

Abstract
Deciphering the evolutionary history and transmission patterns of virulence determinants is necessary to understand the emergence of novel pathogens. The main virulence determinant of most pathogenic proteobacteria is the type three secretion system (T3SS). The Xanthomonas genus includes bacteria responsible for numerous epidemics in agroecosystems worldwide and represents a major threat to plant health. The main virulence factor of Xanthomonas is the Hrp2 family T3SS; however, this system is not conserved in all strains and it has not been previously determined whether the distribution of T3SS in this bacterial genus has resulted from losses or independent acquisitions. Based on comparative genomics of 82 genome sequences representing the diversity of the genus, we have inferred three ancestral acquisitions of the Hrp2 cluster during Xanthomonas evolution followed by subsequent losses in some commensal strains and re‐acquisition in some species. While mutation was the main force driving polymorphism at the gene level, interspecies homologous recombination of large fragments expanding through several genes shaped Hrp2 cluster polymorphism. Horizontal gene transfer of the entire Hrp2 cluster also occurred. A reduced core effectome composed of xopF1, xopM, avrBs2 and xopR was identified that may allow commensal strains overcoming plant basal immunity. In contrast, stepwise accumulation of numerous type 3 effector genes was shown in successful pathogens responsible for epidemics. Our data suggest that capacity to intimately interact with plants through T3SS would be an ancestral trait of xanthomonads. Since its acquisition, T3SS has experienced a highly dynamic evolutionary history characterized by intense gene flux between species that may reflect its role in host adaptation.
Keywords: comparative genomics, homologous recombination, horizontal gene transfer, hrp cluster, pathogen emergence, phylogenomics
1. INTRODUCTION
Unravelling the evolution and transmission of virulence factors is crucial to understanding how pathogens emerge. Type three effectors (T3Es) of Gram‐negative bacteria are major virulence factors in interactions with both plant and animal hosts. These proteins are secreted directly in host cells by the type three secretion system (T3SS), which is a complex protein structure anchored in the bacterial membrane (Diepold & Armitage, 2015). In pathogenic interactions, T3SS may enable the development of disease (via neutralization of plant defences and manipulation of host cellular processes) or trigger host resistance (through recognition inducing hypersensitive response). The T3SS is also widespread in mutualistic and commensal bacteria of protist, fungal and animal hosts (Abby & Rocha, 2012). As the T3SS and T3Es play a crucial role in prokaryote–eukaryote interactions, knowledge of their origin and evolution is likely to be crucial to deepen our understanding of host adaptation.
The origin and evolution of the T3SS has been extensively studied and it has been shown to share a common evolutionary history with the flagellar cluster required for swimming motility. Abby and Rocha (2012) recently proposed that the T3SS evolved by exaptation from the flagellar cluster. Further T3SS diversification then lead to seven distinct families of T3SSs (Ysc, SPI‐1, SPI‐2, Chlamy, Hrp1, Hrp2 and Rhizo) (Troisfontaines & Cornelis, 2005). Diversification of the T3SS is not explained simply by vertical evolution. Indeed, it was shown that the evolution of the T3SS cluster has included numerous horizontal gene transfers (HGTs) (Troisfontaines & Cornelis, 2005). These HGT events are facilitated by localization of T3SS cluster in plasmids or chromosomal pathogenicity islands.
Extensive diversification of the T3SS seems to be driven by bacteria ecology, as T3SS families correlate with the host type. Indeed, Rhizo, Hrp1 and Hrp2 seem to be more frequently involved in interactions with plants, whereas Ysc, Chlamy, SPI‐1 and SPI‐2 appear to be specific to interactions with mammals, insects and amoeba (Abby & Rocha, 2012; Diepold & Armitage, 2015; Troisfontaines & Cornelis, 2005). In general, each bacterial genus harbours a T3SS from a specific family but some bacteria with complex lifestyles harbour several T3SS from different families. These contrasting patterns of T3SS content could be explained by ancient vertical inheritance mixed with T3SS gain and loss events (Kirzinger, Butz, & Stavrinides, 2015). The main T3SS families found in phytopathogens are Hrp1 (in Pseudomonas and Erwinia) and Hrp2 (in Xanthomonas, Ralstonia, Acidovorax and Burkholderia); however, alternative noncanonical T3SS has also been described in some plant‐associated bacteria such as commensal pseudomonads associated with plants (Barret et al., 2013). These two main families of T3SS clusters, Hrp1 and Hrp2, differ in gene content, synteny and transcriptional regulation. Approximately 20 protein‐coding genes, called hrp (hypersensitive reaction and pathogenicity) and hpa (hrp‐associated) genes, are involved in the biogenesis of T3SS. Among them, nine genes, which were renamed hrc (hrp conserved), are highly conserved in plant and animal pathogens, and eight have homologs in the flagellar cluster (Tampakaki et al., 2010).
T3E repertoires are highly diverse within each genus and even within single bacterial species (McCann & Guttman, 2008). They vary both in terms of content and size, for instance a given Xanthomonas axonopodis strain may have any between six and 26 T3E genes (Hajri et al., 2009). It has been suggested that this high variability could be the consequence of the host adaptation process. Indeed, in Pseudomonas and Xanthomonas, the pathogenic strains are highly host specific and the T3E repertoire composition is correlated with host range (Hajri et al., 2009; Sarkar, Gordon, Martin, & Guttman, 2006). The plasticity of T3E repertoire within a species could be explained by frequent HGTs (McCann & Guttman, 2008) as many T3E genes have been found associated with mobile genetic elements. Understanding the diversity and evolution of T3E repertoires in pathogenic bacteria is essential to gain insight into host adaptation mechanisms. However, identification of T3E genes in whole‐genome sequences remains a challenge as T3Es are structurally and functionally highly diverse with more than 50 families identified so far in Xanthomonas and Pseudomonas (Lindeberg, Cunnac, & Collmer, 2012; Ryan et al., 2011). Recently, machine‐learning approaches have been developed. They rely on multiple criteria such as the presence of a secretion signal necessary for recognition by T3SS machinery that is found in N‐terminal region of T3E (McDermott et al., 2011) or specific amino acid composition (Lower & Schneider, 2009). In Xanthomonas, these approaches have enabled the identification of seven novel T3Es in the reference strain 85‐10 (Teper et al., 2016), exhibiting great promise for future discoveries with the exponential growth of genomic data.
Xanthomonas are major plant pathogens, devastating crops worldwide. The major pathogenicity determinants of xanthomonads are the Hrp2 type‐T3SS and its effectors (White, Potnis, Jones, & Koebnik, 2009). Xanthomonas albilineans is an exception in the genus as it has no Hrp2 family gene cluster, but a SPI‐1 T3SS (Marguerettaz et al., 2011). Four Xanthomonas species lacking any Hrp‐T3SSs and associated T3Es, namely Xanthomonas sacchari (Studholme et al., 2011), “Xanthomonas cannabis” (Jacobs, Pesce, Lefeuvre, & Koebnik, 2015), “Xanthomonas pseudalbilineans” (Pieretti et al., 2015) and Xanthomonas maliensis (Triplett et al., 2015) were recently described. Moreover, some Xanthomonas arboricola strains were also found without any T3SS (Cesbron et al., 2015; Merda et al., 2016). The X. arboricola strains lacking any T3SSs are considered commensal, as no pathogenicity on their respective hosts has been observed. The X. arboricola species has an epidemic population structure, where epidemic clones are represented by successful pathovars (defined as pathovars responsible for epidemics worldwide). They infect stone and nut fruit trees. The recombinant network is represented by commensal strains and unsuccessful pathovars (defined by a limited geographical and potentially temporal expansion) (Merda et al., 2016). Epidemic clone emergence seems to be correlated with the acquisition of T3Es, whereas in the recombinant network, strains would have lost T3E‐coding genes and the T3SS cluster.
The recent discovery of several Xanthomonas species and strains that lack the Hrp2 cluster has raised questions about the evolution of virulence and the origin of the T3SS in this genus. Are pathogenicity and Hrp2 clusters ancestral features of Xanthomonas that have been vertically inherited and lost in some species or do they represent more recent acquisitions? Contrasting with our deep knowledge of ancient evolutionary history of T3SS, little is known about recent origin and evolution of T3SS and its role in plant pathogen emergence. Given the pivotal role of T3SS and T3Es in xanthomonads pathogenicity and host specificity, and given their heterogeneous distribution at genus and species scales, the Hrp2 cluster and its effectors in Xanthomonas genus appear to be a good model to study T3SS origin and evolution at a fine evolutionary scale. In this study, we conducted our analyses on a collection of strains representing all valid species from the two phylogenetic groups of the genus (groups 1 and 2) as defined by Young, Park, Shearman, and Fargier (2008). We inferred their phylogenetic relatedness based on the core genome of the whole genus. Moreover, to get insights into T3SS evolution, we studied not only cluster synteny, hrc gene phylogeny and homologous recombination, but we also considered the genomic environment of the T3SS cluster. Finally, to unveil the evolution of T3E repertoires in relation with pathogen emergence, we determined the T3E repertoires in a collection of 44 X. arboricola genomes representing both commensal and pathogenic strains using a machine‐learning approach designed to detect T3E‐coding genes in Xanthomonas genome sequences.
