

Abstract
Manipulation of viral genomes is essential for studying viral gene function and utilizing viruses for therapy. Several techniques for viral genome engineering have been developed. Homologous recombination in virus‐infected cells has traditionally been used to edit viral genomes; however, the frequency of the expected recombination is quite low. Alternatively, large viral genomes have been edited using a bacterial artificial chromosome (BAC) plasmid system. However, cloning of large viral genomes into BAC plasmids is both laborious and time‐consuming. In addition, because it is possible for insertion into the viral genome of drug selection markers or parts of BAC plasmids to affect viral function, artificial genes sometimes need to be removed from edited viruses. Herpes simplex virus (HSV), a common DNA virus with a genome length of 152 kbp, causes labialis, genital herpes and encephalitis. Mutant HSV is a candidate for oncotherapy, in which HSV is used to kill tumor cells. In this study, the clustered regularly interspaced short palindromic repeat‐Cas9 system was used to very efficiently engineer HSV without inserting artificial genes into viral genomes. Not only gene‐ablated HSV but also gene knock‐in HSV were generated using this method. Furthermore, selection with phenotypes of edited genes promotes the isolation efficiencies of expectedly mutated viral clones. Because our method can be applied to other DNA viruses such as Epstein–Barr virus, cytomegaloviruses, vaccinia virus and baculovirus, our system will be useful for studying various types of viruses, including clinical isolates.
Keywords: clustered regularly interspaced short palindromic repeat‐Cas9, genome engineering, herpes simplex virus, large viral genome
List of Abbreviations
- ACV
acyclovir
- BAC
bacterial artificial chromosome
- CRISPR
lustered regularly interspaced short palindromic repeat
- DMEM
Dulbecco’s modified Eagle’s medium
- gB
glycoprotein B
- gE
glycoprotein E
- gI
glycoprotein I
- HEK 293T cell
human embryonic kidney 293T cell
- HSV
Herpes simplex virus
- ORF
open reading frame
- PAM
protospacer adjacent motif
- PEI
polyethylenimine
- sgRNA
single‐guide RNA
- ssODN
single‐stranded oligodeoxynucleotide
- TK
thymidine kinase
Engineering viral genomes is essential not only for the study of viral genes but also for the clinical application of viruses. To date, several techniques for viral genome engineering have been developed. For viruses with small genomes, such as HIV and hepatitis C virus, editing is relatively easy because small viral genomes can be cloned into plasmids. On the other hand, for viruses containing large genomes, including herpesviruses 1, coronaviruses such as severe acute respiratory syndrome coronavirus, and baculoviruses, homologous recombination in virus‐infected cells has traditionally been used to edit viral genomes; however, the frequency of the expected recombination is quite low. Alternatively, large viral genomes can be edited by homologous recombination in Escherichia coli at relatively high frequencies once the viral genome has been successfully cloned into a BAC plasmid 2, 3, 4, 5, 6, 7, 8. However, cloning of large viral genomes into BAC plasmids is both laborious and time‐consuming. In addition, it is possible for insertion into the viral genome of drug selection markers or parts of BAC plasmids to affect viral function. Therefore, artificial genes sometimes need to be removed from edited viruses using recombination systems such as the cre‐loxP or Flp‐FRT recognition target systems.
Invading nucleic acids can be destroyed by the CRISPR microbial defense system 9, 10, 11. A chimeric sgRNA directs Cas9 nuclease from Streptococcus pyogenes to genome sequences that are followed by a 5′‐NGG PAM 12, 13, 14, 15. The CRISPR nuclease system, which has been adapted for use in mammalian cells, facilitates genome editing by co‐expressing bacterial Cas9 nuclease, which is optimized for expression in mammalian cells along with the guide sequence 16, 17, 18. Consequently, the CRISPR‐Cas system has been used to generate genetically engineered mice 19 and induce alterations to the genomes of zebrafish 20, Drosophila 21, Caenorhabditis elegans 22, plants 23 and yeast 24. In this study, we developed an efficient method for editing large viral DNA genomes using the CRISPR‐Cas system.
MATERIALS AND METHODS
Viruses and cells
Herpes simplex virus‐1 (strain F; Gene Bank: GU734771.1) was kindly provided by Dr. Y. Kawaguchi (University of Tokyo, Tokyo, Japan). HEK 293 T and Vero cells were cultured at 37 °C in 5% CO2 in DMEM (Nacalai, Kyoto, Japan) supplemented with 10% or 1% FCS (Hyclone, Logan, UT, USA), 100 U/mL of penicillin, 100 μg/mL of streptomycin and 50 μM 2‐mercaptoethanol (Nacalai).
