G, Wimmer E, Patick AK, Andries K, Rombaut B, De Clercq E, Neyts J. Potential use of antiviral agents in polio eradication. Emerg Infect Dis 2008;14:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Norder H, De Palma AM, Selisko B, Costenaro L, Papageorgiou N, Arnan C, Coutard B, Lantez V, De Lamballerie X, Baronti C, Solà M, Tan J, Neyts J, Canard B, Coll M, Gorbalenya AE, Hilgenfeld R. Picornavirus non‐structural proteins as targets for new anti‐virals with broad activity. Antiviral Res 2011;89:204–218. [DOI] [PubMed] [Google Scholar]
- 154. Su M, Li Y, Leung KT, Cen Y, Li T, Chen R, Ooi VE. Antiviral activity and constituent of Ardisia chinensis benth against coxsackie B3 virus. Phytother Res 2006;20:634–639. [DOI] [PubMed] [Google Scholar]
- 155. Li Y, But PP, Ooi VE. Antiviral activity and mode of action of caffeoylquinic acids from Schefflera heptaphylla (L.) Frodin. Antiviral Res 2005;68:1–9. [DOI] [PubMed] [Google Scholar]
- 156. Liu Q, Wang YF, Chen RJ, Zhang MY, Wang YF, Yang CR, Zhang YJ. Anti‐coxsackie virus B3 norsesquiterpenoids from the roots of Phyllanthus emblica. J Nat Prod 2009;72:969–972. [DOI] [PubMed] [Google Scholar]
- 157. Li Y, Jiang R, Ooi LS, But PP, Ooi VE. Antiviral triterpenoids from the medicinal plant Schefflera heptaphylla. Phytother Res 2007;21:466–470. [DOI] [PubMed] [Google Scholar]
- 158. Zhu H, Zhang Y, Ye G, Li Z, Zhou P, Huang C. In vivo and in vitro antiviral activities of calycosin‐7‐O‐beta‐D‐glucopyranoside against coxsackie virus B3. Biol Pharm Bull 2009;32:68–73. [DOI] [PubMed] [Google Scholar]
- 159. Li Y, Leung KT, Yao F, Ooi LS, Ooi VE. Antiviral flavans from the leaves of Pithecellobium clypearia. J Nat Prod 2006;69:833–835. [DOI] [PubMed] [Google Scholar]
- 160. Tait S, Salvati AL, Desideri N, Fiore L. Antiviral activity of substituted homoisoflavonoids on enteroviruses. Antiviral Res 2006;72:252–255. [DOI] [PubMed] [Google Scholar]
- 161. Sakai A, Watanabe K, Koketsu M, Akuzawa K, Yamada R, Li Z, Sadanari H, Matsubara K, Muroyama T. Anti‐human cytomegalovirus activity of constituents from Sasa albo‐marginata (Kumazasa in Japan). Antivir Chem Chemother 2008;19:125–132. [DOI] [PubMed] [Google Scholar]
- 162. Yazawa K, Tsuchida Y, Yamada R, Sadanari H, Murayama T. Anti‐influenza virus activity by tricin, isolated from Sasa Albo‐marginata in Japan The Twenty‐Third International Conference on Antiviral Research, San Francisco, CA, 25–28 April 2010. Antiviral Res 2010; 86:A49, abstract no 108. [Google Scholar]
- 163. Ninomiya M, Tanaka K, Tsuchida Y, Muto Y, Koketsu M, Watanabe K. Increased bioavailability of tricin‐amino acid derivatives via a prodrug approach. J Med Chem 2011;54:1529–1536. [DOI] [PubMed] [Google Scholar]
- 164. Ogawara H, Akiyama T, Ishida J, Watanabe S, Suzuki K. A specific inhibitor for tyrosine protein kinase from Pseudomonas. J Antibiot 1986;39:606–608. [DOI] [PubMed] [Google Scholar]
- 165. Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine‐specific protein kinases. J Biol Chem 1987;262:5592–5595. [PubMed] [Google Scholar]
- 166. Vela EM, Bowick GC, Herzog NK, Aronson JF. Genistein treatment of cells inhibits arenavirus infection. Antiviral Res 2008;77:153–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Vela EM, Zhang L, Colpitts TM, Davey RA, Aronson JF. Arenavirus entry occurs through a cholesterol‐dependent, non‐caveolar, clathrin‐mediated endocytic mechanism. Virology 2007;369:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Vela EM, Knostman KA, Mott JM, Warren RL, Garver JN, Vela LJ, Stammen RL. Genistein, a general kinase inhibitor, as a potential antiviral for arenaviral hemorrhagic fever as described in the Pirital virus‐Syrian golden hamster model. Antiviral Res 2010;87:318–328. [DOI] [PubMed] [Google Scholar]
- 169. Choi HJ, Lim CH, Song JH, Baek SH, Kwon DH. Antiviral activity of raoulic acid from Raoulia australis against Picornaviruses. Phytomedicine 2009;16:35–39. [DOI] [PubMed] [Google Scholar]
- 170. Pollara JJ, Laster SM, Petty IT. Inhibition of poxvirus growth by Terameprocol, a methylated derivative of nordihydroguaiaretic acid. Antiviral Res 2010;88:287–295. [DOI] [PubMed] [Google Scholar]
