

Abstract
Influenza A and B viruses are highly contagious respiratory pathogens with a considerable medical and socioeconomical burden and known pandemic potential. Current influenza vaccines require annual updating and provide only partial protection in some risk groups. Due to the global spread of viruses with resistance to the M2 proton channel inhibitor amantadine or the neuraminidase inhibitor oseltamivir, novel antiviral agents with an original mode of action are urgently needed. We here focus on emerging options to interfere with the influenza virus entry process, which consists of the following steps: attachment of the viral hemagglutinin to the sialylated host cell receptors, endocytosis, M2‐mediated uncoating, low pH‐induced membrane fusion, and, finally, import of the viral ribonucleoprotein into the nucleus. We review the current functional and structural insights in the viral and cellular components of this entry process, and the diverse antiviral strategies that are being explored. This encompasses small molecule inhibitors as well as macromolecules such as therapeutic antibodies. There is optimism that at least some of these innovative concepts to block influenza virus entry will proceed from the proof of concept to a more advanced stage. Special attention is therefore given to the challenging issues of influenza virus (sub)type‐dependent activity or potential drug resistance.
Keywords: influenza virus, antiviral, hemagglutinin, M2 channel, nucleoprotein
1. INTRODUCTION
Human influenza A and B viruses cause significant morbidity and mortality, particularly in infants and elderly people, or those suffering from preexisting pathology or immunodeficiency.1, 2 The United States Centers for Disease Control and Prevention estimated that, from 1976 to 2000, seasonal influenza epidemics were responsible for >200,000 annual hospitalizations and an annual average of >30,000 influenza‐associated deaths in the USA.3 Approximately 90% of the influenza‐associated deaths occur among adults aged ≥65 years.4
To evade the immune response, the circulating influenza H3N2, H1N1, and B viruses continuously change their antigens, and this explains why current influenza vaccines require annual updating. These vaccines provide inadequate protection in some target populations (particularly the elderly).5 Besides, there is concern that the widely spread and highly pathogenic avian H5N1 influenza virus may acquire human transmissibility and become a potentially disastrous pandemic virus. The human case‐fatality rate of this avian H5N1 is reported to be 59%,6 although some investigators have raised the possibility that subclinical cases of H5N1 infections in humans may remain unnoticed.7 For comparison, the case‐fatality rate of the 1918 influenza virus was estimated >2.5%.8
As shown in Figure 1, the influenza virus replication cycle contains several steps amenable to antiviral intervention. This review focuses on the viral entry pathway, which, given the acute onset of influenza virus infection and the inflammation associated with it, is a particularly attractive process to interfere with. We describe the current insights into the structure and functions of the viral and cellular components involved in this entry process, and the antiviral strategies that are being explored (an overview of the described compounds is given in Table 1). For antiviral approaches affecting other stages in the viral life cycle, the reader is referred to other recent review articles.9, 10, 11, 12
Figure 1.
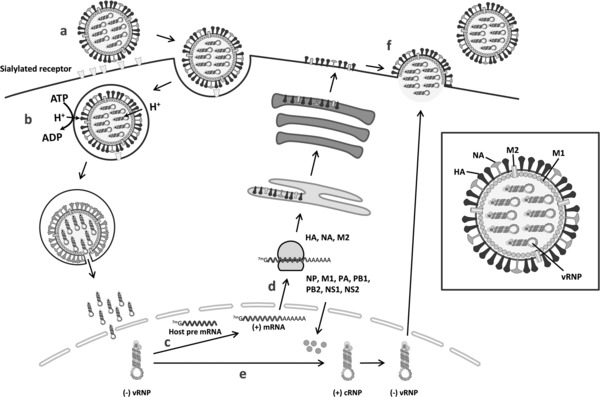
Overview of the influenza virus entry and replication process. In the inset on the right, the different virion components are specified. (a) After binding of the viral HA to sialylated glycans on the host cell surface, the virus is internalized by endocytosis. (b) Acidification of the endosome leads to activation of the M2 proton channel and virion acidification, resulting in virus uncoating (i.e., dissociation of the vRNPs from the M1 capsid protein). The low pH inside the endosome also triggers a conformational change in the HA, leading to fusion of the viral and endosomal membranes. After vRNP release in the cytoplasm and dissociation of residual M1, nuclear localization signals in NP direct the transport of the vRNPs into the nucleus. (c) In the nucleus, the viral polymerase starts mRNA synthesis by cleaving off 5′‐capped RNA fragments from host cell pre‐mRNAs. Then, viral mRNA transcription is initiated from the 3′ end of the cleaved RNA cap. (d) Viral mRNAs are transported to the cytoplasm for translation into viral proteins. HA, M2, and NA are processed in the endoplasmic reticulum and the Golgi apparatus, and subsequently transported to the cell membrane. (e) Besides viral mRNA synthesis, the viral polymerase performs the unprimed replication of vRNAs. The vRNAs are first transcribed into positive‐stranded cRNAs, which then function as the template for the synthesis of new vRNAs. During their synthesis, vRNAs and cRNAs are encapsidated by NPs. Export of the newly formed vRNPs into the cytoplasm is mediated by an M1‐NS2 complex that is bound to the vRNPs. (f) As they reach the cell membrane, the vRNPs associate with viral glycoproteins at the plasma membrane from which new virions bud off. Finally, the NA cleaves the sialic acid termini on viral and cell membrane glycoproteins, thereby releasing the progeny virions from the host cell.
Table 1.
Overview of reported influenza virus entry inhibitors
| Compound code or class | General structure | Proposed mode of actiona | Demonstrated activityb or clinical status | Activity spectrumc | References |
|---|---|---|---|---|---|
| Inhibitors of the HA‐receptor interaction | |||||
| CH65 | Immunoglobulin or Fab | Binding close to HA RBS | In vitro | H1 | 79,80 |
| C05 | Idem | Idem | In vivo | H1, H2, H3, H9, H12 | 82 |
| S139/1 | Idem | Idem | In vivo | H1, H2, H13, H16 | 83,84 |
| 3A2 and 10C4 | Idem | Idem | In vitro | B | 85 |
| CR8033 and CR8071 | Idem | Idem | In vivo | B | 86 |
| Nanobodies | Single‐domain antibody | Idem | In vivo | HA subtype‐specific | 88,89 |
| Surfactant protein D | Lectin | Idem | In vitro / In vivo | A and B | 90,92,93 |
| CL‐43 | Lectin | Idem | In vitro / In vivo | A and B | 94 |
| Cyanovirin‐N | Lectin | Idem | In vitro / In vivo | A and B | 96,97 |
| SPG and GM3 | Sialylated ganglioside | Binding to HA RBS | In vitro | 101,102 | |
| LSTc‐bearing liposomes | Sialylated liposome | Idem | In vivo | 103 | |
| Pentadecapeptides | Peptide | Idem | In vitro | 104 | |
| A22 | Aptamer | Idem | In vivo | 105 | |
| NMSO3 | Sulfated sialyl lipid | Not precisely known | In vitro | 106,107 | |
| c01 and c03 | Acylated peptides | Binding to sialic acid | In vitro | 109 | |
| DAS181 | Sialidase enzyme | Receptor destruction | In vivo / Phase II | A and B | 112,114 |
| Inhibitors of viral endocytosis | |||||
| Fattiviracin | Neutral glycolipid | Membrane fluidity modulator | In vitro | 124 | |
| Glycyrrhizin | Triterpene glycoside | Inhibits virus internalization; immunomodulator | In vivo Natural medicine | 126–128 | |
| LJ001 | Small molecule | Intercalates into viral membranes | In vitro | 129 | |
| Bafilomycin A1 | Small molecule | V‐ATPase inhibitor | In vitro | 135 | |
| Diphyllin | Small molecule | Idem | In vitro | 135 | |
| Chloroquine | 4‐Aminoquinoline | Raises endosomal pH | Not active in humans | 137–139 | |
| SA‐19 | Aglycoristocetin lipoglycopeptide | Intra‐cytoplasmic virus trapping | In vitro | A and B | 107,131 |
| Inhibitors of M2‐mediated proton transport | |||||
| Amantadine | Adamantane | Block the M2 channel | Approved | A | 146,155,159 |
| Rimantadine | Adamantane | Idem | Approved | A | 148 |
| +analogues | Adamantane | Idem | In vitro | A | 161,166–169 |
| Inhibitors of HA‐mediated fusion at low pH | |||||
| TBHQ | Small molecule | Bind to HA stem and inhibit HA refolding | In vitro | H3‐ specific | 55,175 |
| 4c | Small molecule | Idem | In vitro | H3‐ specific | 176 |
| BMY‐27709 | Small molecule | Idem | In vitro | H1‐ and H2‐specific | 177,182 |
| CL 61917 | Small molecule | Idem | In vitro | H1‐ and H2‐specific | 179 |
| Stachyflin | Small molecule | Idem | In vitro | H1‐ and H2‐specific | 180 |
| RO5464466 | Small molecule | Idem | In vitro | H1‐ and H2‐specific | 178 |
| Dextran sulfate | Sulfated polysaccharide | Inhibits membrane mixing? | In vitro | A | 184,187–189 |
| Retrocyclin‐2 | Circular peptide | Immobilizes surface glycoproteins | In vitro | 194 | |
| Arbidol | Small molecule | Inhibitor of HA refolding; membrane fluidity modulator | Approved (Russia and China) | A and B | 196,197,199 |
| F10; CR6261 | Immunoglobulin or Fab | Bind to conserved HA‐stem epitope; inhibit HA refolding | In vivo | Group 1 HAs | 206–208 |
| CR8020 | Immunoglobulin or Fab | Idem | In vivo | Group 2 HAs | 209 |
| FI6v3 | Immunoglobulin or Fab | Idem | In vivo | All A HAs | 210 |
| CR9114 | Immunoglobulin or Fab | Idem | In vivo | A and B | 86 |
| Inhibitors of NP‐mediated viral nuclear import | |||||
| Nucleozin + analogues | Small molecule | Aggregates NP; also inhibits vRNP activity | In vivo | A | 236–239 |
| Inhibitors of entry‐related cellular protein kinases | |||||
| Bisindolylmaleimide I | Small molecule | Broad protein kinase C inhibitor | In vitro | 245 | |
For compounds with broader antiviral activity, only the mode of action relevant for influenza virus is given.
The most advanced level for demonstration of anti‐influenza virus activity is given, in the sequence: in vitro (i.e. in cell culture), in vivo (i.e. animal studies), and in humans.
For most compounds, literature reports are limited to a number of influenza A or B viruses tested, and hence, the activity spectrum was not (yet) specified. The information in this column indicates the clearly defined activity spectrum.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2. CURRENTLY AVAILABLE ANTI‐INFLUENZA VIRUS DRUGS
Effective antiviral drugs to prevent or treat influenza infections should at all times be available. Today, two classes of anti‐influenza virus drugs exist: the M2 proton channel blockers (i.e., the adamantane compounds, amantadine and rimantadine), and the neuraminidase inhibitors (NAIs) (oseltamivir and zanamivir).13 The first two compounds have limited utility, since they are associated with neurological side effects, have no activity against influenza B virus, and the vast majority of circulating strains are adamantane‐resistant.13 A detailed description of their mode of action and resistance mechanisms will be given below. The obviously superior class of anti‐influenza virus drugs are the NAIs oseltamivir and zanamivir that are active against all influenza A and B viruses. These structural analogues of sialic acid bind to the catalytic pocket of the viral NA and inhibit its function in releasing the newly produced virus from the host cells.14, 15 There is a critical difference in the NA binding mode of oseltamivir compared to that of zanamivir, which explains their significantly different resistance profile. Due to its larger hydrophobic side chain, oseltamivir requires rotation of the noncatalytic Glu276 residue within NA to create a binding space for oseltamivir.16 By contrast, the smaller size of zanamivir enables direct binding of this compound to NA. In a mutant N1 NA containing a His to Tyr substitution at position 274, this rotation can no longer occur, rendering the NA resistant to oseltamivir binding. During the 2008–2009 season, oseltamivir‐resistant H1N1 viruses were isolated all over the globe, even from untreated patients.17, 18 In a Japanese study in 2004, nine out of 50 children treated with oseltamivir carried oseltamivir‐resistant H3N2 viruses.19 Fortunately, oseltamivir‐resistant viruses are still sensitive to zanamivir, for which resistance has only scarcely been reported.20, 21 On the other hand, the patient‐unfriendly administration route for zanamivir (i.e., by powder inhalation device) explains why oseltamivir (which is given by oral capsules) is generally preferred in the clinical setting. Inhalation of zanamivir is a priori excluded in patients suffering from severe influenza symptoms with acute respiratory distress, such as patients infected with the highly pathogenic avian H5N1 virus, or severe cases of the 2009 pandemic H1N1 virus. To address this issue, an intravenous formulation of zanamivir is under consideration.22, 23 Besides, new NAIs are being developed. Peramivir, which has to be administered intravenously, has been licensed in Japan and South Korea, while, in the United States, its use was temporarily allowed during the 2009 H1N1 pandemic.24 Unfortunately, the widespread oseltamivir‐resistant H1N1 His274Tyr mutants show intermediate cross‐resistance to peramivir.25 Another NAI, laninamivir (CS‐8958), was approved in Japan in 2010 and is currently in Phase III trials in the United States.26, 27 This promising compound requires only one single intranasal administration (based on its long half‐life), and has a similar NA binding mode and favorable resistance profile as zanamivir.28 Finally, novel NAIs with a sialic acid‐related or unrelated structure have been developed by rational design, but are still in the early experimental stage.29, 30, 31
To face the emerging resistance to NAIs (in particular, oseltamivir), entirely novel anti‐influenza virus drugs are urgently needed. The two products that are most advanced in clinical development are the nucleobase analogue T‐705 (favipiravir) and the receptor destroying protein DAS181. For T‐705, Phase III trials in the United States are pending. Its active ribose‐triphosphate metabolite is recognized by the influenza virus polymerase, causing competitive inhibition of viral RNA synthesis and/or lethal viral mutagenesis.32 T‐705 has broad anti‐RNA virus activity beyond influenza virus and is presumed (based on cell culture data) to have a high barrier for viral resistance.33 The second agent, DAS181, is currently in Phase II trials. This recombinant protein is a sialidase that cleaves the influenza virus receptors in the airway epithelia. More details on DAS181 are provided in Section 3..
3. INHIBITORS OF THE HEMAGGLUTININ‐RECEPTOR INTERACTION
A. Structure of the Viral Hemagglutinin
Within the influenza virus particle, the single‐stranded, negative‐oriented RNA genome is divided over eight viral ribonucleoprotein (vRNP) segments, which are protected by the capsid shell formed by the M1 protein, further surrounded by the viral envelope. Two viral spike proteins protrude from the virion: the hemagglutinin (HA) and NA, which have a leading role in viral entry and release, respectively. The HA and NA glycoproteins are the main antigens against which the host immune response is raised. In the case of influenza A virus, 17 HA and 10 NA subtypes are known, which are all present in aquatic birds, the natural reservoir for influenza A viruses.34 The only exception is H17, which was isolated only recently from bats.35, 36 The emergence of a new pandemic virus is explained by the reassortment of genome segments, which occasionally occurs upon dual infection of an animal species (such as a pig) that carries the avian‐ as well as the human‐type influenza virus receptors.37
The influenza virus HA (Fig. 2A) is a homotrimeric type 1 membrane glycoprotein. Its membrane‐distal globular head domain contains the receptor binding site (RBS), whereas the HA stem structure (which contains the fusion peptide) is responsible for intraendosomal membrane fusion.34 In influenza virus‐infected cells, HA is first synthesized as its precursor protein HA0, which assembles into a noncovalently linked homotrimer38 and is cleaved into two polypeptides (HA1 and HA2 containing, in the case of H3, 328 and 221 amino acids, respectively), which remain covalently attached by a disulfide bond.39 For most HAs, HA0 cleavage occurs at a single arginine residue and is performed by a membrane‐bound or secreted serine protease that is restricted to bronchiolar epithelium, such as tryptase Clara, the human airway trypsin‐like protease or TMPRSS2.40, 41 The HAs from highly pathogenic avian viruses contain a series of basic residues at their cleavage site,42 allowing recognition by furin‐like intracellular proteases that are widely distributed in avian tissues, thus explaining their systemic spread and high virulence.43 Inhibition of the cellular proteases performing HA0 cleavage is an original antiviral strategy, and peptidomimetic furin inhibitors have proven to inhibit the replication of an avian influenza virus in cell culture.44 After HA0 cleavage, minor rearrangements lead to insertion of the fusion peptide (located at the N‐terminus of HA2) into a negatively charged cavity, thus priming the HA for pH‐dependent fusion.40 Posttranslational modifications of HA comprise the addition of acyl chains to the short cytoplasmic tail,45 and N‐glycosylation at several asparagine residues in the ectodomain.39 Besides masking the antigenic epitopes by sterically hindering antibody recognition,46, 47 the N‐linked glycans also function in the correct folding of HA in the endoplasmic reticulum,48, 49 modulation of receptor binding,50 controlling HA0 cleavage,51 and maintaining the HA in its metastable conformation required for fusion activity.52 The N‐glycans that are most conserved among various influenza HAs are located at the N‐terminus of HA0 (or after cleavage, HA1)48 and in the HA stem region.53
Figure 2.