Plasmid and transfection
The targeting plasmids of the CRISPR‐Cas9 system were constructed by introducing synthesized oligo‐DNA primers corresponding to each target sequence into the BbsI restriction sites of the pX330‐U6‐Chimeric_BB‐CBh‐hSpCas9 plasmid (Addgene, Cambridge, MA, USA). Guide sequences were selected by identifying the 20 bp sequence directly upstream of any 5′‐NGG sequence in either gE or TK genes. Twenty nucleotide guide sequences for gE or TK that contained 15 or fewer nucleotides matched to any unrelated genomic sequences were selected. An additional guanine or cytosine was appended in the sense or anti‐sense primer directly at the 5′ or 3′ end of the target sequence to allow guanine to be the first base of the U6 transcript. Human CD8A (AY039664.1) was cloned into the pCIneo expression plasmid (pCIneo‐CD8; Promega, Madison, WI, USA). Plasmids and ssODN were transfected into cells using PEI Max (2 µg/mL in H2O; Polysciences, Warrington, PA, USA).
Mutant virus production
Human embryonic kidney 293 T cells were cultured at a density of 6 × 106 cells/dish in a 100 mm dishes with 12 mL of DMEM containing 10% FCS. Transfection reagents were prepared as described below. For generation of mutant viruses, mock or targeting Cas9/sgRNA constructs and pCIneo‐CD8 (12 μg each) were diluted in 1.5 mL of OPTI‐MEM (Life Technologies, Carlsbad, CA, USA). For generation of revertant or knock‐in viruses, a pair of targeting Cas9/sgRNA constructs, pCIneo‐CD8, and ssODN, containing sequences (6 μg each) for repairing or knock‐in was diluted in 1.5 mL of OPTI‐MEM. The diluted plasmids were mixed with 120 μg of PEI diluted in 1.5 mL of OPTI‐MEM for 20 min at room temperature. Thereafter, the plasmids and PEI mixture were added to HEK 293 T cells. After culture for 2 days, the transfectants were mixed with microbeads labeled with anti‐human CD8 monoclonal antibody (mAb; Miltenyi Biotech, Bergisch Gladbach, Germany) and CD8+ cells isolated using autoMACS or MACS LS columns (Miltenyi), and then infected with HSV‐1. After 2–3 hr of infection, the supernatants were replaced with fresh DMEM containing 1% FCS to remove the original viruses. At the same time, ssODN (0.8 μg/50 μL of OPTI‐MEM) containing sequences for repairing or knock‐in was retransfected using PEI (4 μg/50 μL of OPTI‐MEM) to generate revertant or knock‐in viruses. After 2–3 days of culture, supernatants that were expected to contain mutated viruses were serially diluted 103–105‐fold and then mixed with Vero cells (3 × 104 cells/well in 96‐well tissue culture plates) and kept for 3–5 days. Cell surface phenotypes of Vero cells infected with the recombinant viruses were analyzed by flow cytometry. ACV‐resistant cells were obtained by culturing the infected cells in the presence of ACV (3 μg/mL). The gE and TK genes were amplified from culture supernatants of infected cells and then sequenced.
ssODN
ssODN bearing a wild‐type gE sequence of 150 nt was designed to generate revertant virus (5′‐CAGACACGATTCGTATATTCGCATCTCGGCACACGAGGTCGGCACGTCGAATCTCAACCAGACGACGTCCATGGTGTAGGTCTGGTCGTCGTGCGCGATCGCATGGATCGAGACGTTCGTGCTAAACGCCTCCCCGGGGGAAAACAGGAT‐3′). For generating virus carrying His‐tagged gE, 138 nt ssODN containing a His‐tag sequence between a gE signal sequence and extracellular domain of gE (5′‐ATGGATCGCGGGGCGGTGGTGGGGTTTCTTCTCGGTGTTTGTGTTGTATCGTGCTTAGCGCATCATCACCATCACCATGGAACGCCGAAAACGTCCTGGAGACGGGTGAGTGTCGGCGAGGACGTTTCGTTGCTTCCA‐3′) was used. Silent mutations (italicized) were included in each ssODN sequence so they would not be recognized by the Cas9/sgRNA constructs.
Flow cytometry analysis
Cells were incubated with anti‐gE mAb (clone 0112; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti‐6 × His mAb (clone OGHis; MBL, Nagoya, Japan) as well as anti‐gB mAb (clone 8) followed by Alexa 488‐ or 647‐conjugated anti‐mouse IgG1 or anti‐mouse IgG2a mAb (Life Technologies). Anti‐gB mAb (clone 8) was established by immunizing the N‐terminal gB fragment (amino acid residues 30–150) fused with the human IgG1 Fc region using TiterMax Gold adjuvant (CytRx, Los Angeles, CA, USA) according to the manufacturer's instructions. Anti‐gB mAb produced by B cell hybridoma was generated using a previously described method 25. Expression of HSV‐1 gE, gB and 6xHis was assessed using a flow cytometer, FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA).
