

Abstract
Recent reports have demonstrated that oncogene amplification on extrachromosomal DNA (ecDNA) is a frequent event in cancer, providing new momentum to explore a phenomenon first discovered several decades ago. The direct consequence of ecDNA gains in these cases is an increase in DNA copy number of the oncogenes residing on the extrachromosomal element. A secondary effect, perhaps even more important, is that the unequal segregation of ecDNA from a parental tumor cell to offspring cells rapidly increases tumor heterogeneity, thus providing the tumor with an additional array of responses to microenvironment-induced and therapy-induced stress factors, and perhaps providing an evolutionary advantage. This Perspective discusses the current knowledge and potential implications of oncogene amplification on ecDNA in cancer.
Introduction
Oncogene amplification is one of the most common molecular alterations in cancer, playing a central role in tumorigenesis by providing cancer cells with selective growth advantages through overexpression of oncogenes1 and functional elements, such as enhancers2. Genomic amplification can result from double-strand break events, such as tandem duplication3, breakage–fusion–bridge cycles4, and chromothripsis5, which may result in complex chromosomal rearrangements6. The mechanisms leading up to the very high copy number that oncogenes can achieve in cancer are not well understood. Recently, it was shown that the formation of oncogene-carrying extrachromosomal DNA (ecDNA) is a potent and frequent mechanism by which genes are amplified and that such ecDNA can foment increased intratumoral genetic heterogeneity7,8.
The existence of chromosomal material outside the autosomal genome has been long recognized and also includes neochromosomes, which have been defined as centromere and/or telomere sequence-containing elements ranging in size from 30Mb to 400Mb9,10 and small supernumerary marker chromosomes that are detected in the germlines of 0.5% of the population11. Neochromosomes and supernumerary marker chromosomes may also play important roles in tumor pathogenesis12. In this perspective, we focus on the role of oncogene amplification on ecDNA, highlighting its importance to tumor pathogenesis and accelerated cancer evolution.
Historical notes on ecDNA
The concept that genes can be found on extrachromosomal particles of DNA in eukaryotic cells is not new, but its scale and potential importance has turned out to be a substantial surprise. Extrachromosomal particles of DNA can be found in many eukaryotic species studied to date, including in yeast13, drosophila14, c. elegans15, and humans15–18 (Box 1). These particles, sometimes referred to as extrachromosomal circular DNA (eccDNA), are thought to be circular based on biophysical methods and DNA sequencing, are small – usually less than 1 kb, and are invisible by light microscopy. Various types of these small DNA particles have been described, including telomeric circles19, small polydispersed DNA elements20 and microDNAs16; (reviewed in12). Although these small DNA elements lack full genes and are found in both normal cells and tumor cells, they may play a role in cancer pathogenesis possibly through promoting alternative lengthening of telomeres19 and via enhancement of genomic instability12,20
Box 1. Extrachromosomal DNA in non-cancer cells.
Interesting parallels to ecDNA exist in prokaryotic genetics. Bacterial chromosomes are circular and bacteria often harbor additional small circular DNA molecules or episomes67. Each plasmid has a stretch of DNA, the replication origin, that ensures it gets replicated, and replication happens independently of the bacterial chromosome. Up to hundreds of plasmids may exist within one bacterial cell68. Plasmids contain functionally important genes, including those that confer resistance to antibiotics69. The replication origins found in plasmids have not been observed in human ecDNAs, suggesting that ecDNAs use different replication principles.
In eukaryotes, centromeres are required to ensure proper segregation of chromosomes to each daughter at cell division; because ecDNAs lack a centromere, they may segregate randomly at mitosis (Fig. 2). Small circular DNAs have previously been described in eukaryotes, for both yeast and human cells12,70. However, these appear to be small and non-gene containing, and consist mostly of 200–500bp repetitive stretches of DNA19. These are different from cancer-associated ecDNA, which are often well above 1MB in size and contain genes and non-coding DNA, including regulatory regions.
Contemporaneous with first reports of eccDNAs17, another type of extrachromosomal particles of DNA, the focus of this perspective, was discovered. While examining metaphase spreads of neuroblastoma cells, Spriggs and colleagues noticed very small chromatin bodies21. Since these bodies were often “paired”, they referred to them as double minutes21. Subsequent studies by other groups confirmed the existence of these extrachromosomal particles of DNA in multiple types of cancer cells22–24.
Of note, at that time, researchers focused on the paired chromatin bodies. However, we now know that only about 30% of the extrachromosomal particles of DNA in a tumor cell are actually paired double minutes8. We use the term ecDNA to encompass the full spectrum of large, gene-containing extrachromosomal particles of DNA, including both double minute and single body forms, and lacking centromere or telomere. Unlike the telomeric circles, small polydispersed DNAs and microDNAs, ecDNA differs in the following important ways: 1) ecDNAs are large, often in the 1– 3 MB size range or larger; 2) ecDNAs contain multiple full genes and regulatory regions; 3) ecDNAs are visible by light microscopy.
