

Summary
Swine acute diarrhoea syndrome coronavirus (SADS‐CoV), a novel coronavirus, was first discovered in southern China in January 2017 and caused a large scale outbreak of fatal diarrheal disease in piglets. Here, we conducted a retrospective investigation of 236 samples from 45 swine farms with a clinical history of diarrheal disease to evaluate the emergence and the distribution of SADS‐CoV in pigs in China. Our results suggest that SADS‐CoV has emerged in China at least since August 2016. Meanwhile, we detected a prevalence of SADS‐CoV (43.53%), porcine deltacoronavirus (8.83%), porcine epidemic diarrhoea virus (PEDV) (78.25%), rotavirus (21.77%), and transmissible gastroenteritis virus (0%), and we also found the co‐infection of SADS‐CoV and PEDV occurred most frequently with the rate of 17.65%. We screened and obtained two new complete genomes, five N and five S genes of SADS‐CoV. Phylogenetic analysis based on these sequences revealed that all SADS‐CoV sequences in this study clustered with previously reported SADS‐CoV strains to form a well defined branch that grouped with the bat coronavirus HKU2 strains. This study is the first retrospective investigation for SADS‐CoV and provides the epidemiological information of this new virus in China, which highlights the urgency to develop effective measures to control SADS‐CoV.
Keywords: phylogenetic analysis, prevalence, retrospective detection, Swine Acute Diarrhoea Syndrome Coronavirus
1. INTRODUCTION
Swine acute diarrhoea syndrome coronavirus (SADS‐CoV) is a newly discovered coronavirus which is an enveloped, positive and single‐stranded sense RNA virus with a genome size of approximately 27 kb (Gong et al., 2017; Pan et al., 2017; Zhou et al., 2018). SADS‐CoV belongs to the family Coronaviridae which contains four genera, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus (Woo, Huang, Lau, & Yuen, 2010; Woo et al., 2012). So far, six coronaviruses have been identified from pigs, which include porcine epidemic diarrhoea virus (PEDV), porcine respiratory coronavirus (PRCV), SADS‐CoV and transmissible gastroenteritis virus (TGEV) that all belong to the Alphacoronavirus genus, as well as one betacoronavirus, porcine hemagglutinating encephalomyelitis virus (PHEV) and one deltacoronavirus, porcine deltacoronavirus (PDCoV) (Lin, Saif, Marthaler, & Wang, 2016; Wesley, Woods, & Cheung, 1991; Woo et al., 2010). Among these viruses, SADS‐CoV is the most newly discovered coronavirus, which has been first reported in 2017 in China and is considered to be an HKU2‐related coronavirus with a bat‐origin (Gong et al., 2017; Zhou et al., 2018). In January 2017, SADS‐CoV was detected in a swine farm and subsequently spread rapidly to three other farms in Guangdong Province and caused the fatal swine acute diarrhoea syndrome (SADS) characterized by the clinical signs with severe, acute diarrhoea and rapid weight loss of piglets. The symptoms of SADS‐CoV are similar to those that caused by other swine enteric coronaviruses such as PDCoV and PEDV, but SADS‐CoV is more harmful than these viruses because it has led to the death of almost 25,000 piglets in a short time and resulted in more significant economic losses (Dong et al., 2015; Sun, Wang, Wei, Chen, & Feng, 2016; Zhou et al., 2018). So, it is urgent to investigate the molecular epidemiology and transmission patterns of SADS‐CoV for establishing effective controls for this new coronavirus.
In the present study, we performed the retrospective PCR testing on diarrheal samples from 45 swine farms in Guangdong Province to evaluate the emergence and the distribution of SADS‐CoV in pigs in China. The prevalence and co‐infection information of SADS‐CoV from eleven SADS‐CoV‐positive farms was provided. The sequences of SADS‐CoV, including two complete genomes, five nucleocapsid protein (N) genes and five spike protein (S) genes, were also identified and characterized to investigate the phylogenetic relationships of SADS‐CoV.
2. MATERIALS AND METHODS
2.1. Sample collection
A total of 236 clinical diarrhoea samples including faeces and intestinal contents were collected from piglets and sows from 45 swine farms in the cities Qingyuan and Shaoguan of North Guangdong Province between August 2016 and May 2017 in accordance with the recommendations of National Standards for Laboratory Animals of the People's Republic of China (GB149258‐2010). Samples were preserved at −80 °C from the time of original receipt until use.
2.2. Nucleic acid extraction and molecular diagnosis
Samples were homogenized in phosphate‐buffered saline (PBS) (20% w/v), frozen and thawed three times, then centrifuged for 10 min at 10,000 g. Viral nucleic acid was extracted following the manufacturer's recommendations of AxyPrepTM Body Fluid Viral DNA/RNA Miniprep Kit (Axygen Scientific, Inc). The virus nucleic acid was stored at −80°C until PCR was performed. A pair of primers (forward primer 5′‐GGTCCCTGTGACCGAAGTTTTAG‐3′, reverse primer 5′‐ GCGTTCTGCGATAAAGCTTAAAACTATTA‐3′) was designed to detected SADS‐CoV based on the conserved N gene of this virus. One step RT‐PCR using PrimeScript™ One Step RT‐PCR Kit Ver.2 with Dye Plus (Takara, Biotechnology, Dalian, China) was carried out to amplify the target fragments by the following thermal profile of 50°C for 30 min, 94°C for 3 min, 35 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, an extension at 72°C for 30 s, and a final step of 72°C for 5 min. Four other diarrheal pathogens including PEDV, PDCoV, rotavirus (RV), and TGEV from SADS‐CoV‐positive farms were also tested by RT‐PCR according to the previously described methods (Liu, Zhu, Liao, Xu, & Zhou, 2015; Mai et al., 2017; Stevenson et al., 2013).
2.3. Amplification of the N gene, the S gene, and the complete genome of SADS‐CoV
Specific primer pairs based on reported SADS‐CoV strains (GenBank accession numbers: MF094681‐MF094684) were designed for S genes, N genes and complete genome amplifications, respectively (Table S1). PCR assays were performed with the following thermal profile: 95°C for 5 min, 35 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 1 min 15 s, followed by a final 10 min extension at 72°C. The products were purified following the manufacturer's instructions of Gel Band Purification Kit (Omega Bio‐tek, USA) and then cloned into the PMD19‐T vector (Takara, Biotechnology, Dalian, China) and transformed E. coli DH5α competent cells. The positive clones were screened out and sent to Beijing Genomics Institute (Shenzhen, Guangdong, China) for further sequencing.
2.4. Sequence alignment and phylogenetic analysis
The nucleotide sequences were assembled and aligned using the DNASTAR program (DNAStar V7.1, Madison, WI, USA). Phylogenetic trees were constructed using the neighbour‐joining method in MEGA 7.0 software with bootstrap analysis of 1,000 replicates. Percentages of replicate trees in which the associated taxa clustered are shown as nearby branches (Chenna et al., 2003; Kumar, Stecher, & Tamura, 2016; Tamura, Nei, & Kumar, 2004).
3. RESULTS
3.1. SADS‐CoV detection
Of 236 clinical diarrhoea samples from 45 swine farms, 53 samples collected from eleven swine farms between August 2016 and May 2017 were tested positive for SADS‐CoV and the locations of these eleven farms were indicated in Figure 1. The farms SM, GL, DL, DE, TP, WT, DCD, and SC were in Qingyuan, and the farms LS, ZW, and YX were in Shaoguan. From these eleven SADS‐CoV‐positive farms, a total of 170 archived diarrheal samples including 55 faeces and 115 intestinal contents were collected to further investigate diarrhoeal pathogens including SADS‐CoV, PEDV, RV, PDCoV, and TGEV. The infection rates of these five pathogens in order were 43.5% (74/170), 78.2% (133/170), 21.8% (37/170), 8.8% (15/170), and 0% (0/170). SADS‐CoV was tested positively both in the intestinal and faecal samples and the detection rates were 49.6% (57/115) and 30.9% (17/55), respectively, which were both the second highest among the five tested pathogens. PEDV had the highest positive rate at 84.3% (97/115) for intestinal and 65.5% (36/55) for faecal samples. TGEV was tested negatively in all 170 diarrheal samples (Table S2).
Figure 1.
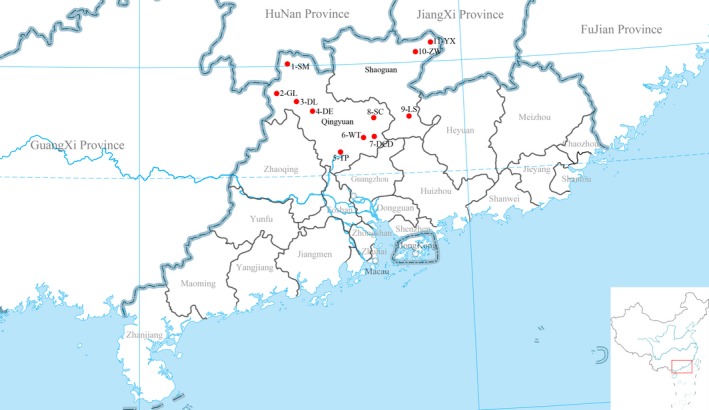
Locations of positive farms for study of SADS‐CoV in Guangdong Province, China [Colour figure can be viewed at http://www.wileyonlinelibrary.com]
The cases of individual infection or co‐infection for SADS‐CoV, PDCoV, PEDV, and RV were also detected. More than half the samples were only infected by one of the four viruses at a rate of 58.82% (100/170), and the individual infection of PEDV had the highest rate of 37.06% (63/170). In the 74 SADS‐CoV‐positive samples, 28 samples were infected alone by SADS‐CoV, and the rest 46 samples were infected by SADS‐CoV combined with one to three other pathogens. The co‐infection of SADS‐CoV and PEDV occurred most frequently with the rate of 17.65% (30/170) (Table 1).
Table 1.
Screening information of SADS‐CoV, and co‐infections of different pathogens detected in north Guangdong Province of China between August 2016 and May 2017
| Months of year | One pathogen positive samples no.(%,n = 170) | Two pathogens positive samples no.(%,n = 170) | Three pathogens positive samples no.(%,n = 170) | Four pathogens positive samples no.(%,n = 170) | SADS‐CoV positive farms (Positive/Total) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SADS‐CoV | PEDV | RV | PDCoV | TGEV | SADS‐CoV+PEDV | PEDV+RV | PEDV+PDCoV | SADS‐CoV+PEDV+RV | SADS‐CoV+PEDV+PDCoV | PEDV+RV+PDCoV | SADS‐CoV+PEDV+RV+PDCoV | ||
| Aug, 2016 | 3 | 4 | 0 | 0 | 0 | 2 | 3 | 1 | 0 | 0 | 1 | 0 | 11/45 |
| Sep, 2016 | 0 | 6 | 0 | 2 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | |
| Oct, 2016 | 0 | 2 | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | |
| Nov, 2016 | 1 | 8 | 1 | 1 | 0 | 1 | 4 | 1 | 1 | 1 | 0 | 2 | |
| Dec, 2016 | 2 | 8 | 0 | 1 | 0 | 3 | 0 | 1 | 3 | 0 | 0 | 0 | |
| Jan, 2017 | 2 | 3 | 2 | 0 | 0 | 10 | 3 | 0 | 2 | 0 | 0 | 0 | |
| Feb, 2017 | 6 | 7 | 0 | 0 | 0 | 4 | 3 | 1 | 2 | 0 | 0 | 0 | |
| Mar, 2017 | 3 | 9 | 0 | 1 | 0 | 4 | 0 | 0 | 4 | 0 | 0 | 0 | |
| Apr, 2017 | 10 | 10 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 1 | 0 | 0 | |
| May,2017 | 1 | 6 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Total | 28 (16.47%) | 63 (37.06%) | 4 (2.35%) | 5 (2.94%) | 0 (0%) | 30 (17.65%) | 18 (10.59%) | 5 (2.94%) | 12 (7.06%) | 2 (1.18%) | 1 (0.59%) | 2 (1.18%) | |
| SADS‐CoV positive samples : 74(43.53%) | |||||||||||||
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The earliest cases with SADS‐CoV‐positive infection were found in the farms LS, TP and ZW on August 2016 according to Table S2. The intestinal contents from these eleven farms were tested positive for SADS‐CoV and the positive rates ranged from 25% to 77.78%. While the faecal samples from two farms ZW and YX were tested negative for SADS‐CoV, but positive for PDCoV, PEDV, and RV infections. The positive rates of SADS‐CoV in the faecal samples of other nine farms varied from 13.33% to 100%. The farm LS had the highest positive rate of 77.8% (7/9) for intestinal samples and the farms DCD and SC had 100% positive rates for faecal samples.
3.2. Sequence identities and phylogenetic analyses
The two complete genomes (accession number MG605090 and MG605091), five N genes (accession number MG605087, MG605088, MG605089, MG775251, and MG775253) and five S genes (accession number MG605084, MG605085, MG605086, MG775250 and MG775252) were sequenced and available in GenBank. The lengths of the complete genome, N gene and S gene were 27,163 bp, 1,128 bp, and 3,390 bp, respectively. The two new complete sequences, five N genes and five S genes sequences each shared high sequence identities of 99.8%–100%, 99.8%–100%, and 99.6%–100% with previous reported complete genomes, N genes and S genes of SADS‐CoV. The two full‐length sequences of SADS‐CoV showed 95%–95.1% nucleotide identities with four bat coronavirus HKU2 strains, and the nucleotide identities between the N genes of SADS‐ CoV studied here and those of bat coronavirus HKU2 strains ranged from 94.2% to 94.3%. The S genes of SADS‐ CoV in this paper shared low nucleotide identities of 79.9%–80% with the S genes of bat coronavirus HKU2 strains. Phylogenetic analyses based on the complete genomes, N genes and S genes indicated that all SADS‐CoV sequences clustered together to form a well‐defined branch and group with four bat coronavirus HKU2 strains In the phylogenetic trees of the full‐length genomes and N genes, all SADS‐CoV strains were phylogenetically located within the AlphaCoVs, while the phylogenetic tree of S genes exhibited that all SADS‐CoV sequences, four bat HKU2 and two BtRf‐AlphaCoV strains separated from other AlpaCoVs sequences and clustered with the BeltaCoV sequences (Figures 2, 3 and 4).
Figure 2.
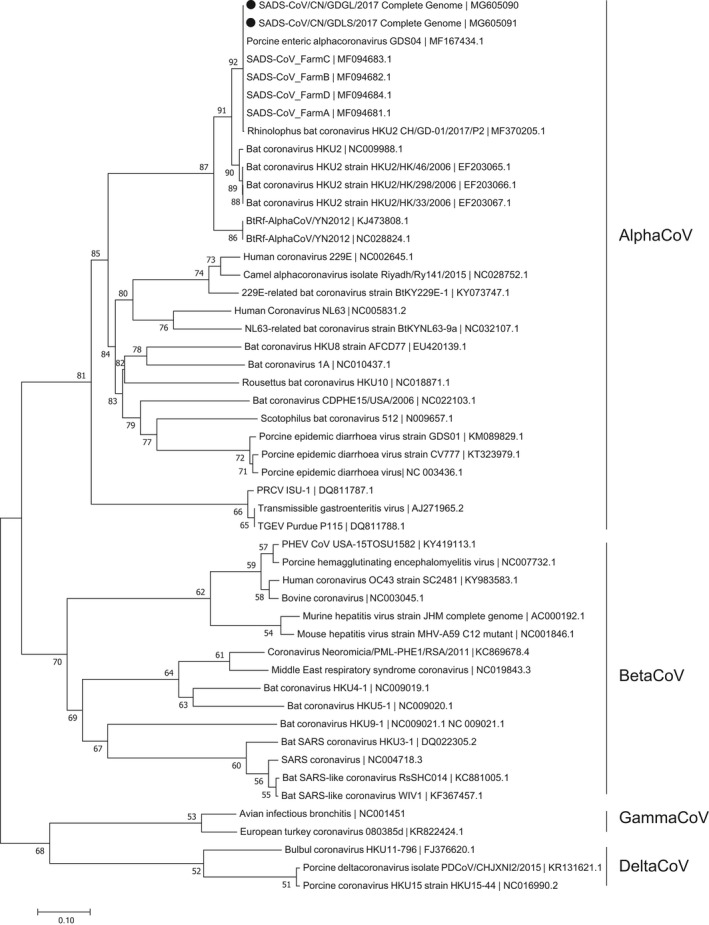
Phylogenetic analysis of the complete genomes of SADS‐CoV and reference coronavirus species. The tree was constructed using MEGA 7.0 software with neighbour‐joining methods and 1,000 replicate sets on bootstrap analysis. Two new complete genomes studied in this work were indicated with “black solid circles”
Figure 3.
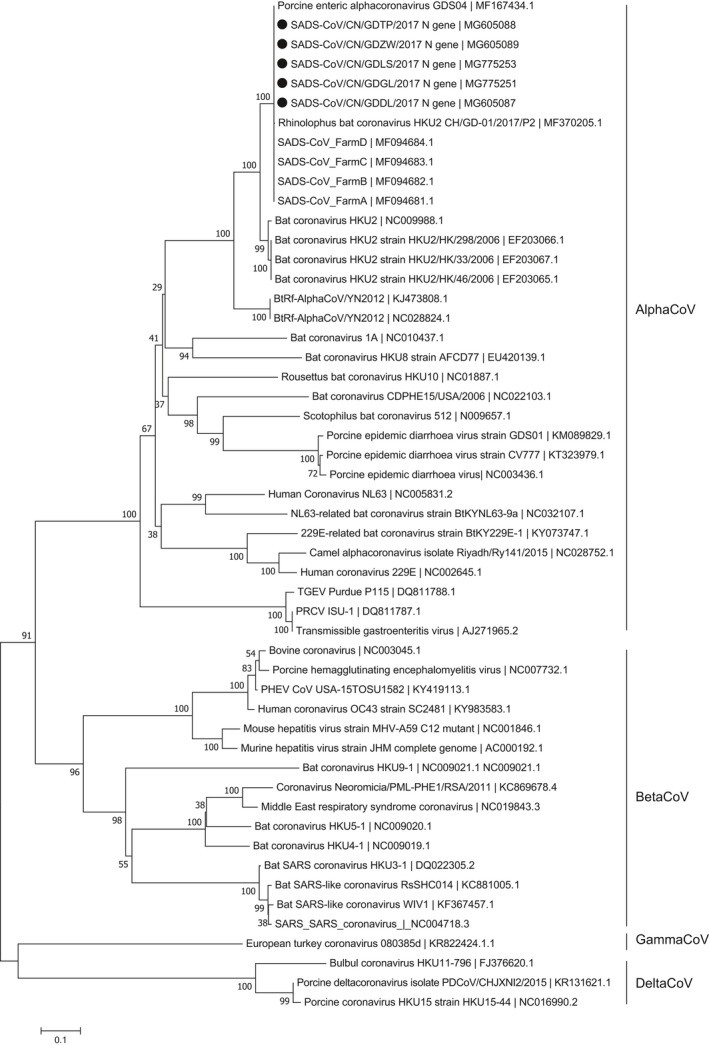
Phylogenetic analysis of the N genes of SADS‐CoV and reference coronavirus species. The tree was constructed as per Figure 2 above. Five new sequences of N genes studied in this work were indicated with “black solid circles”
Figure 4.
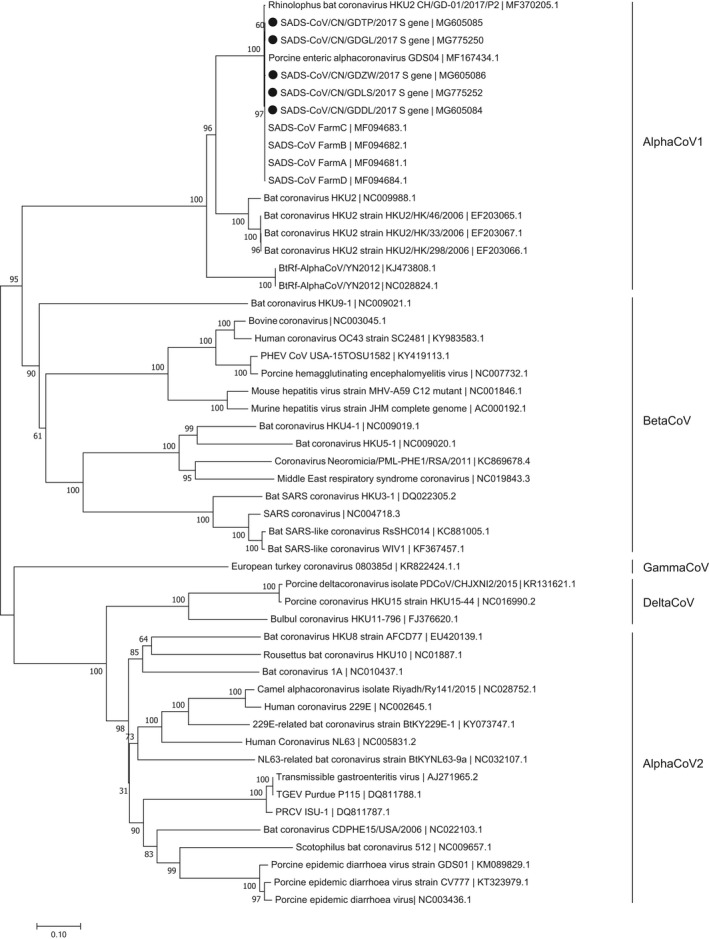
Phylogenetic analysis of the S genes of SADS‐CoV and reference coronavirus species. The tree was constructed as per Figure 2 above. Five new sequences of S genes studied in this work were indicated with “black solid circles”
4. DISCUSSION
In this study, we performed the retrospective investigation of SADS‐CoV in 45 pig farms from Guangdong Province based on 236 diarrhoea samples. Our results showed that the first SADS‐CoV positive sample was collected in August 2016 from the farm LS with a history of diarrhoea, as well as from other two farms TP and ZW, which indicates that SADS‐CoV has emerged in pigs in China at least since August 2016. And this time point is 5 months earlier than the first discovered time reported by our previous study (Zhou et al., 2018). As the same time, clinical signs of SADS‐CoV during the retrospective investigation included sever and acute vomiting and diarrhoea, leading to death in piglets that were less than 5 days of age with a mortality rate of around 50%. These clinical presentations were similar to those signs in the large scale outbreak of SADS‐CoV reported by Zhou et al. (2018), except the mortality rate in piglets later increased to 90%.
Based on the rates of infection documented in our work, it revealed that PEDV (78.25%) was still the primary cause of the porcine diarrhoea, which is consistent with previous studies that PEDV has been considered to be the major pathogen responsible for the porcine diarrhoea epidemic in China since 2010 (Ge et al., 2013; Sun et al., 2012; Zhao et al., 2016). Except PEDV, SADS‐CoV had the second high infection rate (43.53%), which exhibits an evidently prevailing tendency in pigs. Meanwhile, of the SADS‐CoV positive samples, 62.2% were co‐infections with one to four other viruses, revealing a high prevalence of co‐infection in the sampled farms. Notably, our results also showed that SADS‐CoV and PEDV were simultaneously present in all co‐infection samples of SADA‐CoV. Considering the fact that PEDV has caused prior outbreaks of the porcine diarrhoea at the pig farm where Zhou et al. (2018) later reported the occurrence of SADS‐CoV, PEDV seems to be able to contribute to the infection of SADS‐CoV in pigs. Thus, SADS‐CoV may be pathogenic as secondary infection following the infection of PEDV, which needs further studies to better understand the pathogenesis of this novel coronavirus.
The phylogenetic relationships of SADS‐CoV sequences were also identified in this study. The results showed that all SADS‐CoV sequences clustered together to form an independent branch and separated from other viral sequences in the genus Alphacoronavirus. Our results also indicated that both the complete genomes, N genes and S genes of all SADS‐CoV strains shared the highest nucleotides identifies with those corresponding sequences of four bat coronavirus HKU2 strains. In this work, The phylogenetic trees of full length genomes and S genes of SADS‐CoV sequences showed that the SADS‐CoV branch clustered with these four HKU2 strains, which is same to previous results (Gong et al., 2017; Pan et al., 2017; Zhou et al., 2018). Besides the genomes and S genes, the tree of N genes in our study revealed the identical result too. So far, a total of eight full‐length genomes of SADS‐CoV have been reported in Guangdong Province of China (Gong et al., 2017; Pan et al., 2017; Zhou et al., 2018; this study). The two new genomes of SADS‐CoV sequences in this work shared 100% nucleotides identities with the sequence MF167434 published by Gong et al. (2017) and our four previously reported sequences (Zhou et al., 2018), and shared 99.8% nucleotides identities with the sequence MF370205 studied by Pan et al. (2017). The results suggest that these eight SADS‐CoV sequences may come from the same origin. Only the phylogenetic tree of S genes in our work showed that sequences of the AlphaCoV were divided into two sublineages, AlphaCoV1 which contained all SADS‐CoV sequences and AlphaCoV2, clustering together with sequences of the BeltaCoV and the DelatCov, respectively. And this result was consistent with the study of Pan et al. (2017). As a newly discovered coronavirus, the availability of SADS‐CoV sequences data is limited which prevents better understandings of the molecular epidemiology of this virus. Meanwhile, being a RNA virus, SADS‐CoV may mutate rapidly and exhibit high genetic differences (Drummond, Pybus, Rambaut, Forsberg, & Rodrigo, 2003; Kühnert, Wu, & Drummond, 2011). So, new more available sequences data are warranted to give a deep insight of viral genetic origin, evolution, and transmission patterns of SADS‐CoV.
In summary, our retrospective study suggests that SADS‐CoV has emerged in pigs in China at least since August 2016. The severe clinical symptoms and the strong transmission in short term highlights the urgency to develop effective measures to control this new discovered virus. Further studies, such as the epidemiology, virology and pathobiology should be performed for better understanding of SADS‐CoV in China.
CONFLICT OF INTEREST
The authors declare no conflict of interests with any organization.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the National Key Research and Development Program of China (No. 2016YFD0501304). We would acknowledge Guangdong Wen's Foodstuffs Group Co., Ltd. China, for providing us with pigs’ tissue samples.
Zhou L, Sun Y, Lan T, et al. Retrospective detection and phylogenetic analysis of swine acute diarrhoea syndrome coronavirus in pigs in southern China. Transbound Emerg Dis. 2019;66:687–695. 10.1111/tbed.13008
Contributor Information
Xiangbin Zhang, Email: zhangxb@scau.edu.cn.
Jingyun Ma, Email: majy2400@scau.edu.cn.
REFERENCES
- Chenna, R. , Sugawara, H. , Koike, T. , Lopez, R. , Gibson, T. J. , Higgins, D. G. , & Thompson, J. D. (2003). Multiple sequence alignmentwith the Clustal series of programs. Nucleic Acids Research, 31, 3497–3500. 10.1093/nar/gkg500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, N. , Fang, L. , Zeng, S. , Sun, Q. , Chen, H. , & Xiao, S. (2015). Porcine Deltacoronavirus in Mainland China. Emerging Infectious Diseases, 21, 2254–2255. 10.3201/eid2112.150283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. , Pybus, O. , Rambaut, A. , Forsberg, R. , & Rodrigo, A. (2003). Measurably evolving populations. Trends in Ecology & Evolution, 18, 481–488. 10.1016/S0169-5347(03)00216-7 [DOI] [Google Scholar]
- Ge, F. F. , Yang, D. Q. , Ju, H. B. , Wang, J. , Liu, J. , Liu, P. H. , & Zhou, J. P. (2013). Epidemiological survey of porcine epidemic diarrhea virus in swine farms in Shanghai, China. Archives of Virology, 158, 2227–2231. 10.1007/s00705-013-1722-7 [DOI] [PubMed] [Google Scholar]
- Gong, L. , Li, J. , Zhou, Q. , Xu, Z. , Chen, L. , Zhang, Y. , … Cao, Y. (2017). A new bat‐HKU2–like coronavirus in swine, China, 2017. Emerging Infectious Diseases, 23, 1607–1609. 10.3201/eid2309.170915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühnert, D. , Wu, C. , & Drummond, A. J. (2011). Phylogenetic and epidemic modeling of rapidly evolving infectious diseases. Infection, Genetics and Evolution, 11, 1825–1841. 10.1016/j.meegid.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. , Saif, L. J. , Marthaler, D. , & Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Research, 226, 20–39. 10.1016/j.virusres.2016.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Zhu, L. , Liao, S. , Xu, Z. , & Zhou, Y. (2015). The porcine microRNA transcriptome response to transmissible gastroenteritis virus infection. PLoS One, 10(3), e0120377 10.1371/journal.pone.0120377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai, K. , Feng, J. , Chen, G. , Li, D. , Zhou, L. , Bai, Y. , … Ma, J. (2017). The detection and phylogenetic analysis of porcine deltacoronavirus from Guangdong Province in Southern China. Transboundary and Emerging Diseases, 65(1), 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y. , Tian, X. , Qin, P. , Wang, B. , Zhao, P. , Yang, Y. , … Huang, Y. (2017). Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Veterinary Microbiology, 211, 15–21. 10.1016/j.vetmic.2017.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, G. W. , Hoang, H. , Schwartz, K. J. , Burrough, E. R. , Sun, D. , Madson, D. , … Yoon, K. J. (2013). Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. Journal of veterinary Diagnostic Investigation, 25, 649–654. 10.1177/1040638713501675 [DOI] [PubMed] [Google Scholar]
- Sun, R. Q. , Cai, R. J. , Chen, Y. Q. , Liang, P. S. , Chen, D. K. , & Song, C. X. (2012). Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerging Infectious Diseases, 18, 161–163. 10.3201/eid1801.111259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, D. , Wang, X. , Wei, S. , Chen, J. , & Feng, L. (2016). Epidemiology and vaccine of porcine epidemic diarrhea virus in China: A mini‐review. Journal of Veterinary Medical Science, 78, 355–363. 10.1292/jvms.15-0446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Nei, M. , & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor‐joining method. Proceedings of the National Academy of Sciences, 101, 11030–11035. 10.1073/pnas.0404206101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley, R. D. , Woods, R. D. , & Cheung, A. K. (1991). Genetic analysis of porcine respiratory coronavirus, an attenuated variant of transmissible gastroenteritis virus. Journal of Virology, 65, 3369–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Huang, Y. , Lau, S. K. , & Yuen, K. Y. (2010). Coronavirus genomics and bioinformatics analysis. Viruses, 2, 1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau, S. K. , Lam, C. S. , Lau, C. C. , Tsang, A. K. , Lau, J. H. , … Yuen, K. Y. (2012). Discovery of seven novel Mammalian and Avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. Journal of Virology, 86, 3995–4008. 10.1128/JVI.06540-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. P. , Yang, Z. , Lin, W. D. , Wang, W. Y. , Yang, J. , Jin, W. J. , & Qin, A. J. (2016). The rate of co‐infection for piglet diarrhea viruses in China and the genetic characterization of porcine epidemic diarrhea virus and porcine kobuvirus. Acta Virologica, 60, 55–61. 10.4149/av_2016_01_55 [DOI] [PubMed] [Google Scholar]
- Zhou, P. , Fan, H. , Lan, T. , Yang, X. , Zhang, W. , Zhu, Y. , … Ma, J. (2018). Fatal Swine Acute Diarrhea Syndrome caused by an HKU2‐related Coronavirus of Bat Origin. Nature, 556, 255–258. 10.1038/s41586-018-0010-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials