

Abstract
Canine parvovirus type 2 (CPV‐2) emerged suddenly in the late 1970s as pathogen of dogs, causing a severe and often fatal gastroenteric disease. The original CPV‐2 was replaced by three antigenic variants, CPV‐2a, CPV‐2b and CPV‐2c, which to date have gained a worldwide distribution with different relative proportions. All previous studies conducted in Africa were based on partial VP2 gene sequences. The aim of this study was to provide a genome analysis to characterize the CPV strains collected in Nigeria, Africa. Rectal swab samples (n = 320) were collected in 2018 and tested by means of an immunochromatographic assay. Among the 144 positive samples, 59 were selected for further analyses using different molecular assays. The results revealed a high prevalence of CPV‐2c (91.5%) compared to the CPV‐2a variant (8.5%). The VP2 gene sequences showed a divergence from the strains analysed in 2010 in Nigeria and a closer connection with CPV strains of Asian origin. The non‐structural gene analysis evidenced amino acid changes never previously reported. The molecular analysis based on genomic sequences evidenced a geographical pattern of distribution of the analysed strains, suggesting a potential common evolutionary origin with CPV of Asian origin. This study represents the first CPV molecular characterization including all the encoding gene sequences conducted in the African continent and contributes to define the current geographical spread of the CPV variants worldwide.
Keywords: Canine parvovirus, genome analysis, molecular characterization, Nigeria, NS1, protoparvovirus
1. INTRODUCTION
Canine parvovirus (CPV) is a small (about 25 nm in diameter), non‐enveloped DNA virus, which emerged suddenly in the late 1970s as pathogen of dogs, causing severe and often fatal epizootics of gastroenteritis worldwide (Decaro & Buonavoglia, 2012; Decaro, Desario, et al., 2007). CPV was recently included, together with feline panleukopenia virus (FPLV), mink enteritis virus (MEV) and raccoon parvovirus (RaPV), in the unique species Carnivore protoparvovirus 1, within the Protoparvovirus genus (family Parvoviridae, subfamily Parvovirinae) (Cotmore et al., 2019, 2014). Its genome consists of a 5200‐nucleotide (nt) DNA molecule containing two large open reading frames (ORFs) encoding for two non‐structural (NS1 and NS2) and two structural (VP1 and VP2) proteins through alternative splicing of the same mRNAs (Reed at al., 1988).
Soon after its emergence, the original CPV‐2 was replaced by two antigenic variants, CPV‐2a and CPV‐2b (Parrish et al., 1991; Parrish, O’Connell, Evermann, & Carmichael, 1985), and in 2000, a third antigenic variant (CPV‐2c) was described (Decaro & Buonavoglia, 2012). To date, all three CPV variants are worldwide distributed, with different relative proportions according to the year and country of collection (Amrani et al., 2016; Miranda & Thompson, 2016; Woolford, Crocker, Bobrowski, Baker, & Hemmatzadeh, 2017).
In the African continent, CPV has been described in South Africa and Namibia (Dogonyaro, Bosman, Sibeko, Venter, & Vuuren, 2013; Steinel, Venter, Van Vuuren, Parrish, & Truyen, 1998), Zambia (Kapiya et al., 2019), Mozambique (Figuiredo et al., 2017), Ghana (Folitse et al., 2017), Morocco (Amrani et al., 2016), Cape Verde (Costanheira et al., 2014), Nigeria (Chollom et al., 2013) and Tunisia (Touhiri et al., 2009). In Nigeria, only recently the molecular analyses based on the partial VP2 gene sequence of CPV strains described the circulating CPV variants (Apaa, Daly, & Tarlinton, 2016; Dogonyaro et al., 2013; Fagbohun & Omobowale, 2018). Unfortunately, all previous studies conducted in Africa lack a comprehensive sequence analysis including all the viral genome, thus preventing a more in‐depth knowledge on the origin and evolution of the circulating CPV strains. The aim of this study was to characterize the CPV strains recently collected in Nigeria, analysing the nearly complete CPV genome sequence and comparing the obtained sequences with worldwide related sequences available in GenBank.
2. MATERIALS AND METHODS
Samples were collected during the period from January to June 2018 from 320 dogs of different breeds with clinical signs of gastroenteritis and suspected of CPV infection or apparently healthy (tested positive dogs showed gastrointestinal clinical signs in the days after the sampling). Rectal swabs were collected in Nigeria from four cities placed in three states, named Plateau State (Jos; geographical coordinates: 9°55′3.045″N, 8°53′52.584″E; altitude: 1,184 m a.s.l), Benue State (Makurdi; 7°43′52.548″N, 8°32′18.33″E; 90 m a.s.l), Nasarawa State (Lafia: 11°21′23.178″N, 9°23′8.202″E; 455 m a.s.l) and the Federal Capital Territory (Abuja; 9°3′51.59″N, 7°29′21.47″E; 482 m a.s.l). Samples were submitted from eight private veterinary clinics, two from each state, and from kennels/breeders in the same areas. Rectal swabs were tested using an in‐clinic assay for detection of CPV antigen (SensPERT® Canine Parvovirus Test Kit, VetAll Laboratories), according to the manufacturer's instructions. Among the positive samples, 59 rectal swabs were selected and submitted for molecular analyses, where they were stored at −80°C until use. Details are reported in Table S1.
Swab homogenates were obtained as previously described (Purpari et al., 2018). Viral DNA was extracted from 200 µl of swab homogenate using the DNeasy Blood & Tissue Kit (Qiagen S.p.A.), according to the manufacturer's instructions. The presence of CPV DNA was confirmed using a primer pair (Table S2) in a PCR protocol amplifying a 700‐bp fragment of the VP2 gene (Touihri et al., 2009), using the commercial kit GoTaq® G2 DNA Polymerase (Promega Italia s.r.l.), as previously described (Mira, Dowgier, et al., 2018). Amplicons were checked after electrophoresis on a 3% agarose gel supplemented with ethidium bromide.
Ten microlitres of each amplicon was digested with 5 units (U) of restriction endonuclease MboII (New England BioLabs® Inc.) in a 50‐µl reaction mix consisting of 5 µl of NEBuffer and 34 µl of nuclease‐free water, under the following reaction conditions: incubation at 37°C for 2 hr and inactivation at 65°C for 20 min. The profile was determined by electrophoresis on a 3% agarose gel supplemented with ethidium bromide.
Specimens from each city of collection (n = 4 from Makurdi; n = 11 from Jos; n = 8 from Abuja and n = 5 from Lafia), representing dogs with different age, vaccinal and clinical status, were selected to determine the VP2 gene sequence (Table 1). The nearly complete VP2 gene sequence was determined using a primer pair (Table S2), which allows for amplification of a 1,745‐bp fragment (Battilani et al., 2001; Mokizuki et al., 1996), using the commercial kit GoTaq® G2 DNA Polymerase (Promega Italia s.r.l.), as previously described (Purpari et al., 2018) with minor modifications (thermal conditions: 1 min for the annealing step). After electrophoresis on agarose gel supplemented with ethidium bromide, amplicons were purified with Illustra™ GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare Life Sciences) and submitted to BMR Genomics srl for direct Sanger sequencing with 6.4 pmol of the reverse primer used for amplification and of two additional internal primers (Table S2). Sequences were assembled and analysed using BioEdit software (Hall, 1999).
Table 1.