Genetic analysis
The gE and TK ORFs flanked with the 5′ and 3′ untranslated region were amplified by PCR from cultured supernatants of Vero cells infected with mutated HSV‐1 using KOD FX DNA polymerase (Toyobo, Osaka, Japan) and sequence‐specific primer pairs (for gE: sense, 5′‐TGACTTTGGCTCTTCTGGCG‐3′ and anti‐sense, 5′‐ATCGCGATTAGCTCGTCTCC‐3′; for TK: sense, 5′‐TGCAGCGACCCGCTTAACAG‐3′ and anti‐sense, 5′‐CGTCATAGCGCGGGTTCCTT‐3′) according to the manufacturer’s instructions. After purification of amplified DNA using the Wizard SV Gel and PCR Clean‐Up System (Promega), the gE and TK DNA sequences were determined using a 3130×1 genetic analyzer (Applied Biosystems, Waltham, MA, USA).
RESULTS
Ablation of HSV gE and TK gene using the CRISPR‐Cas9 system
Herpes simplex virus 1 contains a large DNA genome of 152 kbp. HSV‐1 gE functions as an Fc receptor to host immunoglobulins and forms a heterodimer with gI 26, 27. To determine whether the CRISPR‐Cas9 system could be used to edit viral genomes, first an attempt was made to edit the gE gene by designing three unique 20 nt guide sequences followed by PAM sequences in the ORF of gE (Fig. 1a) and then cloning them into a plasmid‐expressing codon‐optimized S. pyogenes Cas9 (Cas9) and a chimeric sgRNA 17.These gE‐ or mock‐targeting Cas9/sgRNA constructs were transfected into HEK 293 T cells along with a plasmid expressing human CD8 as a marker of transfection. When CD8 positive cells transfected with gE‐ or mock‐targeting Cas9/sgRNA were enriched and infected with HSV‐1, high proportions of gE‐negative cells in HSV‐infected cells were observed in gE‐targeting Cas9/sgRNA transfectants but not in mock‐transfectants (Fig. 1b). The proportion of gE‐negative cells in HSV‐infected cells was quite low when cells transfected with gE‐targeting Cas9/sgRNA had not been enriched for CD8 positive cells. Therefore, enrichment of sgRNA/Cas9‐expressing cells by using cell surface marker increases efficiency of genome editing by sgRNA/Cas9.
Figure 1.
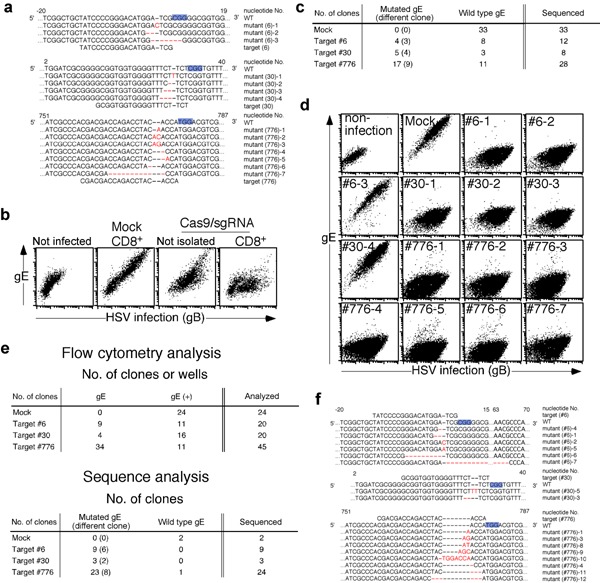
Introduction of high‐frequency gene‐specific mutations into the HSV‐1 genome using the CRISPR‐Cas9 system. (a) Nucleic acid sequences of the targeting sites on the wild‐type HSV‐1 gE gene and the mutated gene sequences induced by gE‐targeting Cas9/sgRNA are shown. Nucleic acid numbers for the gE gene are indicated in the figure. PAM sequences are indicated by blue highlighting and substituted, deleted or inserted nucleotides are in red font. (b) HEK 293T cells were transfected with gE (#776)‐ or mock‐targeting Cas9/sgRNA plasmid along with CD8‐expressing plasmid or only gE (#776)‐targeting Cas9/sgRNA plasmid. CD8 positive cells were isolated with anti‐CD8 mAb labeled with magnetic beads. The isolated transfectants or cells transfected with only gE (#776)‐targeting Cas9/sgRNA were infected or not infected with HSV‐1. Expression of gE on infected and noninfected HEK 293T cells was assess by flow cytometry. Expression of gB was assessed to distinguish infected cells from noninfected cells. (c) Nucleic acid sequences of gE in HSV‐1 clones produced from cells transfected with gE‐ or mock‐targeting Cas9/sgRNA constructs were analyzed. Numbers of gE‐mutated and nonmutated HSV‐1 clones and the total number of clones are shown. (d) Expression of gE on Vero cells infected with the mock‐ or gE‐mutated HSV‐1 and not infected was assessed by flow cytometry. Expression of gB was assessed to distinguish infected cells from noninfected cells. (e) Expression of gE on the surface of HSV‐1 clones produced from cells transfected with gE‐targeting Cas9/sgRNA constructs were assessed by flow cytometry (upper panel). Nucleic acid sequences of gE in HSV‐1 clones that had not expressed gE on the cell surfaces of infected cells were analyzed. Two mock plasmid‐transfected and gE‐expressing clones were analyzed as controls. Numbers of gE‐mutated and nonmutated HSV‐1 clones as well as total number of clones are shown (lower panel). (f) Nucleic acid sequences around the targeting sites of the gE gene are shown. After selection of HSV‐1 clones expressing no gE on the infected cells according to flow cytometry, the sequences of each gE gene of gE negative clones were analyzed. Nucleic acid numbers for the gE gene are indicated in the figure. PAM sequences are indicated by blue highlighting and substituted, deleted or inserted nucleotides are in red font.