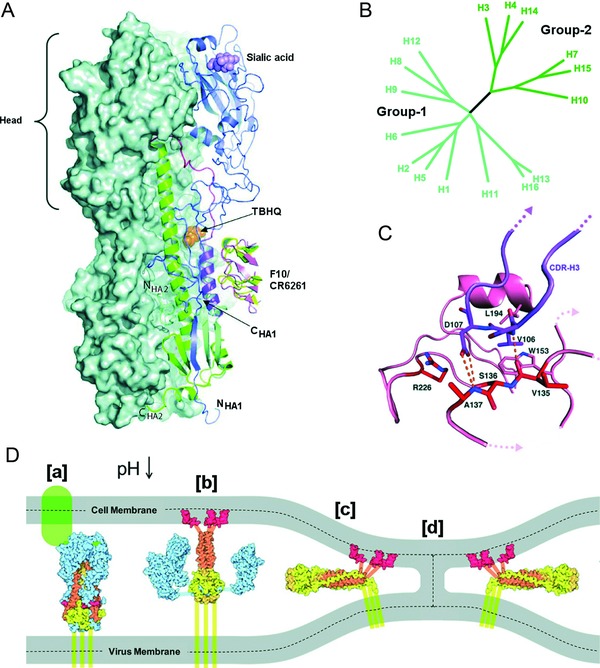
Structure and classification of influenza A HAs. (A) Structure of the viral hemagglutinin, showing the binding site for sialic acid (violet) in the globular head domain (blue ribbon structure), as well as the binding pockets in the HA stem structure for fusion inhibitors reported to prevent the HA conformational change, that is, the small‐molecule inhibitor TBHQ (orange) and the broad‐acting antibodies F10 (pink) and CR6261 (yellow). Two HA subunits are represented by their combined molecular surface, while the third one is shown in a ribbon diagram. [Reprinted by permission from Macmillan Publishers Ltd: Nature Structural & Molecular Biology Ref. Das et al.10 © (2010).] (B) Phylogenetic tree of influenza A HAs. Group 1 (cyan) can be subdivided into three clades (H8, H9, and H12; H1, H2, H5, and H6; H11, H13, and H16). Group 2 (green) is subdivided in two clades (H3, H4, and H14; H7, H10, and H15). The newly identified H17 is classified in the H1 clade of group 1.35 [Taken from Russell et al.,55 Copyright (2008) National Academy of Sciences, USA.] (C) Detail of the HA RBS indicating the binding mode of the CDR‐H3 loop (heavy‐chain complementarity determining region 3) of antibody CH65, which acts as a sialic acid mimic. The HA RBS is colored pink and the CDR‐H3 loop is shown in blue. The residues relevant for the antibody‐HA interaction are labeled; some of these are conserved HA1 residues involved in sialic acid binding (Ser1361, Trp1531, and Leu1941). [Taken, with permission, from Whittle et al.79] (D) Cartoon of the structural changes in HA during the HA‐mediated membrane fusion process. [a] The HA RBS binds to the sialylated cell receptor (in green). [b] The acidic pH in the endosome induces HA refolding, which leads to the exposure of the fusion peptide (in red) and its insertion in the endosomal membrane. [c] As a result of further conformational changes in HA, the viral and endosomal membranes are pulled together. [d] Mixing of the outer membrane leaflets generates the prefusion stalk intermediate. The dashed lines separate the inner and outer membrane leaflets. [Taken from Hamilton et al., 251 with permission]
The 17 influenza HA subtypes are classified into two phylogenetic groups (Fig. 2B). The H1 and H5 HAs belong to the same clade within group 1, whereas H3 HA belongs to group 2.35, 54, 55 Although this phylogenetic classification was primarily based on HA protein sequence, comparison of available HA crystal structures indicates that the regions involved in membrane fusion show striking similarities on a group‐specific basis.54
B. Species‐Specific Virus Binding to Sialylated Glycan Receptors
In the first step of the infection cycle, the HA attaches, via the RBS in its globular head, to sialylated glycoproteins or glycolipids on the host epithelial cells.56 This HA‐receptor interaction is highly specific for sialylglycoconjugates and plays an essential part in the species recognition of avian versus human influenza viruses.57 The HAs from human‐adapted viruses, including the pandemic viruses of the H1N1, H2N2, or H3N2 subtype, preferentially bind to cell‐surface glycans terminating in α2‐6‐linked sialyl‐galactosyl residues [Neu5Ac(α2‐6)Gal], whereas avian influenza A viruses have a preference for α2‐3‐linked sialyl‐galactosyl termini.58, 59, 60, 61, 62 The HAs of influenza B viruses which, in nature, are only detected in humans and seals, show a binding preference for α2‐6‐linked glycans.63, 64, 65 Thus, it is important to underline that the species specificity of the HA–glycan interaction is not based on recognition of the terminal sialic acid itself, but, rather, its linkage to the vicinal galactose and the sugars beyond galactose.66, 67 A correlation between glycan topology and species specificity was established from HA–glycan cocrystal structures as well as glycan array data.58 With regard to the HA residues that are directly involved in sialic acid binding, these are highly conserved across different HA subtypes. These amino acids (Tyr981, Ser1361, Trp1531, His1831, Leu1941) [amino acid numbering based on the H3 HA sequence; the suffixes 1 and 2 denote location in the HA1 and HA2 subunit, respectively] lead to a fixed orientation of the sialic acid relative to the HA RBS.68
Although sialic acid is generally considered to be the primary attachment receptor, influenza virus is able to bind and enter (though considerably less efficiently) into cells of which all surface sialic acids, whether attached to glycolipids or glycoproteins, were removed by treatment with exogenous Micromonospora viridifaciens sialidase.69 Hence, it has been proposed that, besides sialic acid, other receptors may be involved in influenza virus entry, which can work either independently or via a multistep process.69, 70
Which specific amino acid residues in HA govern its avian versus human receptor preference, varies among the different HAs, and is still incompletely understood, although α2‐6 tropism is generally linked to residues Asp1901 and Asp2251 in H1 and Leu2261 in H2 and H3 HAs.71 To cross the avian–human species barrier, acquisition of the human receptor binding preference is not sufficient, since additional amino acid changes are required, particularly in the influenza virus polymerase complex.71 In a recent study in which the avian H5N1 virus was passed in ferrets, four mutations in the head domain of H5 HA, combined with the Glu627Lys hallmark mutation in the PB2 subunit of the polymerase complex, were able to lead to airborne transmission of this virus in ferrets.72 A similar study with a reassortant virus carrying the HA of avian H5N1 also concluded that its avian‐to‐mammalian adaptation requires a combination of HA mutations to not only switch its receptor preference from α2‐3 to α2‐6, but also increase the stability of the HA protein.73
C. Antiviral Strategies to Interfere with HA‐Receptor Binding
When considering the HA‐receptor binding as an antiviral target, the multivalent nature of this interaction may present as a challenge. This binding is highly dynamic and involves an ensemble of sialylated glycans making contact with multiple HA trimers.74 In this manner, the avidity effects of the multivalent interaction compensate for the intrinsically low glycan binding affinity for a single binding site on HA [with a dissociation constant (Kd) in the millimolar range].75
Thus, to develop inhibitors that block the receptor binding of HA, at least three factors need to be taken into account: large sequence variation among HA subtypes and antigenic drift of HA; avian versus human‐specific receptor use; and multivalent nature of the HA‐receptor interaction. An ideal inhibitor would be species‐ and HA subtype‐independent. There are three conceivable strategies for inhibiting attachment of influenza virus to its target cell: (i) an antiviral compound binding to the HA RBS; (ii) an inhibitor blocking the sialic acid‐containing receptors on the epithelial cell membrane; or (iii) a receptor‐destroying agent.
1. HA‐Binding Agents
Virus‐neutralizing antibodies
The first and natural types of binding inhibitors are the virus‐neutralizing antibodies raised during the course of an influenza virus infection. These neutralizing antibodies are predominantly directed toward the surface of the membrane‐distal globular head domain of HA.76 During the 1918 pandemic, some patients were treated with human blood products from recovering influenza patients.77 Eight controlled studies reported between 1918 and 1925 were recently reviewed, and it was concluded that the overall case‐fatality rate was reduced from 37% among control patients to 16% among treated patients. Treatment was most effective when initiated early (i.e., less than 4 days after pneumonia became apparent).77 These historical data demonstrate that passive immunization with anti‐HA antibodies can be considered in case a pandemic occurs. Obviously, safety considerations about the use of patient‐derived materials need to be addressed. An elegant method for the isolation of human antibodies was reported by Simmons et al.,78 who prepared H5N1 neutralizing monoclonal antibodies from the memory B‐cells of patients recovered from an H5N1 infection. Two monoclonal antibodies were effective in a mouse influenza model when administered no later than 72 hr after infection.78 An attractive new concept is the development of monoclonal antibodies that bind to the conserved RBS of HA and, hence, are endowed with heterosubtypic HA neutralizing activity. A first human monoclonal antibody directed against H1 HA, encoded CH65, was derived from plasma cells of a person immunized with the 2007 trivalent influenza vaccine. Cocrystallization of its Fab fragment with H1 HA revealed that this antibody acts as a sialic acid mimic since the tip of its heavy‐chain complementarity determining region 3 (HCDR3) inserts in the RBS of H1 HA (Fig. 2C).79 Since CH65 was shown to neutralize 31 out of 36 H1N1 isolates covering a period of more than 30 years, and to interact with the conserved RBS itself, resistance selection by CH65 may be expected to be rare, unless associated with reduced viral fitness.79, 80 It should however be noted that the RBS of HA is smaller than the interaction site of an antibody81 and, therefore, CH65 forms additional interactions with RBS surrounding residues that are less conserved among the different HAs. The more broadly acting monoclonal antibody C05 binds to H1, H2, H3, H9, and H12 HAs and was isolated from a phage‐display library constructed from bone marrow donated after seasonal influenza infection. Cocrystallization studies demonstrated that the HCDR3 part of C05 forms a loop that inserts into the conserved RBS of HA, while its HCDR1 region makes only minimal contact with RBS surrounding and more variable residues.82 A third cross‐reactive monoclonal antibody, S139/1, neutralizes H1, H2, H3, H13, and H16 virus strains.83 The HCDR2 region of S139/1 was shown to form multiple hydrophobic interactions within the RBS of H3 HA. The rather low affinity of this binding interaction is compensated in the bivalent IgG molecule, and this avidity effect is required to broaden the neutralizing activity of S139/1 to strains of the H1, H2, H13, and H16 subtypes.84 Regarding influenza B viruses, the human monoclonal antibodies 3A2 and 10C4, reactive against B viruses of the Yamagata lineage, recognize the 190‐helix (residues 190–198 in HA1) near the RBS.85 The human monoclonal antibodies CR8033 and CR8071 were shown to neutralize both Yamagata and Victoria lineage B viruses and protect mice after challenge with a lethal dose of influenza virus.86 Although the therapeutic use of an anti‐influenza antibody may appear complicated, some parallel can be seen with the palivizumab antibody that is already in use for the prophylaxis of another respiratory virus, that is, respiratory syncytial virus (RSV).87 An innovative strategy to improve the pharmacokinetics and reduce the production cost of therapeutic antibodies consists of single‐domain antibody fragments (also referred to as Nanobodies) derived from camelid immunoglobulins.88 A Nanobody directed to the globular head of H5 HA was shown to be effective in H5N1‐infected mice. The activity of the monovalent Nanobody was increased by a factor 60 when using a bivalent format, consisting of two paratope containing domains connected by a flexible linker.89
Lectins
Another type of immune proteins capable of catching viruses is the collagenous C‐type lectins (referred to as collectins) such as the lung surfactant proteins. The role of surfactant protein D (SP‐D) in the innate immune response to influenza virus is explained by its capacity to cause virus particle aggregation, thereby preventing virus attachment to the host cells.90 Besides, SP‐D has various immunological effects that account for its ability to limit lung inflammation by respiratory pathogens.91 Regarding potential antiviral use, design of modified forms of the porcine SP‐D lectin (which has higher anti‐influenza virus activity than its human counterpart) is aided by the growing insight into how its carbohydrate recognition domain (CRD) precisely interacts with the high‐mannose glycans attached near the RBS of HA.92, 93 In addition, N‐linked sialoglycans attached to the CRD of SP‐D are considered important, since they may cause additional interactions between the SP‐D and the HA RBS and enhance the antiviral effect.90 A similar action principle, that is, binding to high‐mannose carbohydrates on the viral HA, accounts for the anti‐influenza virus activity of the bovine serum lectin CL‐43.94 Likewise, cyanovirin‐N, a lectin isolated from Escherichia coli, recognizes high‐mannose oligosaccharide structures on diverse viral glycoproteins, explaining its broad activity against unrelated viruses such as influenza virus and HIV.95 Cyanovirin‐N was shown to inhibit influenza virus replication in cell culture as well as mouse and ferret infection models.96, 97 Although SP‐D and cyanovirin‐N manifest broad anti‐influenza A and B virus activity, some virus strains (such as the A/PR/8/34 H1N1 strain) are known to be insensitive, due to the lack of particular Asn‐linked oligosaccharides on the head of their HA.98 The location and number of glycans attached to the head of HA is quite variable, since acquisition of epitope shielding oligosaccharides is part of the viral immune escape.46 In contrast, the glycans attached to the HA stem have a structural function in protein refolding, and the corresponding glycosylation sites are therefore more conserved.52, 53 This implies that antiviral use of lectin compounds directed toward HA head glycans might lead to escape mutants devoid of specific glycans, although the newly exposed antigenic sites might also render the mutated virus susceptible to immunological control.99
Sialyl‐containing macromolecules and sialomimetics
An alternative approach to block the HA RBS makes use of receptor mimics, such as sialyl‐containing macromolecules. The gangliosides sialylparagloboside (SPG) and GM3 (Neu5Acα2‐3Galβ1‐4Glcβ1‐1′ceramide) were proven to bind to HA and inhibit the virus‐induced cytopathic effect,100, 101, 102 and their antiviral activity correlated with their HA binding affinities.101 The hydrophobic ceramide moiety of SPG and GM3 was found essential, since the uncoupled trisaccharides 3′‐sialyllactosamine and 3′‐sialyllactose (which constitute the termini of SPG and GM3, respectively) produced no effect. Micelle formation of these gangliosides in aqueous solution likely causes protrusion of their sialic acid parts toward the outside of the micelles, resulting in high sialic acid density and, hence, a multivalent binding interaction with HA.101
In a recent report, Hendricks et al. described that liposomes bearing sialylneolacto‐N‐tetraose c (LSTc) can form multivalent interactions with influenza virus.103 In contrast to monovalent LSTc, these decoy liposomes are able to competitively bind influenza virus in a hemagglutination inhibition assay, and suppress influenza virus replication in cell culture and mouse models.
Pentadecapeptides binding to H1 and H3 HAs were obtained from phage‐displayed random peptide libraries by serially repeated affinity selection. A docking simulation indicated that these peptides act as sialomimetics. Some showed inhibitory activity against H1 and H3 influenza viruses in cell culture.104 Jeon et al. used a peptide with a sequence derived from the globular head region of HA to screen a DNA library for HA‐binding aptamers. The selected aptamer, A22, was proven to block the RBS of HA and inhibit influenza A viruses in vitro (i.e., cell culture) and in vivo (i.e., animal studies).105
Another macromolecule, the sulfated sialyl lipid NMSO3 (Fig. 3) showed antiviral activity against influenza H3N2, but not against B viruses.106 We recently found that NMSO3 inhibits influenza virus binding to cells at 4°C.107 Although NMSO3 has a strong negative charge and, hence, a direct interaction of NMSO3 with the sialic acid binding residues of the HA RBS can be anticipated, the precise mode of action of this antiviral compound remains to be determined. NMSO3 has broad activity against diverse viruses (in cell culture as well as animal models)106, 108 and presents as a relevant antiviral lead compound.
Figure 3.
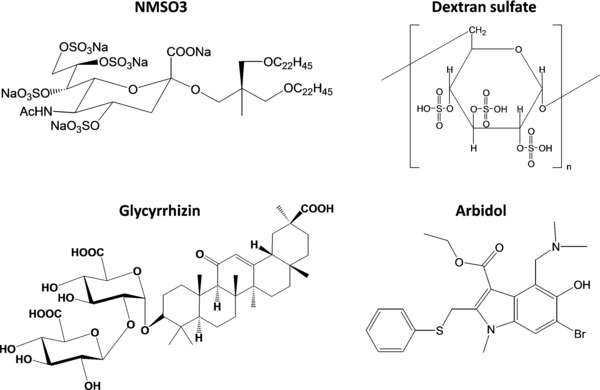
Chemical structures of diverse antiviral agents reported to inhibit the entry of, among others, influenza viruses. The sulfated sialyl lipid NMSO3 may act upon influenza virus binding107; glycyrrhizin may reduce membrane fluidity125, 126; and dextran sulfate184 and arbidol197 probably interfere with the low pH‐induced fusion process (see the text for all details).