2. MATERIALS AND METHODS
2.1. Genome sequencing and annotation
We used a collection of 82 genome sequences (see Data S1) representing the diversity of Xanthomonas genus (36 strains belonging to Xanthomonas spp.) and the known diversity of Xanthomonas arboricola (44 additional strains; 23 strains being commensal and 21 pathogens) (Merda et al., 2016). Genomes were sequenced using the Illumina technology and HiSeq 2500 (Genoscreen, Lille, France) or MiSeq instruments. Libraries of genomic DNA were performed using the Kit NextEra XT (Illumina, USA). Paired‐end reads of 2 × 100 bp were assembled in contigs using soapdenovo 1.05 (Li et al., 2010) and velvet 1.2.02 (Zerbino & Birney, 2008). Annotation was performed using EuGene‐PP (Sallet, Gouzy, & Schiex, 2014).
2.2. Prediction of T3SS cluster and T3E repertoires
The T3SS‐coding genes representing all T3SS families (Ysc, SPI‐1, SPI‐2, Chlamy, Hrp1, Hrp2, Rhizo) and their diversity were identified in genome sequences using BLASTp searches with the query sequences presented in the Data S2. We included in our search T3SS‐encoding genes from Rhizobiales, Burkholderiales, Ralstonia, Bordetella, Xanthomonas, Pseudomonas, Escherichia coli, Erwinia, Salmonella, Shigella, Yersinia and Chlamydia. Candidate T3SS genes were assigned to a T3SS family when the percentage of identity was higher than 80% on at least 80% of the length of the query sequence. Lower thresholds were used to highlight putative pseudogenes. The T3E gene detection was performed in all genomes of X. arboricola by a machine‐learning approach adapted from Teper et al. (2016) (E. Bosis, unpublished data, manuscript in preparation).
2.3. Genomic environments of genes
The genomic environments flanking and encompassing the T3SS cluster were analysed using the r package genoplotr (Guy, Roat Kultima, & Andersson, 2010). BLASTn between contigs encompassing the T3SS cluster was performed, and only BLAST hits with e‐values below 0.01 were used to highlight conserved regions on the plots. First, this analysis was performed only using strains having a T3SS cluster to detect conserved flanking regions upstream and downstream of the cluster between phylogenetic neighbours. For strains lacking T3SS, a BLASTn search was used to find if regions flanking T3SS in T3SS‐positive strains were also present in T3SS‐negative strains; 5‐Kb T3SS‐flanking regions identified in the closest phylogenetic neighbours of each T3SS‐negative strain were used as query sequences to identify the contig to use in further analyses. To study the synteny in the genomic environments of T3SS insertion site, both contigs from strains with and without T3SS cluster were included in the final analysis. Similar genomic environments of T3SS cluster were defined based on synteny and shared conserved regions spreading over at least four CDS in a 20‐Kb window at the left and right sides of T3SS cluster and using X. arboricola CFBP 7179 as a reference (Fig. S1). Similar genomic environments of the T3SS cluster were highlighted with the same colour as shown in Fig. S1. The same approach but using a 200‐Kb window upstream and downstream the cluster was used to define the genomic context of T3SS insertion site. For the genomic environments of T3E genes, these T3E genes were located by their locus tag obtained during the search with the machine‐learning approach. The same strategy as described above was used to study the genomic environment of avrBs2 insertion site.
2.4. Mapping T3E genes on whole‐genome sequences
To locate the T3E genes in the genomes of X. arboricola pathogenic strains of group A, the contigs of genome sequences were ordered using MAUVE (Darling, Mau, Blattner, & Perna, 2004). The sequence of CFBP 2528 was used as reference because, among group A strains of X. arboricola, the number of contigs was the lowest for this strain (8 contigs). For each genome, the contigs were concatenated using Geneious (Kearse et al., 2012) according to the order obtained with MAUVE. The circular representations were obtained using DNAPlotter (Carver, Thomson, Bleasby, Berriman, & Parkhill, 2009). The localization of each T3E gene in pathogenic strains was identified using their locus tag.
2.5. Determination of core proteomes
The core proteome of Xanthomonas was identified with orthoMCL companion (Carrere, Cottret, Rancurel, & Briand, 2015). The core proteome of X. arboricola was identified with orthomcl Version2.0.9 analyses on predicted full‐length proteins (Li, Stoeckert, & Roos, 2003). OrthoMCL clustering analyses were performed using the following parameters: p‐value cut‐off = 1 × 10−5; per cent match cut‐off = 80; MCL inflation = 1.5; maximum weight = 316.
2.6. Phylogenies of core and T3SS‐coding genes
Phylogenies were performed using maximum likelihood in the phyml software package. The phylogeny of the Xanthomonas genus was performed using the concatenated core proteome obtained with orthoMCL companion. For X. arboricola phylogeny, the concatenated orthologous groups were used. Each orthologous group in X. arboricola was aligned using MACSE (Ranwez, Harispe, Delsuc, & Douzery, 2011). Only alignments with more than 75% sequence identity were kept for the phylogeny reconstruction. These alignments were concatenated using Geneious (Kearse et al., 2012). For organism maximum‐likelihood phylogenies, the JTT model was used. For the T3SS‐coding genes phylogeny, each hrc gene (with the exception of hrcL for which some CDSs were truncated) was aligned using MUSCLE (Edgar, 2004), taking into account sequence translation in proteins to conserve the reading frame. The 10 hrc genes were concatenated according to their order in T3SS cluster. The phylogenic analysis was performed with the GTR + I + gamma model, corresponding to the best model identified by jModelTest (Posada, 2008).
2.7. Phylogeny comparisons and recombination analyses
The topology of the concatenated hrc tree was compared to the core proteome tree of Xanthomonas genus with a Shimodaira–Hasegawa test (Shimodaira & Hasegawa, 1999) implemented in r package phangorn (Schliep, 2011). In the same way, topologies of each individual hrc/hrp tree were compared to each other and to the topology of the concatenated hrc/hrp tree. The impact of recombination (r) relative to mutation (m) was analysed with the ρ/θ statistics using rdp v.3.44 (Martin et al., 2010) for each hrc/hrp gene located in the core region of the cluster (16 genes between hrcC and hrpE). The origins of recombinant sequences were identified by examining the concatenated sequences of hrp and hrc genes (concatenated according to their order in T3SS cluster) using RDP 3, GENECONV, BOOTSCAN, MAXIMUM CHI SQUARE, CHIMAERA, SISCAN and 3SEQ implemented in rdp v. 3.44 (Martin et al., 2010). We considered that a recombination event was statistically supported when it was detected by at least two methods (Merda et al., 2016). The recombination event representation was visualized using Circos (Krzywinski et al., 2009).
3. RESULTS
3.1. Organism‐ and T3SS‐evolutionary histories in Xanthomonas
The presence of T3SS genes was investigated in 82 genome sequences of Xanthomonas strains (Data S1) through BLASTp searches of 295 proteins representing the diversity of T3SS families (Data S2). The Hrp2 cluster was detected in 61 genome sequences, SPI‐1 was identified in the genome sequence of Xanthomonas albilineans, and 20 genome sequences were free of any T3SS‐encoding genes whatsoever, with fourteen of which belonging to Xanthomonas arboricola (12 commensal strains and two strains of the pathovar populi). T3SS clusters were also missing in Xanthomonas pisi and Xanthomonas melonis genomes, and as previously shown in some “Xanthomonas cannabis” strains, Xanthomonas maliensis and the group 1 species Xanthomonas sacchari.
A robust phylogenetic tree of the Xanthomonas genus, based on the core proteome, was constructed to provide a reference point to infer evolutionary scenarios of T3SS gains and losses (Figure 1). According to this phylogenetic reconstruction, X. maliensis and Xanthomonas campestris diverged very early from other species in group 2. This isolated phylogenetic position of X. campestris clade was unexpected as previous multilocus sequence analyses on whole genus diversity placed X. campestris in the core of group 2 (Triplett et al., 2015; Young et al., 2008). However, in a genome‐based phylogeny of a limited number of species a similar phylogenetic relationship has been inferred (Naushad & Gupta, 2013; Rodriguez et al., 2012). Three major clades supported by 100% bootstrap values grouped nearly all other group 2 species: (i) clade A encompassing X. arboricola, Xanthomonas gardneri, Xanthomonas cynarae, Xanthomonas hortorum and Xanthomonas populi; Xanthomonas fragariae appeared as an isolated branch at the base of this clade; (ii) clade B encompassing species of the Xanthomonas axonopodis complex (i.e., Xanthomonas alfalfae, Xanthomonas perforans, Xanthomonas euvesicatoria, Xanthomonas axonopodis, Xanthomonas fuscans and Xanthomonas citri), Xanthomonas oryzae, Xanthomonas vasicola and Xanthomonas bromi, and (iii) clade C encompassing “Xanthomonas cannabis,” Xanthomonas codiaei, Xanthomonas cassavae, Xanthomonas melonis, Xanthomonas cucurbitae, Xanthomonas pisi, Xanthomonas dyei and Xanthomonas vesicatoria (Figure 1). Hrp2‐negative strains were interspersed in the phylogenetic tree of the genus, a distribution pattern that could be either explained by ancestral acquisition and subsequent losses or by numerous recent independent Hrp2 acquisitions.
Figure 1.