In order to isolate HSV‐1 clones targeted by the CRISPR‐Cas9 system, culture supernatants containing HSV‐1 produced from the transfectants were sequentially diluted and inoculated onto Vero cells in 96‐well tissue culture plates. Wells containing Vero cells that were monoclonally infected with HSV‐1 were identified by microscopy and the HSV‐1 genome around the gE gene was amplified by PCR from the culture supernatants of the infected cells. Thereafter, each gE gene sequence was determined (Fig. 1a). Mutations in the gE gene were detected in HSV‐1 clones obtained from the transfectants with all three gE‐targeting plasmids but not with mock plasmids (Fig. 1c). In particular, more than 60% of the clones contained the expected mutations in gE when targeting sequences #30 and #776 had been used. Cells infected with viruses possessing frameshift mutations did not express gE on their surfaces (Fig. 1d). On the other hand, cells infected with some HSV‐1 clones carrying in‐frame mutations in gE, such as #6‐3 and #30‐4, still expressed gE on their surfaces (Fig. 1d,f). When expression of gE on cells infected with each HSV‐1 clone was assessed by flow cytometry and the gE gene of each HSV‐1 clone lacking gE expression on the cell surface sequenced, almost all clones were found to contain the expected gE mutations (Fig. 1e,f).
Next, another HSV‐1 gene, TK, which is responsible for susceptibility to ACV, one of the drugs used to treat HSV and varicella‐zoster virus infections, was examined. ACV is phosphorylated by viral TK but not cellular TK, and phosphorylated ACV competitively inhibits viral DNA polymerase for deoxyguanosine triphosphate, resulting in inhibition of viral replication 28, 29. TK‐disrupted viruses were also generated using the CRISPR‐Cas9 system with the same procedure as that used to generate gE‐ablated viruses (Fig. 2a,b). Many ACV‐resistant HSV‐1 mutants were obtained from cells transfected with TK‐targeting Cas9/sgRNA constructs, whereas only one ACV‐resistant HSV‐1 clone was obtained from cells transfected with mock‐targeting Cas9/sgRNA constructs (Fig. 2c). Almost half of the clones obtained from cells transfected with TK‐targeting Cas9/sgRNA constructs contained the expected mutations in the TK gene when construct #515, which targets Cas9/sgRNA, had been used. Some clones (#515‐2 and ‐3) contained in‐frame deletions in the TK gene but were resistant to ACV. Because the region between amino acid residues 161 and 192 in TK is highly conserved among HSVs and is involved in nucleotide binding and homodimerization of TK, an in‐frame deletion in this region might have significantly affected the function of TK. Furthermore, all clones were found to contain mutations in the TK gene after ACV selection (Fig. 2d). In contrast, one ACV‐resistant HSV‐1 clone obtained from cells transfected with mock‐targeting Cas9/sgRNA constructs had a mutation in TK gene that is not related to the targeting site.
Figure 2.
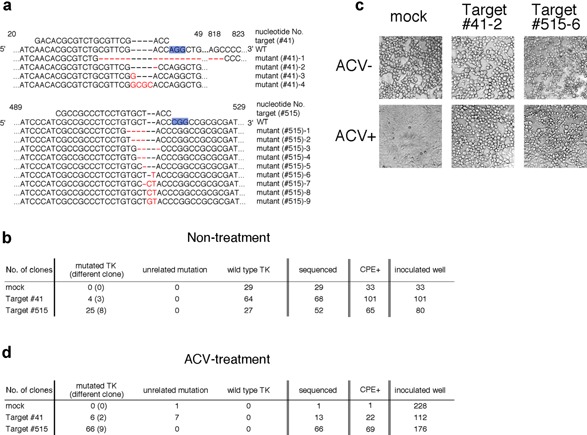
(a) Nucleic acid sequences of targeting sites for the wild‐type HSV‐1 TK gene and mutated gene sequences induced by the CRISPR‐Cas9 system are shown. Nucleic acid numbers for the TK gene are indicated in the figure. PAM sequences are indicated by blue highlighting and substituted, deleted or inserted nucleotides are indicated in red. (b) Nucleic acid sequences of TK in HSV‐1 clones produced from cells transfected with TK‐ or mock‐targeting Cas9/sgRNA constructs were analyzed. (c) Vero cells infected with HSV‐1 produced from cells transfected with TK‐targeting CAS9/sgRNA constructs in the presence or absence of ACV were analyzed by microscopy (200 ×). ACV‐sensitive cells are round. (d) Nucleic acid sequences of TK in HSV‐1 clones that acquired ACV‐resistance were analyzed.