2. Receptor‐Binding Agents
The opposite strategy to block binding of influenza virus to its cell receptor, is the development of sialic acid binding compounds. The HA‐binding and Neu5Acα2‐3Gal‐containing ganglioside GM3 was used to select potential inhibitors from a phage‐displayed random peptide library.109 Two pentadecapeptides, c01 (GWWYKGRARPVSAVA) and c03 (RAVWRHSVATPSHSV), were picked out and acylated to a C18 group, in order to form a molecular assembly and promote multivalent binding. Both C18‐peptides provided inhibition of influenza virus infection in cell culture. Their anti‐influenza virus activity was comparable to that of the wheat‐germ agglutinin lectin,109 which is known to interact with sialoglycoconjugates.110
3. Receptor Destroying Agents
Finally, a third strategy for inhibition of virus binding is destruction of the sialylated glycan receptors. Years ago, it was observed that cells are less susceptible to influenza virus infection after enzymatic removal of sialic acid from the cell surface.111 The new anti‐influenza virus agent DAS181 is a recombinant fusion protein, consisting of a sialidase catalytic domain derived from Actinomyces viscosus and an epithelium‐anchoring domain. DAS181 efficiently removes α2‐3‐ and α2‐6‐linked sialic acids and displays broad activity against influenza A and B viruses as well as parainfluenza viruses.112, 113 Since DAS181 acts on the host cell rather than the virus, it is assumed to have a reduced potential for generating drug resistance. After more than 30 passages in the presence of DAS181, influenza virus mutants were selected with low to moderate resistance to the compound (i.e., three‐ to 18‐fold increase in antiviral EC50 value). The resistant viruses showed an attenuated phenotype compared to the wild‐type virus, yet unchanged virulence in mice. When further passaged in the absence of compound, the viruses quickly regained the wild‐type sensitivity. Sequencing revealed that the resistant mutants contained substitutions in the HA near its RBS, as well as in the NA, leading to altered HA and NA functionality.114 The concern that desialylation of the airway epithelium might unmask certain cryptic receptors and increase the susceptibility to Streptococcus pneumonia, was contradicted by mouse experiments showing that DAS181 treatment does not lead to an increased incidence of secondary pneumonia.115 DAS181 requires topical delivery as an inhalant. It is currently in Phase II clinical trials (at once daily dosing of 10 mg during 3 days) for the treatment and prophylaxis of influenza‐like illness.116
4. INHIBITION OF ENDOCYTIC UPTAKE OR VIRUS TRAFFICKING
A. Different Endocytic Routes Exploited by Influenza Virus
After binding to the sialylated glycans on the cell surface, influenza virions are internalized by endocytosis. In general, viruses can be internalized by clathrin‐mediated endocytosis (CME); caveolin‐mediated endocytosis; macropinocytosis; or other less characterized mechanisms.117
Early electron microscopic analysis of influenza virus‐infected cells showed the presence of virions in clathrin‐coated pits and vesicles, providing direct evidence that influenza virus can enter the cell by CME. However, since virions were also found in smooth pits, the virus is able to follow an alternative clathrin‐independent pathway.118 Further support came from investigations in which dominant negative forms of cellular endocytic regulators were expressed, or by using pharmacological inhibitors, that is, the CME inhibitor chlorpromazine, the cholesterol‐depleting agents nystatin or methyl‐β‐cyclodextrin; or genistein, an inhibitor of caveola formation.119
Additional evidence that influenza virus exploits CME and a clathrin‐ and caveolin‐independent route in parallel, was provided by real‐time imaging. Both routes appear to be equally efficient in supporting the infection once the virus is internalized.120 Only recently, the clathrin‐ and caveolae‐independent influenza virus uptake was shown to have the characteristics of macropinocytosis.121 Influenza virus entry was completely inhibited when cells were simultaneously treated with dynasore and the amiloride derivative EIPA, which inhibit dynamin‐dependent CME and macropinocytosis, respectively.
The sialic acid attachment sites for influenza virus possess no host cell signaling capacity and, hence, additional postattachment factors may be required for efficient viral entry.122 Since the virus fails to enter Lec1 cells, a mutant CHO cell line that is totally deficient in N‐terminal glycosylation, it was suggested that N‐linked glycoproteins may be required for efficient endocytosis of the virus.122 Besides, binding of influenza A virus to cells was found to induce lipid raft rearrangement and activation of signaling molecules, such as the epidermal growth factor receptor (EGFR) or other receptor tyrosine kinases. Also, it was observed that the activated EGFR kinase is involved in promoting the initial virus uptake, and that virus internalization was considerably reduced in the presence of genistein, a broad inhibitor of receptor tyrosine kinases.123
Thus, the precise mechanisms for endocytic uptake of influenza virus are still not fully understood. Whether any of these insights may be translated into a relevant antiviral concept is unsure. Influenza virus appears to exploit endocytic routes and signaling platforms that are intimately linked to normal cell functioning and thus not readily amenable to selective antiviral intervention. For instance, the above‐mentioned pharmacological agents, which were very useful to demonstrate the role of CME or macropinocytosis, only affect the viral uptake at subtoxic concentrations.
B. Antiviral Strategies to Interfere with Endocytic Uptake and Virus Trafficking
One potential approach is the use of membrane fluidity modulators, which restrict the movement of membrane molecules. The neutral glycolipid fattiviracin (FV‐8; isolated from Streptomycetes) interferes with cell–cell fusion in HIV‐infected cells and was also reported to have anti‐influenza virus activity.124 Interference with cell membrane fluidity may also be the principal mode of action of glycyrrhizin (Fig. 3), the main active constituent of licorice root. Glycyrrhizin has broad antiviral activity against diverse enveloped viruses, including influenza virus, herpes simplex virus (HSV), varicella‐zoster virus (VZV), vaccinia virus, vesicular stomatitis virus, measles virus, HIV‐1, and the SARS coronavirus.125, 126 Its anti‐influenza virus activity was already demonstrated in 1983.127 More recently, a flow cytometric internalization assay was used to show that glycyrrhizin inhibits the endocytic uptake of influenza virus.126 Glycyrrhizin was further proven to decrease the fluidity of the cell membrane, an effect that was attributed to its cholesterol‐related chemical structure125 (Fig. 3). Besides its antiviral effect, glycyrrhizin displays anti‐inflammatory and immunomodulatory effects.128 These combined pharmacological effects may be advantageous in the treatment of virus infections with a strong inflammatory component, such as the severe airway inflammation (cytokine storm) caused by the avian H5N1 virus.128 In Japan, glycyrrhizin is already in clinical use since many years, and based on this the compound is considered to have favorable safety with no serious side effects.125, 126
Another broad‐spectrum antiviral agent interfering with membrane fusion is the aryl methyldiene rhodamine derivative LJ001. This compound displays activity against a wide range of unrelated enveloped viruses, including influenza A virus, HIV‐1, yellow fever virus, hepatitis C virus (HCV), vesicular stomatitis virus, and vaccinia virus.129 Time‐of‐addition experiments demonstrated that LJ001 acts upon virus entry, since inhibition was only achieved when the compound was added before or during infection. LJ001 was shown to intercalate into viral as well as cellular membranes. Its potent antiviral activity and low cytotoxicity was explained by the capacity of the host cell for active and rapid biogenic repair, while disruption of the virion envelope is irreversible.
Lipoglycopeptides are lipophilic derivatives of glycopeptide antibiotics such as the widely used antibiotic vancomycin. Some lipoglycopeptides are not only endowed with increased antibacterial activity, but also display activity against diverse viruses such as HIV, herpes viruses, or flaviviruses.130 Regarding influenza virus, we recently described the structure–activity relationship of a series of aglycoristocetin derivatives containing an aryl‐substituted cyclobutenedione.131 The lead compound SA‐19, which carries a phenylbenzyl substituent, displayed strong and consistent activity against all influenza A and B viruses tested.107 No resistance to SA‐19 was observed after 15 virus passages in cell culture. This compound was shown to cause intracytoplasmic trapping of influenza virus prior to its nuclear entry, presumably by disturbing the endocytic uptake of the virus at the site of the plasma membrane. It would be relevant to see whether kistamicin A and B, two ristocetin‐related glycopeptides that were reported to have anti‐influenza virus activity several years ago,132 display a similar mode of action as SA‐19. This is somewhat suggested by the fact that the antiviral activity is higher for kistamicin B, which contains a lipophilic substituent analogous to that of SA‐19.
An alternative strategy would be to interfere with endosome acidification. Upon internalization and entry into early endosomes, influenza viruses undergo an initial acidification step to pH ∼ 6. They then traffic to late endosomes, where further acidification to pH ∼ 5 provides the trigger for fusion of the endosomal and viral membranes.133 Acidification of the endosomes is accomplished by the cellular vacuolar proton ATPase (V‐ATPase), which is potently and selectively inhibited by the macrolide antibiotics bafilomycin A1 and concanamycin A. Both compounds block influenza virus entry when added within the first 10 min after infection.134 A different type of V‐ATPase inhibitor, the natural compound diphyllin, produced surprisingly potent and selective inhibition of influenza virus replication in cell culture.135 Likewise, lysosomotropic weak bases such as ammonium chloride and chloroquine inhibit influenza virus entry by elevating the endosomal pH.118, 136 Chloroquine shows in vitro anti‐influenza virus activity at concentrations that can, based on data from its use for malaria prophylaxis, be reached in humans.137 However, a double‐blind, placebo‐controlled efficacy trial concluded that chloroquine is unable to prevent influenza virus infection,138 and this agrees with its failure to prevent influenza virus infection in mouse and ferret models.139 Possibly, the chloroquine dose used in the clinical study may have been too low. This dose was estimated to produce blood concentrations in the range of the 50% antiviral concentrations in cell culture, and was selected so as to avoid any serious side effects.138 Thus, although bafilomycin A1 and chloroquine represent excellent tools to examine the precise mechanism of influenza virus entry, their relevance for influenza therapy is limited.
As explained in the next part, the adamantane compounds amantadine and rimantadine block influenza A virus entry mainly by inhibiting the M2 proton channel. At higher (∼100 μM) concentrations, they raise the endosomal pH due to their basic character, thereby affecting HA‐mediated fusion at low pH.140 Hence, amantadine‐resistant viruses selected in vitro can contain mutations in either the M2 or HA protein. Most of these HA substitutions render the HA less stable, and thus increase the pH at which fusion occurs. We recently observed that some H1N1 viruses such as the A/PR/8/34 strain are particularly sensitive to a subtle increase in the endosomal pH, as caused by newly synthesized amantadine analogues bearing different scaffold structures (Torres, unpublished data).
5. INHIBITION OF THE VIRAL M2 PROTON CHANNEL
A. Structure of the M2 Ion Channel
The low pH inside the endosomes activates the viral M2 proton channel that is embedded in the viral membrane, leading to transport of proton ions into the interior of the endosomally entrapped virus. As a result, the vRNPs become dissociated from the M1 matrix protein (the so‐called “uncoating” event), and the viral genome is released.141
The M2 of influenza A virus (A/M2) is a short polypeptide of only 97 residues, assembled into a homotetrameric, integral membrane channel protein consisting of (i) a short unstructured N‐terminal ectodomain (residues 1–24); (ii) a pore‐forming transmembrane helix (residues 25–46) responsible for tetramerization and proton translocation; (iii) a cytoplasmic amphipathic helix (residues 47–61), involved in virus assembly and budding; and (iv) a disordered tail (residues 61–97) that interacts with the M1 matrix protein141 (Fig. 4). Activation of the M2 ion channel below pH 6 is caused by protonation of the third His37 residue in the M2 tetramer.142, 143 The protonated imidazole ring of His37 is involved in a cation–π interaction with the indole ring of Trp41.142 These two residues, His37 and Trp41, functioning as a pH sensor and gate, respectively, are critical for M2 proton channel function, and hence invariable among influenza A and B viruses.144, 145 Besides, Val27 forms a valve that controls the entrance of protons, while Asp44 is indirectly hydrogen bonded to the indole nitrogen of Trp41 via a water cluster at the exit of the channel. Thus, Asp44 and Val27 act as gatekeepers at opposite ends of the channel.146, 147 Comparison of the NMR and crystal structures of the A/M2 transmembrane domain obtained at neutral (pH 7.5–8), intermediate (pH 6.5), or acidic (pH 5) pH, provided a detailed insight into the low pH‐induced changes in A/M2 protein conformation.146, 147, 148 At neutral pH, the Val27 valve is open, whereas the Trp basket, formed by the Trp41 residues at the opposing end, has a small hydrophobic opening. When the pH is reduced, the Val27 valve constricts, while the Trp basket opens.147 Two mechanisms for proton transport through the aqueous pore of A/M2 have been proposed. In the wire model, protons are conducted via a continuous column of water molecules. Opening of the pore is achieved by electrostatic repulsion of the protonated His37 residues, which, according to this model, play only a passive role.142, 149 In contrast, in the shuttle model, His37 plays an active role in proton transport by protonation and deprotonation, which is facilitated by imidazole ring reorientations and small‐amplitude backbone fluctuations.150, 151
Figure 4.
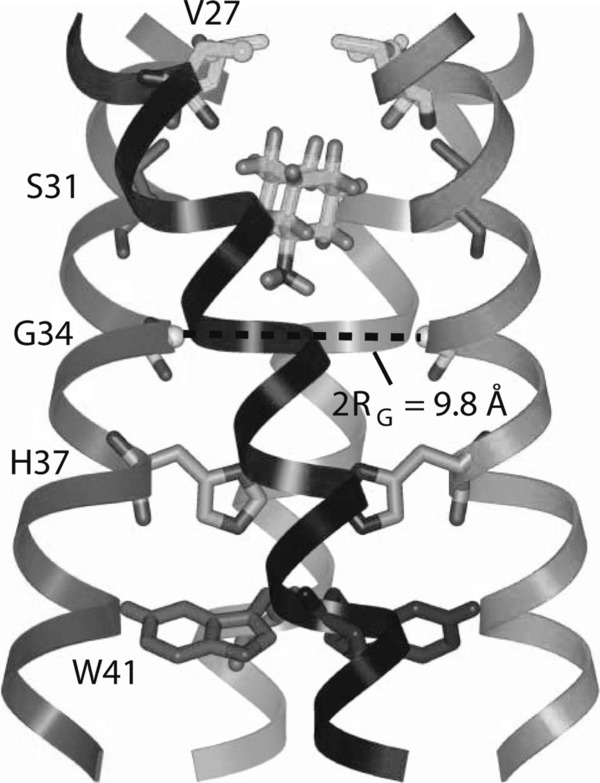
Solid‐state NMR structure of amantadine‐bound A/M2 proton channel in lipid bilayers. Side view showing the luminal site. His37 and Trp41 function as pH sensor and gate, respectively, while Val27 acts as a gatekeeper controlling the entrance of protons. The amantadine binding pocket is formed by Val27, Ala30, Ser31, and Gly34. Substitution of these residues causes amantadine resistance. [Reprinted by permission from Macmillan Publishers Ltd: Nature Ref. Cady et al.155 © (2010).]
In analogy to the A/M2 protein, BM2 (the M2 protein from influenza B virus) forms a homotetrameric integral membrane protein, with characteristic proton channel activity and a pH profile similar to that of its functional homolog A/M2. Due to its coiled‐coil structure, the transmembrane region of BM2 is able to form a stable tetramer by itself, without the C‐terminal amphipathic helix that is necessary for tetramerization of A/M2.152 Except for the HXXXW motif in the transmembrane domain, with the His and Trp acting as key residues for proton channel activation and gating, A/M2 and BM2 share little sequence homology. Furthermore, the BM2 proton channel activity is higher than that of A/M2.144 This higher conductance may in part be explained by two extra serine residues in the channel pore of BM2, which can facilitate proton relay.152
B. Inhibitors of the M2 Ion Channel
The discovery that the adamantane derivatives amantadine and rimantadine (Fig. 5) inhibit influenza A virus replication was made decades ago153 and, in fact, was instrumental in elucidating the function of M2.154 Both amantadine and rimantadine are inactive against influenza B viruses. Cocrystallization of amantadine with the transmembrane domain of A/M2 identified a drug binding site in the N‐terminal channel lumen, that is surrounded by residues that are mutated in amantadine‐resistant viruses (in particular, Val27, Ala30, Ser31, and Gly34). Binding of amantadine apparently leads to occlusion of the channel pore, but may also affect protonation of the critical His37 residue.146 On the other hand, a solution NMR of the A/M2 channel in complex with rimantadine revealed four equivalent binding sites, located on the lipid‐facing side of the channel, between adjacent helices near the Trp41 gate. In this way, binding of rimantadine could stabilize the closed state of the A/M2 tetramer.148 Finally, solid‐state NMR spectroscopy of A/M2 in phospholipid bilayers showed the existence of two amantadine binding sites: a high‐affinity site in the N‐terminal lumen, which is occupied by a single amantadine molecule, and a low‐affinity site at the C‐terminal protein surface, which only becomes occupied at higher amantadine concentrations.155 The presence of both binding sites was confirmed by molecular dynamics simulations, which further indicated that amantadine can bind inside the N‐terminal lumen under low‐ and high‐pH conditions.156 Importantly, the identification of the A/M2 binding sites for amantadine and rimantadine provided an explanation why both compounds lack activity against influenza B viruses. Compared to A/M2, the BM2 pore has two more serine residues, which probably disable binding of the hydrophobic adamantane moiety within the BM2 channel.149, 152 Also, the residues that make up the low‐affinity binding site for amantadine in A/M2 have uncorrelated counterparts in the BM2 protein.152
Figure 5.