The field of ecDNA biology moved beyond a phenomenological description with work from Schimke’s laboratory, which discovered that tumor cells could develop methotrexate resistance through amplification of the DHFR gene25. Further analyses revealed that DHFR amplification could occur either on the paired double minute forms of ecDNA or on aberrant chromosomal regions called homogeneously staining regions (HSRs)26,27. Schimke and colleagues hypothesized that the ecDNA represents an “unstable” form of gene amplification, whereas HSRs represented a “stable” form of amplification24,28.
The first reports that oncogenes may reside on ecDNA elements date back to the 1980s, when Alt and colleagues used cloning methods to demonstrate that sequences resembling MYCN could be mapped to HSRs and double minutes in the IMR-32 neuroblastoma cell line29. Similar observations were made by Bishop and colleagues in neuroendocrine cells derived from a colorectal carcinoma30. Work by others further confirmed that oncogene amplification could occur on ecDNA and that the frequency and amplitude of oncogene amplification could change in response to various stimuli, including treatment with hydroxyurea chemotherapy23,31–33. By studying the kinetics of ecDNA replication, Wahl and colleagues proposed that ecDNA followed the same replication mechanisms as regular chromosomes34.
These studies, which were perceptive and well ahead of their time, suggested that oncogene amplification on ecDNA was important. However, these studies occurred before the sequencing of the human genome. While fluorescence in situ hybridization (FISH) probes and complementary PCR methods have been leveraged to map amplified ecDNA in cancer cells35,36, the advent of DNA and RNA microarrays, and in particular of high-throughput sequencing, profoundly influenced how cancer genomes are interrogated. Computational analysis of sequencing datasets has provided a new view of the landscape of cancer genomes37,38. This approach was so powerful in detecting somatic alterations, that the focus of the field shifted towards genome-wide discovery of somatic variations and away from extrachromosomal amplification. In fact, ecDNA was thought to be a rare event in cancer, occurring in only 1.4% of tumors according to the Mitelman database39.
ecDNA is frequent in cancer cells
This was the state of the field until 2014, when the Mischel lab discovered that resistance to epidermal growth factor receptor (EGFR) inhibitors in glioblastoma—a highly lethal brain cancer—was conferred by reversible loss of the gain of function mutation EGFRvIII on ecDNA40. EGFRvIII amplification in glioblastoma was widely accepted as a source of considerable intratumoral genetic heterogeneity41,42, including via amplification on double minutes43,44, but its implication on EGFR tyrosine kinase inhibitor resistance was not appreciated prior to this study. EGFR inhibitors greatly reduced the fraction of tumor cells bearing high levels of EGFRvIII, in vitro, in mice, and in patients, and upon drug removal the high copy number of EGFRvIII returned40. However, the kinetics were too rapid to be consistent with a classical genetic model of clonal evolution. Further, single cells taken from the tumor, whether they expressed high levels of EGFRvIII, or no detectable EGFRvIII at all, produced identical tumors—in vitro and in mice—that mirrored the heterogeneous compositon of the parent tumor40. Again, this observation would have been virtually impossible to explain by classical genetics. The key to understanding the mechanism that could explain these results was to return to the older way of interrogating tumor genomes, looking at cells in metaphase. EGFRvIII was found to be amplified almost entirely on ecDNA, and the number of EGFRvIII-containing ecDNAs within glioblastoma cells plummeted in response to an EGFR inhibitor, then returned to a high copy number within 1–2 weeks after drug withdrawal40. This supported that the “unstable” nature of ecDNA, first pointed out by Schimke and colleagues27,28, may be more widespread than that seen in methotrexate resistance.
High-throughput short-read DNA sequencing can be used to infer tumor DNA copy number profiles, by comparing sequence read density between a tumor genome and a matching normal human genome reference. This analytic approach assigns amplified genes in cancer cells to the chromosomal loci in which they reside in normal tissue—an assumption that had not actually been tested. Therefore, Mischel, Bafna and colleagues hypothesized that ecDNA could be missed by the standard analytic approaches to high-throughput short-read DNA sequencing. Integrating whole genome sequencing, computational, and cytogenetic image analyses, they developed an unbiased approach to ecDNA detection and discovered that oncogene amplification on native chromosomal loci is relatively infrequent, but that amplification on ecDNA is widespread in cancer8. They further demonstrated that ecDNA-based gene amplification drives elevated copy number and promotes intratumoral genetic heterogeneity, suggesting a pivotal role for ecDNA in cancer evolution8. By developing the AmpliconArchitect tool for fine structural mapping of the amplicons (Fig. 1)45, they were able to show that some ecDNAs can “jump” onto chromosomal HSRs, consistent with the previous finding that oncogenes found on ecDNAs are often also found on chromosomes as HSRs36,48, 45 The mechanism underlying this process is unknown.