Generation of HSV‐1 gE ‐revertant virus using CRISPR‐Cas9 system
Attempts were made to generate gE‐revertant viruses from gE‐mutant HSV‐1 using the CRISPR‐Cas9 system (Fig. 3a). A pair of Cas9/sgRNA constructs targeting the flanking region of the mutated sites, a ssODN bearing a wild‐type gE sequence as the repair template and a CD8‐expressing plasmid, were cotransfected into HEK 293T cells. Silent mutations were included in the repair ssODN sequence so that it would not be recognized by the Cas9/sgRNA constructs. Two days later, CD8 expressing cells were isolated and infected with gE‐mutant viruses (mutants #776‐1,‐2 and ‐9) (Fig. 1a,f). Production of gE‐revertant clones was evaluated by using flow cytometry to assess expression of gE on the cell surfaces as well as sequencing of the gE genes of each clone (Fig. 3a,b). Revertants expressing gE with silent mutations were detected but no clone possessing the wild‐type gE was identified (Fig. 3a). No revertant was obtained when the repair ssODN was transfected without a pair of targeting plasmids. Generation by revertant viruses was less efficient than that by viruses carrying simple gE mutations (Figs. 1c, e and 3c). Some viruses that failed to incorporate the repair ssODN had additional muations in gE that were generated by a pair of gE‐targeting plasmids. The gE gene sequences around the targeted sites of all five revertant clones generated with a pair of plasmids targeting gE (#745c) and gE (#793) along with ssODN were identical. Also, identical gE sequences were obtained from the two revertant clones generated with plasmids pair targeting gE (#754c) and gE (#793) and from all five clones with plasmids targeting gE (#760c) and gE (#793). The gE sequences outside the targeted sites remained identical with the parental sequence (Fig. 3a). HSV‐1 genomic DNA deleted by the pair of Cas9/sgRNA is seemingly repaired more efficiently by ssODN than an undeleted parental genome DNA is.
Figure 3.
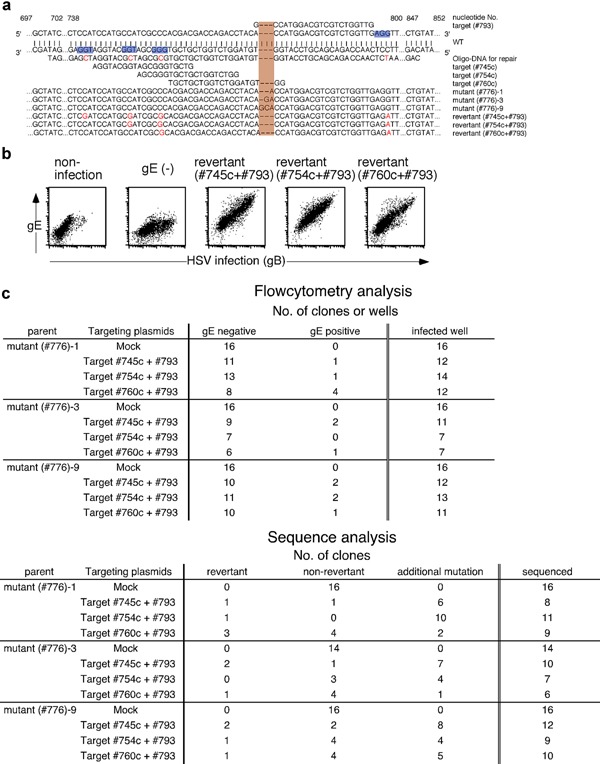
Revertant HSV‐1 was generated from gE‐mutated HSV‐1. (a) Nucleic acid sequences of targeting sites on the HSV‐1 gE gene, sequence of ssODN bearing the wild‐type gE with some silent mutations used as a repair template, and the mutated gene sequences induced by the CRISPR‐Cas9 system are shown. Nucleic acid numbers for the gE gene are indicated in the figure. PAM sequences are indicated by blue highlighting, substituted nucleotides are indicated in red and reverted sequences have orange highlighting. (b) Expression of gE on Vero cells infected with gE‐mutated parental HSV‐1, gE revertant and cells without infection. gB and gE expression on infected Vero cells was assessed by flow cytometry. The expression of gB was analyzed to distinguish infected cells from noninfected cells. Representative data derived from three independent experiments are shown. (c) gE‐reverted HSV‐1 clones were produced from cells transfected with gE‐ or mock‐targeting Cas9/sgRNA constructs and repair ssODN. The gE expression on the surface of cells infected with each HSV‐1 clone was assessed by flow cytometry. The number of HSV‐1 clones that did or did not express gE as well as the total number of HSV‐1 clones analyzed are shown (upper panel). The nucleic acid sequence of gE in each HSV‐1 clone was analyzed. Numbers of gE‐reverted HSV‐1, gE‐mutated parental HSV‐1 and HSV‐1 that acquired additional mutations are shown (lower panel).