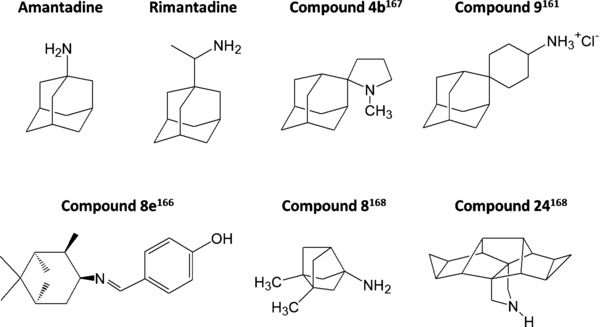
Chemical structures of amantadine, rimantadine, and a selection of published analogues. The codes shown are those used in the original reports. The spiro‐adamantane compound 9161 possesses activity against mutant A/M2 ion channels. The imine compound 8e166 and spiro compound 4b167 are both ∼200‐fold more potent than amantadine. Compounds 8168 and 24168 are ring‐contracted and ring‐expanded polycyclic analogues, respectively.
During many years, amantadine and rimantadine have been successfully used for both prophylaxis and therapy of influenza A virus infections, though amantadine is associated with neurological side effects.13 Nowadays, their clinical utility is limited since most circulating strains are adamantane‐resistant.157, 158, 159 Thirty percent of treated patients shed adamantane‐resistant mutants, which replicate equally well as wild‐type virus, are cross‐resistant to amantadine and rimantadine, and are readily transmitted to contact persons.13, 159 During the 2009–2010 season, 99.9% of H1N1 virus isolates were adamantane‐resistant.157 The resistance mutations are mostly located in the transmembrane region of the A/M2 protein, the most common changes being Leu26Phe, Val27Ala, Ala30Thr, Ser31Asn, Gly34Glu, and Leu38Phe.160
Attempts were made to develop new adamantane derivatives, which are able to interfere with the A/M2 ion channel activity of amantadine‐resistant viruses. Guided by the novel structural insights into the A/M2 binding interaction of amantadine, Wang et al. recently developed spiro‐adamantane inhibitors with potent activity against Val27Ala and Leu26Phe mutant A/M2 proteins.161 These molecules have a larger size than amantadine and are therefore able to fill the upper pore of A/M2, even when its volume is increased by the Val27Ala or Leu26Phe substitution. One compound (9 161; Fig. 5) showed antiviral activity against the wild type as well as the A/M2‐Val27Ala and A/M2‐Leu26Phe mutant viruses, and its EC50 values were similar to that of amantadine against the wild‐type virus.161 An imidazole derivative of pinanamine, synthesized by Zhao et al., showed moderate inhibition of an A/M2‐Ser31Asn mutant virus.162
Several research groups have developed polycyclic amine compounds to achieve more potent inhibitors of A/M2.163, 164, 165 Two fine examples are the imine compound 8e 166 (Fig. 5) and the spiro compound 4b 167 (Fig. 5), which were both reported to be ∼200‐fold more potent than amantadine. Although compound 8e166 was found to be cross‐resistant to amantadine when evaluated against an A/M2 mutant virus, it can serve as a novel scaffold for the design of superior M2 blockers. Another study explored the size limits of polycyclic amine derivatives as potential A/M2 inhibitors.168 Surprisingly, both ring‐contracted (8 168 in Fig. 5) and ring‐expanded (24 168 in Fig. 5) polycyclic compounds were able to bind to wild‐type A/M2, and some analogues showed increased binding affinity compared to amantadine itself. Biochemical studies with mutant A/M2 proteins and molecular docking indicated that compared to amantadine, one of the ring‐expanded derivatives showed a different binding mode to the high‐affinity A/M2 binding site (i.e., the inner channel pore region delineated by Val27, Ala30, and Ser31).
Amantadine not only targets the A/M2 channel, but, as a weak base, also indirectly inhibits HA‐mediated fusion at concentrations at least 100‐fold higher. Thus, an alternative approach is to develop a compound reacting with both targets at similar concentrations. In this case, viral resistance would require the appearance of amino acid changes in two separate viral proteins, which may be expected to be a rare event. Replacement of the primary amino group of amantadine by a more basic secondary or tertiary amino group, and addition of side groups on the adamantane ring system, resulted in compounds interfering with HA at lower concentrations, while the concentration affecting M2 proton channel activity was increased. However, during passage of the virus in the presence of these compounds, the escape rate was still high, yielding drug‐resistant mutants with amino acid substitutions in both the HA and A/M2 proteins.169 A reason for this high escape rate may be that the resistance mutations selected by these compounds can be located at different sites in HA or M2, without any reduction in viral fitness.
6. HA‐MEDIATED MEMBRANE FUSION: AN EMERGING ANTIVIRAL TARGET
A. Low pH‐Induced Fusion Mechanism
The low pH inside the late endosome leads to an extensive and irreversible conformational change of the viral HA, resulting in fusion of the viral and endosomal membranes (Fig. 2D). A key role is played by the fusion peptide (defined as the 23 N‐terminal residues of HA2), which is the most conserved region of HA and contains a series of hydrophobic residues.170, 171 At neutral pH, the fusion peptide is sequestered in a pocket of ionizable residues, but upon acidification, to pH 5–6 for most influenza viruses, the fusion peptide is extruded toward the target membrane. By comparing the X‐ray crystallographic structures of the ectodomain portion of HA, obtained at either neutral or acidic pH, the following rearrangements were noted to occur at low pH: (i) the globular head domain containing the RBS detrimerizes; (ii) the N‐terminus of the central triple‐stranded coiled coil is extended by the interhelical chain and the short α‐helix, hereby releasing the fusion peptide from its buried position; and (iii) in the middle of the long α‐helix two turns undergo a helix‐to‐loop transition to form a 180° reverse turn, positioning the fusion peptide and viral membrane anchor at the same end.55, 172 The actual membrane fusion proceeds through a hemifusion intermediate173 (Fig. 2D). According to the stalk‐pore model, the extruded fusion peptide inserts into the endosomal membrane. At the same time, the C‐terminus of HA2, which is anchored in the viral membrane, is reoriented thereby drawing the endosomal and viral membranes together. After mixing of the outer membrane leaflets (prefusion stalk intermediate), a hemifusion diaphragm is formed. Mixing of the inner and outer membrane leaflets results in the formation of a fusion pore, allowing release of the vRNPs into the cytoplasm.174
B. Inhibitors of HA‐Mediated Membrane Fusion
1. Small Molecules Binding to the HA Stem
A first approach to interfere with the HA‐mediated fusion process is to inhibit the acid‐induced conformational change of HA, using small molecules that bind to and stabilize the neutral pH conformation. One of the first influenza virus fusion inhibitors to be reported was tert‐butyl hydroquinone (TBHQ; Fig. 6), which specifically inhibits H3 viruses.175 Several years later, the binding site of TBHQ within the H3 HA stem structure was identified by crystallization of the TBHQ‐HA complex, and was shown to lie within a hydrophobic pocket, formed at an interface between HA subunits.55 Besides several hydrophobic interactions, TBHQ is hydrogen bonded with the side chain carbonyl of Glu572 and the main chain carbonyl of Arg542 of one monomer, and the main chain carbonyl of Leu982 of another monomer, hereby stabilizing the nonfusogenic HA conformation.55 During the conformational change of HA, a critical role is played by the adjacent Lys582, located at the C‐terminus of the short α‐helix and involved in the loop‐to‐helix transition.55 The relevance of the hydrophobic pocket around Glu572 for the development of fusion inhibitors active against group 2 HAs was further confirmed by our studies with the novel anti‐influenza virus agent 4c (Fig. 6).176 Although 4c and TBHQ have very different chemical structures, we noticed a clear similarity between the HA binding mode of TBHQ and that predicted for the N‐(1‐thia‐4‐azaspiro[4.5]decan‐4‐yl)carboxamide part of 4c. However, the aromatic imidazo[2,1‐b]thiazole ring of 4c allows the formation of several additional hydrophobic interactions within this cavity. The inactivity of the two compounds against group 1 viruses can be explained by analysis of HA crystal structures, which revealed that residues 562–582 in group 1 HAs form an extra turn, resulting in blockage of the TBHQ/4c binding site.55 Unfortunately, the antiviral activity of 4c is restricted to H3N2 viruses, since an H7N2 virus was shown to be insensitive, despite the fact that the H3 and H7 HAs belong to the same phylogenetic group 2. Also, resistance to 4c emerged within only three passages in cell culture.176 Conversely, several fusion inhibitors targeting group 1 HAs have been reported in the literature, that is, BMY‐27709, CL‐385319, RO5464466, and stachyflin (see Fig. 6 for chemical structures), which inhibit the conformational change of H1 (and, when tested, H2) HA but, unfortunately, have no activity against H3 viruses.177, 178, 179, 180 Attempts to override this subtype dependency by synthesizing novel derivatives proved unsuccessful.181 Also, initial predictions of their HA binding pocket using in silico docking did not fully correlate with subsequent data obtained after cocrystallization of the compound with HA or photoaffinity labeling.55, 175, 177, 182 Whatever their virus specificity, these small molecule fusion inhibitors were all found to readily select for resistance, at least in cell culture. Two types of resistance mutations have been identified. The first are amino acid substitutions within the binding pocket, which affect the inhibitor binding to HA. Alternatively, the HA stabilizing effect of the inhibitors can be counteracted by HA mutations that elevate the fusion pH, meaning that the mutant HA acquires its fusogenic conformation at less acidic pH.55, 176
Figure 6.
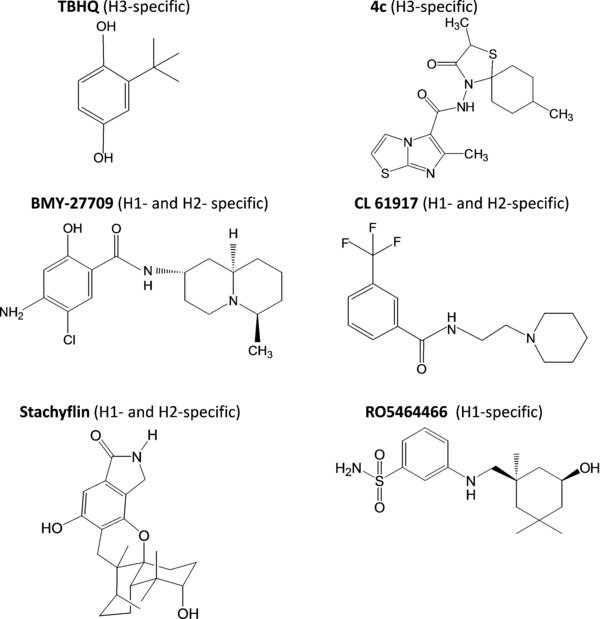
Chemical structures of small‐molecule inhibitors of the HA conformational change. For each compound, the subtype specificity, as far as tested, is given in brackets. See the text for references on individual compounds.
Thus, further development of this type of small molecule fusion inhibitors has been hindered by their subtype‐dependent anti‐influenza virus activities and low barrier for resistance selection. There may, however, be other ways to inhibit the HA‐mediated membrane fusion. Instead of preventing HA refolding, diiodofluorescein induces the irreversible conformational change of HA. These premature rearrangements, resulting in virions with fusion‐inactive HAs, also lead to inhibition of the fusion process.183
2. Antivirals Interfering with Membrane Fusion
Furthermore, it may be possible to interfere with membrane fusion following the HA refolding event. This mode of action has been proposed for dextran sulfate, a sulfated polysaccharide (Fig. 3) with broad‐spectrum antiviral activity. This agent has been reported to inhibit not only influenza A virus, but also HIV, RSV, HSV, and cytomegalovirus.184, 185, 186 The anti‐influenza virus activity of dextran sulfate, which appears to be restricted to influenza A viruses, correlates with its molecular weight, and levels off when the molecular weight increases above 10,000.184 The anionic dextran sulfate can be assumed to form electrostatic interactions with the viral HA, which has a net positive charge at pH 7 or less.187 This is consistent with fluorescence microscopy studies, showing the binding of fluorescein‐labeled dextran sulfate to HA‐expressing cells.188 While dextran sulfate had no effect on virus binding at 4°C,184 it was found to inhibit the low pH‐induced fusion process using a fusion assay based on octadecyl‐rhodamine fluorescence dequenching.187 No direct inhibition of the acid‐induced refolding of HA was noticed.188 However, in order to be active, the compound needed to be present during the fusion process at low pH.187, 188 These combined data suggest that the dextran sulfate binding site might be inaccessible in the low‐pH HA‐membrane complex and that dextran sulfate may interfere with a step subsequent to the conformational rearrangement of HA, for instance by causing steric hindrance of the membrane mixing event.188, 189 It remains to be investigated whether other polysulfated polysaccharides with anti‐influenza virus activity (such as compound pKG‐03 that was isolated from a microalga190) have a similar mode of action as dextran sulfate.
Another high molecular weight molecule, retrocyclin 2, also acts against a wide range of viruses, including influenza virus, HIV, and HSV.191, 192, 193, 194 Retrocyclin 2 is a circular octadecapeptide belonging to the θ‐defensins, which are antimicrobial peptides of the innate immune system.195 A detailed mechanistic study showed that its inhibitory effect on influenza virus replication was based on prevention of the HA‐mediated membrane fusion at low pH.194 However, retrocyclin 2 remained effective when added after the conversion of HA to its fusogenic conformation or after hemifusion, an intermediate state in which the outer membrane leaflets have merged while the inner leaflets are still separated. Thus, retrocyclin 2 was proposed to prevent the subsequent membrane rearrangements by causing cross‐linking and immobilization of surface glycoproteins.194
A similar interference with the membrane fusion process probably accounts for the broad anti‐influenza virus activity of arbidol. This small molecule (Fig. 3) has been licensed in Russia and China for influenza virus prophylaxis and therapy. Besides influenza A and B viruses, its antiviral spectrum encompasses RSV, parainfluenza virus, rhinovirus, hepatitis B virus, and HCV.196 It is well tolerated as a drug and arbidol‐resistant influenza viruses have not (yet) been isolated in the clinic.197 However, arbidol‐resistant viruses, obtained after 14 virus passages in cell culture, were shown to carry mutations in the HA2 subunit associated with an increased fusion pH.197 Arbidol may thus act by stabilizing the prefusogenic HA protein in a similar manner as TBHQ and the other small molecule fusion inhibitors described above, but, unlike the latter compounds, arbidol is less subtype‐specific.197 An alternative mode of action was proposed from biochemical studies with various model membranes, showing that arbidol has membranotropic properties, particularly due to its interaction with negatively charged membrane phospholipids.198, 199 Since this membrane interaction is most pronounced at acidic (fusion) pH, arbidol could alter the membrane fluidity during the fusion process and make the bilayer less fusogenic.196 Likewise, the inhibitory effect of arbidol toward HCV entry was explained by its capacity to dually interact with cell membrane phospholipids and aromatic residues (such as tryptophan) that are present in fusion‐mediating glycoproteins of HCV. This complexation would prevent the conformational changes in the viral glycoprotein required for membrane fusion.196, 198 At this time, a dual interaction of arbidol with membrane phospholipids and the influenza virus HA is merely speculative, but this mode of action would reconcile the biochemical and virological in vitro data outlined above. In the context of in vivo studies, arbidol may also have immunostimulatory properties by inducing interferon‐α, activating phagocytic macrophages, or stimulating the humoral and cell‐mediated immune response.200
3. Broad‐Neutralizing Antibodies
As already explained, several reported fusion inhibitors suffer from subtype‐dependent anti‐influenza virus activity and rapid emergence of resistance. These drawbacks could be avoided by targeting the fusion peptide, which is highly conserved among all HAs and contains the 23 N‐terminal residues of HA2. A monoclonal antibody directed against the first 15 residues of HA2 was selected from mice immunized with an H5N1 virus.201 In vitro, this MAb 1C9 antibody inhibits syncytium formation in HA‐expressing cells, indicating inhibition of the fusion process. When administered to mice, MAb 1C9 provided protection against H5N1, both prophylactically and therapeutically.201 Though highly relevant, cross‐reactivity with other HAs was not yet investigated.