Figure 1. Reconstructing the Architecture of ecDNA using short-read sequencing data.
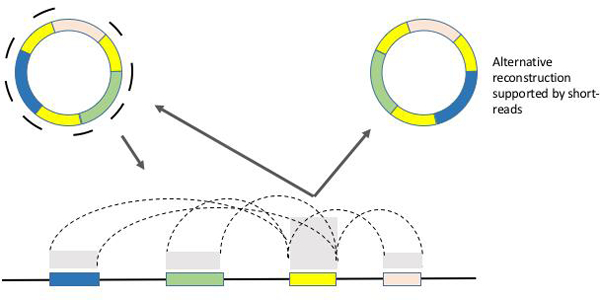
Directed assembly techniques have been developed to reconstruct the architecture of ecDNA. Short-read sequences acquired from ecDNA containing samples are mapped back to a reference genome. The copy number of the reads mapping to ecDNA segments (colored boxes) is proportional to the average number of ecDNA copies per cell, and reveals itself as copy number amplification in the mappings. Reads that span junctions of adjoining segments map back to the reference in a split fashion, revealing breakpoints. These mapping signatures of ecDNA derived complex rearrangements cannot be easily distinguished from chromosomal structural variants, posibly leading to an undercounting of ecDNA. ecDNA reconstruction algorithms explore amplicon graphs where nodes correspond to amplified segments, and edges correspond to breakpoints. Cyclic paths reconstructed by traversing the graph are indicative of ecDNA (Deshpande). Note also that if ecDNA contains large, repeated segments, long reads may be required for unambiguous reconstruction. Improvements in genomic and computational technologies will enhance ecDNA discovery.
Contemporaneous with—and independent of—this discovery, the Verhaak lab identified a central role for oncogene amplification on ecDNA in glioblastoma7. In 2013, they first reported that glioblastoma is characterized by dense patterns of repeated focal amplifications suggesting the frequent presence of ecDNA46. This work resulted in the hypothesis that the majority of oncogenic amplifications in glioblastoma are in fact extrachromosomal, and that ecDNA amplification provides glioblastoma with the ability to rapidly increase intratumoral heterogeneity. The Verhaak group then traced the presence of ecDNA using collections of glioblastoma tumor samples, together with glioma-neurosphere cultures derived from them, and orthotopic xenograft models derived from early passage neurospheres. ecDNA amplification of genes such as MYC, MYCN, EGFR, PDGFRA, MET, the MECOM/PIK3CA/SOX2 gene cluster and the CDK4/MDM2 gene cluster on chr12q14–15 were preserved during in vitro and in vivo tumor growth, suggesting that ecDNA amplification is an important factor of tumor propagation. They leveraged pairs of primary and matched recurrent glioma tumors to show that ecDNA amplifications are longitudinally preserved in patient tumors, which corroborated the importance of ecDNA during disease progression. A key finding from their work was the observation that in both patient tumors and model systems, the evolution of tumor subclones marked by somatic single nucleotide variants diverged from those characterized by ecDNA. This result provides direct evidence for the hypothesis that ecDNA inherits differently than chromosomal material (Fig. 2). Together, these findings support the hypothesis that ecDNA amplification is used by tumors to rapidly increase intratumoral heterogeneity, enabling cancers to adapt to selective pressures in culture or from therapy7.
Figure 2. Inheritance of chromosomal versus extrachromosomal DNA.
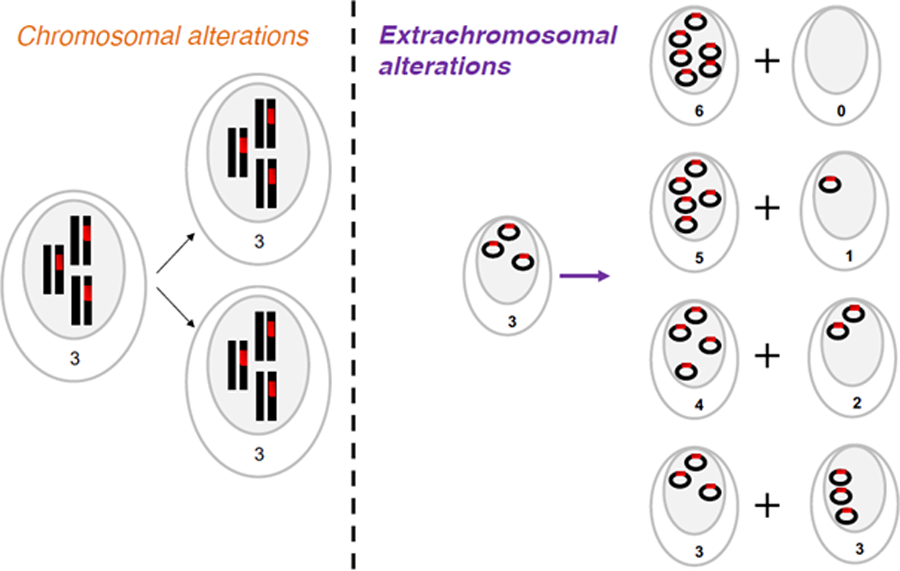
(Left panel) During mitosis, chromosomal DNA is evenly split between two daughter cells. (Right panel) Extrachromosomal DNA lacks centromeres and possibly segregates unevenly. As a result, the number of options for distribution of ecDNA between daughter cells equals n+1, where n is the number of ecDNA elements in the parental cell.
These studies suggested that ecDNA is a powerful and common mechanism by which oncogenes can be amplified in tumor cells at copy numbers that would be difficult to achieve for chromosomal amplifications. Further, the work demonstrated that ecDNA-based amplification enables tumors to rapidly acquire, and maintain intratumoral genetic heterogeneity, implying a central role in accelerated tumor evolution.
The birth of an ecDNA
Currently, the pathogenesis of ecDNA formation is not well understood. A possible step-wise model (Fig. 3) could proceed as follows. First, double strand breaks, chromosomal rearrangements, or possibly chromothriptic events could occur47, resulting in DNA segments that circularize to form ecDNA. Then, in the presence of tumor suppressor losses that enable cells to circumvent the senescence response to damaged DNA, replication of ecDNA elements could occur. Third, because the ecDNA particles lack centromeres, they are subject to non-equal segregation to daughter cells, which could rapidly lead to heterogeneity of ecDNA counts in cells within a tumor, and enable daughter cells to achieve higher ecDNA copy number than mother cells8. Finally, the presence of oncogenes or other proliferative elements could provide a selective advantage to the cell, shaped by environmental conditions. This would enhance the fitness of cells containing ecDNA, resulting in a rapid increase of ecDNA and oncogene copy numbers within the tumor.
Figure 3. Sequence of ecDNA formation and propagation in tumors.
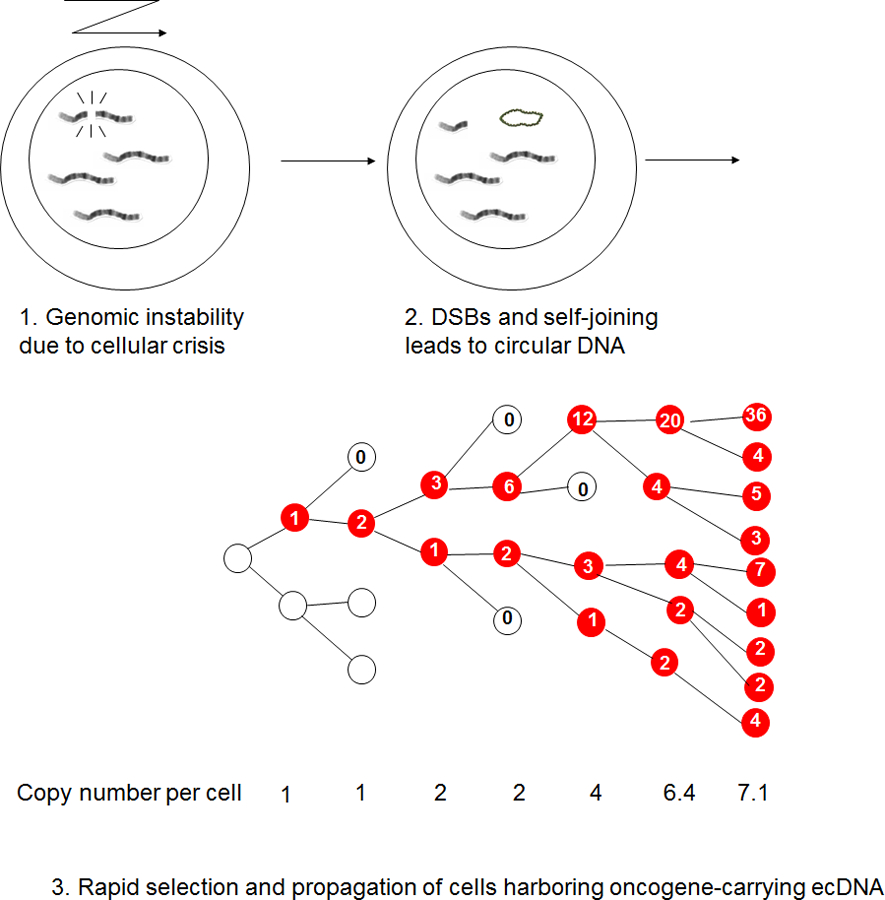
1. Genomic instability causes chromosomal breakage; 2. DNA segments form circular DNA structures through non-homologous recombination. 3. ecDNA elements, lacking centromeres, inherit unevenly from parental to daughter cells. 4. Following the formation of an ecDNA element, cells harboring the ecDNA element (marked in red) are afforded a competitive advantage through the expression of the oncogene contained on the ecDNA. This selection coefficient correlates proportionally with the number of ecDNA elements., resulting in rapid selection of ecDNA-positive cancer cells through increased proliferation rates.