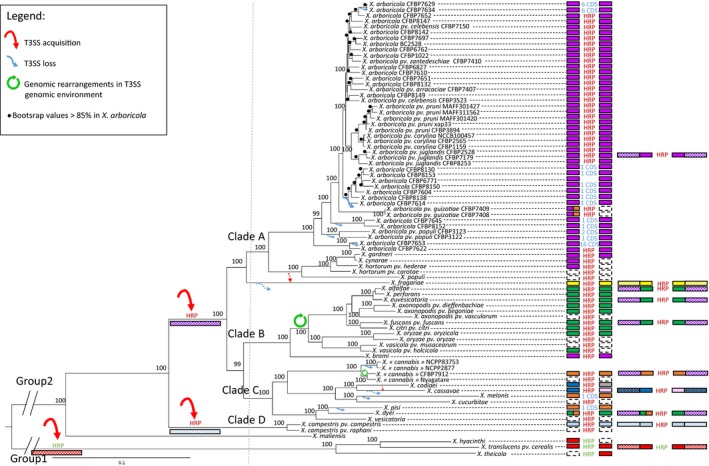
Maximum‐likelihood phylogeny based on the concatenated sequences of the core proteome (993 proteins) of 80 strains representing the entire Xanthomonas genus and schematic representation of T3SS genomic environments. The T3SS cluster is represented by the letters HRP, its genomic environments (20 kb on each side) by coloured rectangles and its genomic contexts (200 kb on each side) by hatched rectangles in the right column. Different colours correspond to different genomic environments or contexts (Fig. S1 and S2). The colour of the letters HRP represents the different cluster organizations; HRP written in red represents the cluster organization found in group 2 xanthomonads and HRP written in green represents the one found in group 1. Dotted line represents absence of information due to contig interruption. In Hrp‐negative strains, HRP letters are replaced by the number of CDS found in place of T3SS cluster at the putative T3SS cluster insertion site. Genomic environments of the insertion site are represented as described above. Probable T3SS acquisition events are represented by red arrow, loss events by blue arrow, and genomic rearrangements by green circled arrow. A dotted arrow represents hypothesis of T3SS loss and re‐acquisition. Bootstrap scores (100 bootstraps) higher than 85% are displayed at each node [Colour figure can be viewed at http://wileyonlinelibrary.com]
A comparison of upstream and downstream genomic environments (20 kb on each side) of the T3SS cluster in the different Xanthomonas species allowed us to define similar genomic environments based on synteny and similarities of DNA fragments (Fig. S1). Strains exhibiting similar flanking regions around the T3SS cluster were considered to have vertically inherited a T3SS cluster following a single acquisition event in their common ancestor. This analysis revealed three ancestral acquisitions of this cluster. One of these acquisitions would have occurred in the ancestor of the three group 2 clades (A, B, C). The same genomic environments of Hrp2 clusters were highlighted in strains belonging to clade A and X. bromi (clade B) (Figures 1 and S1). Given the divergence between these strains, it is tempting to speculate that Hrp2 cluster was acquired through a single acquisition event in a common ancestor. However, except in the case of clade A and X. bromi, the hrp cluster was retrieved in several different genomic environments in clades B and C (Figures 1 and S1). Two scenarios could explain this situation: the first scenario involves the loss of the ancestral Hrp2 cluster and re‐acquisition at a different genomic context, and the second scenario includes rearrangements in the 20‐kb flanking regions of the ancestral Hrp2 cluster without affecting the genomic context of T3SS insertion site. To decipher which scenario is the most probable, genomic contexts of the T3SS insertion site (i.e., broader genomic environments spreading over 200 kb upstream and downstream) were compared in clades B and C strains selected for the quality of their genome assembly (Figures 1 and S2). We showed that rearrangements occurred in the direct flanking regions of T3SS cluster of these strains but that the genomic context of T3SS cluster was similar to those of clade A strains and X. bromi. Insertions of large fragments (80 kb for X. euvesicatoria, X. alfalfae and X. fuscans and 50 kb for “X. cannabis” strain CFBP 7912) in T3SS flanking regions broke synteny in the 20‐kb window but this synteny with the direct genomic environment of X. bromi and clade A was observed further away from T3SS cluster in the clade B and C genome sequences. Thus, genomic rearrangement events and gene insertions affected the 20‐Kb genomic environment of the T3SS cluster but not its location in the genome; the genomic contexts of T3SS cluster remained similar. These rearrangements differed between different clades and were sometimes supported by the presence of insertion sequences (ISs) and tRNAs (Figs S1 and S2). Altogether, these results support the hypothesis that there was an ancestral acquisition of the T3SS cluster in the common ancestor of clades A, B and C and subsequent rearrangements in flanking regions of the Hrp2 cluster (Figure 1).
The second acquisition most likely occurred in X. campestris ancestor. In this clade, the T3SS cluster was found in a different genomic context as no synteny could be found even when flanking regions as broad as 200 kb were compared with those of other Xanthomonas species (Figures 1 and S2). The phylogenetic tree of T3SS‐encoding genes (Figure 2b) showed that X. campestris T3SS genes were phylogenetically related to those of clade A strains. Thus, the common ancestor of X. campestris might have acquired the hrp cluster by HGT from a clade A strain. However, the T3SS‐encoding genes underwent homologous recombination (see below) that altered the phylogenetic signal of the hrp genes. Thus, we cannot exclude the possibility that an ancestral X. campestris hrp cluster would have been replaced by homologous recombination with a clade A strain. Thus, we favour the most parsimonious scenario of an independent acquisition in X. campestris ancestor as opposed to an acquisition by the common ancestor of all group 2 strains followed by the loss of this T3SS cluster in X. campestris and its re‐acquisition in a different genomic context.
Figure 2.
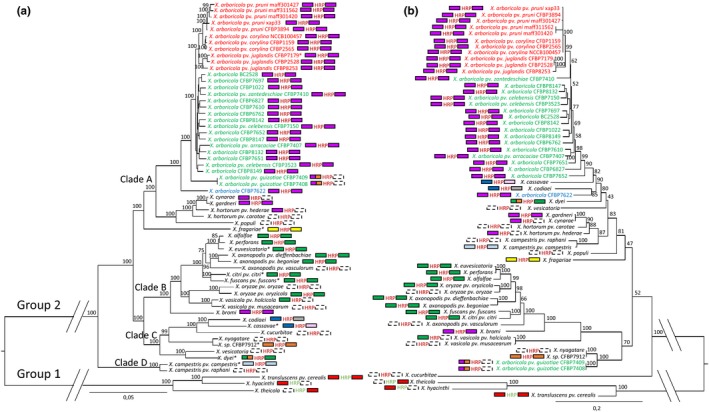
Comparison of organism and T3SS phylogenies and schematic representation of T3SS genomic environments. These two phylogenies were constructed in maximum likelihood. (a) The organism phylogeny is based on the concatenated sequences of the core proteome (1135 proteins) of 61 Xanthomonas spp. strains harbouring a T3SS cluster. (b) The T3SS phylogeny is based on the concatenated sequences of 10 hrc genes. For strains belonging to Xanthomonas arboricola, a colour code was used to represent the three genetic groups previously defined (Merda et al., 2016). Group A strains are indicated in red, group B strains in green and group C strains in blue. Strains of all other species of Xanthomonas are indicated in black. Bootstrap scores (100 bootstraps) higher than 85% are displayed at each node. T3SS clusters and their genomic environments (20 kb on each side of T3SS cluster) are represented as explained in the legend of Figure 1 [Colour figure can be viewed at http://wileyonlinelibrary.com]
The third acquisition of the T3SS cluster would have occurred in group 1. In this case, the T3SS cluster included the genes encoding the master regulators HrpX and HrpG. In contrast, in group 2 species, these two genes were located outside the T3SS cluster. Moreover, all T3SS‐encoding genes are highly divergent from those of group 2 strains (Figures 2b and S3). While the genomic environments of the T3SS clusters among group 1 strains shared similarities, the T3SS genomic context had no similarity to those of group 2 strains (Figures 1, S1 and S2). Altogether, this suggests an independent acquisition event of the T3SS in the common ancestor of Xanthomonas translucens, Xanthomonas hyacinthi and Xanthomonas theicola.
Ancestral acquisitions of T3SS clusters imply that the absence of Hrp2 clusters in the 19 strains of Xanthomonas group 2 was the result of multiple loss events. The X. arboricola strains lacking T3SSs were dispersed in five monophyletic lineages indicating at least five loss events during diversification of this species (Figure 1). One loss event could have occurred in the common ancestor of the two strains belonging to “X. cannabis” and two independent loss events could have occurred in X. melonis and X. pisi. As X. maliensis was the most divergent species of group 2, it was impossible to hypothesize any event responsible for absence of the T3SS cluster, and this species might have never had any T3SS cluster. In the 19 strains without T3SSs, the entire T3SS cluster was missing. No traces of pseudogenization, which could be identified by a weak homology with T3SS‐coding genes with a BLASTn approach, were detected. BLASTn searches with the flanking regions of the Hrp2 cluster in strains lacking it and synteny analysis allowed us to identify the probable excision sites of the DNA fragment containing the Hrp2‐encoding genes (Fig. S1). Excision would have occurred between trpG and ltaE. Nevertheless, we did not find any mobile genetic elements between these loci.