Identification code, origin, age, vaccination and clinical status, strain and sequence information of the dogs selected for molecular investigation
| ID code | City | Age | Vaccination status | Remarks | Strain | Sequence | Acc. no. |
|---|---|---|---|---|---|---|---|
| YV 8 | Makurdi | 12 weeks | Vaccinated | Clinical | CPV‐2a | ORF1/2 | MK895484 |
| UV 1 | Abuja | 6 months | Not vaccinated | Subclinical | CPV‐2a | ORF1/2 | MK895483 |
| UV 6 | Abuja | 10 weeks | Not vaccinated | Subclinical | CPV‐2a | ORF1/2 | MK895485 |
| RKB 2 | Abuja | 5 weeks | Not vaccinated | Clinical | CPV‐2a | VP2 | ‐ |
| LTG 3 | Lafia | 12 weeks | Not vaccinated | Apparently healthy | CPV‐2a | VP2 | ‐ |
| PB 3 | Makurdi | 6 weeks | Vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| YV 2 | Makurdi | 6 weeks | Vaccinated | Clinical | CPV‐2c | ORF1/2 | MK895486 |
| JOE 2 | Makurdi | 7 weeks | Vaccinated | Apparently healthy | CPV‐2c | ORF1/2 | MK895488 |
| NV 19 | Jos | 5 months | Vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| NC | Jos | 10 weeks | Vaccinated | Clinical | CPV‐2c | ORF1/2 | MK895489 |
| YA 3 | Jos | 6 weeks | Vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| NV 10 | Jos | 10 weeks | Vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| NV 4 | Jos | 10 weeks | Vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| CVC 14 | Jos | 6 months | Not vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| PSV 21 | Jos | 11 weeks | Not vaccinated | Clinical | CPV‐2c | ORF1/2 | MK895490 |
| CVC 1 | Jos | 2 years | Not vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| CVC 21 | Jos | 12 weeks | Not vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| PSV 15 | Jos | 8 weeks | Not vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| PSV 5 | Jos | 8 weeks | Not vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| VC 5 | Abuja | 8 weeks | Vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| EV 8 | Abuja | 5 months | Vaccinated | Clinical | CPV‐2c | ORF1/2 | MK895487 |
| MV 1 | Abuja | 8 weeks | Vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| EV 3 | Abuja | 7 weeks | Vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| EV 5 | Abuja | 9 weeks | Not vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| RF 3 | Lafia | 7 weeks | Vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| N 1 | Lafia | 9 weeks | Vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
| LO 11 | Lafia | 2 years | Vaccinated | Apparently healthy | CPV‐2c | VP2 | ‐ |
| V 4 | Lafia | 18 weeks | Not vaccinated | Clinical | CPV‐2c | VP2 | ‐ |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
By excluding the VP2 gene sequences with complete nt identities, 8 CPV DNAs from different cities of collection were further selected for a further sequence analysis by amplifying a long genome sequence encompassing both major ORFs, without the 5′ and 3′ flanking sequences (Table 1). Sequence analyses were carried out using primer pairs described by Pérez et al. (2014), using the commercial kit GoTaq® G2 DNA Polymerase (Promega Italia s.r.l.), as previously described (Mira, Dowgier, et al., 2018) with minor modifications (Mira, Canuti, et al., 2019). After electrophoresis on agarose gel, amplicons were purified and submitted for direct Sanger sequencing with a set of primers, as previously described (Mira, Purpari, Lorusso, et al., 2018). Sequences were assembled, analysed and submitted to nBLAST program (Zhang, Schwartz, Wagner, & Miller, 2000) to search related sequences into public domain databases. These sequence data were submitted to the DDBJ/EMBL/GenBank databases under accession numbers MK895483‐90.
Phylogenetic analyses based on the full‐length NS1 and VP2 gene sequences and on the whole genome encompassing the two ORFs were conducted using the best‐fit model of nt substitution with MEGA version X software (Kumar, Stecher, Li, Knyaz, & Tamura, 2018), inferred with the maximum‐likelihood method based on the Tamura 3‐parameter (T92) and Hasegawa–Kishino–Yano (HKY) models, with discrete gamma distribution (five rate categories) (G) and invariant sites (I) (bootstrap 1,000 replicates), the best‐fitting models after the model test analyses (VP2: T92 + G; NS1: HKY + G; whole genome: HKY + G+I).
RNA was extracted from samples using the QIAamp Viral RNA Mini Kit (Qiagen S.p.A.), according to the manufacturer's instructions. Extracted DNA/RNA was amplified using a set of gel‐based or real‐time (RT‐)PCR assays useful for the detection of canine distemper virus (CDV) (Elia et al., 2006), canine adenovirus (CAdV) types 1 and 2 (Dowgier et al., 2016), canine coronavirus (CCoV) (Decaro et al., 2004) and canine rotavirus (CRoV) (Freeman, Kerin, Hull, McCaustland, & Gentsch, 2008).
3. RESULTS
Among the collected 320 samples, 144 rectal swabs tested positive for CPV by in‐clinic assay (45%). The prevalence of the positive samples for each city of collection is reported in Table 2. The presence of CPV DNA was confirmed in the selected samples (n = 59) using the conventional PCR assay. The same samples tested negative for CDV, CAdVs, CCoV and CRoV by gel‐based or real‐time (RT)‐PCR assays. Based on the RFLP analysis, 54 CPV‐positive samples (91.5%) were typed as CPV‐2c (yielding 3 fragments of 56, 275 and 369 bp, respectively) and 5 (8.5%) as a different CPV type (CPV‐2, CPV‐2a or CPV‐2b) (yielding 2 fragments of 331 and 369 bp, respectively).
Table 2.
Number and prevalence of CPV‐positive samples
| State | City | Total samples | Positive samples | Negative samples | Prevalence (%) |
|---|---|---|---|---|---|
| Plateau | Jos | 80 | 54 | 26 | 67.50 |
| Benue | Makurdi | 80 | 35 | 45 | 43.75 |
| Nasarawa | Lafia | 80 | 23 | 57 | 28.75 |
| Federal Capital Territory | Abuja | 80 | 32 | 48 | 40.00 |
| Total | 320 | 144 | 176 | 45 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The RFLP untyped CPV strains (n = 5) and the CPV‐2c strains (n = 23) from different geographical areas were subjected to sequence analyses to screen the nearly complete VP2 gene sequence. According to aa residues 297 and 426, the sequence analysis confirmed the RFPL assay results for samples typed as CPV‐2c (426‐Glu) and typed the other five samples as CPV‐2a (297‐Ala, 426‐Asn). The VP2 sequences showed 99.77%–100% and 99.98%–100% nt identities among the CPV‐2a and CPV‐2c strains, respectively. Four synonymous changes were observed in the VP2 sequence of the CPV‐2a strains (nts: a639g, c969t, a1074t, a1275g). One non‐synonymous (nt: c14g; aa: A5G) and four synonymous (nts: a318g, t1014c, g1083t, t1629c) changes were observed in the CPV‐2c strains. VP2 synonymous changes are reported in Table S3.
Compared to CPV sequences previously analysed in Nigeria, the VP2 sequences of the analysed CPV‐2a strains showed 99.60%–99.25% (calculated on 1,750 nts), 99.87% (on 787 nts) and 99.34%–98.72% (on 459 and 470 nts) nt identities with strains collected in 2010, 2014 and 2017 (Apaa et al., 2016; Dogonyaro et al., 2013; Fagbohun & Omobowale, 2018), respectively. Compared to the CPV‐2a strains collected in 2010, three non‐synonymous changes (F267Y, Y324I and T440A) were observed (Table 3). Compared to the CPV‐2c sequence previously analysed in Nigeria (acc. no. MH795059), the VP2 sequences of CPV‐2c strains showed 99.36% of nt identity (on 466 nts).
Table 3.