There are many molecular steps that will need to be fully delineated and proven. It is formally possible, if not even likely, that there are multiple routes by which ecDNAs can be generated. The striking differences between genomic composition of ecDNAs in different cancer types8 may suggest that tumors can rely on multiple formational processes to derive circular DNAs, including a multistep process during which ecDNA elements evolve from single-segment to a complex multi-segment structure48. However, we note that the ecDNA model we have proposed only requires relatively plausible and simple events, including as little as a single random breakage event in the right cellular context, and it does not impose a requirement of fragility at the breakpoint. In contrast, when an oncogene or other proliferative element is present, the expression of that element may provide a fitness advantage which, due to the disjoint nature of ecDNA replication, can result in rapid accumulation of ecDNA copy numbers7,8. Additional DNA segments may be inserted into ecDNA elements which could provide increased mitotic potential33. This may happen progressively or though punctuated bursts49 and result in the highly complex DNA sequences that have been reported7,50.
It is interesting to note that ecDNA elements may be lost over time when patient-derived tumor cells are brought into culture, suggesting a fitness cost to the cell for maintaining the ecDNA51,52. Analysis of glioma samples at diagnosis and relapse have shown that ecDNA oncogene amplification can be maintained over time, apparently sufficiently benefiting the tumor to overcome the fitness cost7. Multiple and possibly competing subclones carrying unique ecDNAs with the same oncogene target have been reported with variable frequencies at the different time points50 suggesting a process of ongoing converging evolution. Multiple parallel ecDNAs carrying different oncogenes, i.e. MYC and EGFR, have also been described7. It is unclear whether an individual cell may harbor multiple ecDNAs or whether these reflect co-existing subclones.
Taken together, a common theme of ecDNA biology is emerging. Cancers that have ecDNA may be more capable of responding quickly to changing environments, including therapy40,53. Tumor cells are subject to constantly changing environmental stresses in competitive microenvironments54 – they compete for nutrients, aim to evade immune surveillance55–57, and are usually subject to an array of chemotherapeutic, targeted, and radiological therapies. The differential mechanism of ecDNA inheritance relative to chromosomal DNA and its effectiveness in maintinaing intratumoral genetic heterogeneity, promotes rapid genomic diversification and enhances the likelihood of selecting for cells that are preferentially fit for changing environments. Future studies will be needed to better understand whether ecDNA-driven cancers are more able to evade treatments, and if so, by what molecular mechanisms.
The impact of ecDNA
Clearly, ecDNA-based oncogene amplification can play an important role in accelerated cancer evolution through its unique mechanisms of inheritance that enable it to change quickly in changing environments. But does ecDNA also function differently than chromosomal DNA? The spatial organization of the genome is important for determining its function58–60. In human cells, DNA is segmented into 23 pairs of chromosomes, which are subdivided in the nucleus into distinct territories, and hierarchically organized. The DNA is wound around histone octamers that are organized into nucleosomes, whose position determines chromatin accessibility to the transcriptional and regulatory machinery of the cell. This dynamic process is further organized into chromatin loops, generating insulated domains in which regulatory and transcription factor interactions determine which genes will be expressed. In cancer, alterations in the spatial architecture, including disruption of topologically associated domain boundaries61,62, is implicated in tumor pathogenesis and a recent pioneering study has highlighted major alterations of the chromatin landscape in cancer63. Understanding the shape of ecDNA and its impact on epigenetic regulation, chromatin accessibility, and gene transcription is a fundamental question that must be answered.
A recent study focusing on the smaller circular DNA fragments similar to the small circular eccDNAs described above, shows transcriptional silencing of a gene encoding an enhanced green fluroescent protein on kilobase sized circular DNA generated by CRISPR/Cas9 editing64. However, this model is very different from the large (105–107 bp) oncogene and regulatory element containing ecDNAs that are the focus of this article, for which higher expression levels of oncogene transcription have already been shown7,8. New methods are being developed to probe the chromatin structure, transcriptional landscape, and functional contribution of ecDNAs in cancer pathogenesis, which will greatly enhance our understanding of their pathogenic role in cancer, and enlighten us as to how chromosomal or extrachromosomal context affects the impact of an oncogene on tumor development, progression, and drug resistance.