Mutant viruses expressing His‐tagged gE from wild‐type HSV‐1 were then generated using a procedure similar to that used to generate the gE‐revertant viruses. A pair of Cas9/sgRNA constructs was designed to target the flanking region of the signal sequence cleavage site. ssODN co‐transfected with the pair of Cas9/sgRNA constructs was designed to include a His‐tag downstream of the gE signal sequence (Fig. 4a). Vero cells infected with the His‐tagged gE mutant HSV‐1 generated by this technique expressed His‐tags on their surfaces (Fig. 4b,c upper panel). Sequencing of gE genes amplified by PCR from the culture supernatants of His‐tag expressing HSV‐1‐infected cells confirmed that the His‐tag was inserted within the gE gene sequence as designed (Fig. 4a). The overall production frequency of His‐tagged knock‐in viruses was approximately 10% (Fig. 4c lower panel).
Figure 4.
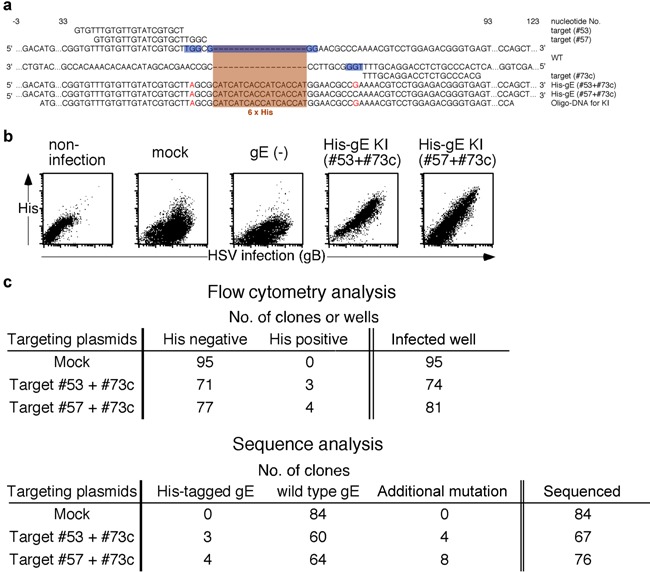
A His‐tag was inserted into the gE sequence of wild‐type HSV‐1. (a) Nucleic acid sequences of targeting sites on the HSV‐1 gE gene, sequence of ssODN bearing a His‐tag sequence downstream of gE signal sequence with some silent mutations used as a repair template, and the mutated gene sequences induced by the CRISPR‐Cas9 system are shown. Nucleic acid numbers for the gE gene are indicated in the figure. PAM sequences are indicated by blue highlighting, substituted nucleotides are in red font and inserted sequences have orange highlighting. (b) The His‐tagged gE expression on Vero cells infected with HSV‐1 mutated with mock‐targeting Cas9/sgRNA along with ssODN, mutated with gE‐targeting Cas9/sgRNA along with ssODN and cells without infection are shown. Cells infected with HSV‐1 that acquired additional mutations are indicated as gE(−). Expression of gB and His‐tagged gE on infected Vero cells was assessed by flow cytometry. The expression of gB was analyzed to distinguish infected cells from noninfected cells. Representative data derived from three independent experiments are shown. (c) HSV‐1 clones expressing His‐tagged gE were produced from cells transfected with gE‐ or mock‐targeting Cas9/sgRNA constructs and ssODN for His‐tag insertion. The expression of His‐tagged gE on the surface of cells infected with each HSV‐1 clone was analyzed by flow cytometry. The number of HSV‐1 clones that did or did not express His‐tagged gE as well as the total number of HSV‐1 clones analyzed are shown (upper panel). The nucleic acid sequence of gE in each HSV‐1 clone was analyzed. Numbers of viruses expressing the His‐tagged gE, wild‐type gE and additionally mutated gE are shown (lower panel).