VP2 non‐synonymous changes of CPV strains analysed in this study and of CPV strains from Africa previously analysed
| Strain | Acc. No. | Type | Country | Year | VP2 amino acids | References | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 265 | 267 | 297 | 324 | 370 | 401 | 413 | 424 | 426 | 440 | ||||||
| IZSSI_PA1464/19_idUV1 | MK895483 | CPV‐2a | Nigeria | 2018 | T | Y | A | I | Q | I | D | V | N | A | This study |
| IZSSI_PA1464/19_idYV8 | MK895484 | CPV‐2a | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| IZSSI_PA1464/19_idUV6 | MK895485 | CPV‐2a | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| IZSSI_PA1464/19_idYV2 | MK895486 | CPV‐2c | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | R | ‐ | ‐ | ‐ | E | T | |
| IZSSI_PA1464/19_idEV8 | MK895487 | CPV‐2c | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | R | ‐ | ‐ | ‐ | E | T | |
| IZSSI_PA1464/19_idJOE2 | MK895488 | CPV‐2c | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | R | ‐ | ‐ | ‐ | E | T | |
| IZSSI_PA1464/19_idNC | MK895489 | CPV‐2c | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | R | ‐ | ‐ | ‐ | E | T | |
| IZSSI_PA1464/19_idPSV21 | MK895490 | CPV‐2c | Nigeria | 2018 | ‐ | ‐ | ‐ | ‐ | R | ‐ | ‐ | ‐ | E | T | |
| 3‐10NGR2010 | HQ602990 | CPV‐2a | Nigeria | 2010 | ‐ | F | ‐ | Y | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | Dogonyaro et al., 2013 |
| 19‐10NGR2010 | HQ602992 | CPV‐2a | Nigeria | 2010 | ‐ | F | ‐ | Y | ‐ | ‐ | ‐ | ‐ | ‐ | T | |
| 23‐10NGR2010 | HQ602994 | CPV‐2a | Nigeria | 2010 | ‐ | F | ‐ | Y | ‐ | ‐ | ‐ | ‐ | ‐ | T | |
| 15‐10NGR2010 | HQ602995 | CPV‐2a | Nigeria | 2010 | ‐ | F | ‐ | Y | ‐ | ‐ | ‐ | ‐ | ‐ | T | |
| NG21 | KX192407 | CPV‐2a | Nigeria | 2014 | ‐ | ‐ | ‐ | ‐ | ‐ | Apaa et al., 2016 | |||||
| NG32 | KX192416 | CPV‐2a | Nigeria | 2014 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ||||
| NG35 | KX192418 | CPV‐2a | Nigeria | 2014 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ||||
| NGA1‐2° | MH795055 | CPV‐2a | Nigeria | 2017 | ‐ | ‐ | ‐ | ‐ | ‐ | Fagbohun & Omobowale, 2018 | |||||
| NGA7‐2° | MH795061 | CPV‐a | Nigeria | 2017 | ‐ | ‐ | ‐ | ‐ | ‐ | ||||||
| NGA2‐2b | MH795056 | CPV‐2b | Nigeria | 2017 | K | N | ‐ | D | T | ||||||
| NGA5‐2c | MH795059 | CPV‐2c | Nigeria | 2017 | ‐ | ‐ | ‐ | ‐ | E | T | |||||
| CPV‐Africa 3 | AJ007498 | CPV‐2a | South Africa | 1996 | ‐ | F | S | Y | ‐ | ‐ | ‐ | ‐ | ‐ | T | Steinel et al., 1998 |
| CPV‐Africa 9 | AJ007500 | CPV‐2b | South Africa | 1996 | ‐ | F | S | Y | ‐ | ‐ | ‐ | ‐ | D | T | |
| 63‐10SA 2010 | HQ602978 | CPV‐2a | South Africa | 2010 | K | F | N | Y | ‐ | ‐ | ‐ | A | ‐ | ‐ |
Dogonyaro et al., 2013 |
| 100‐10SA 2010 | HQ602977 | CPV‐2a | South Africa | 2010 | ‐ | F | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | T | |
| 72‐10SA 2010 | HQ602976 | CPV‐2b | South Africa | 2010 | ‐ | F | N | Y | ‐ | ‐ | ‐ | ‐ | D | T | |
| MZ_CPV36/2010 | KU523909 | CPV‐2a | Mozambique | 2010 | ‐ | A | ‐ | ‐ | Figueiredo et al., 2017 | ||||||
| MZ_CPV38/2010 | KU523911 | CPV‐2b | Mozambique | 2010 | ‐ | ‐ | D | T | |||||||
| CPV‐2c‐Lus2 | LC409276 | CPV‐2a | Zambia | 2017 | ‐ | ‐ | ‐ | ‐ | Kapiya et al., 2019 | ||||||
| CPV‐2c‐Lus3 | LC409277 | CPV‐2b | Zambia | 2017 | ‐ | ‐ | D | T | |||||||
| CPV‐2c‐Lus11 | LC409284 | CPV‐2c | Zambia | 2017 | ‐ | ‐ | E | T | |||||||
Dark grey cells indicate that fragment of encoding sequences is not available. ‘‐’ same amino acid as in the first row.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Compared to CPV sequences previously analysed in other African countries, the VP2 sequences of CPV‐2a strains showed 99.81% (on 529 nts), 99.61% (on 515 nts) and 99.85%–99.05% (on 1,745 and 1,583 nts) sequence identities with strains collected in 2017 in Zambia (GenBank accession number LC409284), 2010 in Mozambique (acc. no. KU523911), 2010 and 1996 in South Africa (acc. no. HQ602976‐8 and AJ007500), respectively. Compared to the CPV‐2a strains previously collected in other African countries, seven non‐synonymous changes (K265T, F267Y, S/N297A, Y324I, A424V and T440A) were observed (Table 3). Compared to the CPV‐2c sequence previously analysed in other African countries (Zambia, 2017; acc. no. LC409284; Kapiya et al., 2019), the VP2 sequences of CPV‐2c strains showed 100% (on 529 nts).
By the further sequence amplifications, the nearly complete genome sequences including both ORFs (4,269 nt) of 8 CPV strains were obtained. The complete genome sequences of CPV‐2a strains showed 99.27%–99.34% nt identities with CPV strains collected in China in 2017 (acc. no. MH476580, MH476590, MH476591) and in Uruguay in 2011 (acc. no. KM457139). The complete genome sequences of CPV‐2c strains showed 99.88%–99.92% nt identities with CPV strains of Asian origin (China, 2017, acc. no. MG013488; Thailand, 2016, acc. no. MH711902; Italy, 2017, acc. no. MF510157).
Sequence analysis revealed aa changes previously described mainly in CPV‐2a/2c strains of Asian origin (NS1: 60V, 544F, 545V, 630P; NS2: 60V, 151N, 152V; VP2: 5A/G, 267Y, 297A, 324I, 370R) (Tables 4 and 5). Additional changes at aa residues 164L, 351K, 584K, 586V, 596A and 517I of the CPV‐2a and CPV‐2c NS1 protein, respectively, were also found (Table 5). While changes 351K and 596A have been detected in CPV sequences mainly from South and North Americas, changes 164L, 584K, 586V and 517I have not been previously reported.
Table 4.
VP2 non‐synonymous changes of analysed CPV strains described in this study
| CPV variant | Strain | Country (year) | Acc. no. | VP2 amino acids | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 87 | 101 | 267 | 297 | 300 | 305 | 324 | 370 | 375 | 426 | 440 | ||||
| CPV‐2a | CPV‐b | USA (1978) | M38245 | A | M | I | F | S | A | D | Y | Q | N | N | T |
| CPV‐2a | CPV‐15 | USA (1984) | M24003 | ‐ | L | T | ‐ | ‐ | G | Y | ‐ | ‐ | D | ‐ | ‐ |
| CPV‐2ba | CPV‐39 | USA (1984) | M74849 | ‐ | L | T | ‐ | ‐ | G | Y | ‐ | ‐ | D | D | ‐ |
| CPV‐2ca | 288‐01 | Italy (2001) | MF177239 | ‐ | L | T | ‐ | A | G | Y | ‐ | ‐ | D | E | ‐ |
| CPV‐2aa | TN/CPV2a/2018 | India (2018) | MH545963 | ‐ | L | T | Y | A | G | Y | I | ‐ | D | ‐ | A |
| CPV‐2ca | Canine/China/23/2017 | China (2017) | MH476592 | G | L | T | Y | A | G | Y | I | R | D | E | ‐ |
| CPV‐2a | IZSSI_PA1464/19_idUV1 | Nigeria (2018) | MK895483 | ‐ | L | T | Y | A | G | Y | I | ‐ | D | ‐ | A |
| CPV‐2a | IZSSI_PA1464/19_idYV8 | Nigeria (2018) | MK895484 | ‐ | L | T | Y | A | G | Y | I | ‐ | D | ‐ | A |
| CPV‐2a | IZSSI_PA1464/19_idUV6 | Nigeria (2018) | MK895485 | ‐ | L | T | Y | A | G | Y | I | ‐ | D | ‐ | A |
| CPV‐2c | IZSSI_PA1464/19_idYV2 | Nigeria (2018) | MK895486 | ‐ | L | T | Y | A | G | Y | I | R | D | E | ‐ |
| CPV‐2c | IZSSI_PA1464/19_idEV8 | Nigeria (2018) | MK895487 | ‐ | L | T | Y | A | G | Y | I | R | D | E | ‐ |
| CPV‐2c | IZSSI_PA1464/19_idJOE2 | Nigeria (2018) | MK895488 | G | L | T | Y | A | G | Y | I | R | D | E | ‐ |
| CPV‐2c | IZSSI_PA1464/19_idNC | Nigeria (2018) | MK895489 | G | L | T | Y | A | G | Y | I | R | D | E | ‐ |
| CPV‐2c | IZSSI_PA1464/19_idPSV21 | Nigeria (2018) | MK895490 | ‐ | L | T | Y | A | G | Y | I | R | D | E | ‐ |
Reference strains retrieved from GenBank. ‘‐’ same amino acid as in the first row.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 5.