A recent strategy with high clinical relevance comes from the discovery of broad neutralizing antibodies directed against relatively conserved pockets in the HA stem structure.202 Already in 1993, the first antibody reacting with different HA subtypes was selected.203 This mouse monoclonal antibody, designated C179, was shown to neutralize the H1, H2, and H5 HAs, all belonging to group 1.203, 204 Identification of the resistance mutations in C179‐resistant viruses, obtained by virus passaging in the presence of this antibody, allowed to locate its binding site in the middle of the HA stem. C179 was proven to inhibit HA‐mediated fusion in a polykaryon assay in influenza virus‐infected cells,203 and was shown to be effective in H1N1‐ or H2N2‐infected mice.205 The first human antibodies to be identified were specific for either group 1 or group 2 HAs, and were obtained by systematic screening of a wide array of B‐cells from influenza‐vaccinated or influenza‐infected individuals, or by constructing combinatorial libraries. The antibodies F10 and CR6261 show broad neutralizing activity against group 1 HAs, and a partially overlapping binding pocket within the HA stem.206, 207, 208 Crystallization of F10 and CR6261 in complex with H1 or H5 HA revealed that a conserved hydrophobic tip on their HCDR2 region inserts into a hydrophobic pocket adjacent to the short α‐helix in the HA stem, thereby allowing interactions of the antibody with the fusion peptide.206, 207 Another monoclonal antibody, encoded CR8020, interacts with H3 and H7 HAs, which belong to group 2. Cocrystallization of CR8020 with H3 HA identified its binding pocket lower down the HA stalk, thus in closer proximity to the viral membrane compared to CR6261.209 Stabilization of the HA prefusogenic conformation by CR6261 and CR8020 was corroborated by the finding that both antibodies prevented exposure of protease‐susceptible sites in HA when the virus was incubated at low pH.206, 209
Broad coverage of all influenza A viruses could be achieved by combining a group 1 and group 2 specific antibody. Further significant progress was made by Corti et al., who successfully isolated a pan‐influenza A neutralizing antibody that recognizes all group 1 and group 2 HAs, by interrogating a large number (about 100,000) of donor plasma cells.210 Cocrystallization of this FI6 antibody with an H1 or H3 HA protein revealed its interaction with a conserved epitope in the F (fusion) subdomain.210 Hence, binding of FI6 or the optimized FI6v3 antibody is assumed to increase the stability of the F subdomain, thus preventing the conformational change of HA that is required for membrane fusion. This mode of action accords with the inhibitory effect of FI6 on syncytium formation in HA‐positive cells. Alternatively, prevention of HA0 cleavage (at least for viruses requiring extracellular cleavage) or cross‐linking of HA subunits, have been implicated in the virus‐neutralizing activity of FI6. Passive immunization with FI6 was shown to confer prophylactic and therapeutic protection to influenza virus‐infected mice and ferrets.210 Recently, Dreyfus et al. isolated the human monoclonal antibody CR9114, which neutralizes influenza A and B viruses.86 CR9114 recognizes an epitope in the HA stem that is nearly identical to that of the group 1‐specific antibody CR6261. However, subtle conformational differences explain why the CR9114 antibody has a much broader anti‐HA reactivity.
Another HA stem‐binding antibody, PN‐SIA28, showed antiviral activity against all group 1 viruses tested (i.e., H1N1, H2N2, H5N1, and H9N2 viruses), as well as some isolates of the H3N2 virus, which belongs to group 2.211 However, H3N2 strains isolated after 1982 and H7N2 viruses were not inhibited. In order to localize the binding epitope for PN‐SIA28 in the HA stem, the authors selected escape mutants by repeated passaging of the virus in the presence of the antibody. Similar attempts to generate escape mutants with some of the other broad neutralizing antibodies suggest that viruses with mutations in the corresponding HA stem regions do not readily emerge in cell culture. For instance, with the CR6261 antibody, ten virus passages were required,208 while in other studies, no escape mutants were detected.212 These observations seem to indicate that the conserved HA stem epitopes targeted by these broad acting antibodies are less prone to mutations due to fitness constraints. This hypothesis, however, still remains to be verified by mutational analysis. It is clear that the discovery of these broad neutralizing anti‐HA stem antibodies offers entirely new perspectives for passive or active immunization against influenza A viruses. Also, peptides directed against the conserved epitopes in the HA stem region have been developed, such as the HB36 peptide that interacts with the CR6261‐binding epitope and recognizes several group 1 HAs (i.e., H1, H2, H5, and H6).213 The concept of a therapeutic peptide used to inhibit virus fusion is validated in the HIV field by the clinical use of enfuvirtide, a 36 amino acid peptide that binds to the HIV gp41 protein.214
7. INHIBITION OF NP‐MEDIATED VIRAL NUCLEAR IMPORT
After disruption of the vRNPs from the matrix M1 protein and fusion pore formation, the vRNPs are released in the cytoplasm and transported into the nucleus.215, 216 How the vRNPs are released from M1 is only partially understood. In the intact virion, the M1 protein forms the capsid shell located underneath the envelope, and is tightly associated with the vRNPs.217, 218 The current insights into the protein structure of M1 and its crucial role in organizing virion structure were recently reviewed.219 Once inside the endosome, the M2‐mediated acidification of the virion interior leads to vRNP uncoating, possibly by inducing a conformational change in M1.220, 221 Recent studies indicate that the oligomerization state of M1 is pH‐dependent and that oligomers of intact M1 dissociate into stable dimers at acidic pH.222 The disappearance of a visible M1 layer in virions exposed to pH 5 was imaged by cryoelectron tomography.223 After their transport through the fusion pore, the vRNPs appear to be associated with some residual M1 protein, which, inside the cytoplasm, dissociates from the vRNPs to finally allow their nuclear entry.215, 224 This second dissociation process may depend on cytosolic M1 modifications, such as phosphorylation or zinc binding.220, 225 A peptide derived from the zinc finger domain of M1 was reported to display broad and potent anti‐influenza virus activity in cell culture when added within 1 hr after infection, classifying this “peptide 6” as an entry inhibitor.226 As far as we know, no other attempts have been reported in which M1 was explored as an antiviral target. Development of a potent M1 inhibitor might be challenging, due to the abundant presence of this protein in the virion.
At last, the free vRNPs are imported in the nucleus via the nuclear pores. Each vRNP contains one of the eight vRNA genome segments, which is associated with a single copy of the viral polymerase (the heterotrimer of PB1, PB2, and PA), and multiple copies of the nucleoprotein (NP).227 Although these four viral proteins all contain at least one nuclear localization signal (NLS), the vRNP nuclear import appears to be primarily dependent on the NLS in the N‐terminus of NP.228, 229, 230 Due to this NLS, the vRNP is recognized as a cargo by the cellular importin‐α protein, and after formation of a ternary complex with importin‐β, is transported into the nucleus.231 The specificity of NP (and PB2) for the different isoforms of importin‐α differs for avian and human viruses, implicating a role in influenza virus adaptation.231, 232
The viral NP has both structural and regulatory functions in influenza virus replication. Besides being the main structural component of the vRNPs, NP has a crucial role in the consecutive replicative stages, by regulating vRNP nuclear import; viral RNA transcription; and nuclear export (via interaction of NP with M1).233 The conserved protein sequence of NP further adds to its attractiveness as an antiviral target, since this implicates that NP inhibitors could be broadly active across the different virus subtypes.233 This is illustrated by the small molecule ingavirin, which inhibits influenza A and B viruses in vitro and in vivo.234 Ingavirin was reported to inhibit NP oligomerization and subsequent nuclear import of newly synthesized NP.235 This mechanism of action is distinct from that of the NP aggregating agents nucleozin and its structural analogues 3061 and “compound 3”, which were independently identified by several groups236, 237, 238, 239 (Fig. 7A).
Figure 7.
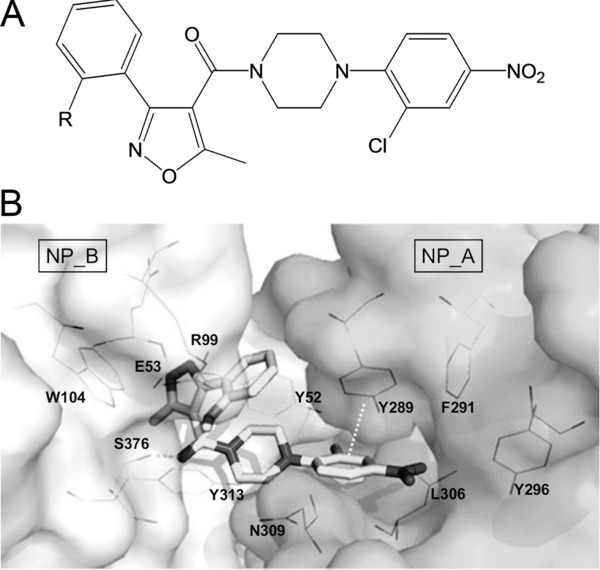
Chemical structure and NP‐binding site of nucleozin. (A) Chemical structure of nucleozin (R = H),236 3061 (R = Cl),238 and compound 3 (R = OMe).239 (B) X‐ray structure of the oligomeric complex of compound 3 with influenza virus NP. Six molecules of compound 3 bridge two NP trimers (NP trimer A and NP trimer B) to form a hexamer. [Taken from Gerritz et al.,239 with permission.] Critical interactions made by compound 3 include a hydrogen bond with Ser376 and a π‐stacking interaction with Tyr289.
These NP binding agents display anti‐influenza virus activity against all influenza A viruses tested, including H1N1, H3N2, and H5N1 viruses.238, 239 In the first report, nucleozin was proven, by fluorescence microscopy, to cause NP aggregation and trap the vRNPs in a peri‐nuclear halo.236 Nucleozin was also active in the cellular vRNP reconstitution assay, which directly measures the transcriptional activity of vRNP.236 This indicates that nucleozin not only interferes with nuclear entry, but also with other replicative processes in which NP is involved. The NP aggregating activity of nucleozin was confirmed by experiments in which NP was cocrystallized with the related “compound 3”.239 Formation of higher order NP oligomers was observed, in which two NP trimers are linked to each other through six molecules of “compound 3”, each interacting with two antiparallel binding pockets.239 The nitrophenyl moiety of “compound 3” interacts with one binding pocket (close to the NP residues Tyr289 and Asn309) in an NP from one trimer, while the isoxazole heterocycle binds to the other binding pocket (around the Tyr52 residue) of an NP in the other trimer, and vice versa239 (Fig. 7B). These data nicely agree with the resistance mutations identified in NP (i.e., Tyr52Cys/His, Tyr289His, and Asn309Lys) after virus passaging in the presence of these NP binding agents.236, 238, 239 Nucleozin showed a rather modest in vivo activity, protecting 50% of H5N1‐infected mice.236 However, full protection of H5N1‐infected mice was obtained with “compound 5”, a derivative of “compound 3” with improved solubility and metabolic stability.239
8. INTERFERING WITH CELLULAR FACTORS INVOLVED IN INFLUENZA VIRUS ENTRY
Although most available antiviral strategies are directed toward a viral protein, the possibility to block a cellular component with a critical role in virus replication receives increasing attention.240 An antiviral targeting a host cell factor can be assumed to have reduced selectivity (i.e., window between cytotoxicity and antiviral efficacy). On the other hand, its resistance barrier could (in theory) be higher when compared to a direct antiviral compound.241 For a virus with a high mutation rate such as influenza virus, this appears a considerable advantage.242
Two studies using genome‐wide RNA interference screening identified several host cell factors necessary for influenza virus replication.135, 243 Further analyses, based on a pseudotyped particle entry assay, allowed the selection of cellular factors that regulate the low pH‐dependent and HA‐mediated entry. Among them are proteins involved in the IP3‐protein kinase C (PKC) or phosphatidylinositol‐3‐kinase (PI3K)‐Akt signaling pathways; COPI components (involved in endosomal trafficking); vacuolar ATPases; fibroblast growth factor receptor135; and SON DNA binding protein (important for influenza virus trafficking to late endosomes).243 These intriguing data create new opportunities for designing antiviral concepts toward host cell factors.242 In a proof‐of‐concept study, compounds such as sirolimus, podophyllotoxin, or other inhibitors of any of these host cell factors, were found to inhibit virus replication with quite remarkable selectivity.135
The bisindolylmaleimide compounds specifically inhibit all PKC isoenzymes with a similar potency, by blocking the ATP‐binding site on the catalytic domain of PKC.244 Bisindolylmaleimide I has been shown to interfere with influenza A and B virus replication early in infection, probably by affecting endocytosis or vesicle transport.245 The PKCβII isoform was proven to be critical for influenza virus entry, since accumulation of the virus in late endosomes was observed in cells expressing a phosphorylation‐deficient form of PKCβII.246
The PI3K and its downstream effector Akt/protein kinase B are signaling mediators induced by influenza A virus, and their role in virus replication seems to be multifaceted.247 Upon virus attachment to the cell, PI3K is activated in a short and transient manner, promoting a step during virus entry that precedes early and late endosomal transport.123, 247 At later stages of infection, the influenza A virus nonstructural NS1 protein induces a second phase of sustained PI3K activation to prevent premature apoptosis during viral propagation. The influenza B virus NS1 protein apparently lacks this function.248 In contrast, accumulation of vRNA leads to PI3K activation in cells infected with influenza A or B viruses. Finally, PI3K signaling is also essential for efficient IRF‐3 activation during type I interferon (IFN) induction.249 The antiviral effect of IFN is mediated by the interferon inducible transmembrane (IFITM) protein 3 and results in inhibition of viral genome release into the cytoplasm.250 Notwithstanding this complex function of PI3K, it was shown that inhibition of PI3K results in decreased influenza virus replication,247 which makes this pathway a potential antiviral target.
9. PERSPECTIVES
During the past years, significant advances have been made in unraveling the structure of the influenza virus proteins involved in virus entry, from its initial attachment to the sialylated receptors until its nuclear entry. The available crystal structures of HA, M2, and NP enable a rational and computer‐aided design of directly acting antivirals. A few M2 inhibitors with activity against amantadine‐resistant viruses have already been reported. The old paradigm that the HA appears too variable to be a valid antiviral target is challenged by broad‐acting macromolecule inhibitors, such as lectins or antibodies interacting with a conserved site in the HA RBS or stem region. Whereas small molecules inhibiting the conformational change of HA have a restricting subtype dependency, this is not the case for compounds (such as arbidol), which interfere with the membrane fusion event itself. The recently discovered NP aggregating agent nucleozin represents an entirely novel class of anti‐influenza agents with clinical relevance.
Complementary to these structural studies, much attention is currently given to the complex cell biology of the influenza virus entry pathway, with a particular interest in the host cell factors involved. Diverse compounds interfering with any of these cellular factors have been reported, setting the stage for a new type of indirectly acting antivirals with a higher barrier for resistance selection. In times of increasing resistance to oseltamivir, agents with a more favorable resistance profile are indeed urgently required.
ACKNOWLEDGMENTS
The authors acknowledge a grant from the Geconcerteerde Onderzoeksacties (GOA/10/014). They wish to thank W. van Dam, S. Stevens, L. Persoons, and F. De Meyer for their dedicated contribution to their research work.
Biographies
Evelien Vanderlinden obtained her Master in Pharmaceutical Sciences (2006) at the KU Leuven, Belgium and her Ph.D. as Doctor in Pharmaceutical Sciences (2012) at the Rega Institute for Medical Research, KU Leuven. She was holder of a Ph.D. grant as a teaching assistant in practical courses in virology at the KU Leuven. Her current research is focused on the development of novel inhibitors of the influenza virus entry process or the influenza virus polymerase complex.
Lieve Naesens obtained her degree of Master in Pharmaceutical Sciences (1987) at the KU Leuven, Belgium, and her Ph.D. as Doctor in Pharmaceutical Sciences (1993) at the Rega Institute for Medical Research, KU Leuven. In 2003, she was appointed as assistant and later associate professor at the Faculty of Medicine of the KU Leuven to teach courses in virology and immunology. She is currently a senior investigator and project leader in influenza virus research at the Laboratory of Virology and Chemotherapy, Rega Institute, KU Leuven. Her research is focused on the design and mechanism of action of novel antiviral compounds, with a particular interest in inhibitors targeting the processes of viral entry or genome replication of influenza virus.
REFERENCES
- 1. Monto AS. Epidemiology of influenza. Vaccine 2008;26(Suppl 4):D45–8.:D45–D48. [DOI] [PubMed] [Google Scholar]
- 2. Schanzer DL, Langley JM, Tam TW. Hospitalization attributable to influenza and other viral respiratory illnesses in Canadian children. Pediatr Infect Dis J 2006;25:795–800. [DOI] [PubMed] [Google Scholar]
- 3. Thompson WW, Comanor L, Shay DK. Epidemiology of seasonal influenza: use of surveillance data and statistical models to estimate the burden of disease. J Infect Dis 2006;194(Suppl 2):S82–S91. [DOI] [PubMed] [Google Scholar]
- 4. Cocoros N, Hernandez R, Harrington N, Rausch‐Phung E, Schulte CR, Blog D, Gallagher K, Klevos A, Kim C, Marano N, Alvarado‐Ramy. Estimates of deaths associated with seasonal influenza—United States, 1976–2007. MMWR Morb Mortal Wkly Rep 2010;59:1057–1062. [PubMed] [Google Scholar]
- 5. Osterhaus A, Fouchier R, Rimmelzwaan G. Towards universal influenza vaccines? Philos Trans R Soc Lond B Biol Sci 2011;366:2766–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. WHO . Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO, 2003–2012. http://www.who.int/influenza/human_animal_interface/consulted on 7‐6‐2012.