- 171. Myskiw C, Piper J, Huzarewich R, Booth TF, Cao J, He R. Nigericin is a potent inhibitor of the early stage of vaccinia virus replication. Antiviral Res 2010;88:304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Alonso‐Caplen FV, Compans RW. Modulation of glycosylation and transport of viral membrane glycoproteins by a sodium ionophore. J Cell Biol 1983;97:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Irurzun A, Sánchez‐Palomino S, Novoa I, Carrasco L. Monensin and nigericin prevent the inhibition of host translation by poliovirus, without affecting p220 cleavage. J Virol 1995;69:7453–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Naesens L, Vanderlinden E, Roth E, Jeko J, Andrei G, Snoeck R, Pannecouque C, Illyés E, Batta G, Herczegh P, Sztaricskai F. Anti‐influenza virus activity and structure‐activity relationship of aglycoristocetin derivatives with cyclobutenedione carrying hydrophobic chains. Antiviral Res 2009;82:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Balzarini J, Pannecouque C, De Clercq E, Pavlov AY, Printsevskaya SS, Miroshnikova OV, Reznikova MI, Preobrazhenskaya MN. Antiretroviral activity of semisynthetic derivatives of glycopeptide antibiotics. J Med Chem 2003;46:2755–2764. [DOI] [PubMed] [Google Scholar]
- 176. Balzarini J, Keyaerts E, Vijgen L, Egberink H, De Clercq E, Van Ranst M, Printsevskaya SS, Olsufyeva EN, Solovieva SE, Preobrazhenskaya MN. Inhibition of feline (FIPV) and human (SARS) coronavirus by semisynthetic derivatives of glycopeptide antibiotics. Antiviral Res 2006;72:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Wachsman MB, López EM, Ramirez JA, Galagovsky LR, Coto CE. Antiviral effect of brassinosteroids against herpes virus and arenaviruses. Antivir Chem Chemother 2000;11:71–77. [DOI] [PubMed] [Google Scholar]
- 178. Wachsman MB, Ramirez JA, Galagovsky LR, Coto CE. Antiviral activity of brassinosteroids derivatives against measles virus in cell cultures. Antivir Chem Chemother 2002;13:61–66. [DOI] [PubMed] [Google Scholar]
- 179. Castilla V, Larzábal M, Sgalippa NA, Wachsman MB, Coto CE. Antiviral mode of action of a synthetic brassinosteroid against Junin virus replication. Antiviral Res 2005;68:88–95. [DOI] [PubMed] [Google Scholar]
- 180. Tanaka N, Okasaka M, Ishimaru Y, Takaishi Y, Sato M, Okamoto M, Oshikawa T, Ahmed SU, Consentino LM, Lee KH. Biyouyanagin A, an anti‐HIV agent from Hypericum chinense L. var. salicifolium. Org Lett 2005;7:2997–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Nicolaou KC, Sanchini S, Sarlah D, Lu G, Wu TR, Nomura DK, Cravatt BF, Cubitt B, de la Torre JC, Hessell AJ, Burton DR. Design, synthesis, and biological evaluation of a biyouyanagin compound library. Proc Natl Acad Sci USA 2011;108:6715–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Droebner K, Pleschka S, Ludwig S, Planz O. Antiviral activity of the MEK‐inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res 2011;92:195–203. [DOI] [PubMed] [Google Scholar]
- 183. Lorusso PM, Adjei AA, Varterasian M, Gadgeel S, Reid J, Mitchell DY, Hanson L, DeLuca P, Bruzek L, Piens J, Asbury P, Van Becelaere K, Herrera R, Sebolt‐Leopold J, Meyer MB. Phase I and pharmacodynamic study of the oral MEK inhibitor CI‐1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–5293. [DOI] [PubMed] [Google Scholar]
- 184. Haura EB, Ricart AD, Larson TG, Stella PJ, Bazhenova L, Miller VA, Cohen RB, Eisenberg PD, Selaru P, Wilner KD, Gadgeel SM. A phase II study of PD‐0325901, an oral MEK inhibitor, in previously treated patients with advanced non‐small cell lung cancer. Clin Cancer Res 2010;16:2450–2457. [DOI] [PubMed] [Google Scholar]
- 185. Pinto R, Herold S, Cakarova L, Hoegner K, Lohmeyer J, Planz O, Pleschka S. Inhibition of influenza virus‐induced NF‐kappaB and Raf‐MEK‐ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antiviral Res 2011, 92:45–56. [DOI] [PubMed] [Google Scholar]
- 186. Ludwig S, Wolff T, Ehrhardt C, Wurzer WJ, Reinhardt J, Planz O, Pleschka S. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett 2004;561:37–43. [DOI] [PubMed] [Google Scholar]
- 187. Ludwig S, Ehrhardt C, Neumeier ER, Kracht M, Rapp UR, Pleschka S. Influenza virus‐induced AP‐1‐dependent gene expression requires activation of the JNK signaling pathway. J Biol Chem 2001;276:10990–10998. [PubMed] [Google Scholar]