Developing an ecDNA toolbox
EcDNA is different from chromosomal DNA in its location, its topology, and likely in key aspects of its biology. Consequently, new tools will need to be developed in order to better understand the role of oncogene amplification on ecDNA in cancer pathogenesis. Currently, defining the nucleotide sequence of ecDNAs by computational analysis is challenged by short-read sequencing, which makes it difficult to capture more than a single breakpoint at a time. When multiple ecDNAs are derived from the same locus within a given sample, or when a genome shattering event has taken place, multiple breakpoints are found within short distance of each other and standard chromosomal rearrangement analysis pipelelines of short-read sequencing data are not designed to recognize these possibilities. As a consequence, current catalogues of chromosomal structural rearrangements in cancer such as those derived by The Cancer Genome Atlas do not label structural variants as being part of an ecDNA. We are actively working to expand this capability and integrate it with long-readsequencing tools, to better understand and map the structure of ecDNAs. Long-read sequences, such as the 5–10kb sequence reads that can be as derived using Nanopore or Pacific Bioscience sequencing platforms or optical map technologies (e.g. BioNano), may be able to capture multiple breakpoints and provide more conclusive evidence on ecDNA frequency and structure7,45,50 Ultimately, library enrichment approaches for ecDNA sequencing or methods that can determine full ecDNA structure in one read or map are needed to completely elucidate individual circular DNA molecules. Further, we are in the process of developing a tool kit that will enable the better resolution of ecDNAs from next generation sequencing data. Achievement of this goal will permit a deeper understanding of the scale, scope, and contents of ecDNA in cancer, and its association with clinical features.
In addition to the computational tool kit, it is necessary to expand the cell biology tool box to interrogate the activity of key components of the replication, transcription, and repair machinery with respect to ecDNA relative to chromosomal DNA. The toolbox for studying cells at the most detailed level is ever expanding. For example, the development of genome-editing techniques such as CRISPR-Cas and advanced cell imaging may be leveraged to further our understanding of the basic biology that creates and maintains ecDNA.
Conclusions and perspective
The rediscovery that oncogenes can be amplified on ecDNA, and its new found importance in cancer, has forced the reconsideration of spatial information contained in current cancer genome maps, and provides an exciting new dimensions to modern cancer genomics. Where genes are amplified matters. How can we use this information for the design of new rational therapies, for the benefit of cancer patients? Basic questions will need to be addressed in order to get to targeted ecDNA inhibition for treatment: What are the underlying mechanisms that drive ecDNA formation and maintenance? What determines if and where it re-integrates in chromosomes? How can other forms of rearrangements seen in cancer, such as breakage–fusion–bridge, chromothripsis, chromoplexy, and neochromosome formation be reconciled with ecDNA? Does extrachromosomal amplification provide a sufficiently unique cancer cell-specific trait that it may be exploited for cancer therapy? Can we learn from the behavior of circular DNA found elsewhere in nature? The conversation about oncogene amplification has started to refocus, recognizing that intranuclear location and spatial organization of amplified genes may have important consequences for the development and progression of cancer, and ultimately for its treatment.
Acknowledgements
We thank Christine Beck (Jackson Laboratory for Genomic Medicine), Sihan Wu and Kristen M. Turner (Mischel laboratory), for feedback on the manuscript content and Sara Cassidy (Jackson Laboratory for Genomic Medicine) for support in manuscript writing. RGWV is supported by grants from the US National Institutes of Health R01 CA190121 and Cancer Center Support Grant P30CA034196, and grants from the Musella Foundation and the B*CURED Foundation. VB is supported in part by grants from the NIH (GM114362,HG004962) and NSF (DBI-1458557). PSM is supported in part by grants from the NIH (NS73831), the Defeat GBM program of the National Brain Tumor Society, The Ben and Catherine Ivy Foundation, an award from the Sharpe/National Brain Tumor Society Research Program, and a Compute for the Cure Award from the Nvidia Foundation.
Glossary
- Tandem duplication
Repeated segments of DNA inserted in the genome, that disrupt expression of important tumor suppressor genes or amplify tumor promoter genes
- Breakage–fusion–bridge cycles
A mechanism of chromosomal instability initiated by a telomeric loss and multiple cycles of anaphase bridge formation followed by unequal breakage.
- Chromothripsis
A phenomenon marked by shattering of a chromosome and re-ligation of some of the fragments, causing massive rearrangement in a single catastrophic event.
- Supernumerary marker chromosomes
An additional copy of an autosomal chromosome that is infrequently found in individuals.
- Telomeric circles
A structure based on circularization of telomeric tandem repeats that allows for rolling circle amplification and synthesis of longer repeat elements that help with lengthening of telomeres, and stabilizing the chromosome.
- Alternative lengthening of telomeres
One or more mechanisms that are frequently observed in tumors lacking telomerase activity and manifested by long but highly variable telomeres.
- MicroDNAs
A form of short, extrachromosomal, circular DNA elements that are up to 400bp long, and non-repetitive, and putatively formed due to excision and replication of short DNA.