A loss followed by a re‐acquisition of the whole T3SS cluster should have taken place in the evolutionary history of X. fragariae and X. cassavae. Despite their phylogenetic positions in Xanthomonas group 2, the genomic context of T3SS insertion site was different. Indeed, a lack of synteny in T3SS flanking regions was observed among X. fragariae, X. cassavae and other group 2 Xanthomonas species, even when flanking regions as large as 200 kb were considered (Figs S1 and S2). Because these species were localized in two different phylogenetic positions within group 2, this suggests independent re‐acquisition events (Figure 1). However, for X. fragariae, another hypothesis could be put forth. The position of this species is similar in the organism‐ and T3SS phylogenies indicating a probable ancestral T3SS acquisition. In this hypothesis, the different genomic context of T3SS could be the result of a transposition of the cluster within X. fragariae genome. Four ISs were found upstream and downstream of the T3SS cluster in X. fragariae genome that corroborated either transposition or re‐acquisition via HGT as a mechanism. As for strains devoid of the T3SS cluster, we looked at the insertion site of the ancestral T3SS acquisition in clades A, B and C and no remnants of the ancestral T3SS cluster were detected at this location. Xanthomonas codiaei shared the same left border of T3SS cluster as X. cassavae, its sister species, suggesting that the loss and re‐acquisition of T3SS cluster might have occurred in their common ancestor, but unfortunately contig interruption in X. codiaei precluded the analysis of a large genomic environment to confirm this hypothesis.
3.2. Homologous recombination in the T3SS‐coding genes
To determine if the T3SS cluster follows the same evolutionary history as the species, a phylogeny based on concatenated hrc‐coding genes was compared to the organism phylogeny based on the core proteome of Hrp2‐positive strains (Figure 2a and b). Numerous incongruences were observed and confirmed by the SH test (p‐value = .00092). For instance, while most X. arboricola strains exhibited a monophyly for the T3SS‐coding genes, T3SS‐coding genes from the X. arboricola pv. guizotiae diverged from those of other X. arboricola strains (Figure 2b) and they were closely related to those of “X. cannabis” strains CFBP 7912 and Nyagatare. Similar incongruences were observed for X. campestris, X. cassavae, X. codiaei, X. dyei and X. vesicatoria whose T3SS‐coding genes were phylogenetically related to those of clade A despite the high divergence among these species in the organism phylogeny. These incongruences between hrc phylogeny and organism phylogeny can be explained by homologous recombination occurring during T3SS evolution.
To determine if recombination events affected the whole cluster or only some genes, individual phylogenies were built for each hrc/hrp gene coding for the T3SS (Fig. S3). In all phylogenies, X. arboricola pv. guizotiae strains (CFBP 7408 and CFBP 7409) did not group with other X. arboricola strains, but with “X. cannabis” strains CFBP 7912 and Nyagatare, suggesting that their whole T3SS cluster was acquired through a single homologous recombination event with these phylogenetically distant strains. Individual hrc/hrp phylogenies were compared in pairs using a SH test (Table S1). For half of the comparisons, the p‐values were below 0.05, indicating that most topologies of these trees were significantly different and that recombination occurred between hrc genes. For instance, T3SS‐coding genes from X. campestris clustered with genes from clades A and C (Figure 2b and Fig. S3) that did not reflect the intermediate position of X. campestris between group 1 and group 2 in the organism phylogeny (Figure 2a). In contrast to clade B strains, which clustered together in most hrc phylogenies, strains from clades A and C were interspersed. This suggested numerous HGTs between these two latter clades.
To characterize the gene flow affecting the T3SS cluster in Xanthomonas spp. strains, potential recombinant sequences and their likely parental sequences were detected based on phylogenetic incongruences (Martin et al., 2010). Identification of the likely origin of the recombinant fragment can be achieved if at least one sequence resembling the donor sequence is present in the data set. The identified exchanges concerned the entire sequence of one or two adjacent genes in the concatenated hrc genes. The two genes for which the number of exchanges were the highest were hrpE and hrpB2 (32 and 26 events, respectively) and the two genes for which the smallest number of exchange events were observed were hrcC and hrcT (3 and 0 events, respectively). Xanthomonas arboricola strains were the main recipients of recombination events (Figure 3a). They mostly received genes from X. dyei and X. hortorum pv. hederae. Notably, most exchanges were detected within the recombinant network of X. arboricola and epidemic clones gave hrp/hrc alleles to strains belonging to this network, but no reverse events were detected. In contrast, only two X. arboricola strains were donors for other species (CFBP 1022 was donor for X. cassavae and CFBP 8149 for X. hortorum, X. gardneri and X. cynarae). T3SS gene exchanges were also detected between strains of the X. axonopodis species complex but remarkably no gene flow occurred between this clade and other clades.
Figure 3.
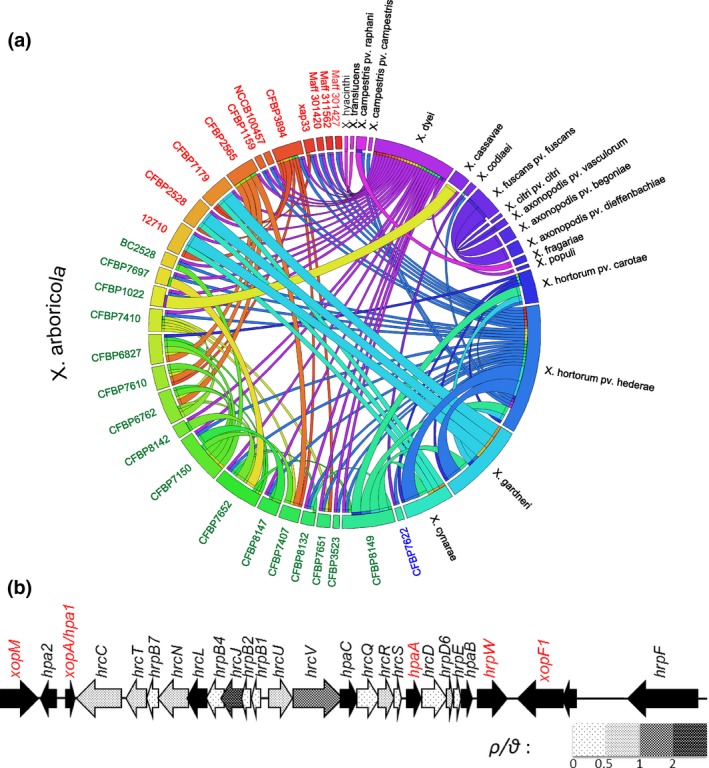
Gene flow affecting the T3SS cluster. (a) Representation of recombination events affecting 16 hrc/hrp genes of the T3SS cluster in Xanthomonas genus. The donor and recipient strains were identified with rdp software, and the representation was obtained using Circos. Each strain is represented by a rectangle of a different colour. The recombination events are represented by a link between donor and recipient strains. The colour of the link corresponds to the colour of the donor and indicates the direction of recombination event. The width of the links represents the number of genes involved in the recombination events. For strains belonging to Xanthomonas arboricola a colour code was used to represent the three groups previously defined (Merda et al., 2016). Group A strains are indicated in red, group B strains in green, and group C strains in blue. (b) Representation of ratios of recombination rate vs. mutation rate (ρ/θ) along the T3SS cluster using 61 genomes representing the genus diversity. Ratios were calculated for the 16 hrc/hrp genes of the core region of T3SS cluster using rdp software. Arrows are shaded according to the shading scale which indicates the range of the ρ/θ value. The black arrows represent hpa and T3E genes for which ρ/θ was not calculated. Genetic organization of the cluster is based on the sequence of CFBP 2528. In red are represented the five core T3E genes located in the T3SS cluster of X. arboricola strains [Colour figure can be viewed at http://wileyonlinelibrary.com]
For each individual T3SS‐coding gene, we estimated the evolutionary force responsible for observed polymorphism using the ρ/θ ratio. For most genes (14 of 16), mutation had more impact than recombination on generating new alleles (ρ/θ < 1) (Figure 3b). Only two genes had a ρ/θ ratio above one, hrcJ (ρ/θ > 2) and hrcV (1 > ρ/θ > 2). In conclusion, within hrc/hrp genes, mutation was the major evolutionary force that have brought polymorphism and generated allelic variants at gene scale. This polymorphism was disseminated across the genus through homologous recombination of entire genes or contiguous genes.
3.3. T3E repertoires in X. arboricola
To decipher the diversity of T3E repertoires in X. arboricola, T3E‐coding genes were predicted for all genome sequences belonging to this species by a machine‐learning approach dedicated to Xanthomonas organisms. Briefly, T3E‐encoding genes were searched for based on criteria referring to type three N‐terminal secretion signal, structural disorder, regulation by HrpX/HrpG, GC content, codon usage, amino acid properties and homology to known and validated T3Es. Based on this prediction, a set of seven ancestral core T3E genes was observed. The predicted T3E repertoires were highly variable with some strains having no T3Es and others having up to 34 predicted T3Es (Figure 4). Eight of the 14 strains lacking the T3SS cluster were also deprived of T3E‐coding genes. In contrast, between one and two T3E genes (avrBs2 and xopR) were identified in the remaining six strains (Data S3). While synteny in the xopR genomic environments has previously been shown (Merda et al., 2016), here we observed that genomic environments of avrBs2 were also highly syntenic between all strains, with the exception of strains CFBP 7408 and CFBP 7409 (Fig. S4), favouring an ancestral acquisition and subsequent losses of these T3Es during X. arboricola evolution. The two strains of pathovar guizotiae, CFBP 7408 and CFBP 7409, probably lost and reacquired avrBs2. It has to be noted that avrBs2 was systematically accompanied by three CDSs, namely xylR, a TonB‐dependent receptor and a hypothetical protein that presented the same distribution in our collection whatever the genomic context (see Fig. S4). In addition to xopR and avrBs2, five other T3E genes (already described in Xanthomonas strains) were found in all X. arboricola strains having a T3SS. These five T3E genes were located within the T3SS cluster (Figure 3b) and their distribution strictly followed the distribution of the T3SS cluster. However, among them were XopA, HpaA and HrpW, which, while listed as T3Es in some studies (Hajri et al., 2009; Merda et al., 2016), are secreted regulator (HpaA) or hairpin‐like proteins and their effector function remains unclear (Lorenz et al., 2008; White et al., 2009). Synteny in the flanking regions of avrBs2, xopR, and of the five T3E genes associated with the T3SS cluster suggested that they were most probably acquired by an ancient X. arboricola strain; thus, these seven T3E genes will be designated as the ancestral repertoire thereafter.