- 7. Palese P, Wang TT. H5N1 influenza viruses: facts, not fear. Proc Natl Acad Sci USA 2012;109:2211–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taubenberger JK, Morens DM. 1918. Influenza: the mother of all pandemics. Emerg Infect Dis 2006;12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov 2007;6:967–974. [DOI] [PubMed] [Google Scholar]
- 10. Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol 2010;17:530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krug RM, Aramini JM. Emerging antiviral targets for influenza A virus. Trends Pharmacol Sci 2009;30:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee SM, Yen HL. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res 2012;96:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moscona A. Medical management of influenza infection. Annu Rev Med 2008;59:397–413.:397–413. [DOI] [PubMed] [Google Scholar]
- 14. von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW. Rational design of potent sialidase‐based inhibitors of influenza virus replication. Nature 1993;363:418–423. [DOI] [PubMed] [Google Scholar]
- 15. Eisenberg EJ, Bidgood A, Cundy KC. Penetration of GS4071, a novel influenza neuraminidase inhibitor, into rat bronchoalveolar lining fluid following oral administration of the prodrug GS4104. Antimicrob Agents Chemother 1997;41:1949–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collins PJ, Haire LF, Lin YP, Liu J, Russell RJ, Walker PA, Skehel JJ, Martin SR, Hay AJ, Gamblin SJ. Crystal structures of oseltamivir‐resistant influenza virus neuraminidase mutants. Nature 2008;453:1258–1261. [DOI] [PubMed] [Google Scholar]
- 17. Moscona A. Global transmission of oseltamivir‐resistant influenza. N Engl J Med 2009;360:953–956. [DOI] [PubMed] [Google Scholar]
- 18. van der Vries E, van den Berg B, Schutten M. Fatal oseltamivir‐resistant influenza virus infection. N Engl J Med 2008;359:1074–1076. [DOI] [PubMed] [Google Scholar]
- 19. Kiso M, Mitamura K, Sakai‐Tagawa Y, Shiraishi K, Kawakami C, Kimura K, Hayden FG, Sugaya N, Kawaoka Y. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet 2004;364:759–765. [DOI] [PubMed] [Google Scholar]
- 20. van der V, Stelma FF, Boucher CA. Emergence of a multidrug‐resistant pandemic influenza A (H1N1) virus. N Engl J Med 2010;363:1381–1382. [DOI] [PubMed] [Google Scholar]
- 21. Thorlund K, Awad T, Boivin G, Thabane L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect Dis 2011;19;11:134.:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shelton MJ, Lovern M, Ng‐Cashin J, Jones L, Gould E, Gauvin J, Rodvold KA. Zanamivir pharmacokinetics and pulmonary penetration into epithelial lining fluid following intravenous or oral inhaled administration to healthy adult subjects. Antimicrob Agents Chemother 2011;55:5178–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fraaij PL, van der Vries E, Beersma MF, Riezebos‐Brilman A, Niesters HG, van der Eijk AA, de Jong MD, Reis MD, Horrevorts AM, Ridwan BU, Wolfhagen MJ, Houmes RJ, van Dissel JT, Fouchier RA, Kroes AC, Koopmans MP, Osterhaus AD, Boucher CA. Evaluation of the antiviral response to zanamivir administered intravenously for treatment of critically ill patients with pandemic influenza A (H1N1) infection. J Infect Dis 2011;204:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Birnkrant D, Cox E. The Emergency Use Authorization of peramivir for treatment of 2009 H1N1 influenza. N Engl J Med 2009;361:2204–2207. [DOI] [PubMed] [Google Scholar]
- 25. Chairat K, Tarning J, White NJ, Lindegardh N. Pharmacokinetic Properties of Anti‐Influenza Neuraminidase Inhibitors. J Clin Pharmacol 2013;53:119–139. [DOI] [PubMed] [Google Scholar]
- 26. Watanabe A, Chang SC, Kim MJ, Chu DW, Ohashi Y. Long‐acting neuraminidase inhibitor laninamivir octanoate versus oseltamivir for treatment of influenza: A double‐blind, randomized, noninferiority clinical trial. Clin Infect Dis 2010;51:1167–1175. [DOI] [PubMed] [Google Scholar]
- 27. Katsumi Y, Otabe O, Matsui F, Kidowaki S, Mibayashi A, Tsuma Y, Ito H. Effect of a single inhalation of laninamivir octanoate in children with influenza. Pediatrics 2012;129:e1431–e1436. [DOI] [PubMed] [Google Scholar]
- 28. Vavricka CJ, Li Q, Wu Y, Qi J, Wang M, Liu Y, Gao F, Liu J, Feng E, He J, Wang J, Liu H, Jiang H, Gao GF. Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog 2011;7:e1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mohan S, McAtamney S, Haselhorst T, von Itzstein M, Pinto BM. Carbocycles related to oseltamivir as influenza virus group‐1‐specific neuraminidase inhibitors. Binding to N1 enzymes in the context of virus‐like particles. J Med Chem 2010;53:7377–7391. [DOI] [PubMed] [Google Scholar]
- 30. Kim JH, Resende R, Wennekes T, Chen HM, Bance N, Buchini S, Watts AG, Pilling P, Streltsov VA, Petric M, Liggins R, Barrett S, McKimm‐Breschkin JL, Niikura M, Withers SG. Mechanism‐Based Covalent Neuraminidase Inhibitors with Broad Spectrum Influenza Antiviral Activity. Science 2013;340:71–75. [DOI] [PubMed] [Google Scholar]
- 31. An J, Lee DC, Law AH, Yang CL, Poon LL, Lau AS, Jones SJ. A novel small‐molecule inhibitor of the avian influenza H5N1 virus determined through computational screening against the neuraminidase. J Med Chem 2009;52:2667–2672. [DOI] [PubMed] [Google Scholar]
- 32. Baranovich T, Wong SS, Armstrong J, Marjuki H, Webby RJ, Webster RG, Govorkova EA. T‐705 (Favipiravir) Induces Lethal Mutagenesis in Influenza A H1N1 Viruses In Vitro. J Virol 2013;87:3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. T‐705 (favipiravir) and related compounds: Novel broad‐spectrum inhibitors of RNA viral infections. Antiviral Res 2009;82:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gamblin SJ, Skehel JJ. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J Biol Chem 2010;285:28403–28409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu X, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci USA 2012;109:4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu X, Yang H, Guo Z, Yu, W , Carney PJ, Li Y, Chen LM, Paulson JC, Donis RO, Tong S, Stevens J, Wilson IA. Crystal structures of two subtype N10 neuraminidase‐like proteins from bat influenza A viruses reveal a diverged putative active site. Proc Natl Acad Sci USA 2012;109:18903–18908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salomon R, Webster RG. The influenza virus enigma. Cell 2009;136:402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Copeland CS, Doms RW, Bolzau EM, Webster RG, Helenius A. Assembly of influenza hemagglutinin trimers and its role in intracellular transport. J Cell Biol 1986;103:1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981;289:366–373. [DOI] [PubMed] [Google Scholar]
- 40. Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998;95:409–417. [DOI] [PubMed] [Google Scholar]
- 41. Bertram S, Glowacka I, Steffen I, Kuhl A, Pohlmann S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 2010;20:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perdue ML, Garcia M, Senne D, Fraire M. Virulence‐associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res 1997;49:173–186. [DOI] [PubMed] [Google Scholar]
- 43. Stieneke‐Grober A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin‐like endoprotease. EMBO J 1992;11:2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Becker GL, Lu Y, Hardes K, Strehlow B, Levesque C, Lindberg I, Sandvig K, Bakowsky U, Day R, Garten W, Steinmetzer T. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J Biol Chem 2012;287:21992–22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kordyukova LV, Serebryakova MV, Baratova LA, Veit M. S acylation of the hemagglutinin of influenza viruses: mass spectrometry reveals site‐specific attachment of stearic acid to a transmembrane cysteine. J Virol 2008;82:9288–9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kobayashi Y, Suzuki Y. Evidence for N‐glycan shielding of antigenic sites during evolution of human influenza A virus hemagglutinin. J Virol 2012;86:3446–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Das SR, Puigbo P, Hensley SE, Hurt DE, Bennink JR, Yewdell JW. Glycosylation focuses sequence variation in the influenza A virus H1 hemagglutinin globular domain. PLoS Pathog 2010;6:e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Daniels R, Kurowski B, Johnson AE, Hebert DN. N‐linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol Cell 2003;11:79–90. [DOI] [PubMed] [Google Scholar]
- 49. Gallagher PJ, Henneberry JM, Sambrook JF, Gething MJ. Glycosylation requirements for intracellular transport and function of the hemagglutinin of influenza virus. J Virol 1992;66:7136–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohuchi M, Ohuchi R, Feldmann A, Klenk HD. Regulation of receptor binding affinity of influenza virus hemagglutinin by its carbohydrate moiety. J Virol 1997;71:8377–8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deshpande KL, Fried VA, Ando M, Webster RG. Glycosylation affects cleavage of an H5N2 influenza virus hemagglutinin and regulates virulence. Proc Natl Acad Sci USA 1987;84:36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ohuchi R, Ohuchi M, Garten W, Klenk HD. Oligosaccharides in the stem region maintain the influenza virus hemagglutinin in the metastable form required for fusion activity. J Virol 1997;71:3719–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wagner R, Heuer D, Wolff T, Herwig A, Klenk HD. N‐Glycans attached to the stem domain of haemagglutinin efficiently regulate influenza A virus replication. J Gen Virol 2002;83:601–609. [DOI] [PubMed] [Google Scholar]
- 54. Russell RJ, Gamblin SJ, Haire LF, Stevens DJ, Xiao B, Ha Y, Skehel JJ. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 2004;325:287–296. [DOI] [PubMed] [Google Scholar]
- 55. Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, Gamblin SJ, Skehel JJ. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci USA 2008;105:17736–17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 1983;304:76–78. [DOI] [PubMed] [Google Scholar]
- 57. Imai M, Kawaoka Y. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr Opin Virol 2012;2:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilks S, de Graaf M, Smith DJ, Burke DF. A review of influenza haemagglutinin receptor binding as it relates to pandemic properties. Vaccine 2012;19;30:4369–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Matrosovich MN, Gambaryan AS, Teneberg S, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Karlsson KA. Avian influenza A viruses differ from human viruses by recognition of sialyloligosaccharides and gangliosides and by a higher conservation of the HA receptor‐binding site. Virology 1997;233:224–234. [DOI] [PubMed] [Google Scholar]
- 60. Xu R, McBride R, Nycholat CM, Paulson JC, Wilson IA. Structural characterization of the hemagglutinin receptor specificity from the 2009 H1N1 influenza pandemic. J Virol 2012;86:982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol.Biol 2006;355:1143–1155. [DOI] [PubMed] [Google Scholar]
- 62. Xu R, McBride R, Paulson JC, Basler CF, Wilson IA. Structure, receptor binding, and antigenicity of influenza virus hemagglutinins from the 1957 H2N2 pandemic. J Virol 2010;84:1715–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gambaryan AS, Robertson JS, Matrosovich MN. Effects of egg‐adaptation on the receptor‐binding properties of human influenza A and B viruses. Virology 1999;258:232–239. [DOI] [PubMed] [Google Scholar]
- 64. Wang Q, Tian X, Chen X, Ma J. Structural basis for receptor specificity of influenza B virus hemagglutinin. Proc Natl Acad Sci USA 2007;104:16874–16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Q, Cheng F, Lu M, Tian X, Ma J. Crystal structure of unliganded influenza B virus hemagglutinin. J Virol 2008;82:3011–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM, Sasisekharan V, Sasisekharan R. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol 2008;26:107–113. [DOI] [PubMed] [Google Scholar]
- 67. Gambaryan AS, Tuzikov AB, Piskarev VE, Yamnikova SS, Lvov DK, Robertson JS, Bovin NV, Matrosovich MN. Specification of receptor‐binding phenotypes of influenza virus isolates from different hosts using synthetic sialylglycopolymers: non‐egg‐adapted human H1 and H3 influenza A and influenza B viruses share a common high binding affinity for 6’‐sialyl(N‐acetyllactosamine). Virology 1997;232:345–350. [DOI] [PubMed] [Google Scholar]
- 68. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000;69:531–69.:531–569. [DOI] [PubMed] [Google Scholar]
- 69. Stray SJ, Cummings RD, Air GM. Influenza virus infection of desialylated cells. Glycobiology 2000;10:649–658. [DOI] [PubMed] [Google Scholar]
- 70. Nicholls JM, Chan RW, Russell RJ, Air GM, Peiris JS. Evolving complexities of influenza virus and its receptors. Trends Microbiol 2008;16:149–157. [DOI] [PubMed] [Google Scholar]
- 71. Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010;7:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 2012;336:1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kawaoka Y, Neumann G. Influenza viruses: an introduction. Methods Mol Biol 2012;865:1–9.:1–9. [DOI] [PubMed] [Google Scholar]
- 74. Sieben C, Kappel C, Zhu R, Wozniak A, Rankl C, Hinterdorfer P, Grubmuller H, Herrmann A. Influenza virus binds its host cell using multiple dynamic interactions. Proc Natl Acad Sci USA 2012;109:13626–13631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Takemoto DK, Skehel JJ, Wiley DC. A surface plasmon resonance assay for the binding of influenza virus hemagglutinin to its sialic acid receptor. Virology 1996;217:452–458. [DOI] [PubMed] [Google Scholar]
- 76. Knossow M, Skehel JJ. Variation and infectivity neutralization in influenza. Immunology 2006;119:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta‐analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann Intern Med 2006;145:599–609. [DOI] [PubMed] [Google Scholar]
- 78. Simmons CP, Bernasconi NL, Suguitan AL, Mills K, Ward JM, Chau NV, Hien TT, Sallusto F, Ha, do Q , Farrar J, de Jong MD, Lanzavecchia A, Subbarao K. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med 2007;4:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC. Broadly neutralizing human antibody that recognizes the receptor‐binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci USA 2011;108:14216–14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schmidt AG, Xu H, Khan AR, O'Donnell T, Khurana S, King LR, Manischewitz J, Golding H, Suphaphiphat P, Carfi A, Settembre EC, Dormitzer PR, Kepler TB, Zhang R, Moody MA, Haynes BF, Liao HX, Shaw DE, Harrison SC. Preconfiguration of the antigen‐binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci USA 2012;110:264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bizebard T, Gigant B, Rigolet P, Rasmussen B, Diat O, Bosecke P, Wharton SA, Skehel JJ, Knossow M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995;376:92–94. [DOI] [PubMed] [Google Scholar]
- 82. Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O'Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. Cross‐neutralization of influenza A viruses mediated by a single antibody loop. Nature 2012;489:526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. Cross‐protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog 2009;5:e1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lee PS, Yoshida R, Ekiert DC, Sakai N, Suzuki Y, Takada A, Wilson IA. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc Natl Acad Sci USA 2012;109:17040–17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yasugi M, Kubota‐Koketsu R, Yamashita A, Kawashita N, Du A, Sasaki T, Nishimura M, Misaki R, Kuhara M, Boonsathorn N, Fujiyama K, Okuno Y, Nakaya T, Ikuta K. Human Monoclonal Antibodies Broadly Neutralizing against Influenza B Virus. PLoS Pathog 2013;9:e1003150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, van der V, Lamrani M, Korse HJ, Geelen E, Sahin O, Sieuwerts M, Brakenhoff JP, Vogels R, Li OT, Poon LL, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RH. Highly conserved protective epitopes on influenza B viruses. Science 2012;337:1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shadman KA, Wald ER. A review of palivizumab and emerging therapies for respiratory syncytial virus. Expert Opin Biol Ther 2011;11:1455–1467. [DOI] [PubMed] [Google Scholar]
- 88. Vanlandschoot P, Stortelers C, Beirnaert E, Ibanez LI, Schepens B, Depla E, Saelens X. Nanobodies(R): new ammunition to battle viruses. Antiviral Res 2011;92:389–407. [DOI] [PubMed] [Google Scholar]
- 89. Ibanez LI, De Filette M, Hultberg A, Verrips T, Temperton N, Weiss RA, Vandevelde W, Schepens B, Vanlandschoot P, Saelens X. Nanobodies with in vitro neutralizing activity protect mice against H5N1 influenza virus infection. J Infect Dis 2011;203:1063–1072. [DOI] [PubMed] [Google Scholar]
- 90. van Eijk M, Bruinsma L, Hartshorn KL, White MR, Rynkiewicz MJ, Seaton BA, Hemrika W, Romijn RA, van Balkom BW, Haagsman HP. Introduction of N‐linked glycans in the lectin domain of surfactant protein D: impact on interactions with influenza A viruses. J Biol Chem 2011;286:20137–20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nayak A, Dodagatta‐Marri E, Tsolaki AG, Kishore U. An Insight into the Diverse Roles of Surfactant Proteins, SP‐A and SP‐D in Innate and Adaptive Immunity. Front Immunol 2012;3:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Crouch E, Nikolaidis N, McCormack FX, McDonald B, Allen K, Rynkiewicz MJ, Cafarella TM, White M, Lewnard K, Leymarie N, Zaia J, Seaton BA, Hartshorn KL. Mutagenesis of surfactant protein D informed by evolution and x‐ray crystallography enhances defenses against influenza A virus in vivo. J Biol Chem 2011;286:40681–40692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. van Eijk M, Rynkiewicz MJ, White MR, Hartshorn KL, Zou X, Schulten K, Luo D, Crouch EC, Cafarella TR, Head JF, Haagsman HP, Seaton BA. A unique sugar‐binding site mediates the distinct anti‐influenza activity of pig surfactant protein D. J Biol Chem 2012;287:26666–26677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hartshorn KL, Holmskov U, Hansen S, Zhang P, Meschi J, Mogues T, White MR, Crouch EC. Distinctive anti‐influenza properties of recombinant collectin 43. Biochem J 2002;366:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Francois KO, Balzarini J. Potential of carbohydrate‐binding agents as therapeutics against enveloped viruses. Med Res Rev 2012;32:349–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. O'Keefe BR, Smee DF, Turpin JA, Saucedo CJ, Gustafson KR, Mori T, Blakeslee D, Buckheit R, Boyd MR. Potent anti‐influenza activity of cyanovirin‐N and interactions with viral hemagglutinin. Antimicrob Agents Chemother 2003;47:2518–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Smee DF, Bailey KW, Wong MH, O'Keefe BR, Gustafson KR, Mishin VP, Gubareva LV. Treatment of influenza A (H1N1) virus infections in mice and ferrets with cyanovirin‐N. Antiviral Res 2008;80:266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tate MD, Brooks AG, Reading PC. Specific sites of N‐linked glycosylation on the hemagglutinin of H1N1 subtype influenza A virus determine sensitivity to inhibitors of the innate immune system and virulence in mice. J Immunol 2011;187:1884–1894. [DOI] [PubMed] [Google Scholar]
- 99. Balzarini J. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat Rev Microbiol 2007;5:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sato T, Ishii M, Ohtake F, Nagata K, Terabayashi T, Kawanishi Y, Okahata Y. Binding affinity of GM3 lactone for influenza virus. Glycoconj J 1999;16:223–227. [DOI] [PubMed] [Google Scholar]
- 101. Terabayashi T, Morita M, Ueno M, Nakamura T, Urashima T. Inhibition of influenza‐virus‐induced cytopathy by sialylglycoconjugates. Carbohydr Res 2006;341:2246–2253. [DOI] [PubMed] [Google Scholar]
- 102. Suzuki Y, Nakao T, Ito T, Watanabe N, Toda Y, Xu G, Suzuki T, Kobayashi T, Kimura Y, Yamada A. Structural determination of gangliosides that bind to influenza A, B, and C viruses by an improved binding assay: strain‐specific receptor epitopes in sialo‐sugar chains. Virology 1992;189:121–131. [DOI] [PubMed] [Google Scholar]
- 103. Hendricks GL, Weirich KL, Viswanathan K, Li J, Shriver ZH, Ashour J, Ploegh HL, Kurt‐Jones EA, Fygenson DK, Finberg RW, Comolli JC, Wang JP. Sialylneolacto‐N‐tetraose c (LSTc)‐bearing liposomal decoys capture influenza A virus. J Biol Chem 2013;288:8061–8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Matsubara T, Onishi A, Saito T, Shimada A, Inoue H, Taki T, Nagata K, Okahata Y, Sato T. Sialic acid‐mimic peptides as hemagglutinin inhibitors for anti‐influenza therapy. J Med Chem 2010;53:4441–4449. [DOI] [PubMed] [Google Scholar]
- 105. Jeon SH, Kayhan B, Ben Yedidia T, Arnon R. A DNA aptamer prevents influenza infection by blocking the receptor binding region of the viral hemagglutinin. J Biol Chem 2004;279:48410–48419. [DOI] [PubMed] [Google Scholar]
- 106. Kimura K, Mori S, Tomita K, Ohno K, Takahashi K, Shigeta S, Terada M. Antiviral activity of NMSO3 against respiratory syncytial virus infection in vitro and in vivo. Antiviral Res 2000;47:41–51. [DOI] [PubMed] [Google Scholar]
- 107. Vanderlinden E, Vanstreels E, Boons E, Ter Veer W, Huckriede A, Daelemans D, Van Lommel A, Roth E, Sztaricskai F, Herczegh P, Naesens L. Intracytoplasmic trapping of influenza virus by a lipophilic derivative of aglycoristocetin. J Virol 2012;86:9416–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Terada M, Fujita S, Suda I, Mastico R. Polysulfated sialic acid derivatives as anti‐human immunodeficiency virus. Biomed Pharmacother 2005;59:423–429. [DOI] [PubMed] [Google Scholar]
- 109. Matsubara T, Sumi M, Kubota H, Taki T, Okahata Y, Sato T. Inhibition of influenza virus infections by sialylgalactose‐binding peptides selected from a phage library. J Med Chem 2009;52:4247–4256. [DOI] [PubMed] [Google Scholar]
- 110. Bhavanandan VP, Katlic AW. The interaction of wheat germ agglutinin with sialoglycoproteins. The role of sialic acid. J Biol Chem 1979;254:4000–4008. [PubMed] [Google Scholar]
- 111. Gottschalk A. On the mechanism underlying initiation of influenza virus infection. Ergeb Mikrobiol Immunitatsforsch Exp Ther 1959;32:1–22.:1–22. [DOI] [PubMed] [Google Scholar]
- 112. Malakhov MP, Aschenbrenner LM, Smee DF, Wandersee MK, Sidwell RW, Gubareva LV, Mishin VP, Hayden FG, Kim DH, Ing A, Campbell ER, Yu M, Fang F. Sialidase fusion protein as a novel broad‐spectrum inhibitor of influenza virus infection. Antimicrob Agents Chemother 2006;50:1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Moscona A, Porotto M, Palmer S, Tai C, Aschenbrenner L, Triana‐Baltzer G, Li QX, Wurtman D, Niewiesk S, Fang F. A recombinant sialidase fusion protein effectively inhibits human parainfluenza viral infection in vitro and in vivo. J Infect Dis 2010;202:234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Triana‐Baltzer GB, Sanders RL, Hedlund M, Jensen KA, Aschenbrenner LM, Larson JL, Fang F. Phenotypic and genotypic characterization of influenza virus mutants selected with the sialidase fusion protein DAS181. J Antimicrob Chemother 2011;66:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hedlund M, Aschenbrenner LM, Jensen K, Larson JL, Fang F. Sialidase‐based anti‐influenza virus therapy protects against secondary pneumococcal infection. J Infect Dis 2010;201:1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ison MG. Expanding the armamentarium against respiratory viral infections: DAS181. J Infect Dis 2012;206:1806–1808. [DOI] [PubMed] [Google Scholar]
- 117. Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem 2010;79:803–33.:803–833. [DOI] [PubMed] [Google Scholar]
- 118. Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 1981;91:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin‐mediated endocytosis. J Virol 2002;76:10455–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rust MJ, Lakadamyali M, Zhang F, Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 2004;11:567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. de Vries E, Tscherne DM, Wienholts MJ, Cobos‐Jimenez V, Scholte F, Garcia‐Sastre A, Rottier PJ, de Haan CA. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 2011;7:e1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chu VC, Whittaker GR. Influenza virus entry and infection require host cell N‐linked glycoprotein. Proc Natl Acad Sci USA 2004;101:18153–18158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog 2010;6:e1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Harada S, Yokomizo K, Monde K, Maeda Y, Yusa K. A broad antiviral neutral glycolipid, fattiviracin FV‐8, is a membrane fluidity modulator. Cell Microbiol 2007;9:196–203. [DOI] [PubMed] [Google Scholar]
- 125. Harada S. The broad anti‐viral agent glycyrrhizin directly modulates the fluidity of plasma membrane and HIV‐1 envelope. Biochem J 2005;392:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wolkerstorfer A, Kurz H, Bachhofner N, Szolar OH. Glycyrrhizin inhibits influenza A virus uptake into the cell. Antiviral Res 2009;83:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Pompei R, Paghi L, Ingianni A, Uccheddu P. Glycyrrhizic acid inhibits influenza virus growth in embryonated eggs. Microbiologica 1983;6:247–250. [PubMed] [Google Scholar]
- 128. Michaelis M, Geiler J, Naczk P, Sithisarn P, Ogbomo H, Altenbrandt B, Leutz A, Doerr HW, Cinatl J Jr. Glycyrrhizin inhibits highly pathogenic H5N1 influenza A virus‐induced pro‐inflammatory cytokine and chemokine expression in human macrophages. Med Microbiol Immunol 2010;199:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wolf MC, Freiberg AN, Zhang T, Akyol‐Ataman Z, Grock A, Hong PW, Li J, Watson NF, Fang AQ, Aguilar HC, Porotto M, Honko AN, Damoiseaux R, Miller JP, Woodson SE, Chantasirivisal S, Fontanes V, Negrete OA, Krogstad P, Dasgupta A, Moscona A, Hensley LE, Whelan SP, Faull KF, Holbrook MR, Jung ME, Lee B. A broad‐spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci USA 2010;107:3157–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Preobrazhenskaya MN, Olsufyeva EN. Polycyclic peptide and glycopeptide antibiotics and their derivatives as inhibitors of HIV entry. Antiviral Res 2006;71:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Naesens L, Vanderlinden E, Roth E, Jeko J, Andrei G, Snoeck R, Pannecouque C, Illyes E, Batta G, Herczegh P, Sztaricskai F. Anti‐influenza virus activity and structure‐activity relationship of aglycoristocetin derivatives with cyclobutenedione carrying hydrophobic chains. Antiviral Res 2009;82:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Naruse N, Tenmyo O, Kobaru S, Hatori M, Tomita K, Hamagishi Y, Oki T. New antiviral antibiotics, kistamicins A and B. I. Taxonomy, production, isolation, physico‐chemical properties and biological activities. J Antibiot (Tokyo) 1993;46:1804–1811. [DOI] [PubMed] [Google Scholar]
- 133. Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microbes Infect 2004;6:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Guinea R, Carrasco L. Requirement for vacuolar proton‐ATPase activity during entry of influenza virus into cells. J Virol 1995;69:2306–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia‐Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. Human host factors required for influenza virus replication. Nature 2010;463:813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yoshimura A, Kuroda K, Kawasaki K, Yamashina S, Maeda T, Ohnishi S. Infectious cell entry mechanism of influenza virus. J Virol 1982;43:284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ooi EE, Chew JS, Loh JP, Chua RC. In vitro inhibition of human influenza A virus replication by chloroquine. Virol J 2006;3:39.:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Paton NI, Lee L, Xu Y, Ooi EE, Cheung YB, Archuleta S, Wong G, Wilder‐Smith A. Chloroquine for influenza prevention: a randomised, double‐blind, placebo controlled trial. Lancet Infect Dis 2011;11:677–683. [DOI] [PubMed] [Google Scholar]
- 139. Vigerust DJ, McCullers JA. Chloroquine is effective against influenza A virus in vitro but not in vivo. Influenza Other Respi Viruses 2007;1:189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML, Wiley DC. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 1985;40:431–439. [DOI] [PubMed] [Google Scholar]
- 141. Wang J, Qiu JX, Soto C, DeGrado WF. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr Opin Struct Biol 2011;21:68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Okada A, Miura T, Takeuchi H. Protonation of histidine and histidine‐tryptophan interaction in the activation of the M2 ion channel from influenza a virus. Biochemistry 2001;40:6053–6060. [DOI] [PubMed] [Google Scholar]
- 143. Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci USA 2006;103:6865–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Mould JA, Paterson RG, Takeda M, Ohigashi Y, Venkataraman P, Lamb RA, Pinto LH. Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev Cell 2003;5:175–184. [DOI] [PubMed] [Google Scholar]
- 145. Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, DeGrado WF, Pinto LH. Functional studies and modeling of pore‐lining residue mutants of the influenza a virus M2 ion channel. Biochemistry 2010;49:696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008;451:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc Natl Acad Sci USA 2010;107:15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008;451:591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J Biol Chem 2006;281:8997–9000. [DOI] [PubMed] [Google Scholar]
- 150. Hu F, Schmidt‐Rohr K, Hong M. NMR detection of pH‐dependent histidine‐water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J Am Chem Soc 2012;134:3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hu F, Luo W, Hong M. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid‐state NMR. Science 2010;330:505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wang J, Pielak RM, McClintock MA, Chou JJ. Solution structure and functional analysis of the influenza B proton channel. Nat Struct Mol Biol 2009;16:1267–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Davies WL, Grunert RR, Haff RF, Mcgahen JW, Neumayer EM, Paulshock M, Watts JC, Wood TR, Hermann EC, Hoffmann CE. Antiviral activity of 1‐adamantanamine (amantadine). Science 1964;144:862–863. [DOI] [PubMed] [Google Scholar]
- 154. Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell 1992;69:517–528. [DOI] [PubMed] [Google Scholar]
- 155. Cady SD, Schmidt‐Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010;463:689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Khurana E, Devane RH, Dal Peraro M, Klein ML. Computational study of drug binding to the membrane‐bound tetrameric M2 peptide bundle from influenza A virus. Biochim Biophys Acta 2011;1808:530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gubareva LV, Trujillo AA, Okomo‐Adhiambo M, Mishin VP, Deyde VM, Sleeman K, Nguyen HT, Sheu TG, Garten RJ, Shaw MW, Fry AM, Klimov AI. Comprehensive assessment of 2009 pandemic influenza A (H1N1) virus drug susceptibility in vitro. Antivir Ther 2010;15:1151–1159. [DOI] [PubMed] [Google Scholar]
- 158. Belshe RB, Burk B, Newman F, Cerruti RL, Sim IS. Resistance of influenza A virus to amantadine and rimantadine: results of one decade of surveillance. J Infect Dis 1989;159:430–435. [DOI] [PubMed] [Google Scholar]
- 159. Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis 2007;196:249–257. [DOI] [PubMed] [Google Scholar]
- 160. Pielak RM, Chou JJ. Flu channel drug resistance: a tale of two sites. Protein Cell 2010;1:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Klein ML, DeGrado WF. Molecular dynamics simulation directed rational design of inhibitors targeting drug‐resistant mutants of influenza A virus M2. J Am Chem Soc 2011;133:12834–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zhao X, Jie Y, Rosenberg MR, Wan J, Zeng S, Cui W, Xiao Y, Li Z, Tu Z, Casarotto MG, Hu, W . Design and synthesis of pinanamine derivatives as anti‐influenza A M2 ion channel inhibitors. Antiviral Res 2012;96:91–99. [DOI] [PubMed] [Google Scholar]
- 163. Hu W, Zeng S, Li C, Jie Y, Li Z, Chen L. Identification of hits as matrix‐2 protein inhibitors through the focused screening of a small primary amine library. J Med Chem 2010;53:3831–3834. [DOI] [PubMed] [Google Scholar]
- 164. Torres E, Duque MD, Lopez‐Querol M, Taylor MC, Naesens L, Ma C, Pinto LH, Sureda FX, Kelly JM, Vazquez S. Synthesis of benzopolycyclic cage amines: NMDA receptor antagonist, trypanocidal and antiviral activities. Bioorg Med Chem 2012;20:942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Zoidis G, Kolocouris N, Kelly JM, Prathalingam SR, Naesens L, De Clercq E. Design and synthesis of bioactive adamantanaminoalcohols and adamantanamines. Eur J Med Chem 2010;45:5022–5030. [DOI] [PubMed] [Google Scholar]
- 166. Zhao X, Li C, Zeng S, Hu W. Discovery of highly potent agents against influenza A virus. Eur J Med Chem 2011;46:52–57. [DOI] [PubMed] [Google Scholar]
- 167. Kolocouris N, Kolocouris A, Foscolos GB, Fytas G, Neyts J, Padalko E, Balzarini J, Snoeck R, Andrei G, De Clercq E. Synthesis and antiviral activity evaluation of some new aminoadamantane derivatives. 2. J Med Chem 1996;39:3307–3318. [DOI] [PubMed] [Google Scholar]
- 168. Duque MD, Ma C, Torres E, Wang J, Naesens L, Juarez‐Jimenez J, Camps P, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J Med Chem 2011;54:2646–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Scholtissek C, Quack G, Klenk HD, Webster RG. How to overcome resistance of influenza A viruses against adamantane derivatives. Antiviral Res 1998;37:83–95. [DOI] [PubMed] [Google Scholar]
- 170. Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DC. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus‐mediated membrane fusion. Proc Natl Acad Sci USA. 1982;79:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Carr CM, Kim PS. A spring‐loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993;73:823–832. [DOI] [PubMed] [Google Scholar]