- Homogeneously staining region (HSR)
A region of the chromosome that has duplicated many times and shows up as large homogenously stained region when painted with a FISH probe unique to the region. While tandem duplications are usually implicated in HSR formation, replication of ecDNA and their reintegration into the genome may also cause HSRs.
- High-throughput short-read DNA sequencing
The term given to a variety of sequencing technologies that generate short (100–300bp) reads in an unbiased and massively parallel manner allowing for inexpensive, and redundant sampling of a genome.
- Optical map technologies
Genomic technologies that construct ordered maps of restriction site locations in large genomic fragments (150–400kbp). The maps serve as a unique fingerprint for the fragment, and are useful in validating large structural variations, including insertions.
- Topologically associated domain
A region of the genome that is characterized by extensive interactions within due to the spatial organization of the genome, and reduced
Footnotes
Competing interests
RGWV, VB, and PSM are co-founders of and have equity interest in Pretzel Therapeutics, Inc. (PT). PSM serves as a consultant to PT. VB is a co-founder, and has equity interest in Digital Proteomics, LLC (DP), and receives income from DP. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. PT and DP were not involved in the research presented here.
References
- 1.Beroukhim R et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905, doi: 10.1038/nature08822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weischenfeldt J et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet 49, 65–74, doi: 10.1038/ng.3722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menghi F et al. The Tandem Duplicator Phenotype Is a Prevalent Genome-Wide Cancer Configuration Driven by Distinct Gene Mutations. Cancer Cell 34, 197–210 e195, doi: 10.1016/j.ccell.2018.06.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gisselsson D et al. Generation of trisomies in cancer cells by multipolar mitosis and incomplete cytokinesis. Proc Natl Acad Sci U S A 107, 20489–20493, doi: 10.1073/pnas.1006829107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korbel JO & Campbell PJ Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–1236, doi: 10.1016/j.cell.2013.02.023 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Solomon E, Borrow J & Goddard AD Chromosome aberrations and cancer. Science 254, 1153–1160 (1991). [DOI] [PubMed] [Google Scholar]
- 7.deCarvalho AC et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet 50, 708–717, doi: 10.1038/s41588-018-0105-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner KM et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125, doi: 10.1038/nature21356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garsed DW et al. The architecture and evolution of cancer neochromosomes. Cancer Cell 26, 653–667, doi: 10.1016/j.ccell.2014.09.010 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Macchia G et al. The Hidden Genomic and Transcriptomic Plasticity of Giant Marker Chromosomes in Cancer. Genetics 208, 951–961, doi: 10.1534/genetics.117.300552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liehr T, Claussen U & Starke H Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res 107, 55–67, doi: 10.1159/000079572 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Paulsen T, Kumar P, Koseoglu MM & Dutta A Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet 34, 270–278, doi: 10.1016/j.tig.2017.12.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moller HD, Parsons L, Jorgensen TS, Botstein D & Regenberg B Extrachromosomal circular DNA is common in yeast. Proc Natl Acad Sci U S A 112, E3114–3122, doi: 10.1073/pnas.1508825112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanfield SW & Lengyel JA Small circular DNA of Drosophila melanogaster: chromosomal homology and kinetic complexity. Proc Natl Acad Sci U S A 76, 6142–6146 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shoura MJ et al. Intricate and Cell Type-Specific Populations of Endogenous Circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3 (Bethesda) 7, 3295–3303, doi: 10.1534/g3.117.300141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibata Y et al. Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science 336, 82–86, doi: 10.1126/science.1213307 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotta Y & Bassel A Molecular Size and Circularity of DNA in Cells of Mammals and Higher Plants. Proc Natl Acad Sci U S A 53, 356–362 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moller HD et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun 9, 1069, doi: 10.1038/s41467-018-03369-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henson JD et al. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol 27, 1181–1185, doi: 10.1038/nbt.1587 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Cohen S, Regev A & Lavi S Small polydispersed circular DNA (spcDNA) in human cells: association with genomic instability. Oncogene 14, 977–985, doi: 10.1038/sj.onc.1200917 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Cox D, Yuncken C & Spriggs AI Minute Chromatin Bodies in Malignant Tumours of Childhood. Lancet 1, 55–58 (1965). [DOI] [PubMed] [Google Scholar]
- 22.Fan Y et al. Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J Appl Genet 52, 53–59, doi: 10.1007/s13353-010-0007-z (2011). [DOI] [PubMed] [Google Scholar]
- 23.Stark GR, Debatisse M, Giulotto E & Wahl GM Recent progress in understanding mechanisms of mammalian DNA amplification. Cell 57, 901–908 (1989). [DOI] [PubMed] [Google Scholar]
- 24.Schimke RT Gene amplification in cultured animal cells. Cell 37, 705–713 (1984). [DOI] [PubMed] [Google Scholar]
- 25.Alt FW, Kellems RE, Bertino JR & Schimke RT Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem 253, 1357–1370 (1978). [PubMed] [Google Scholar]
- 26.Haber DA, Beverley SM, Kiely ML & Schimke RT Properties of an altered dihydrofolate reductase encoded by amplified genes in cultured mouse fibroblasts. J Biol Chem 256, 9501–9510 (1981). [PubMed] [Google Scholar]
- 27.Haber DA & Schimke RT Unstable amplification of an altered dihydrofolate reductase gene associated with double-minute chromosomes. Cell 26, 355–362 (1981). [DOI] [PubMed] [Google Scholar]