- 188. Kujime K, Hashimoto S, Gon Y, Shimizu K, Horie T. p38 mitogen‐activated protein kinase and c‐jun‐NH2‐terminal kinase regulate RANTES production by influenza virus‐infected human bronchial epithelial cells. J Immunol 2000;164:3222–3228. [DOI] [PubMed] [Google Scholar]
- 189. Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol 2001;3:301–305. [DOI] [PubMed] [Google Scholar]
- 190. Jacqué JM, Mann A, Enslen H, Sharova N, Brichacek B, Davis RJ, Stevenson M. Modulation of HIV‐1 infectivity by MAPK, a virion‐associated kinase. EMBO J 1998;17:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Popik W, Pitha PM. Inhibition of CD3/CD28‐mediated activation of the MEK/ERK signaling pathway represses replication of X4 but not R5 human immunodeficiency virus type 1 in peripheral blood CD4(+) T lymphocytes. J Virol 2000;74:2558–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Luo H, Yanagawa B, Zhang J, Luo Z, Zhang M, Esfandiarei M, Carthy C, Wilson JE, Yang D, McManus BM. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal‐regulated kinase (ERK) signaling pathway. J Virol 2002;76:3365–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Cai Y, Liu Y, Zhang X. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol 2007;81:446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhang H, Feng H, Luo L, Zhou Q, Luo Z, Peng Y. Distinct effects of knocking down MEK1 and MEK2 on replication of herpes simplex virus type 2. Virus Res 2010;150:22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Planz O, Pleschka S, Ludwig S. MEK‐specific inhibitor U0126 blocks spread of Borna disease virus in cultured cells. J Virol 2001;75:4871–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Shyu HW, Lin YY, Chen LC, Wang YF, Yeh TM, Su SJ, Cheng WC, Chen CY, Lin KH, Chou MC. he dengue virus envelope protein induced PAI‐1 gene expression via MEK/ERK pathways. Thromb Haemost 2010;104:1219–1227. [DOI] [PubMed] [Google Scholar]
- 197. Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen‐activated protein kinase kinase in vitro and in vivo. J Biol Chem 1995;270:27489–27494. [DOI] [PubMed] [Google Scholar]
- 198. Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen‐activated protein kinase cascade. Proc Natl Acad Sci USA 1995;92:7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Barber SA, Bruett L, Douglass BR, Herbst DS, Zink MC, Clements JE. Visna virus‐induced activation of MAPK is required for virus replication and correlates with virus‐induced neuropathology. J Virol 2002;76:817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Michaelis M, Paulus C, Löschmann N, Dauth S, Stange E, Doerr HW, Nevels M, Cinatl J, Jr . The multi‐targeted kinase inhibitor sorafenib inhibits human cytomegalovirus replication. Cell Mol Life Sci 2011;68:1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Soroceanu L, Akhavan A, Cobbs CS. Platelet‐derived growth factor‐alpha receptor activation is required for human cytomegalovirus infection. Nature 2008;455:391–395. [DOI] [PubMed] [Google Scholar]
- 202. Schleiss M, Eickhoff J, Auerochs S, Leis M, Abele S, Rechter S, Choi Y, Anderson J, Scott G, Rawlinson W, Michel D, Ensminger S, Klebl B, Stamminger T, Marschall M. Protein kinase inhibitors of the quinazoline class exert anti‐cytomegaloviral activity in vitro and in vivo. Antiviral Res 2008;79:49–61. [DOI] [PubMed] [Google Scholar]
- 203. Rodems SM, Spector DH. Extracellular signal‐regulated kinase activity is sustained early during human cytomegalovirus infection. J Virol 1998;72:9173–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Wolf MC, Freiberg AN, Zhang T, Akyol‐Ataman Z, Grock A, Hong PW, Li J, Watson NF, Fang AQ, Aguilar HC, Porotto M, Honko AN, Damoiseaux R, Miller JP, Woodson SE, Chantasirivisal S, Fontanes V, Negrete OA, Krogstad P, Dasgupta A, Moscona A, Hensley LE, Whelan SP, Faull KF, Holbrook MR, Jung ME, Lee B. A broad‐spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci USA 2010;107:3157–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. St Vincent MR, Colpitts CC, Ustinov AV, Muqadas M, Joyce MA, Barsby NL, Epand RF, Epand RM, Khramyshev SA, Valueva OA, Korshun VA, Tyrrell DL, Schang LM. Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc Natl Acad Sci USA 2010;107:17339–17344. [DOI] [PMC free article] [PubMed] [Google Scholar]