- 172. Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994;371:37–43. [DOI] [PubMed] [Google Scholar]
- 173. Kemble GW, Danieli T, White JM. Lipid‐anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 1994;76:383–391. [DOI] [PubMed] [Google Scholar]
- 174. Cross KJ, Langley WA, Russell RJ, Skehel JJ, Steinhauer DA. Composition and functions of the influenza fusion peptide. Protein Pept Lett 2009;16:766–778. [DOI] [PubMed] [Google Scholar]
- 175. Bodian DL, Yamasaki RB, Buswell RL, Stearns JF, White JM, Kuntz ID. Inhibition of the fusion‐inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 1993;32:2967–2978. [DOI] [PubMed] [Google Scholar]
- 176. Vanderlinden E, Goktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. Novel inhibitors of influenza virus fusion: structure‐activity relationship and interaction with the viral hemagglutinin. J Virol 2010;84:4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Luo G, Torri A, Harte WE, Danetz S, Cianci C, Tiley L, Day S, Mullaney D, Yu KL, Ouellet C, Dextraze P, Meanwell N, Colonno R, Krystal M. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J Virol 1997;71:4062–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhu L, Li Y, Li S, Li H, Qiu Z, Lee C, Lu H, Lin X, Zhao R, Chen L, Wu JZ, Tang G, Yang W. Inhibition of influenza A virus (H1N1) fusion by benzenesulfonamide derivatives targeting viral hemagglutinin. PLoS One 2011;6:e29120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Plotch SJ, O'Hara B, Morin J, Palant O, LaRocque J, Bloom JD, Lang SA Jr, DiGrandi MJ, Bradley M, Nilakantan R, Gluzman Y. Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin. J Virol 1999;73:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Yoshimoto J, Kakui M, Iwasaki H, Fujiwara T, Sugimoto H, Hattori N. Identification of a novel HA conformational change inhibitor of human influenza virus. Arch Virol 1999;144:865–878. [DOI] [PubMed] [Google Scholar]
- 181. Deshpande MS, Wei J, Luo G, Cianci C, Danetz S, Torri A, Tiley L, Krystal M, Yu KL, Huang S, Gao Q, Meanwell NA. An approach to the identification of potent inhibitors of influenza virus fusion using parallel synthesis methodology. Bioorg Med Chem Lett 2001;11:2393–2396. [DOI] [PubMed] [Google Scholar]
- 182. Cianci C, Yu KL, Dischino DD, Harte W, Deshpande M, Luo G, Colonno RJ, Meanwell NA, Krystal M. pH‐dependent changes in photoaffinity labeling patterns of the H1 influenza virus hemagglutinin by using an inhibitor of viral fusion. J Virol 1999;73:1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hoffman LR, Kuntz ID, White JM. Structure‐based identification of an inducer of the low‐pH conformational change in the influenza virus hemagglutinin: irreversible inhibition of infectivity. J Virol 1997;71:8808–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hosoya M, Balzarini J, Shigeta S, De Clercq E. Differential inhibitory effects of sulfated polysaccharides and polymers on the replication of various myxoviruses and retroviruses, depending on the composition of the target amino acid sequences of the viral envelope glycoproteins. Antimicrob Agents Chemother 1991;35:2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Baba M, Snoeck R, Pauwels R, De Clercq E. Sulfated polysaccharides are potent and selective inhibitors of various enveloped viruses, including herpes simplex virus, cytomegalovirus, vesicular stomatitis virus, and human immunodeficiency virus. Antimicrob Agents Chemother 1988;32:1742–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Ito M, Baba M, Sato A, Pauwels R, De Clercq E, Shigeta S. Inhibitory effect of dextran sulfate and heparin on the replication of human immunodeficiency virus (HIV) in vitro. Antiviral Res 1987;7:361–367. [DOI] [PubMed] [Google Scholar]
- 187. Luscher‐Mattli M, Gluck R. Dextran sulfate inhibits the fusion of influenza virus with model membranes, and suppresses influenza virus replication in vivo. Antiviral Res 1990;14:39–50. [DOI] [PubMed] [Google Scholar]
- 188. Krumbiegel M, Dimitrov DS, Puri A, Blumenthal R. Dextran sulfate inhibits fusion of influenza virus and cells expressing influenza hemagglutinin with red blood cells. Biochim Biophys Acta 1992;1110:158–164. [DOI] [PubMed] [Google Scholar]
- 189. Ramalho‐Santos J, de Lima MC. Fusion and infection of influenza and Sendai viruses as modulated by dextran sulfate: a comparative study. Biosci Rep 2001;21:293–304. [DOI] [PubMed] [Google Scholar]
- 190. Kim M, Yim JH, Kim SY, Kim HS, Lee WG, Kim SJ, Kang PS, Lee CK. In vitro inhibition of influenza A virus infection by marine microalga‐derived sulfated polysaccharide p‐KG03. Antiviral Res 2012;93:253–259. [DOI] [PubMed] [Google Scholar]
- 191. Yasin B, Wang W, Pang M, Cheshenko N, Hong T, Waring AJ, Herold BC, Wagar EA, Lehrer RI. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J Virol 2004;78:5147–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Liang QL, Zhou K, He HX. Retrocyclin 2: a new therapy against avian influenza H5N1 virus in vivo and vitro. Biotechnol Lett 2010;32:387–392. [DOI] [PubMed] [Google Scholar]
- 193. Cole AM, Hong T, Boo LM, Nguyen T, Zhao C, Bristol G, Zack JA, Waring AJ, Yang OO, Lehrer RI. Retrocyclin: a primate peptide that protects cells from infection by T‐ and M‐tropic strains of HIV‐1. Proc Natl Acad Sci USA 2002;19(99):1813–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Leikina E, Delanoe‐Ayari H, Melikov K, Cho MS, Chen A, Waring AJ, Wang W, Xie Y, Loo JA, Lehrer RI, Chernomordik LV. Carbohydrate‐binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat Immunol 2005;6:995–1001. [DOI] [PubMed] [Google Scholar]
- 195. Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol 2005;6:551–557. [DOI] [PubMed] [Google Scholar]
- 196. Teissier E, Zandomeneghi G, Loquet A, Lavillette D, Lavergne JP, Montserret R, Cosset FL, Bockmann A, Meier BH, Penin F, Pecheur EI. Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS One 2011;6:e15874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Leneva IA, Russell RJ, Boriskin YS, Hay AJ. Characteristics of arbidol‐resistant mutants of influenza virus: implications for the mechanism of anti‐influenza action of arbidol. Antiviral Res 2009;81:132–140. [DOI] [PubMed] [Google Scholar]
- 198. Pecheur EI, Lavillette D, Alcaras F, Molle J, Boriskin YS, Roberts M, Cosset FL, Polyak SJ. Biochemical mechanism of hepatitis C virus inhibition by the broad‐spectrum antiviral arbidol. Biochemistry 2007;46:6050–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Villalain J. Membranotropic effects of arbidol, a broad anti‐viral molecule, on phospholipid model membranes. J Phys Chem B 2010;114:8544–8554. [DOI] [PubMed] [Google Scholar]
- 200. Liu MY, Wang S, Yao WF, Wu HZ, Meng SN, Wei MJ. Pharmacokinetic properties and bioequivalence of two formulations of arbidol: an open‐label, single‐dose, randomized‐sequence, two‐period crossover study in healthy Chinese male volunteers. Clin Ther 2009;31:784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Prabhu N, Prabakaran M, Ho HT, Velumani S, Qiang J, Goutama M, Kwang J. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J Virol 2009;83:2553–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Mancini N, Solforosi L, Clementi N, De Marco D, Clementi M, Burioni R. A potential role for monoclonal antibodies in prophylactic and therapeutic treatment of influenza. Antiviral Res 2011;92:15–26. [DOI] [PubMed] [Google Scholar]
- 203. Okuno Y, Isegawa Y, Sasao F, Ueda S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol 1993;67:2552–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Smirnov YA, Lipatov AS, Gitelman AK, Okuno Y, van Beek R, Osterhaus AD, Claas EC. An epitope shared by the hemagglutinins of H1, H2, H5, and H6 subtypes of influenza A virus. Acta Virol 1999;43:237–244. [PubMed] [Google Scholar]
- 205. Okuno Y, Matsumoto K, Isegawa Y, Ueda S. Protection against the mouse‐adapted A/FM/1/47 strain of influenza A virus in mice by a monoclonal antibody with cross‐neutralizing activity among H1 and H2 strains. J Virol 1994;68:517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. Antibody recognition of a highly conserved influenza virus epitope. Science 2009;324:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, Wan H, Murakami A, Yammanuru A, Han T, Cox NJ, Bankston LA, Donis RO, Liddington RC, Marasco WA. Structural and functional bases for broad‐spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol 2009;16:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Throsby M, van den BE., Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, Guan Y, Cinatl J, ter Meulen J, Lasters I, Carsetti R, Peiris M, de Kruif J, Goudsmit J. Heterosubtypic neutralizing monoclonal antibodies cross‐protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 2008;3(12):e3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 2011;333:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez‐Rodriguez BM, Agatic G, Bianchi S, Giacchetto‐Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011;333:850–856. [DOI] [PubMed] [Google Scholar]
- 211. Clementi N, De Marco D, Mancini N, Solforosi L, Moreno GJ, Gubareva LV, Mishin V, Di Pietro A, Vicenzi E, Siccardi AG, Clementi M, Burioni R. A human monoclonal antibody with neutralizing activity against highly divergent influenza subtypes. PLoS One 2011;6:e28001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Han T, Marasco WA. Structural basis of influenza virus neutralization. Ann N Y Acad Sci 2011;1217:178–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 2011;332:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Yu F, Lu L, Du L, Zhu X, Debnath AK, Jiang S. Approaches for identification of HIV‐1 entry inhibitors targeting gp41 pocket. Viruses 2013;5:127–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Martin K, Helenius A. Transport of incoming influenza virus nucleocapsids into the nucleus. J Virol 1991;65:232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hutchinson EC, Fodor E. Nuclear import of the influenza A virus transcriptional machinery. Vaccine 2012;30:7353–7358. [DOI] [PubMed] [Google Scholar]
- 217. Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, Digard P. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol 2007;88:2280–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Whittaker G, Bui M, Helenius A. The role of nuclear import and export in influenza virus infection. Trends Cell Biol 1996;6:67–71. [DOI] [PubMed] [Google Scholar]
- 219. Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology 2011;411:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Bui M, Whittaker G, Helenius A. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J Virol 1996;70:8391–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Helenius A. Unpacking the incoming influenza virus. Cell 1992;69:577–578. [DOI] [PubMed] [Google Scholar]
- 222. Zhang K, Wang Z, Liu X, Yin C, Basit Z, Xia B, Liu W. Dissection of influenza A virus M1 protein: pH‐dependent oligomerization of N‐terminal domain and dimerization of C‐terminal domain. PLoS One 2012;7:e37786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. Structural changes in Influenza virus at low pH characterized by cryo‐electron tomography. J Virol 2012;86:2919–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Bukrinskaya AG, Vorkunova NK, Kornilayeva GV, Narmanbetova RA, Vorkunova GK. Influenza virus uncoating in infected cells and effect of rimantadine. J Gen Virol 1982;60:49–59. [DOI] [PubMed] [Google Scholar]
- 225. Elster C, Fourest E, Baudin F, Larsen K, Cusack S, Ruigrok RW. A small percentage of influenza virus M1 protein contains zinc but zinc does not influence in vitro M1‐RNA interaction. J Gen Virol 1994;75:37–42. [DOI] [PubMed] [Google Scholar]
- 226. Nasser EH, Judd AK, Sanchez A, Anastasiou D, Bucher DJ. Antiviral activity of influenza virus M1 zinc finger peptides. J Virol 1996;70:8639–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, Kawaoka Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006;439:490–492. [DOI] [PubMed] [Google Scholar]
- 228. Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006;444:1078–1082. [DOI] [PubMed] [Google Scholar]
- 229. Wu WW, Weaver LL, Pante N. Ultrastructural analysis of the nuclear localization sequences on influenza A ribonucleoprotein complexes. J Mol Biol 2007;374:910–916. [DOI] [PubMed] [Google Scholar]
- 230. Cros JF, Garcia‐Sastre A, Palese P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 2005;6:205–213. [DOI] [PubMed] [Google Scholar]
- 231. O'Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem 1995;270:22701–22704. [DOI] [PubMed] [Google Scholar]
- 232. Gabriel G, Klingel K, Otte A, Thiele S, Hudjetz B, Arman‐Kalcek G, Sauter M, Shmidt T, Rother F, Baumgarte S, Keiner B, Hartmann E, Bader M, Brownlee GG, Fodor E, Klenk HD. Differential use of importin‐alpha isoforms governs cell tropism and host adaptation of influenza virus. Nat Commun 2011;2:156.:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional RNA‐binding protein pivotal to virus replication. J Gen Virol 2002;83:723–734. [DOI] [PubMed] [Google Scholar]
- 234. Zarubaev V, Garshinina A, Kalinina N, Shtro A, Belyaevskaya S, Slita A, Nebolsin V, Kiselev O. Activity of Ingavirin (6‐[2‐(1H‐Imidazol‐4‐yl)ethylamino]‐5‐oxohexanoic Acid) Against Human Respiratory Viruses in in Vivo Experiments. Pharmaceuticals 2011;4:1518–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Semenova NP, Prokudina EN, Livov DK, Nebol'sin VE. Effect of the antiviral drug Ingavirin on intracellular transformations and import into the nucleus of influenza A virus nucleocapsid protein. Vopr Virusol 2010;55:17–20. [PubMed] [Google Scholar]
- 236. Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol 2010;28:600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Cheng H, Wan J, Lin MI, Liu Y, Lu X, Liu J, Xu Y, Chen J, Tu Z, Cheng YS, Ding K. Design, synthesis, and in vitro biological evaluation of 1H‐1,2,3‐triazole‐4‐carboxamide derivatives as new anti‐influenza A agents targeting virus nucleoprotein. J Med Chem 2012;55:2144–2153. [DOI] [PubMed] [Google Scholar]
- 238. Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin‐Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC, Wu YT, Tsai MD, Cheng YS, Wong CH. High‐throughput identification of compounds targeting influenza RNA‐dependent RNA polymerase activity. Proc Natl Acad Sci USA 2010;107:19151–19156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mortl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva‐Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. Inhibition of influenza virus replication via small molecules that induce the formation of higher‐order nucleoprotein oligomers. Proc Natl Acad Sci USA 2011;108:15366–15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 2010;7:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Ludwig S. Targeting cell signalling pathways to fight the flu: towards a paradigm change in anti‐influenza therapy. J Antimicrob Chemother 2009;64:1–4. [DOI] [PubMed] [Google Scholar]
- 242. Shaw ML. The host interactome of influenza virus presents new potential targets for antiviral drugs. Rev Med Virol 2011;21:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Maurer AP, Muller E, Wolff T, Rudel T, Meyer TF. Genome‐wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010;463:818–822. [DOI] [PubMed] [Google Scholar]
- 244. Toullec D, Pianetti P, Coste H, Bellevergue P, Grand‐Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 1991;266:15771–15781. [PubMed] [Google Scholar]
- 245. Root CN, Wills EG, McNair LL, Whittaker GR. Entry of influenza viruses into cells is inhibited by a highly specific protein kinase C inhibitor. J Gen Virol 2000;81:2697–2705. [DOI] [PubMed] [Google Scholar]
- 246. Sieczkarski SB, Brown HA, Whittaker GR. Role of protein kinase C betaII in influenza virus entry via late endosomes. J Virol 2003;77:460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Ehrhardt C, Marjuki H, Wolff T, Nurnberg B, Planz O, Pleschka S, Ludwig S. Bivalent role of the phosphatidylinositol‐3‐kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol 2006;8:1336–1348. [DOI] [PubMed] [Google Scholar]
- 248. Ehrhardt C, Wolff T, Pleschka S, Planz O, Beermann W, Bode JG, Schmolke M, Ludwig S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol 2007;81:3058–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Hrincius ER, Dierkes R, Anhlan D, Wixler V, Ludwig S, Ehrhardt C. Phosphatidylinositol‐3‐kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig‐I to promote efficient type I interferon production. Cell Microbiol 2011;13:1907–1919. [DOI] [PubMed] [Google Scholar]
- 250. Feeley EM, Sims JS, John SP, Chin CR, Pertel T, Chen LM, Gaiha GD, Ryan BJ, Donis RO, Elledge SJ, Brass AL. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog 2011;7:e1002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Hamilton BS, Whittaker GR, Daniel S. Influenza virus‐mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 2012;4:1144–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]