DISCUSSION
In this study, we exploited the CRISPR‐Cas9 system to edit a large 152 kb genome of HSV‐1. Our procedure enabled us to generate HSV‐1‐containing specific gene mutations at a high frequency in less than 2 weeks (Figs. 1c and 2b). More than 50% of the HSV‐1 clones generated by this system had the expected mutations when appropriate target sequences had been used. Because of the high efficiency of HSV‐1 genome editing by our Cas9/sgRNA system, we could obtain the specific mutants apparently without unexpected mutations. However, not all targeting plasmids were highly efficient at HSV‐1 genome editing. Indeed, gene‐ablation efficiency was 33% and 5.8% when plasmids targeting gE #6 and TK #41, respectively, were used. Efficiency of editing viral genomes seems to be affected by the target sequence. Use of certain phenotypic changes induced by mutations enhances efficiency, as we have shown for mutations of gE or TK. Indeed, almost 100% of the viral clones had the expected mutations when the mutant viruses were phenotypically enriched by gE expression or ACV sensitivity. Therefore, generation of mutant HSV‐1 using the CRISPR‐Cas9 system is much more efficient than that using the conventional method, in which mutant viruses are generated by spontaneous homologous recombination in the presence of an extrinsically introduced gene. Although we did not edit essential genes that are involved in viral survival or infectivity, it may be possible to edit the essential genes by using HEK 293T cells and Vero cells transfected with the wild‐type gene of interest. Although undesired “off‐target” mutations have been of concern with the CRISPR‐Cas9 system, off‐target mutations might be decreased by using a mutated Cas9 nuclease that does not cause double‐strand breaks but introduces a single‐strand nick 30. Use of mutant Cas9 might be useful for decreasing the off‐target effect in viral genome editing. Recently, engineering of genes of adenovirus and HSV‐1 by the CRISPR‐Cas9 system was reported 31. However, in the current study, we generated not only gene‐ablated virus but also revertant virus from the gene gene‐ablated virus, which is close to essential for studying specific viral genes. In addition, we assessed cells infected with mutant virus by flow cytometry to directly ascertain the effects of gene editing. Therefore, this study provides important information required for editing viral genes with the CRISPR‐Cas9 system.
Use of the CRISPR‐Cas9 system enabled us to rapidly and efficiently generate mutant viruses without constructing BAC plasmids containing the viral genomes. In addition, this method will allow functional analysis of viral genes directly derived from the field or patients. Because we detected knock‐in viruses in approximately 10% of randomly chosen clones without the use of selection markers, this system is useful not only for introducing genetic ablation into viruses but also for generating knock‐in viruses. The CRISPR‐Cas9 system is reportedly useful for editing HIV genome integrated into host cell genome 32. However, these authors' technique differed from ours in that viral DNA genome was directly edited by the CRISPR‐Cas9 system. Because HSV‐1 mutants are candidates for clinical trials of oncolytic virotherapy, editing large viral genomes using the CRISPR‐Cas9 system could contribute to generating mutant viruses suitable for therapeutic applications. In addition, this system can be further applied to other large DNA viruses belonging to the Herpesviridae, Poxviridae and Baculoviridae families, such as cytomegalovirus, varicella‐zoster virus, Epstein–Barr virus and vaccinia virus. Furthermore, our technique will enable engineering of viruses for therapeutic usage such as viral vector and tumor lysis more readily than conventional methods.
DISCLOSURE
The authors declare they have no competing financial interests.
Acknowledgements
We thank Ms. K. Furuno, K. Shida and S. Matsuoka for technical assistance, C. Kita for secretarial assistance, and M. Ikawa for advice on gene targeting. This study was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Science and Culture, Japan (T.S. and H.A.).
REFERENCES
- 1. Post L.E., Roizman B. ( 1981) A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell 25: 227–32. [DOI] [PubMed] [Google Scholar]
- 2. Almazan F., Gonzalez J.M., Penzes Z., Izeta A., Calvo E., Plana‐Duran J., Enjuanes L. ( 2000) Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci USA 97: 5516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Domi A., Moss B. ( 2002) Cloning the vaccinia virus genome as a bacterial artificial chromosome in Escherichia coli and recovery of infectious virus in mammalian cells. Proc Natl Acad Sci USA 99: 12415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luckow V.A., Lee S.C., Barry G.F., Olins P.O. ( 1993) Efficient generation of infectious recombinant baculoviruses by site‐specific transposon‐mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli . J Virol 67: 4566–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Messerle M., Crnkovic I., Hammerschmidt W., Ziegler H., Koszinowski U.H. ( 1997) Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci USA 94: 14759–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'connor M., Peifer M., Bender W. ( 1989) Construction of large DNA segments in Escherichia coli . Science 244: 1307–12. [DOI] [PubMed] [Google Scholar]
- 7. Tanaka M., Kagawa H., Yamanashi Y., Sata T., Kawaguchi Y. ( 2003) Construction of an excisable bacterial artificial chromosome containing a full‐length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild‐type properties in vitro and in vivo . J Virol 77: 1382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y., Buchholz F., Muyrers J.P., Stewart A.F. ( 1998) A new logic for DNA engineering using recombination in Escherichia coli . Nat Genet 20: 123–8. [DOI] [PubMed] [Google Scholar]
- 9. Garneau J.E., Dupuis M.E., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadan A.H., Moineau S. ( 2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468: 67–71. [DOI] [PubMed] [Google Scholar]
- 10. Horvath P., Barrangou R. ( 2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327: 167–70. [DOI] [PubMed] [Google Scholar]
- 11. Wiedenheft B., Sternberg S.H., Doudna J.A. ( 2012) RNA‐guided genetic silencing systems in bacteria and archaea. Nature 482: 331–8. [DOI] [PubMed] [Google Scholar]
- 12. Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E. ( 2011) CRISPR RNA maturation by trans‐encoded small RNA and host factor RNase III. Nature 471: 602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deveau H., Garneau J.E., Moineau S. ( 2010) CRISPR/Cas system and its role in phage–bacteria interactions. Annu Rev Microbiol 64: 475–93. [DOI] [PubMed] [Google Scholar]
- 14. Gasiunas G., Barrangou R., Horvath P., Siksnys V. ( 2012) Cas9‐crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109: E2579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. ( 2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cho S.W., Kim S., Kim J.M., Kim J.S. ( 2013) Targeted genome engineering in human cells with the Cas9 RNA‐guided endonuclease. Nat Biotechnol 31: 230–2. [DOI] [PubMed] [Google Scholar]
- 17. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. ( 2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mali P., Yang L., Esvelt K.M., Aach J., Guell M., Dicarlo J.E., Norville J.E., Church G.M. ( 2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. ( 2013) One‐step generation of mice carrying mutations in multiple genes by CRISPR/Cas‐mediated genome engineering. Cell 153: 910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang N., Sun C., Gao L., Zhu D., Xu X., Zhu X., Xiong J.W., Xi J.J. ( 2013) Genome editing with RNA‐guided Cas9 nuclease in zebrafish embryos. Cell Res 23: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gratz S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., O'connor‐Giles K.M. ( 2013) Genome engineering of Drosophila with the CRISPR RNA‐guided Cas9 nuclease. Genetics 194: 1029–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friedland A.E., Tzur Y.B., Esvelt K.M., Colaiacovo M.P., Church G.M., Calarco J.A. ( 2013) Heritable genome editing in C. elegans via a CRISPR‐Cas9 system. Nat Methods 10: 741–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nekrasov V., Staskawicz B., Weigel D., Jones J.D., Kamoun S. ( 2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA‐guided endonuclease. Nat Biotechnol 31: 691–3. [DOI] [PubMed] [Google Scholar]
- 24. Dicarlo J.E., Norville J.E., Mali P., Rios X., Aach J., Church G.M. ( 2013) Genome engineering in Saccharomyces cerevisiae using CRISPR‐Cas systems. Nucleic Acids Res 41: 4336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Satoh T., Arii J., Suenaga T., Wang J., Kogure A., Uehori J., Arase N., Shiratori I., Tanaka S., Kawaguchi Y., Spear P.G., Lanier L.L., Arase H. ( 2008) PILRalpha is a herpes simplex virus‐1 entry coreceptor that associates with glycoprotein B. Cell 132: 935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson D.C., Feenstra V. ( 1987) Identification of a novel herpes simplex virus type 1‐induced glycoprotein which complexes with gE and binds immunoglobulin. J Virol 61: 2208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson D.C., Frame M.C., Ligas M.W., Cross A.M., Stow N.D. ( 1988) Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J Virol 62: 1347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elion G.B., Furman P.A., Fyfe J.A., De Miranda P., Beauchamp L., Schaeffer H.J. ( 1977) Selectivity of action of an antiherpetic agent, 9‐(2‐hydroxyethoxymethyl) guanine. Proc Natl Acad Sci USA 74: 5716–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schaeffer H.J., Beauchamp L., De Miranda P., Elion G.B., Bauer D.J., Collins P. ( 1978) 9‐(2‐hydroxyethoxymethyl) guanine activity against viruses of the herpes group. Nature 272: 583–5. [DOI] [PubMed] [Google Scholar]
- 30. Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. ( 2013) Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bi Y., Sun L., Gao D., Ding C., Li Z., Li Y., Cun W., Li Q. ( 2014) High‐efficiency targeted editing of large viral genomes by rna‐guided nucleases. PLoS Pathog 10: e1004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ebina H., Misawa N., Kanemura Y., Koyanagi Y. ( 2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV‐1 provirus. Sci Rep 3: 2510. [DOI] [PMC free article] [PubMed] [Google Scholar]