Figure 4.
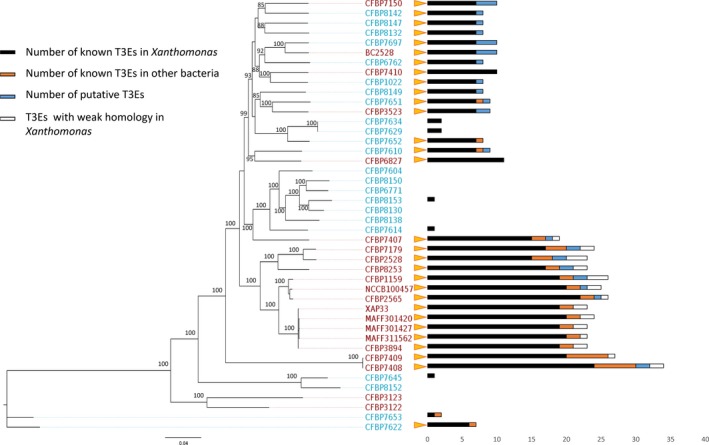
Representation of T3E repertoires in Xanthomonas arboricola strains. The phylogeny was performed in maximum likelihood using the concatenated sequences of 1705 CDS composing the core genome of these 44 strains. Bootstrap values (100 bootstraps) higher than 85% are indicated at each node. Pathogenic strains are represented in red and commensal ones in blue. At the tip of each branch, the orange triangles represent the presence of T3SS cluster and bars represent the composition of the T3E repertoire according to the legend [Colour figure can be viewed at http://wileyonlinelibrary.com]
Pathogenic strains had a higher number of predicted T3E genes than commensal strains (Figure 4). However, pathogenic strains CFBP 3122 and CFBP 3123 of pathovar populi lacked T3SS and T3E genes and hence represented an exception. Therefore, the pathogenicity of these bacteria, previously qualified as opportunistic pathogens (Haworth & Spiers, 1992), may rely on different virulence factors. The T3E repertoire of pathogenic strains encompassed the ancestral repertoire and a large number of additional predicted T3E genes. Indeed, the pan‐T3 effectome of X. arboricola was composed of 57 predicted T3Es, and among them, only 11 were found in both pathogenic and commensal strains, with the 46 others present exclusively in pathogenic strains. Most putative T3E genes identified in X. arboricola were already described in other Xanthomonas spp. This is the case for the seven T3E genes of the ancestral repertoire and 31 other T3E genes (Data S3). Among the 19 remaining T3E genes composing the pan‐effectome, seven were known in other bacteria (Ralstonia and Pseudomonas) and 12 were putative novel T3E genes. Among these putative novel T3Es, six (T3E_14 to T3E_19) had a weak similarity (less than 30% of sequence similarity) to T3E genes known in Xanthomonas (xopAH, xopJ1, xopAO, xopAV, xopG and xopM, respectively) and thus are not considered as orthologous of these genes, but they could share a common ancestor.
The three successful pathovars (pvs. pruni, corylina and juglandis) shared ten T3E genes that were sequentially acquired in their common ancestor. These ten T3E genes were xopX, xopV, xopL, xopK, xopN, xopAV, xopQ, avrXccA2, xopZ and T3E_16, which shared 27.2% sequence similarity with xopJ1. To determine if their ancestral acquisition resulted from one or several events, we analysed their genomic context. Contig alignment using CFBP 2528 as reference revealed that these T3E genes were dispersed along the chromosome, except xopAV and xopQ, which were colocalized (Figs S5 and S6). The dispersal of these T3E genes along the genome sequences suggested that they were acquired following several acquisition events. Given the synteny observed in the flanking regions of each of these T3E genes in the genomes of the successful pathovars (Fig. S5), it is likely that these independent acquisition events probably occurred in their ancestor before separation into three distinct pathovars.
4. DISCUSSION
The acquisition and evolution of the T3SS have played major roles in ecological adaptation of pathogens, and HGT has been a driving force in T3SS evolutionary history at multiple evolutionary timescales. We investigated T3SS evolution in the Xanthomonas genus, a major clade of plant pathogens. Comparative genomic analyses of a collection of 82 strains allowed us to infer three ancestral acquisitions of the Hrp2 gene cluster during Xanthomonas evolution, two in group 2 strains and one in group 1 strains. Indeed, we highlighted an ancestral acquisition in the common ancestor of all group 2 species excluding Xanthomonas campestris. This species, which diverged early in group 2, has a T3SS cluster at a different chromosomal location, supporting an independent acquisition event. The third ancestral T3SS acquisition occurred in group 1. A different genetic organization of the T3SS cluster, a high divergence among T3SS‐coding genes from the group 2 species, and the different genomic contexts of the T3SS clusters all indicate that group 1 strains probably acquired a different Hrp2 cluster independently as previously proposed by Jacobs et al. (2015). Before this study, Xanthomonas translucens was the only group 1 species known to harbour a Hrp2 cluster (Wichmann et al., 2013). Our results indicate that this atypical Hrp2 cluster is shared with Xanthomonas theicola and Xanthomonas hyacinthi and that it was probably acquired by their common ancestor. T3SS cluster acquisitions occurred before speciation of most xanthomonads; capacity to interact with plants through translocation of T3Es would thus be an ancient trait of xanthomonads as it is the case for other important plant bacterial pathogens (Diallo et al., 2012; Kirzinger et al., 2015).
After ancestral acquisition, the T3SS was lost in some strains and species scattered throughout in the Xanthomonas phylogenetic tree (Figure 1). The scattering of strains without T3SS in the tree, the conservation of similar genomic environments in the T3SS‐positive strains, and a similar genome sequence in strains without T3SS are three lines of evidence in favour of the T3SS loss hypothesis. Absence of pseudogenes or remnants of T3SS‐encoding genes might be surprising, but a similar observation was made in nonpathogenic Pseudomonas syringae strains, from which the entire cluster has been excised (Mohr et al., 2008). The loss hypothesis in commensal strains of Xanthomonas arboricola species was previously proposed (Merda et al., 2016) based on Bayesian inference of gene gains and losses. Such complex scenarios with ancestral acquisition, losses and regains have also been proposed in Pantoea genus (Kirzinger et al., 2015) and P. syringae (Clarke, Cai, Studholme, Guttman, & Vinatzer, 2010).
Losses of T3SS could be explained by a loss of function (Abby & Rocha, 2012). Indeed, it could be beneficial to lose this energetically costly machinery if it does not enhance bacterial fitness (Gophna, Ron, & Graur, 2003). Thus, for commensal strains colonizing various plant hosts and with a limited set of T3Es (like X. arboricola group B strains) (Merda et al., 2016), the fitness cost provided by T3SS might be high and consequently it could be lost. T3SS‐negative strains may also act as profiteers and benefit from the presence of T3SS‐positive strains colonizing the same niche as demonstrated in murine infections by Pseudomonas aeruginosa (Czechowska, McKeithen‐Mead, Al Moussawi, & Kazmierczak, 2014).
Once acquired, we showed that T3SS‐coding genes were prone to homologous recombination events leading to replacement of large fragments encompassing one complete gene, adjacent genes or even the entire cluster. The two genes for which the number of recombination events was the highest were hrpE and hrpB2, which encode the Hrp pilin and the putative inner rod, respectively (Hartmann et al., 2012). These two proteins correspond to the early substrates of the secretion machinery. Weber and Koebnik (2006) observed positive diversifying selection in the hrpE sequence corresponding to the surface exposed part of the protein and interpreted it as an adaptative mechanism of the pathogen to escape recognition by the host. Homologous replacement of the hrpE gene by recombination could be an alternative mechanism to generate diversity and to escape host recognition. This latter mechanism has been extensively described in the mammalian pathogen Neisseria gonorrhoeae where it drives antigenic variation of the type IV pilus and avoidance of the host immune system (Obergfell & Seifert, 2015). The two genes that showed the fewest exchanges, hrcC and hrcT, encode highly conserved proteins located in the basal structure of the secretion system and embedded in the bacterial envelope. Within each gene, allelic polymorphism is mostly generated by mutation, except for hrcJ and hrcV (Figure 3b). The study of genomic environment of the T3SS allowed us to distinguish two mechanisms of HGT: acquisition of a new cluster in a different chromosomal context as previously discussed and homologous recombination within T3SS cluster. One homologous recombination event leading to entire T3SS cluster replacement was shown between X. arboricola pv. guizotiae and the phylogenetically distant strains CFBP 7912 and Nyagatare. Interestingly, all these strains originated from south‐east Africa and were isolated from two crops (Niger seed and bean) used in mixed crop‐livestock farming systems and as intercrops in maize production (Abera, Feyisa, & Friesen, 2009), making their co‐occurrence plausible. Beside this single whole Hrp2 cluster replacement, numerous localized homologous recombination events between strains of different species have perturbed the vertical inheritance signal. Similarly, conflicts between phylogenies of some hrp/hrc genes and of housekeeping genes were observed in pseudomonads and enterobacterial plant pathogens (Sarris et al., 2013; Tegli, Gori, Cerboneschi, Cipriani, & Sisto, 2011) and a HGT event expanding through several hrp genes was previously suggested in pseudomonads (Sarris et al., 2013).
Understanding gene flow within and between populations sheds light on bacterial ecology. The study of “donor” and “recipient” strains of recombinant fragments showed that X. arboricola strains were the main “recipient,” particularly in commensal strains, and Xanthomonas dyei and Xanthomonas hortorum were the two main donors (Figure 3a). This suggests that commensal strains are found in sympatry with a large number of different Xanthomonas species because genetic material exchanges can only take place when individuals colonize the same niche. X. dyei and X. arboricola strains were isolated from the endemic species Dysoxylum spectabile in New Zealand (Young, Wilkie, Park, & Watson, 2010). These observations reinforce the hypothesis proposed by Merda et al. (2016) that the commensal strains in X. arboricola are generalist organisms colonizing many different plants. In contrast, no gene flow at T3SS locus occurred between clade B strains and other clades. Divergent evolution of the T3SS cluster in this clade may have led to an optimized allelic combination. This result reinforces the major role of T3SS in this important clade of devastating host specialized pathogens within which the presence of the T3SS is conserved.
The function of T3SS is to deliver T3Es into host cells. In most strains devoid of T3SSs, no T3E genes could be detected in their genomes using machine‐learning approach and BLASTp (data not shown). Indeed, some T3E genes are housed in the T3SS cluster (Figure 3b) and thus were lost with it. xopR and avrBs2, which are not located in T3SS cluster, were found in the genomes of some commensal Xanthomonas strains lacking T3SS. Their conserved genomic environments, when compared to strains with T3SSs, suggest that they are remnants of an ancestral T3E repertoire (Fig. S4) (Merda et al., 2016). A recent loss of the T3SS could explain why the T3Es were present despite the lack of the T3SS. Alternatively, xopR and avrBs2 secretion might be mediated by the flagellum apparatus as demonstrated for some nonflagellar proteins (Journet, Hughes, & Cornelis, 2005). They might also have an additional function independent of the T3SS.
We have highlighted an extremely reduced ancestral core repertoire and stepwise acquisition of numerous additional T3Es in pathogenic strains of X. arboricola. Five of the seven core T3E genes were located in the T3SS cluster as previously observed in other Xanthomonas species (Noel, Thieme, Nennstiel, & Bonas, 2002; Potnis et al., 2011; da Silva et al., 2002; Teper et al., 2016). Among them, XopA, HpaA and HrpW should be better considered as accessory or translocation proteins that help the translocation process (Lorenz et al., 2008; Roux et al., 2015; White et al., 2009). Taking this into account, the X. arboricola core effectome comprises only four T3E genes (xopF1, xopM, avrBs2 and xopR) and is comparable in size to that of X. campestris (Roux et al., 2015). Together, these results challenge the list of ten core T3E genes (avrBs2, xopF1, xopK, xopL, xopN, xopP, xopQ, xopR, xopX, xopZ) previously proposed (Ryan et al., 2011; White et al., 2009). xopM, missing in this list, was recently shown to be a T3E gene of Xanthomonas euvesicatoria strain 85‐10 (Schulze et al., 2012; Teper et al., 2016). Our BLASTp searches showed that it is present in most group 2 Xanthomonas species (data not shown). Considering that xopR and avrBs2 were missing in only one and two strains, respectively, of 13 Xanthomonas campestris (Roux et al., 2015), we propose a list of four putative core Xanthomonas T3Es: AvrBs2, XopF1, XopM and XopR. Interestingly, AvrBs2 contributes to bacterial fitness in field conditions, including epiphytic survival (Wichmann & Bergelson, 2004). It is required for full aggressiveness both in dicots and monocots and was shown to inhibit pathogen‐associated molecular pattern‐triggered immune (PTI) responses in rice (Li et al., 2015; Zhao, Dahlbeck, Krasileva, Fong, & Staskawicz, 2011). Similarly, XopM inhibits immunity‐associated cell death mediated by MAP kinase cascades (Teper, Sunitha, Martin, & Sessa, 2015) and XopR inhibits plant basal defences (Akimoto‐Tomiyama et al., 2012).
Besides the reduced ancestral core T3E repertoire, stepwise accumulation of additional T3Es has occurred in pathogenic strains and particularly in successful pathovars of X. arboricola. This accumulation appears to be a long‐term evolutionary process as many T3Es were acquired before the radiation of the three successful pathovars. At the basal steps of pathogen emergence, accumulation of numerous T3Es including XopL, XopN, XopQ, XopX and XopZ occurred. These were shown to target PTI in addition to the ancestral T3Es, which are also involved in PTI suppression. This reinforces the idea that PTI suppression is crucial for pathogenic strains to achieve successful infection (Macho & Zipfel, 2015).
In conclusion, we showed three ancestral acquisitions of the Hrp2 cluster demonstrating that an intimate interaction with plants is an ancestral trait of xanthomonads. During radiation, most species retained this ancestral T3SS, but some lost it and subsequently it was re‐acquired in some strains. Mutation is the main evolutionary force generating new hrc/hrp alleles. In group 2, Xanthomonas, the inter‐ and intraspecies homologous recombination of large fragments expanding through one or more genes shuffles this polymorphism generating new allelic combinations in Hrp2 clusters. A set of four ancestral core T3E genes is found in commensal strains and pathogens in X. arboricola that may approximate the Xanthomonas ancestral core effectome. We propose that these may allow the strains to overcome basal plant immunity under specific environmental conditions, but could have a fitness cost explaining why they were lost in some strains. In contrast, some strains experienced a different evolutionary pathway with stepwise accumulation of T3Es that probably accounts for their efficacy to overcome plant immunity and could explain the high aggressiveness. X. arboricola represents the archetype of this evolutionary scenario, which seems to share similarities with the one proposed for P. syringae (Lindeberg et al., 2012) and culminates in a narrow host range.
DATA ACCESSIBILITY
The whole‐genome sequences obtained for this project have been deposited in GenBank. Accession nos. are available in Data S1. Most bacterial strains used in this study are available at the microbial resource centre CIRM‐CFBP (Beaucouzé, INRA, France; http://www6.inra.fr/cirm_eng/CFBP-Plant-Associated-Bacteria).
AUTHOR CONTRIBUTIONS
This study is part of the doctoral research of D.M. supervised by M.A.J. and M.F.L.S. Funds for genome sequencing were obtained by M.A.J., M.F.L.S and P.P. M.Ba. supervised genome sequencing in ANAN platform (Angers, France). D.M. conducted bioinformatics analyses in collaboration with M.Br., C.R. and E.B. E.B. designed the machine‐learning method. D.M., M.F.L.S and M.A.J. analysed, interpreted the data and wrote the manuscript with input from all co‐authors. All authors read and approved the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank Geraldine Taghouti for DNA extraction, ANAN platform for genome sequencing and Jérôme Gouzy and Sébastien Carrère for genome assembling and annotations. We thank CIRM‐CFBP (Beaucouzé, INRA, France; http://www6.inra.fr/cirm_eng/CFBP-Plant-Associated-Bacteria) for strain preservation and supply. Inn‐Shik Myung is acknowledged for providing a strain from Korea and Lionel Gagnevin for providing unpublished data. Jason Shiller is acknowledged for editing the English. This work was supported by the French Agence Nationale de la Recherche (grant number ANR‐2010‐GENM‐013); Institut National de la Recherche Agronomique (INRA) (AIP Bioresources project “Taxomic”); and INRA Plant Health and Environment Division and the regional government of the Pays de la Loire (doctoral fellowship of Déborah Merda). We thank Ralf Koebnik for his comments on the manuscript and members of the French Network of xanthomonads (FNX) for fruitful discussions. This work benefited from interactions promoted by COST Action FA 1208 (https://www.cost-sustain.org).
Merda D, Briand M, Bosis E, et al. Ancestral acquisitions, gene flow and multiple evolutionary trajectories of the type three secretion system and effectors in Xanthomonas plant pathogens. Mol Ecol. 2017;26:5939–5952. 10.1111/mec.14343
Contributor Information
Marie‐Agnès Jacques, Email: marie-agnes.jacques@inra.fr.
Marion Fischer‐Le Saux, Email: marion.le-saux@inra.fr.
REFERENCES
- Abby, S. S. , & Rocha, E. P. C. (2012). The non‐flagellar type III secretion system evolved from the bacterial flagellum and diversified into host‐cell adapted systems. Plos Genetics, 8(9), e1002983 10.1371/journal.pgen.1002983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abera, T. , Feyisa, D. , & Friesen, D. K. (2009). Effects of crop rotation and N‐P fertilizer rate on grain yield and related characteristics of maize and soil fertility at Bako, Western Oromia, Ethiopia. East African Journal of Sciences, 3(1), 70–79. [Google Scholar]
- Akimoto‐Tomiyama, C. , Furutani, A. , Tsuge, S. , Washington, E. J. , Nishizawa, Y. , Minami, E. , & Ochiai, H. (2012). XopR, a type III effector secreted by Xanthomonas oryzae pv. oryzae, suppresses microbe‐associated molecular pattern‐triggered immunity in Arabidopsis thaliana . Molecular Plant‐Microbe Interactions, 25(4), 505–514. 10.1094/MPMI-06-11-0167 [DOI] [PubMed] [Google Scholar]
- Barret, M. , Egan, F. , Moynihan, J. , Morrissey, J. P. , Lesouhaitier, O. , & O'Gara, F. (2013). Characterization of the SPI‐1 and Rsp type three secretion systems in Pseudomonas fluorescens F113. Environmental Microbiology Reports, 5(3), 377–386. 10.1111/1758-2229.12039 [DOI] [PubMed] [Google Scholar]
- Carrere, S. , Cottret, L. , Rancurel, C. , & Briand, M. (2015). Orthomcl‐companion: A user friendly tool to analyze protein families [version 1; not peer reviewed]. F1000Research, 4, 489 10.7490/f1000research.1110226.1 [DOI] [Google Scholar]
- Carver, T. , Thomson, N. , Bleasby, A. , Berriman, M. , & Parkhill, J. (2009). DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics, 25(1), 119–120. 10.1093/bioinformatics/btn578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesbron, S. , Briand, M. , Essakhi, S. , Gironde, S. , Boureau, T. , Manceau, C. , … Jacques, M.‐A. (2015). Comparative genomics of pathogenic and nonpathogenic strains of Xanthomonas arboricola unveil molecular and evolutionary events linked to pathoadaptation. Frontiers in Plant Science, 6, 1126 10.3389/fpls.2015.01126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, C. R. , Cai, R. M. , Studholme, D. J. , Guttman, D. S. , & Vinatzer, B. A. (2010). Pseudomonas syringae strains naturally lacking the classical P. syringae hrp/hrc locus are common leaf colonizers equipped with an atypical type III secretion system. Molecular Plant‐Microbe Interactions, 23(2), 198–210. 10.1094/mpmi-23-2-0198 [DOI] [PubMed] [Google Scholar]
- Czechowska, K. , McKeithen‐Mead, S. , Al Moussawi, K. , & Kazmierczak, B. I. (2014). Cheating by type 3 secretion system‐negative Pseudomonas aeruginosa during pulmonary infection. Proceedings of the National Academy of Sciences, 111(21), 7801–7806. 10.1073/pnas.1400782111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling, A. C. , Mau, B. , Blattner, F. R. , & Perna, N. T. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Research, 14(7), 1394–1403. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo, M. D. , Monteil, C. L. , Vinatzer, B. A. , Clarke, C. R. , Glaux, C. , Guilbaud, C. , … Morris, C. E. (2012). Pseudomonas syringae naturally lacking the canonical type III secretion system are ubiquitous in nonagricultural habitats, are phylogenetically diverse and can be pathogenic. ISME Journal, 6(7), 1325–1335. 10.1038/ismej.2011.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepold, A. , & Armitage, J. P. (2015). Type III secretion systems: The bacterial flagellum and the injectisome. Philosophical Transactions of the Royal Society B: Biological Sciences, 370(1679), 10.1098/rstb.2015.0020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gophna, U. , Ron, E. Z. , & Graur, D. (2003). Bacterial type III secretion systems are ancient and evolved by multiple horizontal‐transfer events. Gene, 312, 151–163. 10.1016/S0378-1119(03)00612-7 [DOI] [PubMed] [Google Scholar]
- Guy, L. , Roat Kultima, J. , & Andersson, S. G. E. (2010). genoPlotR: Comparative gene and genome visualization in R. Bioinformatics, 26(18), 2334–2335. 10.1093/bioinformatics/btq413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajri, A. , Brin, C. , Hunault, G. , Lardeux, F. , Lemaire, C. , Manceau, C. , … Poussier, S. (2009). A “Repertoire for Repertoire” hypothesis: Repertoires of type three effectors are candidate determinants of host specificity Xanthomonas . PLoS ONE, 4(8), 10.1371/journal.pone.0006632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, N. , Schulz, S. , Lorenz, C. , Fraas, S. , Hause, G. , & Büttner, D. (2012). Characterization of HrpB2 from Xanthomonas campestris pv. vesicatoria identifies protein regions that are essential for type III secretion pilus formation. Microbiology, 158(5), 1334–1349. 10.1099/mic.0.057604-0 [DOI] [PubMed] [Google Scholar]
- Haworth, R. H. , & Spiers, A. G. (1992). Isolation of Xanthomonas campestris pv. populi from stem lesions on Salix matsudana X alba ‘Aokautere’ in New Zealand. European Journal of Forest Pathology, 22(4), 247–251. 10.1111/j.1439-0329.1992.tb00789.x [DOI] [Google Scholar]
- Jacobs, J. M. , Pesce, C. , Lefeuvre, P. , & Koebnik, R. (2015). Comparative genomics of a cannabis pathogen reveals insight into the evolution of pathogenicity in Xanthomonas . Frontiers in Plant Science, 6(431), 10.3389/fpls.2015.00431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Journet, L. , Hughes, K. T. , & Cornelis, G. R. (2005). Type III secretion: A secretory pathway serving both motility and virulence (Review). Molecular Membrane Biology, 22(1–2), 41–50. 10.1080/09687860500041858 [DOI] [PubMed] [Google Scholar]
- Kearse, M. , Moir, R. , Wilson, A. , Stones‐Havas, S. , Cheung, M. , Sturrock, S. , … Drummond, A. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28(12), 1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirzinger, M. W. , Butz, C. J. , & Stavrinides, J. (2015). Inheritance of Pantoea type III secretion systems through both vertical and horizontal transfer. Molecular Genetics and Genomics, 290(6), 2075–2088. 10.1007/s00438-015-1062-2 [DOI] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , … Marra, M. A. (2009). Circos: An information aesthetic for comparative genomics. Genome Research, 19(9), 1639–1645. 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C. J. Jr , & Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Research, 13(9), 2178–2189. 10.1101/gr.1224503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Wang, Y. , Wang, S. , Fang, A. , Wang, J. , Liu, L. , … Sun, W. (2015). The type III effector AvrBs2 in Xanthomonas oryzae pv. oryzicola suppresses rice immunity and promotes disease development. Molecular Plant‐Microbe Interactions, 28(8), 869–880. 10.1094/mpmi-10-14-0314-r [DOI] [PubMed] [Google Scholar]
- Li, R. , Zhu, H. , Ruan, J. , Qian, W. , Fang, X. , Shi, Z. , … Wang, J. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Research, 20(2), 265–272. 10.1101/gr.097261.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeberg, M. , Cunnac, S. , & Collmer, A. (2012). Pseudomonas syringae type III effector repertoires: Last words in endless arguments. Trends in Microbiology, 20(4), 199–208. 10.1016/j.tim.2012.01.003 [DOI] [PubMed] [Google Scholar]
- Lorenz, C. , Kirchner, O. , Egler, M. , Stuttmann, J. , Bonas, U. , & Büttner, D. (2008). HpaA from Xanthomonas is a regulator of type III secretion. Molecular Microbiology, 69(2), 344–360. 10.1111/j.1365-2958.2008.06280.x [DOI] [PubMed] [Google Scholar]
- Lower, M. , & Schneider, G. (2009). Prediction of type III secretion signals in genomes of gram‐negative bacteria. PLoS ONE, 4(6), e5917 10.1371/journal.pone.0005917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macho, A. P. , & Zipfel, C. (2015). Targeting of plant pattern recognition receptor‐triggered immunity by bacterial type‐III secretion system effectors. Current Opinion in Microbiology, 23, 14–22. 10.1016/j.mib.2014.10.009 [DOI] [PubMed] [Google Scholar]
- Marguerettaz, M. , Pieretti, I. , Gayral, P. , Puig, J. , Brin, C. , Cociancich, S. , … Royer, M. (2011). Genomic and evolutionary features of the SPI‐1 type III secretion system that is present in Xanthomonas albilineans but is not essential for xylem colonization and symptom development of sugarcane leaf scald. Molecular Plant‐Microbe Interactions, 24(2), 246–259. 10.1094/MPMI-08-10-0188 [DOI] [PubMed] [Google Scholar]
- Martin, D. P. , Lemey, P. , Lott, M. , Moulton, V. , Posada, D. , & Lefeuvre, P. (2010). RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics, 26(19), 2462–2463. 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann, H. C. , & Guttman, D. S. (2008). Evolution of the type III secretion system and its effectors in plant‐microbe interactions. New Phytologist, 177(1), 33–47. 10.1111/j.1469-8137.2007.02293.x [DOI] [PubMed] [Google Scholar]
- McDermott, J. E. , Corrigan, A. , Peterson, E. , Oehmen, C. , Niemann, G. , Cambronne, E. D. , … Heffron, F. (2011). Computational prediction of type III and IV secreted effectors in gram‐negative bacteria. Infection and Immunity, 79(1), 23–32. 10.1128/iai.00537-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merda, D. , Bonneau, S. , Guimbaud, J.‐F. , Durand, K. , Brin, C. , Boureau, T. , … Fischer‐Le Saux, M. (2016). Recombination‐prone bacterial strains form a reservoir from which epidemic clones emerge in agroecosystems. Environmental Microbiology Reports, 8, 572–581. 10.1111/1758-2229.12397 [DOI] [PubMed] [Google Scholar]
- Mohr, T. J. , Liu, H. , Yan, S. , Morris, C. E. , Castillo, J. A. , Jelenska, J. , & Vinatzer, B. A. (2008). Naturally occurring nonpathogenic isolates of the plant pathogen Pseudomonas syringae lack a type III secretion system and effector gene orthologues. Journal of Bacteriology, 190(8), 2858–2870. 10.1128/jb.01757-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naushad, H. S. , & Gupta, R. S. (2013). Phylogenomics and molecular signatures for species from the plant pathogen‐containing order xanthomonadales. PLoS ONE, 8(2), e55216 10.1371/journal.pone.0055216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel, L. , Thieme, F. , Nennstiel, D. , & Bonas, U. (2002). Two novel type III‐secreted proteins of Xanthomonas campestris pv. vesicatoria are encoded within the hrp pathogenicity island. Journal of Bacteriology, 184(5), 1340–1348. 10.1128/JB.184.5.1340-1348.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obergfell, K. P. , & Seifert, H. S. (2015). Mobile DNA in the pathogenic Neisseria . Microbiology Spectrum, 3(1), MDNA3‐0015‐2014 10.1128/microbiolspec.MDNA3-0015-2014 [DOI] [PubMed] [Google Scholar]
- Pieretti, I. , Cociancich, S. , Bolot, S. , Carrere, S. , Morisset, A. , Rott, P. , & Royer, M. (2015). Full genome sequence analysis of two isolates reveals a novel Xanthomonas Species Close to the sugarcane pathogen Xanthomonas albilineans . Genes, 6(3), 714–733. 10.3390/genes6030714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D. (2008). jModelTest: Phylogenetic model averaging. Molecular Biology and Evolution, 25(7), 1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- Potnis, N. , Krasileva, K. , Chow, V. , Almeida, N. F. , Patil, P. B. , Ryan, R. P. , … Jones, J. B. (2011). Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics, 12, 146 10.1186/1471-2164-12-146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez, V. , Harispe, S. , Delsuc, F. , & Douzery, E. J. (2011). MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE, 6(9), e22594 10.1371/journal.pone.0022594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, R. L. , Grajales, A. , Arrieta‐Ortiz, M. L. , Salazar, C. , Restrepo, S. , & Bernal, A. (2012). Genomes‐based phylogeny of the genus Xanthomonas . BMC Microbiology, 12(43), 1471–2180. 10.1186/12-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B. , Bolot, S. , Guy, E. , Denance, N. , Lautier, M. , Jardinaud, M.‐F. , … Noel, L. D. (2015). Genomics and transcriptomics of Xanthomonas campestris species challenge the concept of core type III effectome. BMC Genomics, 16, 975 10.1186/s12864-015-2190-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, R. P. , Vorholter, F. J. , Potnis, N. , Jones, J. B. , Van Sluys, M. A. , Bogdanove, A. J. , & Dow, J. M. (2011). Pathogenomics of Xanthomonas: Understanding bacterium‐plant interactions. Nature Reviews Microbiology, 9(5), 344–355. 10.1038/nrmicro2558 [DOI] [PubMed] [Google Scholar]
- Sallet, E. , Gouzy, J. , & Schiex, T. (2014). EuGene‐PP: A next‐generation automated annotation pipeline for prokaryotic genomes. Bioinformatics, 30(18), 2659–2661. 10.1093/bioinformatics/btu366 [DOI] [PubMed] [Google Scholar]
- Sarkar, S. F. , Gordon, J. S. , Martin, G. B. , & Guttman, D. S. (2006). Comparative genomics of host‐specific virulence in Pseudomonas syringae . Genetics, 174(2), 1041–1056. 10.1534/genetics.106.060996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarris, P. F. , Trantas, E. A. , Baltrus, D. A. , Bull, C. T. , Wechter, W. P. , Yan, S. , … Goumas, D. E. (2013). Comparative genomics of multiple strains of Pseudomonas cannabina pv. alisalensis, a potential model pathogen of both monocots and dicots. PLoS ONE, 8(3), e59366 10.1371/journal.pone.0059366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep, K. P. (2011). phangorn: Phylogenetic analysis in R. Bioinformatics, 27(4), 592–593. 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze, S. , Kay, S. , Büttner, D. , Egler, M. , Eschen‐Lippold, L. , Hause, G. , … Bonas, U. (2012). Analysis of new type III effectors from Xanthomonas uncovers XopB and XopS as suppressors of plant immunity. New Phytologist, 195(4), 894–911. 10.1111/j.1469-8137.2012.04210.x [DOI] [PubMed] [Google Scholar]
- Shimodaira, H. , & Hasegawa, M. (1999). Multiple comparisons of log‐likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution, 16(8), 1114–1116. [Google Scholar]
- da Silva, A. C. , Ferro, J. A. , Reinach, F. C. , Farah, C. S. , Furlan, L. R. , Quaggio, R. B. , … Kitajima, J. P. (2002). Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature, 417, 459–463. 10.1038/417459a [DOI] [PubMed] [Google Scholar]
- Studholme, D. J. , Wasukira, A. , Paszkiewicz, K. , Aritua, V. , Thwaites, R. , Smith, J. , & Grant, M. (2011). Draft genome sequences of Xanthomonas sacchari and two banana‐associated xanthomonads reveal insights into the Xanthomonas group 1 clade. Genes, 2(4), 1050–1065. 10.3390/genes2041050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampakaki, A. P. , Skandalis, N. , Gazi, A. D. , Bastaki, M. N. , Sarris, P. F. , Charova, S. N. , … Panopoulos, N. J. (2010). Playing the “Harp”: Evolution of our understanding of hrp/hrc genes. Annual Review of Phytopathology, 48(1), 347–370. 10.1146/annurev-phyto-073009-114407 [DOI] [PubMed] [Google Scholar]
- Tegli, S. , Gori, A. , Cerboneschi, M. , Cipriani, M. G. , & Sisto, A. (2011). Type three secretion system in Pseudomonas savastanoi pathovars: Does timing matter? Genes, 2(4), 957–979. 10.3390/genes2040957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teper, D. , Burstein, D. , Salomon, D. , Gershovitz, M. , Pupko, T. , & Sessa, G. (2016). Identification of novel Xanthomonas euvesicatoria type III effector proteins by a machine‐learning approach. Molecular Plant Pathology, 17(3), 398–411. 10.1111/mpp.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teper, D. , Sunitha, S. , Martin, G. B. , & Sessa, G. (2015). Five Xanthomonas type III effectors suppress cell death induced by components of immunity‐associated MAP kinase cascades. Plant Signaling & Behavior, 10(10), e1064573 10.1080/15592324.2015.1064573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triplett, L. R. , Verdier, V. , Campillo, T. , Van Malderghem, C. , Cleenwerck, I. , Maes, M. , … Leach, J. E. (2015). Characterization of a novel clade of Xanthomonas isolated from rice leaves in Mali and proposal of Xanthomonas maliensis sp nov. Antonie Van Leeuwenhoek International Journal of General and Molecular Microbiology, 107(4), 869–881. 10.1007/s10482-015-0379-5 [DOI] [PubMed] [Google Scholar]
- Troisfontaines, P. , & Cornelis, G. R. (2005). Type III secretion: More systems than you think. Physiology, 20, 326–339. 10.1152/physiol.00011.2005 [DOI] [PubMed] [Google Scholar]
- Weber, E. , & Koebnik, R. (2006). Positive selection of the Hrp Pilin HrpE of the plant pathogen Xanthomonas . Journal of Bacteriology, 188(4), 1405–1410. 10.1128/jb.188.4.1405-1410.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, F. F. , Potnis, N. , Jones, J. B. , & Koebnik, R. (2009). The type III effectors of Xanthomonas . Molecular Plant Pathology, 10(6), 749–766. 10.1111/j.1364-3703.2009.00590.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann, G. , & Bergelson, J. (2004). Effector genes of Xanthamonas axonopodis pv. vesicatoria promote transmission and enhance other fitness traits in the field. Genetics, 166(2), 693–706. 10.1534/genetics.166.2.693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann, F. , Vorhölter, F.‐J. , Hersemann, L. , Widmer, F. , Blom, J. , Niehaus, K. , … Kölliker, R. (2013). The noncanonical type III secretion system of Xanthomonas translucens pv. graminis is essential for forage grass infection. Molecular Plant Pathology, 14(6), 576–588. 10.1111/mpp.12030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J. M. , Park, D. C. , Shearman, H. M. , & Fargier, E. (2008). A multilocus sequence analysis of the genus Xanthomonas . Systematic and Applied Microbiology, 31(5), 366–377. 10.1016/j.syapm.2008.06.004 [DOI] [PubMed] [Google Scholar]
- Young, J. M. , Wilkie, J. P. , Park, D. C. , & Watson, D. R. W. (2010). New Zealand strains of plant pathogenic bacteria classified by multi‐locus sequence analysis; proposal of Xanthomonas dyei sp. nov. Plant Pathology, 59(2), 270–281. 10.1111/j.1365-3059.2009.02210.x [DOI] [Google Scholar]
- Zerbino, D. R. , & Birney, E. (2008). Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research, 18(5), 821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, B. , Dahlbeck, D. , Krasileva, K. V. , Fong, R. W. , & Staskawicz, B. J. (2011). Computational and biochemical analysis of the Xanthomonas effector AvrBs2 and its role in the modulation of Xanthomonas type three effector delivery. Plos Pathogens, 7(12), e1002408 10.1371/journal.ppat.1002408 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole‐genome sequences obtained for this project have been deposited in GenBank. Accession nos. are available in Data S1. Most bacterial strains used in this study are available at the microbial resource centre CIRM‐CFBP (Beaucouzé, INRA, France; http://www6.inra.fr/cirm_eng/CFBP-Plant-Associated-Bacteria).