NS1 and NS2 non‐synonymous changes of analysed CPV strains described in this study
| CPV variant | Strain | Country (year) | Acc. no. | NS1 amino acids | NS2 amino acids | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 60 | 164 | 351 | 517 | 544 | 545 | 584 | 586 | 596 | 630 | 60 | 93 | 94 | 151 | 152 | ||||
| CPV‐2a | CPV‐b | USA (1978) | M38245 | I | S | N | V | Y | E | T | A | V | L | I | D | T | D | M |
| CPV‐2aa | CPV‐15 | USA (1984) | AY787926 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| CPV‐2ba | CPV‐39 | USA (1984) | AY787930 | ‐ | ‐ | ‐ | ‐ | F | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | V |
| CPV‐2ca | 288‐01 | Italy (2001) | MF177239 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | A | N | V |
| CPV‐2aa | TN/CPV2a/2018 | India (2018) | MH545963 | ‐ | ‐ | ‐ | ‐ | F | V | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | N | V |
| CPV‐2ca | Canine/China/23/2017 | China (2017) | MH476592 | V | ‐ | ‐ | ‐ | F | V | ‐ | ‐ | ‐ | P | V | ‐ | ‐ | N | V |
| CPV‐2a | UV1 | Nigeria (2018) | MK895483 | ‐ | ‐ | K | ‐ | F | ‐ | K | V | A | ‐ | ‐ | E | A | N | V |
| CPV‐2a | YV8 | Nigeria (2018) | MK895484 | ‐ | ‐ | K | ‐ | F | ‐ | K | V | A | ‐ | ‐ | E | A | N | V |
| CPV‐2a | UV6 | Nigeria (2018) | MK895485 | ‐ | L | K | ‐ | F | ‐ | K | V | A | ‐ | ‐ | E | A | N | V |
| CPV‐2c | YV2 | Nigeria (2018) | MK895486 | V | ‐ | ‐ | ‐ | F | V | ‐ | ‐ | ‐ | P | V | ‐ | ‐ | N | V |
| CPV‐2c | EV8 | Nigeria (2018) | MK895487 | V | ‐ | ‐ | ‐ | F | V | ‐ | ‐ | ‐ | P | V | ‐ | ‐ | N | V |
| CPV‐2c | JOE2 | Nigeria (2018) | MK895488 | V | ‐ | ‐ | I | F | V | ‐ | ‐ | ‐ | P | V | ‐ | ‐ | N | V |
| CPV‐2c | NC | Nigeria (2018) | MK895489 | V | ‐ | ‐ | ‐ | F | V | ‐ | ‐ | ‐ | P | V | ‐ | ‐ | N | V |
| CPV‐2c | PSV21 | Nigeria (2018) | MK895490 | V | ‐ | ‐ | ‐ | F | V | ‐ | ‐ | ‐ | P | V | ‐ | ‐ | N | V |
Reference strains retrieved from GenBank. ‘‐’ same amino acid as in the first row.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Amino acid change I60V in NS1 also lies at the same residue in the NS2‐encoding sequence. Additional four amino acid changes in the NS2‐encoding sequences were observed: D93E, T94A, D151N and M152V (Table 5). These changes resulted in silent mutations in the corresponding encoded NS1 protein.
The aa change K116R in the VP1 gene sequence is added to the aa changes of the VP2 gene sequence lying in the corresponding encoded VP1 protein (A148G, M230L, I244T, F410Y, S440A, A443G, D448Y, Y467I, Q513R, N518D, T583A).
Phylogenetic analysis inferred from VP2 sequences showed that the analysed strains are more closely related to the CPV strains of Asian origin rather than to strains collected in Europe or in North/South America, clustering in separate branches within the clades (Figure 1). Phylogenetic tree inferred from NS1 sequences (Figure 2) indicated that the CPV‐2a strains cluster separately within the clade of CPV strains of Asian origin. Although with a low bootstrap support, the almost complete genome sequence phylogeny showed that the analysed strains clustered according to the geographical origin and the year of collection rather than to the CPV variant (Figure 3).
Figure 1.
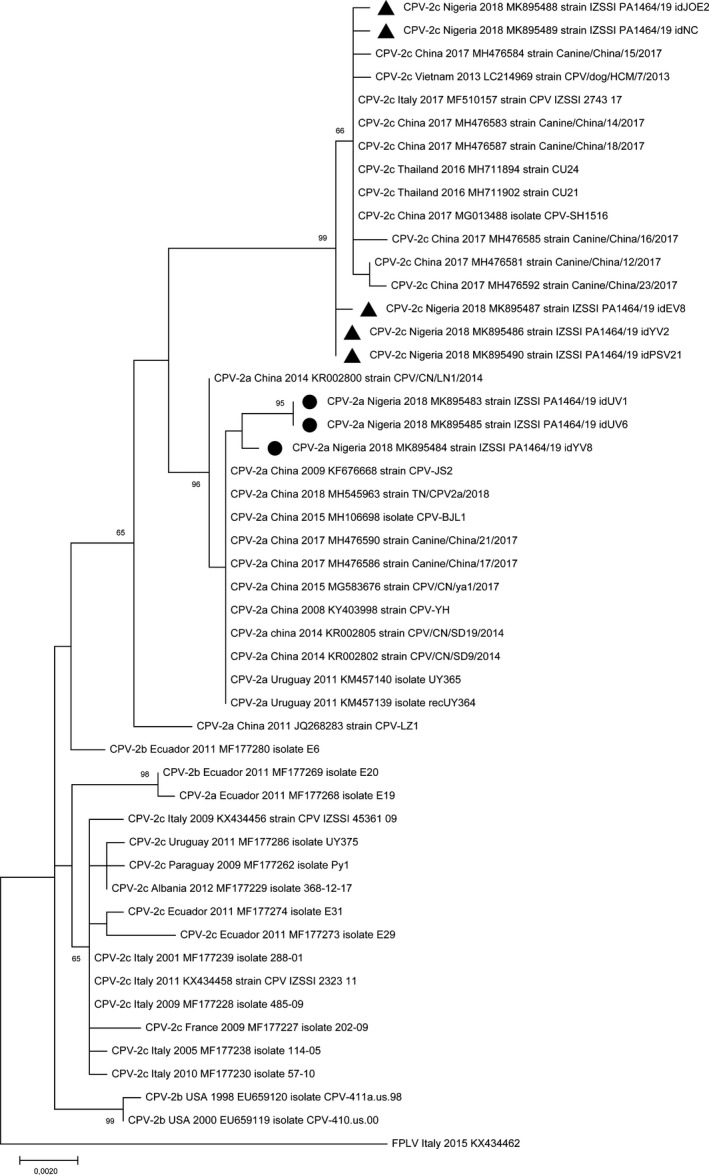
Maximum‐likelihood tree based on 49 full‐length VP2 gene sequences of canine parvovirus type 2 strains (bootstrap 1,000 replicates; bootstrap values greater than 65 are shown). Black dot (●) and black triangle (▲) markings indicate CPV‐2a and CPV‐2c strains from Nigeria, respectively, analysed in this study. Each sequence is indicated with virus type (FPLV, feline panleukopenia virus; CPV, canine parvovirus) or variant (CPV‐2, CPV‐2a, CPV‐2b, CPV‐2c), country and year of collection, accession number and strain/isolate name. The scale bar indicates the estimated numbers of nucleotide substitutions per site
Figure 2.
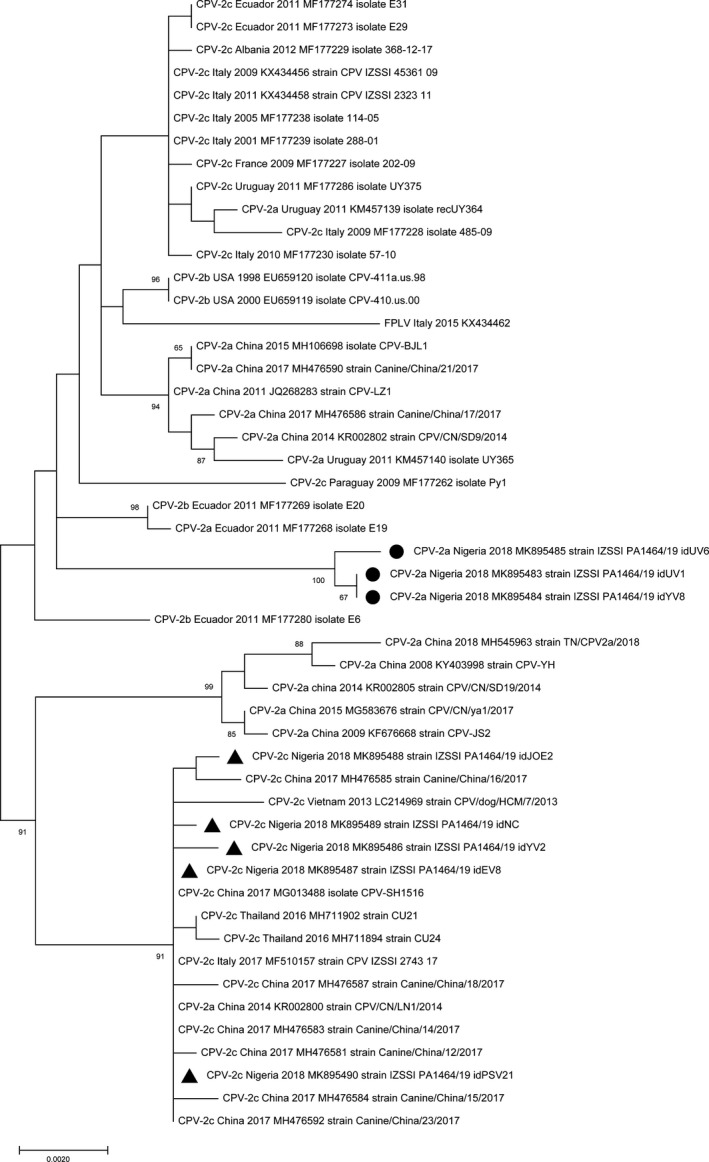
Maximum‐likelihood tree based on 49 full‐length NS1 gene sequences of canine parvovirus type 2 strains (bootstrap 1,000 replicates; bootstrap values greater than 65 are shown). Black dot (●) and black triangle (▲) markings indicate CPV‐2a and CPV‐2c strains from Nigeria, respectively, analysed in this study. Each sequence is indicated with virus type (FPLV: feline panleukopenia virus; CPV: canine parvovirus) or variant (CPV‐2, CPV‐2a, CPV‐2b, CPV‐2c), country and year of collection, accession number and strain/isolate name. The scale bar indicates the estimated numbers of nucleotide substitutions per site
Figure 3.