- 28.Beverley SM, Coderre JA, Santi DV & Schimke RT Unstable DNA amplifications in methotrexate-resistant Leishmania consist of extrachromosomal circles which relocalize during stabilization. Cell 38, 431–439 (1984). [DOI] [PubMed] [Google Scholar]
- 29.Kohl NE et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35, 359–367 (1983). [DOI] [PubMed] [Google Scholar]
- 30.Alitalo K, Schwab M, Lin CC, Varmus HE & Bishop JM Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc Natl Acad Sci U S A 80, 1707–1711 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Von Hoff DD et al. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci U S A 89, 8165–8169 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Von Hoff DD et al. Hydroxyurea accelerates loss of extrachromosomally amplified genes from tumor cells. Cancer Res 51, 6273–6279 (1991). [PubMed] [Google Scholar]
- 33.Carroll SM et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol 8, 1525–1533 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz JC, Choi KH, von Hoff DD, Roninson IB & Wahl GM Autonomously replicating episomes contain mdr1 genes in a multidrug-resistant human cell line. Mol Cell Biol 9, 109–115 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Storlazzi CT et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet 15, 933–942, doi: 10.1093/hmg/ddl010 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Storlazzi CT et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome research 20, 1198–1206, doi: 10.1101/gr.106252.110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garraway LA & Lander ES Lessons from the cancer genome. Cell 153, 17–37, doi: 10.1016/j.cell.2013.03.002 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558, doi: 10.1126/science.1235122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitelman F, Johansson B & Mertens F The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7, 233–245, doi: 10.1038/nrc2091 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Nathanson DA et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–76, doi: 10.1126/science.1241328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snuderl M et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20, 810–817, doi: 10.1016/j.ccr.2011.11.005 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Szerlip NJ et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 109, 3041–3046, doi: 10.1073/pnas.1114033109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogt N et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A 101, 11368–11373, doi: 10.1073/pnas.0402979101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bigner SH et al. Relationship between gene amplification and chromosomal deviations in malignant human gliomas. Cancer Genet Cytogenet 29, 165–170 (1987). [DOI] [PubMed] [Google Scholar]
- 45.Deshpande V et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 10, 392, doi: 10.1038/s41467-018-08200-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng S et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev 27, 1462–1472, doi: 10.1101/gad.213686.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ly P & Cleveland DW Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol 27, 917–930, doi: 10.1016/j.tcb.2017.08.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.L’Abbate A et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res 42, 9131–9145, doi: 10.1093/nar/gku590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malhotra A et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome research 23, 762–776, doi: 10.1101/gr.143677.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu K et al. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol, doi: 10.1007/s00401-018-1912-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nikolaev S et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun 5, 5690, doi: 10.1038/ncomms6690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yost SE et al. High-resolution mutational profiling suggests the genetic validity of glioblastoma patient-derived pre-clinical models. PLoS One 8, e56185, doi: 10.1371/journal.pone.0056185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xue Y et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med 23, 929–937, doi: 10.1038/nm.4369 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gatenby RA & Gillies RJ A microenvironmental model of carcinogenesis. Nat Rev Cancer 8, 56–61, doi: 10.1038/nrc2255 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Zhang AW et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 173, 1755–1769 e1722, doi: 10.1016/j.cell.2018.03.073 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Grasso CS et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov 8, 730–749, doi: 10.1158/2159-8290.CD-17-1327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McGranahan N et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271 e1211, doi: 10.1016/j.cell.2017.10.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319, doi: 10.1016/j.cell.2013.03.035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380, doi: 10.1038/nature11082 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shachar S, Voss TC, Pegoraro G, Sciascia N & Misteli T Identification of Gene Positioning Factors Using High-Throughput Imaging Mapping. Cell 162, 911–923, doi: 10.1016/j.cell.2015.07.035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hnisz D et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458, doi: 10.1126/science.aad9024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114, doi: 10.1038/nature16490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corces MR et al. The chromatin accessibility landscape of primary human cancers. Science 362, doi: 10.1126/science.aav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moller HD et al. CRISPR-C: circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res 46, e131, doi: 10.1093/nar/gky767 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grohmann E, Muth G & Espinosa M Conjugative plasmid transfer in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67, 277–301 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Figurski DH & Helinski DR Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl Acad. Sci. USA 76, 1648–1652 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bennett PM Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol. 153 (Suppl. 1), 347–357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]