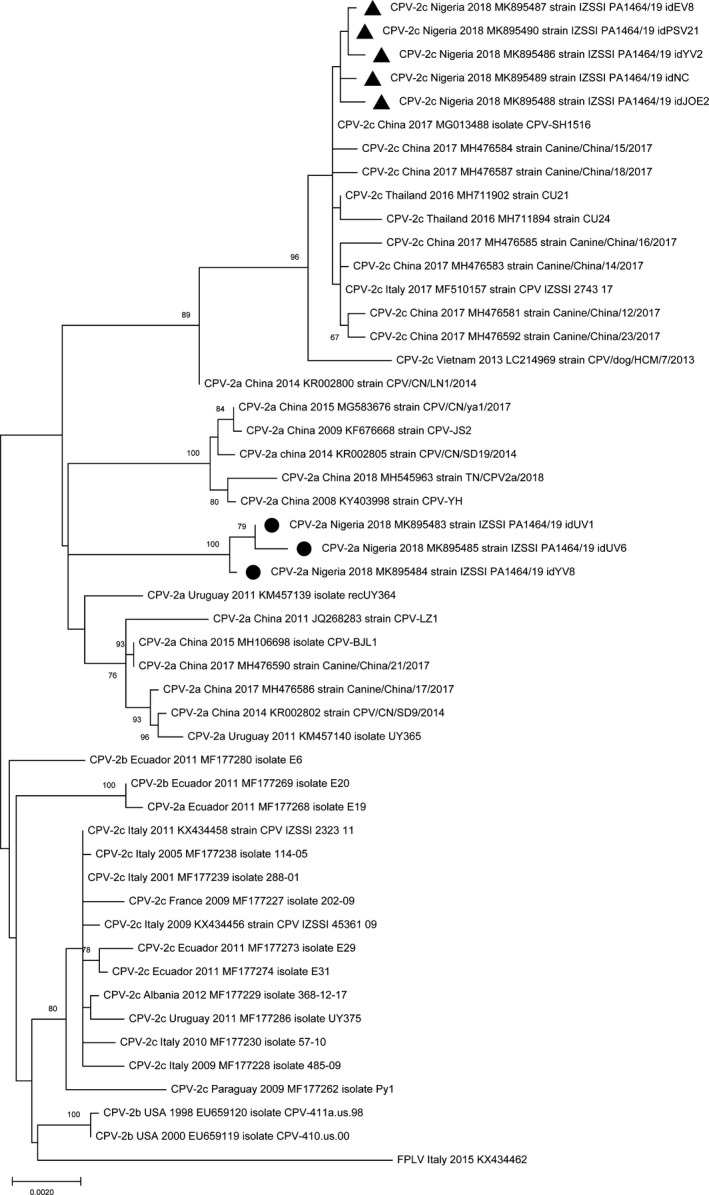
Maximum‐likelihood tree based on 49 nearly full‐length genome sequences of canine parvovirus type 2 strains (bootstrap 1,000 replicates; bootstrap values greater than 65 are shown). Black dot (●) and black triangle (▲) markings indicate CPV‐2a and CPV‐2c strains from Nigeria, respectively, analysed in this study. Each sequence is indicated with virus type (FPLV, feline panleukopenia virus; CPV, canine parvovirus) or variant (CPV‐2, CPV‐2a, CPV‐2b, CPV‐2c), country and year of collection, accession number and strain/isolate name. The scale bar indicates the estimated numbers of nucleotide substitutions per site
4. DISCUSSION
Canine parvovirus has still been playing a main role in inducing severe and often fatal gastroenteritis in young or non‐immunized dogs. During years, CPV spread and evolution have been well documented in North and South America, Europe and Asia (Miranda & Thompson, 2016; Zhou, Zeng, Zhang, & Li, 2017). More recently, data about its spread were also obtained from Australia and Africa (Amrani et al., 2016; Castanheira et al., 2014; Chollom et al., 2013; Dogonyaro et al., 2013; Figuiredo et al., 2017; Folitse et al., 2017; Kapiya et al., 2019; Touhiri et al., 2009; Woolford et al., 2017). Most of these studies were based on the partial or complete VP2 gene sequence, due to the involvement of the VP2 capsid protein in host switch and to its fast evolutionary rate (Hueffer et al., 2003; Nelson, Palermo, Hafenstein, & Parrish, 2007; Shackelton, Parrish, Truyen, & Holmes, 2005), with limited information on other CPV encoding gene sequences. With the aim to investigate the epidemiology and evolution of CPV in Nigeria, the VP2 gene or nearly complete genome sequences including both ORFs of CPV representative strains were analysed and compared to the related strains retrieved from the GenBank database.
Early studies on detection of CPV in Nigeria were based on serological or PCR assays (Babalola, Ijaopo, & Okonko, 2016; Chollom et al., 2013), and only recently, the analysis of the partial VP2 gene sequence was included in the studies (Apaa et al., 2016; Dogonyaro et al., 2013; Fagbohun & Omobowale, 2018). These analyses conducted in Nigeria evidenced firstly the circulation of CPV‐2a strains (Apaa et al., 2016; Dogonyaro et al., 2013) and only recently of CPV‐2b/2c types (Fagbohun & Omobowale, 2018). In the present study, based on the analysis of 59 CPV‐positive samples, 5 CPV‐2a and 54 CPV‐2c strains have been found. These data showed a high prevalence of CPV‐2c (91.5%) compared to the CPV‐2a variant (8.5%), without any evidence of CPV‐2b. Moreover, the divergence in the VP2 gene sequence of the CPV strains compared to those previously circulating in Nigeria suggests the potential circulation or introduction of different strains during years. Indeed, despite the limits of the length of available sequences, the CPV strains analysed in this study are prone to be related to a different origin than the previous ones. All CPV strains collected in 2010 (Dogonyaro et al., 2013) displayed divergent amino acids at specific VP2 residues (267, 324) with a closer phylogenetic connection with the CPV‐2a strains previously collected in other continents such as North/South America and Europe. Differently, since 2014 the analysed circulating CPV strains showed a closer connection with CPV strains of Asian origin. Despite some previously analysed CPV sequences lack of informative VP2 aa residues (5, 267, 324, 370), the comparison with our sequences suggested that both CPV‐2a and CPV‐2c strains collected in Nigeria after 2014 have a different phylogenetic origin with respect to previous analyses (Apaa et al., 2016; Fagbohun & Omobowale, 2018), suggesting a most recent introduction and spreading.
The analysis included both ORFs and allowed to gain additional information. Indeed, in this study the whole VP gene sequences were analysed, showing the VP1 and VP2 aa changes K116R and A5G, respectively. The VP1 aa change was due to a nt change in the second base of the codon (VP1 a347g) and was common to CPV strains of different origin, irrespective of the CPV antigenic variant. Moreover, the VP2 aa change was due to a nt change in the second base of the codon (VP2 c14g), but this change was common only to CPV strains of Asian origin (Mira, Purpari, et al., 2019; Mira, Purpari, Lorusso, et al., 2018; Wang et al., 2016; Zhuang et al., 2019). The potential biological relevance of these changes has not been described yet and needs to be assessed in further studies.
As most recent Asian CPVs, the Nigerian strains displayed other three aa substitutions in the VP2 sequence (F267Y, Y324I and Q370R). While change at aa residue 324 is predominant in all three CPV variants in Asia (Geng et al., 2015; Yi, Tong, Cheng, Song, & Cheng, 2016; Zhao et al., 2017; Zhou et al., 2017), the other changes have been less frequently observed, mainly in China since 2013, and change Q370R has been detected only in CPV‐2c strains (Geng et al., 2015; Guo et al., 2013; Mira, Purpari, et al., 2019; Mira, Purpari, Lorusso, et al., 2018; Wang et al., 2016; Zhuang et al., 2019). These aa substitutions are located in the greatest variable VP2 GH loop, comprised between aa 267 and 498, but while residue 267 is not exposed on the capsid surface (Chiang, Wu, Chiou, Chang, & Lin, 2016) and may not affect the antigenicity of CPV (Xu et al., 2015), residues 324 and 370 could have immunological implications or biological relevance. Indeed, residue 324 is subject to positive selection (Hoelzer, Shackelton, Parrish, & Holmes, 2008) and is adjacent to residue 323, which affects binding to the canine transferrin receptor (Hueffer & Parrish, 2003). Residue 370 is close to residues associated with the ability of CPV to haemagglutinate, altering the pH dependence of haemagglutination or affecting the canine transferrin receptor (TfR) binding that determines the canine host range (Guo et al., 2013; Kaelber et al., 2012; Tsao et al., 1991).
Among the synonymous substitutions observed in the CPV‐2a VP2 gene sequences, nt change a1275g has been previously described in CPV‐2c strains (Amrani et al., 2016; Decaro, Desario, Amorisco, et al., 2013; Decaro et al., 2009). This change, observed in the strains UV1 and UV6, was detected in the binding region of the type‐2a and type‐2c specific probes of the minor groove binder (MGB) probe assay (Decaro et al., 2005, 2006). Although this change potentially accounts for the absence of VIC fluorescence in the 2a/2b and 2b/2c assays, specific additional studies are necessary to evaluate its real implication in the characterization of this CPV‐2a mutant by the MGB probe assay, as previously done for the same substitution in the CPV‐2c mutants (Decaro, Desario, Billi, et al., 2013). The in‐clinic assay used in this study was able to detect the CPV‐2a showing this substitution, as previously observed for another rapid assay used to test also the CPV‐2c mutants (Decaro, Desario, Billi, et al., 2013).
Limited studies are available on the CPV non‐structural genes (Hoelzer et al., 2008; Pérez et al., 2014), and, only recently, the analysis of the NS1 gene sequence was included in the CPV phylogenies from several countries (Canuti, Rodrigues, Whitney, & Lang, 2017; Grecco et al., 2018; Mira, Canuti, et al., 2019; Mira, Purpari, et al., 2019; Zhuang et al., 2019). In this study, sequence analysis revealed aa changes previously described mainly in NS1/NS2 gene sequences of CPV‐2a/2c strains of Asian origin. Additional changes were also evident, some previously reported in South/North America and others never previously observed. This divergence may suggest the same ancestral origin with the CPV strains of Asian origin but a separate evolution, as well as a continuous adaptive process of the virus in separate environments. Indeed, some of these changes lay in the potential encoding sequence of functional domains (Mira, Canuti, et al., 2019) and, particularly, residues 351, 517 and 545 are located between the α5‐ and α6‐helices, between the β5‐ and α11‐helices and just close to the α11‐helix of the helicase domain protein sequence, respectively, as illustrated in Niskanen, Ihalainen, Kalliolinna, Häkkinen, and Vihinen‐Ranta (2010). Therefore, their role needs to be further evaluated.
The molecular analysis based on long genome sequences evidenced the geographical origin of the analysed strains rather than the clustering based only on the CPV antigenic variant. Therefore, this study supports further studies aimed to track the viral spread and elucidate the CPV evolution. Indeed, the recent evidence of specific aa changes, as well as the divergence from previous circulating strains, does not allow to rule out the possible introduction of these strains from other countries, highlighting the need for further studies on CPV whole genome in different geographical areas. This suggestion is supported by the evidence of genetic signatures typical of CPV or other canine viruses with different origins (Decaro, Campolo, et al., 2007; Martella et al., 2006; Mira, Purpari, Di Bella, et al., 2018; Mira, Purpari, Lorusso, et al., 2018), probably connected with the trading and transport of dogs between countries and continents.
In this study, with the aim to investigate the prevalence of the most frequent canine enteric viruses, all collected samples were analysed for selected pathogens. CPV was the only enteric viral pathogen detected in this study and this result confirmed the correlation between CPV infection and development of enteric signs, with a limited role of other viral enteric pathogens (Dowgier et al., 2017). Nevertheless, further studies, based on a wider sampling also including the wild potential susceptible species, are necessary to better elucidate the effective spread of the other viruses in Nigeria.
This study represents the first CPV molecular characterization including all the encoding gene sequences conducted in the African continent and contributes to define the current geographical spread of the CPV variants worldwide. The evidence of mutations that have not been detected before suggests the need for further investigations in order to determine any biological consequences and underlines the continue evolution of CPV.
CONFLICT OF INTEREST
The authors of this manuscript declare that there are no conflicts of interest.
ETHICAL APPROVAL
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to.
Supporting information
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Ijeoma Chekwube Chukwudi, Dr. Pam Dachung Luka, Dr. Emmanuel Tumininu Obishakin and Dr. Ukamaka Uchenna Eze for their skilful technical assistance and also practitioners, private veterinary clinics and dog breeders for sample collection.
Ogbu KI, Mira F, Purpari G, et al. Nearly full‐length genome characterization of canine parvovirus strains circulating in Nigeria. Transbound Emerg Dis. 2020;67:635–647. 10.1111/tbed.13379
REFERENCES
- Amrani, N. , Desario, C. , Kadiri, A. , Cavalli, A. , Berrada, J. , Zro, K. , … Decaro, N. (2016). Molecular epidemiology of canine parvovirus in Morocco. Infection, Genetics and Evolution, 41, 201–206. 10.1016/j.meegid.2016.04.005 [DOI] [PubMed] [Google Scholar]
- Apaa, T. T. , Daly, J. M. , & Tarlinton, R. E. (2016). Canine parvovirus (CPV‐2) variants circulating in Nigerian dogs. Veterinary Record Open, 3(1), e000198 10.1136/vetreco-2016-000198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babalola, E. T. , Ijaopo, O. K. , & Okonko, I. O. (2016). Evaluation of Immunity and Seropositivity of IgG Antibodies to Canine Parvoviruses in Vaccinated and Unvaccinated Dogs in Abeokuta, Nigeria. Journal of Immunoassay & Immunochemistry, 37(1), 16–28. 10.1080/15321819.2015.1040159 [DOI] [PubMed] [Google Scholar]
- Battilani, M. , Scagliarini, A. , Tisato, E. , Turilli, C. , Jacoboni, I. , Casadio, R. , & Prosperi, S. (2001). Analysis of canine parvovirus sequences from wolves and dogs isolated in Italy. The Journal of General Virology, 82(Pt 7), 1555–1560. 10.1099/0022-1317-82-7-1555 [DOI] [PubMed] [Google Scholar]
- Canuti, M. , Rodrigues, B. , Whitney, H. G. , & Lang, A. S. (2017). Introduction of canine parvovirus 2 into wildlife on the Island of Newfoundland, Canada. Infection, Genetics and Evolution, 55, 205–208. 10.1016/j.meegid.2017.09.018 [DOI] [PubMed] [Google Scholar]
- Castanheira, P. , Duarte, A. , Gil, S. , Cartaxeiro, C. , Malta, M. , Vieira, S. , & Tavares, L. (2014). Molecular and serological surveillance of canine enteric viruses in stray dogs from Vila do Maio, Cape Verde. BMC Veterinary Research, 10, 91 10.1186/1746-6148-10-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, S.‐Y. , Wu, H.‐Y. , Chiou, M.‐T. , Chang, M.‐C. , & Lin, C.‐N. (2016). Identification of a novel canine parvovirus type 2c in Taiwan. Virology Journal, 13(1), 160 10.1186/s12985-016-0620-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chollom, S. C. , Fyaktum, E. J. , Okwori, A. E. J. , Agada, G. O. A. , Hashimu, G. , Akele, R. Y. , … Egah, D. Z. (2013). Molecular detection of canine parvovirus in Jos, Nigeria. Journal of Veterinary Medicine and Animal Health, 5, 57–59. [Google Scholar]
- Cotmore, S. F. , Agbandje‐McKenna, M. , Canuti, M. , Chiorini, J. A. , Eis‐Hubinger, A.‐M. , Hughes, J. , … Tijssen, P. (2019). ICTV virus taxonomy profile: Parvoviridae. The Journal of General Virology, 100(3), 367–368. 10.1099/jgv.0.001212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotmore, S. F. , Agbandje‐McKenna, M. , Chiorini, J. A. , Mukha, D. V. , Pintel, D. J. , Qiu, J. , … Davison, A. J. (2014). The family Parvoviridae. Archives of Virology, 159(5), 1239–1247. 10.1007/s00705-013-1914-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro, N. , & Buonavoglia, C. (2012). Canine parvovirus–a review of epidemiological and diagnostic aspects, with emphasis on type 2c. Veterinary Microbiology, 155(1), 1–12. 10.1016/j.vetmic.2011.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro, N. , Campolo, M. , Elia, G. , Buonavoglia, D. , Colaianni, M. L. , Lorusso, A. , … Buonavoglia, C. (2007). Infectious canine hepatitis: An ‘old’ disease reemerging in Italy. Research in Veterinary Science, 83(2), 269–273. 10.1016/j.rvsc.2006.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro, N. , Desario, C. , Addie, D. D. , Martella, V. , Vieira, M. J. , Elia, G. , … Buonavoglia, C. (2007). The study molecular epidemiology of canine Parvovirus, Europe. Emerging Infectious Diseases, 13(8), 1222–1224. 10.3201/eid1308.070505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro, N. , Desario, C. , Amorisco, F. , Losurdo, M. , Elia, G. , Parisi, A. , Buonavoglia, C. (2013). Detection of a canine parvovirus type 2c with a non‐coding mutation and its implications for molecular characterisation. Veterinary Journal (London, England: 1997), 196(3), 555–557. 10.1016/j.tvjl.2012.12.017 [DOI] [PubMed] [Google Scholar]
- Decaro, N. , Desario, C. , Billi, M. , Lorusso, E. , Colaianni, M. L. , Colao, V. , Buonavoglia, C. (2013). Evaluation of an in‐clinic assay for the diagnosis of canine parvovirus. Veterinary Journal (London, England: 1997), 198(2), 504–507. 10.1016/j.tvjl.2013.08.032 [DOI] [PubMed] [Google Scholar]
- Decaro, N. , Desario, C. , Parisi, A. , Martella, V. , Lorusso, A. , Miccolupo, A. , … Buonavoglia, C. (2009). Genetic analysis of canine parvovirus type 2c. Virology, 385(1), 5–10. 10.1016/j.virol.2008.12.016 [DOI] [PubMed] [Google Scholar]
- Decaro, N. , Elia, G. , Campolo, M. , Desario, C. , Lucente, M. S. , Bellacicco, A. L. , & Buonavoglia, C. (2005). New approaches for the molecular characterization of canine parvovirus type 2 strains. Journal of Veterinary Medicine. B, Infectious Diseases and Veterinary Public Health, 52(7–8), 316–319. 10.1111/j.1439-0450.2005.00869.x [DOI] [PubMed] [Google Scholar]
- Decaro, N. , Elia, G. , Martella, V. , Campolo, M. , Desario, C. , Camero, M. , … Buonavoglia, C. (2006). Characterisation of the canine parvovirus type 2 variants using minor groove binder probe technology. Journal of Virological Methods, 133(1), 92–99. 10.1016/j.jviromet.2005.10.026 [DOI] [PubMed] [Google Scholar]
- Decaro, N. , Pratelli, A. , Campolo, M. , Elia, G. , Martella, V. , Tempesta, M. , & Buonavoglia, C. (2004). Quantitation of canine coronavirus RNA in the faeces of dogs by TaqMan RT‐PCR. Journal of Virological Methods, 119(2), 145–150. 10.1016/j.jviromet.2004.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogonyaro, B. B. , Bosman, A.‐M. , Sibeko, K. P. , Venter, E. H. , & van Vuuren, M. (2013). Genetic analysis of the VP2‐encoding gene of canine parvovirus strains from Africa. Veterinary Microbiology, 165(3–4), 460–465. 10.1016/j.vetmic.2013.04.022 [DOI] [PubMed] [Google Scholar]
- Dowgier, G. , Lorusso, E. , Decaro, N. , Desario, C. , Mari, V. , Lucente, M. S. , … Elia, G. (2017). A molecular survey for selected viral enteropathogens revealed a limited role of Canine circovirus in the development of canine acute gastroenteritis. Veterinary Microbiology, 204, 54–58. 10.1016/j.vetmic.2017.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowgier, G. , Mari, V. , Losurdo, M. , Larocca, V. , Colaianni, M. L. , Cirone, F. , … Decaro, N. (2016). A duplex real‐time PCR assay based on TaqMan technology for simultaneous detection and differentiation of canine adenovirus types 1 and 2. Journal of Virological Methods, 234, 1–6. 10.1016/j.jviromet.2016.03.011 [DOI] [PubMed] [Google Scholar]
- Elia, G. , Decaro, N. , Martella, V. , Cirone, F. , Lucente, M. S. , Lorusso, E. , … Buonavoglia, C. (2006). Detection of canine distemper virus in dogs by real‐time RT‐PCR. Journal of Virological Methods, 136(1–2), 171–176. 10.1016/j.jviromet.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Fagbohun, O. A. , & Omobowale, T. O. (2018). Sequence and phylogenetic analysis of canine parvovirus‐2 isolates in dogs revealed circulation of three subtypes in Nigeria. Virusdisease, 29(3), 411–415. 10.1007/s13337-018-0475-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo, J. , Miranda, C. , Souto, R. , Silva, E. , Fafetine, J. , & Thompson, G. (2017). Genetic characterization of canine parvovirus type 2 subtypes in Maputo, Mozambique. Archives of Microbiology, 199(4), 543–549. 10.1007/s00203-016-1320-7 [DOI] [PubMed] [Google Scholar]
- Folitse, R. D. , Kodie, D. O. , Amemor, E. , Dei, D. , Tasiame, W. , Burimuah, V. , & Emikpe, B. O. (2017). Detection of canine parvovirus antigen in dogs in Kumasi, Ghana. African Journal of Infectious Diseases, 12(1), 28–32. 10.21010/ajid.v12i1.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, M. M. , Kerin, T. , Hull, J. , McCaustland, K. , & Gentsch, J. (2008). Enhancement of detection and quantification of rotavirus in stool using a modified real‐time RT‐PCR assay. Journal of Medical Virology, 80(8), 1489–1496. 10.1002/jmv.21228 [DOI] [PubMed] [Google Scholar]
- Geng, Y. , Guo, D. , Li, C. , Wang, E. , Wei, S. , Wang, Z. , … Sun, D. (2015). Co‐circulation of the rare CPV‐2c with unique Gln370Arg substitution, new CPV‐2b with unique Thr440Ala substitution, and new CPV‐2a with high prevalence and variation in Heilongjiang Province, Northeast China. PLoS ONE, 10(9), e0137288 10.1371/journal.pone.0137288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grecco, S. , Iraola, G. , Decaro, N. , Alfieri, A. , Alfieri, A. , Gallo Calderón, M. , … Pérez, R. (2018). Inter‐ and intracontinental migrations and local differentiation have shaped the contemporary epidemiological landscape of canine parvovirus in South America. Virus Evolution, 4(1), vey011 10.1093/ve/vey011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L. , Yang, S. , Chen, S. , Zhang, Z. , Wang, C. , Hou, R. , … Yan, Q. (2013). Identification of canine parvovirus with the Q370R point mutation in the VP2 gene from a giant panda (Ailuropoda melanoleuca). Virology Journal, 10, 163 10.1186/1743-422X-10-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- Hoelzer, K. , Shackelton, L. A. , Parrish, C. R. , & Holmes, E. C. (2008). Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. The Journal of General Virology, 89(Pt 9), 2280–2289. 10.1099/vir.0.2008/002055-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueffer, K. , Parker, J. S. L. , Weichert, W. S. , Geisel, R. E. , Sgro, J.‐Y. , & Parrish, C. R. (2003). The natural host range shift and subsequent evolution of canine parvovirus resulted from virus‐specific binding to the canine transferrin receptor. Journal of Virology, 77(3), 1718–1726. 10.1128/jvi.77.3.1718-1726.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueffer, K. , & Parrish, C. R. (2003). Parvovirus host range, cell tropism and evolution. Current Opinion in Microbiology, 6(4), 392–398. 10.1016/S1369-5274(03)00083-3 [DOI] [PubMed] [Google Scholar]
- Kaelber, J. T. , Demogines, A. , Harbison, C. E. , Allison, A. B. , Goodman, L. B. , Ortega, A. N. , … Parrish, C. R. (2012). Evolutionary reconstructions of the transferrin receptor of Caniforms supports canine parvovirus being a re‐emerged and not a novel pathogen in dogs. PLoS Path, 8(5), e1002666 10.1371/journal.ppat.1002666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapiya, J. , Nalubamba, K. S. , Kaimoyo, E. , Changula, K. , Chidumayo, N. , Saasa, N. , … Simulundu, E. (2019). First genetic detection and characterization of canine parvovirus from diarrheic dogs in Zambia. Archives of Virology, 164(1), 303–307. 10.1007/s00705-018-4068-3 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella, V. , Cirone, F. , Elia, G. , Lorusso, E. , Decaro, N. , Campolo, M. , … Buonavoglia, C. (2006). Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy. Veterinary Microbiology, 116(4), 301–309. 10.1016/j.vetmic.2006.04.019 [DOI] [PubMed] [Google Scholar]
- Mira, F. , Canuti, M. , Purpari, G. , Cannella, V. , Di Bella, S. , Occhiogrosso, L. , … Guercio, A. (2019). Molecular characterization and evolutionary analyses of carnivore protoparvovirus 1 NS1 gene. Viruses, 11(4), 308 10.3390/v11040308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira, F. , Dowgier, G. , Purpari, G. , Vicari, D. , Di Bella, S. , Macaluso, G. , … Guercio, A. (2018). Molecular typing of a novel canine parvovirus type 2a mutant circulating in Italy. Infection, Genetics and Evolution, 61, 67–73. 10.1016/j.meegid.2018.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira, F. , Purpari, G. , Di Bella, S. , Colaianni, M. L. , Schirò, G. , Chiaramonte, G. , … Guercio, A. (2019). Spreading of canine parvovirus type 2c mutants of Asian origin in southern Italy. Transboundary and Emerging Diseases, 10.1111/tbed.13283 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira, F. , Purpari, G. , Di Bella, S. , Vicari, D. , Schirò, G. , Di Marco, P. , … Guercio, A. (2018). Update on canine distemper virus (CDV) strains of Arctic‐like lineage detected in dogs in Italy. Veterinaria Italiana, 54(3), 225–236. 10.12834/VetIt.1455.7862.2 [DOI] [PubMed] [Google Scholar]
- Mira, F. , Purpari, G. , Lorusso, E. , Di Bella, S. , Gucciardi, F. , Desario, C. , … Guercio, A. (2018). Introduction of Asian canine parvovirus in Europe through dog importation. Transboundary and Emerging Diseases, 65(1), 16–21. 10.1111/tbed.12747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda, C. , & Thompson, G. (2016). Canine parvovirus: The worldwide occurrence of antigenic variants. The Journal of General Virology, 97(9), 2043–2057. 10.1099/jgv.0.000540 [DOI] [PubMed] [Google Scholar]
- Mochizuki, M. , Horiuchi, M. , Hiragi, H. , San Gabriel, M. C. , Yasuda, N. , & Uno, T. (1996). Isolation of canine parvovirus from a cat manifesting clinical signs of feline panleukopenia. Journal of Clinical Microbiology, 34(9), 2101–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C. D. S. , Palermo, L. M. , Hafenstein, S. L. , & Parrish, C. R. (2007). Different mechanisms of antibody‐mediated neutralization of parvoviruses revealed using the Fab fragments of monoclonal antibodies. Virology, 361(2), 283–293. 10.1016/j.virol.2006.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niskanen, E. A. , Ihalainen, T. O. , Kalliolinna, O. , Häkkinen, M. M. , & Vihinen‐Ranta, M. (2010). Effect of ATP binding and hydrolysis on dynamics of canine parvovirus NS1. Journal of Virology, 84(10), 5391–5403. 10.1128/JVI.02221-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, C. R. , Aquadro, C. F. , Strassheim, M. L. , Evermann, J. F. , Sgro, J. Y. , & Mohammed, H. O. (1991). Rapid antigenic‐type replacement and DNA sequence evolution of canine parvovirus. Journal of Virology, 65(12), 6544–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish, C. R. , O’Connell, P. H. , Evermann, J. F. , & Carmichael, L. E. . (1985). Natural variation of canine parvovirus. Science, 230(4729), 1046–1048. 10.1126/science.4059921 [DOI] [PubMed] [Google Scholar]
- Pérez, R. , Calleros, L. , Marandino, A. , Sarute, N. , Iraola, G. , Grecco, S. , … Panzera, Y. (2014). Phylogenetic and genome‐wide deep‐sequencing analyses of canine parvovirus reveal co‐infection with field variants and emergence of a recent recombinant strain. PLoS ONE, 9(11), e111779 10.1371/journal.pone.0111779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpari, G. , Mira, F. , Di Bella, S. , Di Pietro, S. , Giudice, E. , & Guercio, A. (2018). Investigation on canine parvovirus circulation in dogs from Sicily (Italy) by biomolecular assay. Acta Veterinaria (Beograd), 68(1), 80–94. [Google Scholar]
- Reed, A. P. , Jones, E. V. , & Miller, T. J. (1988). Nucleotide sequence and genome organization of canine parvovirus. Journal of Virology, 62(1), 266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton, L. A. , Parrish, C. R. , Truyen, U. , & Holmes, E. C. (2005). High rate of viral evolution associated with the emergence of carnivore parvovirus. Proceedings of the National Academy of Sciences of the United States of America, 102(2), 379–384. 10.1073/pnas.0406765102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinel, A. , Venter, E. H. , Van Vuuren, M. , Parrish, C. R. , & Truyen, U. (1998). Antigenic and genetic analysis of canine parvoviruses in southern Africa. The Onderstepoort Journal of Veterinary Research, 65(4), 239–242. [PubMed] [Google Scholar]
- Touihri, L. , Bouzid, I. , Daoud, R. , Desario, C. , El Goulli, A. F. , Decaro, N. , … Bahloul, C. (2009). Molecular characterization of canine parvovirus‐2 variants circulating in Tunisia. Virus Genes, 38(2), 249–258. 10.1007/s11262-008-0314-1 [DOI] [PubMed] [Google Scholar]
- Truyen, U. , Müller, T. , Heidrich, R. , Tackmann, K. , & Carmichael, L. E. (1998). Survey on viral pathogens in wild red foxes (Vulpes vulpes) in Germany with emphasis on parvoviruses and analysis of a DNA sequence from a red fox parvovirus. Epidemiology and Infection, 121(2), 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao, J. , Chapman, M. S. , Agbandje, M. , Keller, W. , Smith, K. , Wu, H. , Compans, R. W. (1991). The three‐dimensional structure of canine parvovirus and its functional implications. Science, 251(5000), 1456–1464. 10.1126/science.2006420 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Lin, P. , Zhao, H. , Cheng, Y. , Jiang, Z. , Zhu, H. , … Cheng, S. (2016). Continuing evolution of canine parvovirus in China: Isolation of novel variants with an Ala5Gly mutation in the VP2 protein. Infection, Genetics and Evolution, 38, 73–78. 10.1016/j.meegid.2015.12.009 [DOI] [PubMed] [Google Scholar]
- Woolford, L. , Crocker, P. , Bobrowski, H. , Baker, T. , & Hemmatzadeh, F. (2017). Detection of the canine parvovirus 2c subtype in australian dogs. Viral Immunology, 30(5), 371–376. 10.1089/vim.2017.0019 [DOI] [PubMed] [Google Scholar]
- Xu, J. , Guo, H. C. , Wei, Y. Q. , Shu, L. , Wang, J. , Li, J. S. , Cao, S. Z. , & Sun, S. Q. (2015). Phylogenetic analysis of canine parvovirus isolates from Sichuan and Gansu provinces of China in 2011. Transbound Emerg Dis., 62(1), 91–5. 10.1111/tbed.12078 [DOI] [PubMed] [Google Scholar]
- Yi, L. , Tong, M. , Cheng, Y. , Song, W. , & Cheng, S. (2016). Phylogenetic analysis of canine parvovirus VP2 gene in China. Transboundary and Emerging Diseases, 63(2), e262–269. 10.1111/tbed.12268 [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Schwartz, S. , Wagner, L. , & Miller, W. (2000). A greedy algorithm for aligning DNA sequences. Journal of Computational Biology, 7(1–2), 203–214. 10.1089/10665270050081478 [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Wang, J. , Jiang, Y. , Cheng, Y. , Lin, P. , Zhu, H. , … Cheng, S. (2017). Typing of canine parvovirus strains circulating in North‐East China. Transboundary and Emerging Diseases, 64(2), 495–503. 10.1111/tbed.12390 [DOI] [PubMed] [Google Scholar]
- Zhou, P. , Zeng, W. , Zhang, X. , & Li, S. (2017). The genetic evolution of canine parvovirus ‐ A new perspective. PLoS ONE, 12(3), e0175035 10.1371/journal.pone.0175035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, Q.‐Y. , Qiu, Y. , Pan, Z.‐H. , Wang, S.‐C. , Wang, B. , Wu, W.‐K. , … Wang, K.‐C. (2019). Genome sequence characterization of canine parvoviruses prevalent in the Sichuan province of China. Transboundary and Emerging Diseases, 66(2), 897–907. 10.1111/tbed.13100 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials