

Abstract
Bovine coronavirus (BCoV) is the causative agent of diarrhoea in newborn calves, winter dysentery in adult cattle and respiratory tract illnesses in cattle across the world. In this study, a total of 190 faecal samples from dairy calves with diarrhoea were collected from 14 farms in six Chinese provinces, and BCoV was detected in 18.95% (36/190) of the samples by reverse transcriptase polymerase chain reaction. Full‐length spike, hemagglutinin/esterase (HE), nucleocapsid and transmembrane genes were simultaneously cloned from 13 clinical samples (eight farms in four provinces), and most of the BCoV strains showed a unique evolutionary pattern based on the phylogenetic analysis of these genes. Interesting, 10 of the 13 strains were identified as HE recombinant strains, and these strains had experienced the same recombination event and carried the same recombination sites located between the esterase and lectin domain. They also shared an identical aa variant (F181V) in the R2‐loop. Moreover, 9/10 strains displayed another identical aa variant (P, S158A) in the adjacent R1‐loop of the HE gene, which differs from the other available BCoV HE sequences in the GenBank database. Our results showed that BCoV is widely circulating in dairy cattle in China, contributing to the diagnosis and control of dairy calves diarrhoea. Furthermore, a BCoV strain that carries a recombinant HE gene has spread in dairy calves in China. To the best of our knowledge, this is the first description of an HE recombination event occurring in BCoV; this is also the first description of the molecular prevalence of BCoV in China. Our findings will enhance current understanding about the genetic evolution of BCoV.
Keywords: bovine coronavirus, China, dairy calves, hemagglutinin/esterase gene, prevalence, recombination
1. INTRODUCTION
Bovine coronavirus (BCoV) is a lineage A member of the betacoronavirus genus. Other members include human OC43 coronavirus (HCoV‐OC43), mouse hepatitis virus (MHV), equine coronavirus, porcine hemagglutinating encephalomyelitis virus and canine respiratory coronavirus. BCoV, which causes diarrhoea in newborn calves, winter dysentery in adult calves and respiratory tract illnesses in calves and adult cattle, inflicts in severe economic losses on the global farming industry (Azizzadeh, Shooroki, Kamalabadi, & Stevenson, 2012; BOK et al., 2015; Johnson & Pendell, 2017).
Bovine coronavirus possesses five major structural proteins: the spike (S), hemagglutinin/esterase (HE), nucleocapsid (N), transmembrane (M) and the small membrane (E) (Lai & Cavanagh, 1997). The S protein is involved in receptor recognition and carries distinct functional domains near its amino (S1) and carboxy (S2) termini, while the N‐terminal S1 domains recognize sugar receptors, and the S2 subunit is a transmembrane protein that mediates viral and cellular membrane fusion during cell invasion (Fang Li, 2016). S1 and S2 contain several antigenic domains, but S1 appears to be the most efficient at inducing antibodies with high neutralizing activities in its host (Yoo & Deregt, 2001). The HE protein contains two important functional domains: the lectin domain and the esterase domain. The lectin domain recognizes sugar receptors in the cell, whereas the esterase domain possesses a receptor‐destroying enzyme activity capable of removing cellular receptors from the surfaces of the targeted cells. The receptor‐binding (lectin) and receptor‐destroying (esterase) domains may be important for virus entry (Kienzle, Abraham, Hogue, & Brian, 1990; Schultze, Wahn, Klenk, & Herrler, 1991). Therefore, in addition to the S protein, the HE protein serves as a second viral attachment protein for infection initiation (Groot, 2006). The primary role of BCoV N protein is to package the viral genome into long, flexible, helical ribonucleoprotein (RNP) complexes, protect the genome and ensure its timely replication and reliable transmission, as well as playing a role in viral transcription and translation (Hurst, Ye, Goebel, Jayaraman, & Masters, 2010). In contrast, the M protein plays a crucial role in BCoV assembly (Oostra, Haan, Groot, & Rottier, 2006).
The high genetic diversity in coronaviruses is attributable to the high mutation rates associated with RNA replication, the high recombination frequencies within the coronavirus family and the large coronavirus genomes (Woo, Lau, Huang, & Yuen, 2009). Recombination in coronaviruses plays an important role in virus evolution and can result in the emergence of new pathotypes (Menachery, Graham, & Baric, 2017; Wang et al., 2015) as well as changing the host ranges and ecological niches (Bakkers et al., 2017). Thus far, recombination regions in coronaviruses have been extensively reported for the S gene (Kin et al., 2015; Lau et al., 2011; Minami et al., 2016), a finding also applicable to BCoV (Martínez et al., 2012). Recombination events in M (Herrewegh, Smeenk, Horzinek, Rottier, & Groot, 1998), N (Kin et al., 2015), RP3 (Lau et al., 2010) and the ORF1 gene (Chen et al., 2017; Kin et al., 2015) have also been reported. However, to date, recombination events in HE have only been reported in MHV, a betacoronavirus, and this situation may act as a strong force for generating strains with new genotypes, host spectra and tissue tropisms (Groot, 2006; Luytjes, Bredenbeek, Noten, Horzinek, & Spaan, 1988; Smits et al., 2005).
The presence of BCoV has been confirmed in Chinese dairy cows (GenBank accession number FJ556872), but the prevalence and molecular characteristics of BCoV are still largely unknown. Therefore, we sought to investigate the prevalence of BCoV in dairy calves with diarrhoea in China. Unexpectedly, our results reveal that a BCoV containing a recombinant HE gene has emerged and spread in dairy calves in China.
2. MATERIALS AND METHODS
2.1. Faecal samples
A total of 190 faecal samples were collected from dairy calves (≤3 months of age) with obvious diarrhoea at 14 farms from six provinces in China during September 2017 and May 2018 (Table 1). The samples were shipped on ice and stored at −80°C.
Table 1.
The result of BCoV detection in diarrhoea faecal samples
| Province | Number of farms | Numbers of samples | The positive rate% |
|---|---|---|---|
| Sichuan | 4 | 56 | 16 |
| Shandong | 3 | 23 | 8.7 |
| Shanxi | 2 | 40 | 5.0 |
| Henan | 3 | 15 | 46.7 |
| Liaoning | 1 | 18 | 77.8 |
| Jilin | 1 | 38 | 5.3 |
| Total | 14 | 190 | 18.95 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.2. RNA extraction and cDNA synthesis
The faecal samples were fully resuspended in phosphate‐buffered saline (1:5 w/v) and centrifuged at 10,000 × g for 10 min. Viral RNA was extracted from 300 μl of the faecal suspension using RNAios Plus (TaKaRa Bio Inc) according to the manufacturer's instructions. The cDNA was synthesized using the PrimeScript™ RT Reagent Kit according to the manufacturer's instructions (TaKaRa Bio Inc.) and then stored at −20°C until required.
2.3. BCoV detection using polymerase chain reaction
Bovine coronavirus nucleic acids in the faecal samples were identified using a PCR assay established in our laboratory that targets the BCoV polymerase gene. After validating the specificity and stability of the assay, the detection limit for the viral nucleic acid in the assays was determined to be 1 × 10−2 pg per μL‐1. In detail, a primer pair (F: 5′‐ CGAGTTGAACACCC AGAT‐3′, R: 5′‐GAGACGGGCATCTACACT‐3′)were used to amplify a 230 nucleotide (nt) region of the polymerase gene (position 13,144–13,373 bp) in the BCoV Mebus genome sequence, GenBank accession: U00735.2). The PCR mixture (25 μl) contained 1 μl cDNA, 1 μl forward primer (10 μM), 1 μl reverse primer (10 μM), 12.5 μl PremixTaq (TakaRa Taq version 2.0 plus dye) and 9.5 μl nuclease‐free water. The mixtures were PCR‐amplified with 35 cycles at 94°C for 5 min, 94°C for 30 s, 49°C for 30 s and 72°C for 30 s, with a final extension step at 72°C for 10 min in an automated thermal cycler (BIOER, TC‐96/G/H(b)c).
2.4. PCR amplification of S, HE, N and M genes
The complete S, HE, N and M genes were PCR‐amplified from samples already known to be BCoV‐positive based on RT‐PCR assays previously reported (Gélinas, Boutin, Sasseville, & Dea, 2001; Lau et al., 2011; Martínez et al., 2012; Park et al., 2006). All PCR products were purified using the Omega Gel Kit (Omega) following the manufacturer's instructions, after which they were ligated to the pMD19‐T vector (TaKaRa Bio Inc.) and transformed into DH5α competent Escherichia coli cells (Yeasen) for sequencing. The S and N gene sequences were assembled using SeqMansoftware (version 7.0; DNASTAR Inc).
2.5. Sequence, phylogenetic, and recombination analyses
The homologies of the nt and deduced amino acid (aa) sequences were determined using the MegAlign program in DNASTAR 7.0 software (DNASTAR Inc). MEGA 7.0 was used for multiple sequence alignment and to subsequently build the maximum‐likelihood phylogenetic tree with bootstrap testing (1,000 replicates). Recombination events were assessed using SimPlot software (version 3.5.1) and the Recombination Detection Program RDP 4.0 (version 4.9.5) with the RDP, GeneConv, Chimaera, MaxChi, BootScan, SiScan and 3Seq methods (Martin, Murrell, Golden, Khoosal, & Muhire, 2015).
3. RESULTS
3.1. BCoV detection in samples of faecal diarrhoea from dairy calves
Of the 190 faecal samples from the calves with diarrhoea, 36(18.95%) were found to be BCoV‐positive, which revealed that the virus was distributed in 13/14 farms across the six provinces (Table 1).
3.2. PCR amplification of the S, HE, N and M genes from BCoV
Full‐length S, HE, N and M genes were successfully cloned out of 13 positive samples from eight farms in four different Chinese provinces (Shanxi, two strains; Henan, three strains; Liaoning, five strains; and Sichuan, three strains).
3.3. Molecular characterization of the S genes
The 13 S genes, at 4,092 ‐bp each, encode a protein of 1,363 aa, the cleavage site of which is located at aa 768 in all 13. Sequence comparisons revealed that all 13 S genes share 98.6%–100% nt identity and 98.5%–100% aa identity with each other. They also share 96.8%–100% nt identity and 95.3%–100% aa identity with all 163 full‐length BCoV S genes available in the GenBank database. A phylogenetic tree based on the complete S gene sequences using the maximum‐likelihood method showed that 12 of the 13 S genes from this study together with 13 other BCoV S genes from China (one strain from cattle, GenBank accession number KU886291; 12 strains from Yaks, Bos grunniens, submitted by our team, GenBank accession number MH810151–MH810162) clustered on an independent large branch. The remaining S genes clustered with three North American BCoV strains (GenBank accession number MH043952, MH043954 and MH043955) on a small independent branch of the tree (Figure 1). Compared with the other BCoV S genes, 9/13 sequences from this study and the 13 other Chinese BCoV sequences motioned above, which were located in the independent large branch, each had an identical aa variant (N1192Y) in the S2 subunit. Additionally, 4/13 sequences from this study and the above‐mentioned 12 sequences from Chinese Yaks, which are located in the large independent branch, have an identical aa variant (E121V) in the S1 subunit. Compared with the BCoV Mebus prototype strain, these BCoV S genes have a total of 13 aa changes in the S1 subunit and 3 aa changes in the S2 subunit (Figure 2). No frame shifts, deletions, insertions or recombination events were observed in the S gene sequences from all the strains in this study.
Figure 1.
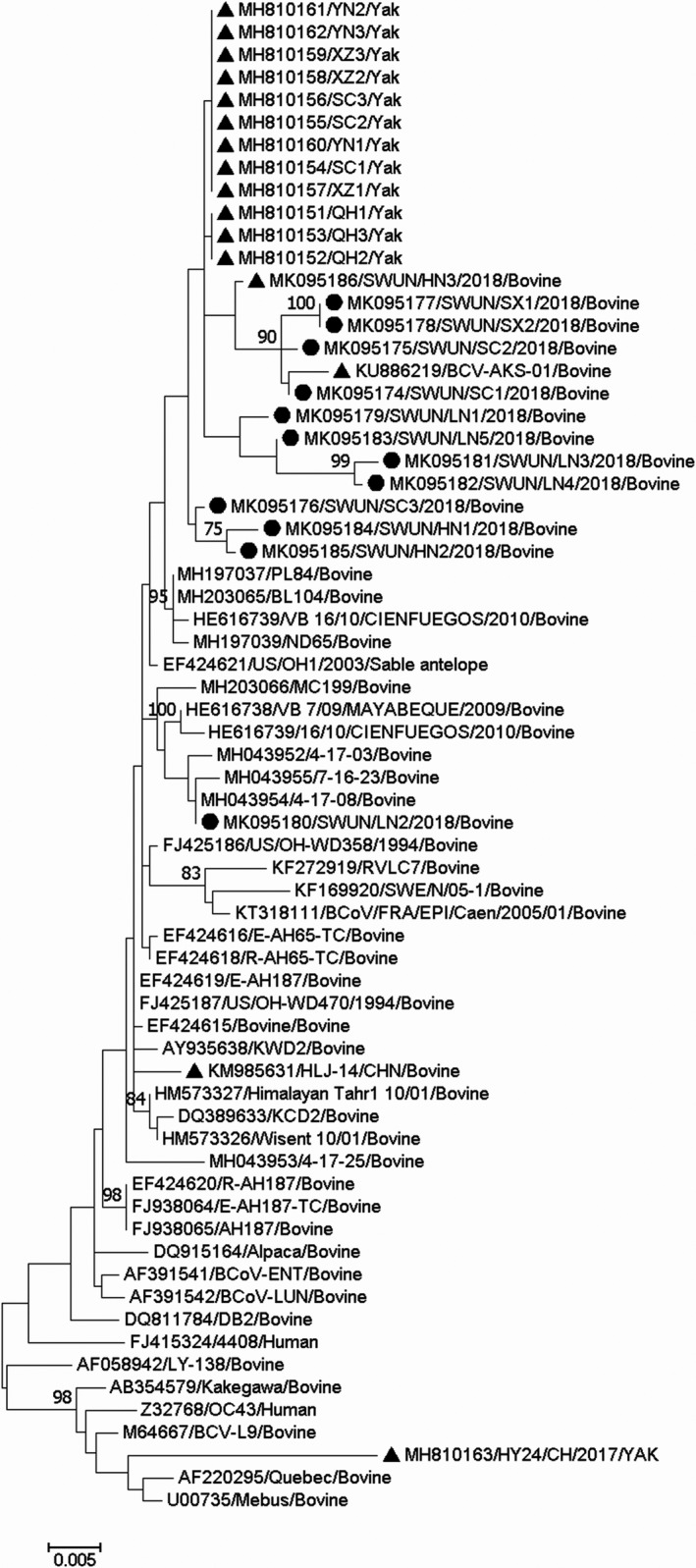
Phylogenetic tree based on the deduced 1,363 aa sequences of the complete S gene. Sequence alignments and clustering were performed by ClustalW in MEGA 7.0 software. The tree was constructed by the maximum‐likelihood method with bootstrap values calculated for 1,000 replicates. The strains in this study were marked with a circle, and the other Chinese BCoV strains were marked with a triangle
Figure 2.
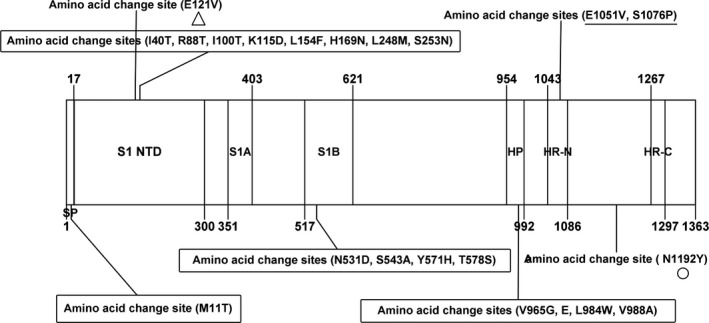
Amino acid variants of the 13 complete S genes in this study. The figures in the box indicate the identical amino acid change sites in all 13 strains in this study compared with the BCoV prototype strain Mebus S sequences; the figure marked with triangle was an unique aa variant in the four sequences in this study and 12 sequences from Chinese Yaks; the figure marked with circular was an unique aa variant in the nine sequences in this study and 13 sequences (12 from Chinese Yaks and one from Chinese cattle); the figure marked with line was an unique aa variant in Shanxi strains in this study; which compared with the other available BCoV S sequences in the GenBank database. HP, the first hydrophobic domain of the S2 subunit; HR‐N and HR‐C, the heptad repeats; S1A and S1B, the immune reactive domain; S1‐NTD, receptor‐binding domain; SP, signal peptide
3.4. Molecular characterization of the HE genes
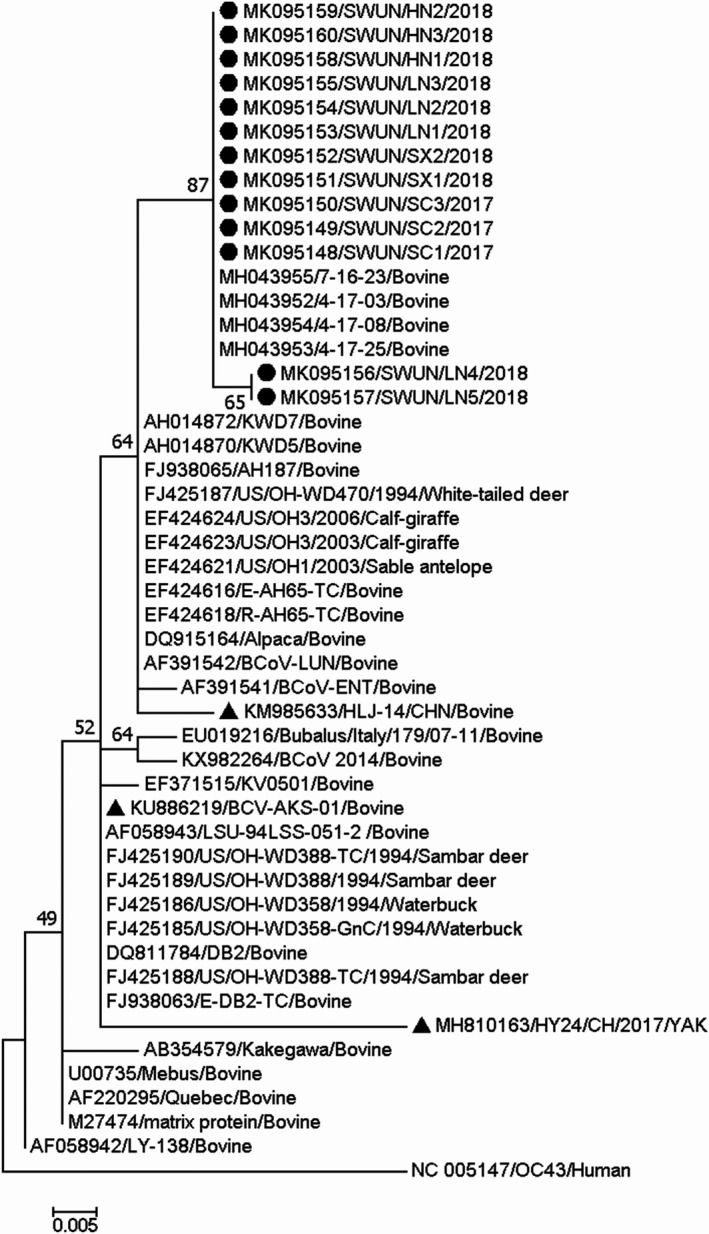
All 13 HE genes were 1,275 ‐bp long, and the protein they encode is 424 aa residues in length. FGDS, the putative esterase active site in all HE proteins, was located at aa positions 37–40, and nine N‐glycosylation sites were located at 54, 89, 104,153, 236, 301, 316, 358 and 417 in the protein. Sequence comparisons of the HE genes revealed that the 13 strains shared 97.3%–100% nt sequence identity and 98.8%–100% aa identity between each other, and shared 96.1%–99.3% nt sequence identity and 93.2%–99.1% aa sequence identity with all 115 of the complete BCoV HE genes available in the GenBank database. A phylogenetic tree based on the full‐length HE genes and the maximum‐likelihood methodology showed that 10/13 of these genes together with two Chinese BCoV HE genes (one strain from cattle, GenBank accession number KU886291; one strain from Yak, Bos grunniens, submitted by our team, GenBank accession number MH810163) clustered into a large independent branch. The remaining three HE genes clustered with HE genes of three North American BCoV strains (GenBank accession number FJ938065, AF230528 and AF391542) into a small independent branch (Figure 3).
Figure 3.
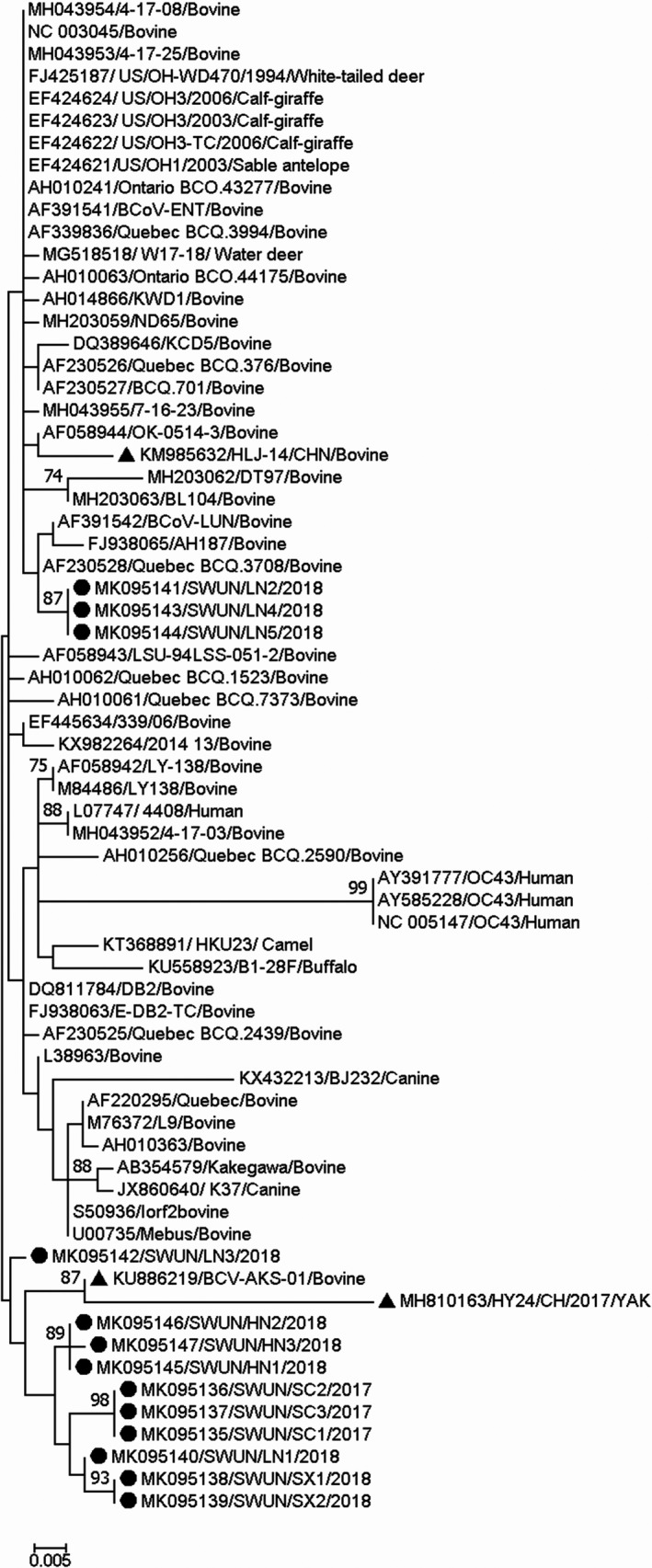
Phylogenetic tree based on the deduced 424 aa sequences of the complete HE gene. Sequence alignments and clustering were performed by ClustalW in MEGA 7.0 software. The tree was constructed by the maximum‐likelihood method with bootstrap values calculated for 1,000 replicates. The strains in this study were marked with a circle, and the other Chinese BCoV strains were marked with a triangle
Interestingly, recombination analysis of the 13 complete HE genes showed that recombination events had occurred in 10 of the strains (GenBank accession number MK095135–MK095140, MK095142 and MK095145–MK095147) from eight farms in four provinces, as based on the RDP 4.0 (six method) and SimPlot 3.5.1. The recombination breakpoint based on RDP 4.0 (six method: RDP, GeneConv, Chimaera, MaxChi, SiScan and 3Seq; recombinant score: 0.613) identified the beginning of the breakpoint at nt 150 in the fragment (breakpoint 99% confidence intervals: nt position 1–524 in the fragment) and nt 724 at the end of the breakpoint (breakpoint 99% confidence intervals: nt position 544–869 in the fragment), in the putative major parental strain, KCD1 (GenBank accession number DQ389642) and the possible minor parental strain, LY‐138 (GenBank accession number M84486). However, using SimPlot 3.5.1, the crossover site of the putative parental strains mainly mapped to nt 168 at the beginning of the breakpoint and out nt 702 at the end of the breakpoint in the 1,275 bp sequence (Figure 4). Although the recombination breakpoints predicted by RDP 4.0 and SimPlot differ, both programs showed that the recombination breakpoint was located between the esterase and lectin domain in HE (at nt position 70 bp–1,024 bp in the full‐length BCoV HE gene). Sequence comparisons of the 10 strains in which the HE gene showed evidence of recombination revealed that the strains shared 98.4%–100% nt identity and 97.9%–100% aa identity between each other. In addition, two China strains (one strain from cattle, GenBank accession number KU886291; one strain from Yak, GenBank accession number MH810163) that clustered with the 10 recombinant strains based on phylogenetic tree were identified as recombinant strains also, with the recombination event the same as that in this study.
Figure 4.
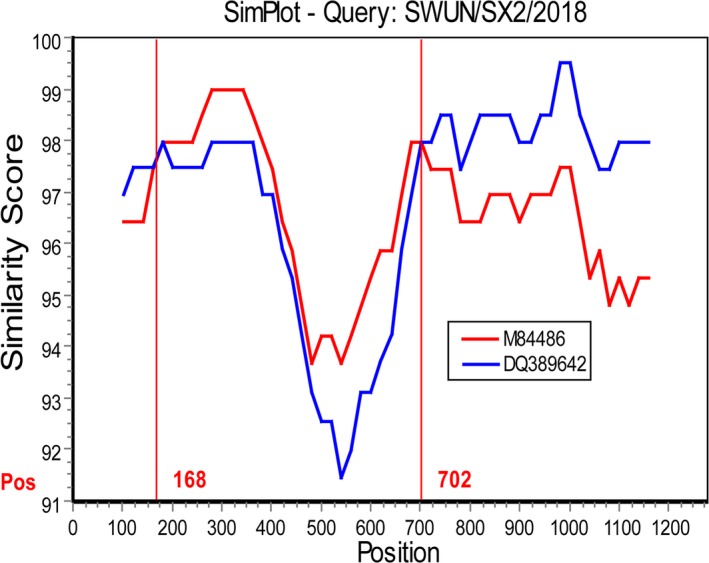
The recombination analysis of strain SWUN/SX2/2018 using SimPlot 3.5.1. A nucleotide (nt) identity plot comparing a 1,275 bp fragment of strain SWUN/SX2/2018 with BCoV strains KCD1 (GenBank accession number DQ389642) and LY138 (GenBank accession number M84486) is shown. The putative recombination region is located at the 168‐702 bp. The vertical axis indicates the similarity (%) of nucleotide sequences between the query strain and other reference strains. The horizontal axis indicates the nucleotide positions. SimPlot analysis was performed using a window size of 200 nt and step size of 20 nt (only one recombinant strain was showed due to typewritten pages requirement)
Compared with the Mebus prototype strain, the 13 HE genes cloned in the present study each have an aa substitution (L5P) in the signal peptide, an aa substitution (N49T) in the putative esterase domain and an aa substitution in the membrane‐proximal domain (S367P; Figure 5). No frame shifts, deletions or insertions were observed in the HE gene sequences in this study. Compared with all other available BCoV HE genes in the GenBank database, we found that the recombinant HE genes in the 10 strains and the two Chinese BCoV HE genes (GenBank accession number KU886219 and MH810163) that were also identified as HE recombination strains in this study each have an identical aa variant (F181V) located in the R2‐loop of the HE gene. Moreover, 9/10 strains with recombinant HE genes carried an identical aa variant (P, S158A) located adjacent to the R1‐loop in the HE gene; surprisingly, the aa residue at this site is identical to that in the HCoV‐OC43 (Figure 5).
Figure 5.
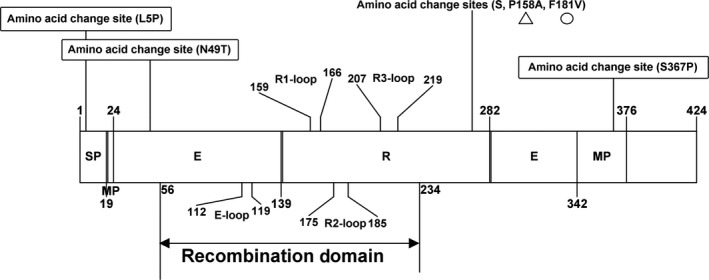
Amino acid variants of the 13 complete HE genes in this study. The figures in the box indicate the identical amino acid change sites in all 13 strains in this study compared with the BCoV prototype strain Mebus HE sequence; the figure marked with triangle was an unique aa variant in the 9/10 recombinant HE sequences, and the figure marked with circular was an unique aa variant in the 10 recombinant HE sequences in this study and two other Chinese strains (GenBank accession number KU886219, MH810163), which compared with the other available BCoV HE sequences in the GenBank database. Recombination domain is located at E‐loop, R1‐loop, R2‐loop and R3‐loop between esterase and lectin domain of HE. E, esterase domain; MP, membrane‐proximal domain; R, lectin domain; SP, signal peptide
3.5. Molecular characterization of N and M genes
All of the 13 N genes were 1,347 ‐bp in length, each encoding a protein of 230 aa residues. Sequence comparison of these genes revealed that they share 99.8%–100% nt sequence identity and 99.3%–100% aa identity between each other, and share 96.4%–99.6% nt sequence identity and 96.8%–99.9% aa identity with all 81 of the full‐length BCoV N genes available in the GenBank database. The phylogenetic tree based on the complete N gene sequences and the maximum‐likelihood method showed that all 13 N genes, together with one Chinese strain from Yak (GenBank accession number MH810163), clustered on an independent large branch (Figure 6). Compared with other BCoV N genes, all 13 strains in this study and the Chinese strain (GenBank accession number MH810163) carry an identical aa variant (S416I) in the N gene. Compared with the Mebus prototype strain, the 13 strains have six other aa changes (F15S, L53Q, M386T, M387I, S423I and Y441F) in the N gene. No frame shifts, deletions or insertions were observed in the N gene sequences from all strains in this study.
Figure 6.
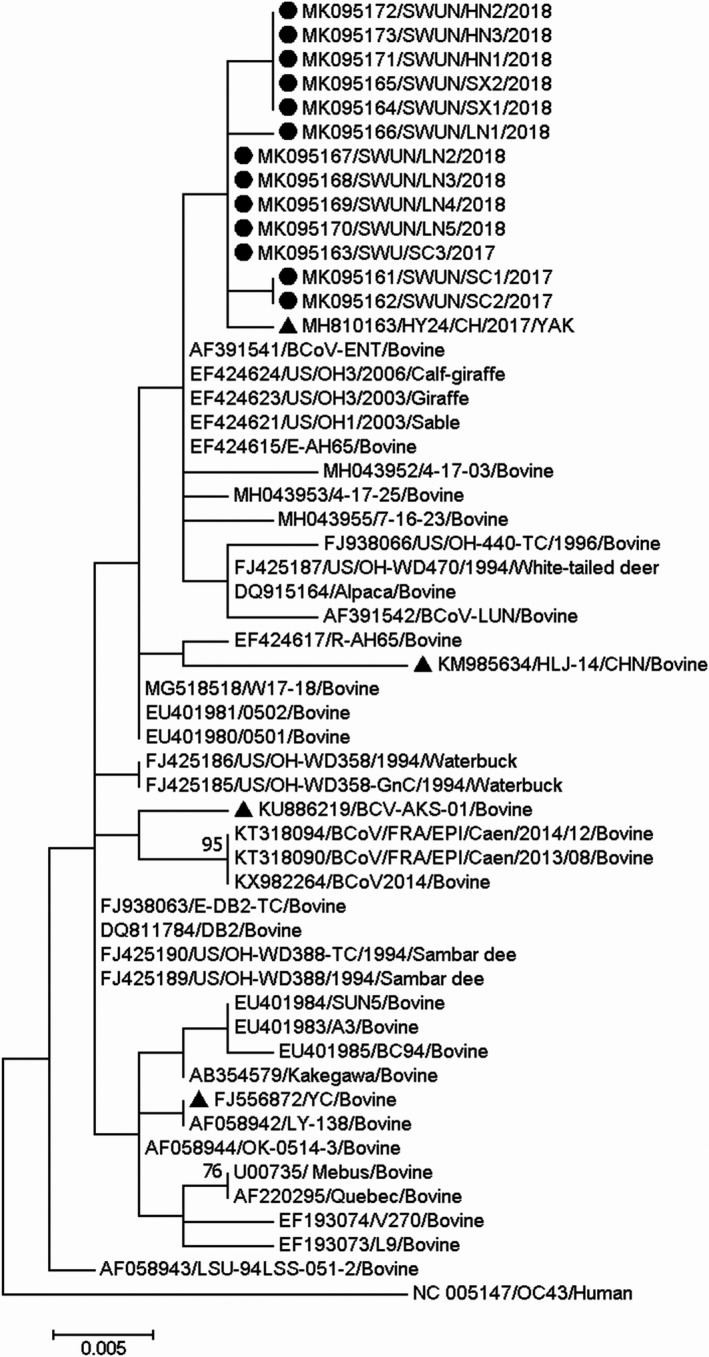
Phylogenetic tree based on the deduced 448 aa sequences of the complete N gene. Sequence alignments and clustering were performed by ClustalW in MEGA 7.0 software. The tree was constructed by the maximum‐likelihood method with bootstrap values calculated for 1,000 replicates. The strains in this study were marked with a circle, and the other Chinese BCoV strains were marked with a triangle
In contrast, all 13 M genes were 690 ‐bp in length, each encoding a protein of 230 aa residues. Sequence comparisons of the M genes revealed that the 13 M genes shared 99.4%–100% nt identity and 99.6%–100% aa identity between each other, and shared 94.1%–100% nt identity and 94.4%–100% aa identity with all 75 complete BCoV M genes available in the GenBank database. The phylogenetic tree based on the complete M gene sequences and the maximum‐likelihood method showed that all 13 M genes, together with four recently reported M genes from North American strains (GenBank accession number MH043952, MH043953, MH043954 and MH04955) fell into an independent large branch (Figure 7). Compared with the other BCoV M genes, all the strains in this study and the four North American strains (GenBank accession number MH043952, MH043953, MH043954 and MH04955) share two identical aa variants (C126S, T175S) in the M gene. Compared with the M gene from the Mebus prototype strain, the 13 strains have two other aa changes (I38V, M221L) in the N gene. No frame shifts, deletions or insertions were observed in the N gene sequences from all the strains examined in this study.
Figure 7.
Phylogenetic tree based on the deduced 230 aa sequences of the complete M gene. Sequence alignments and clustering were performed by ClustalW in MEGA 7.0 software. The tree was constructed by the maximum‐likelihood method with bootstrap values calculated for 1,000 replicates. The strains in this study were marked with a circle, and the other Chinese BCoV strains were marked with a triangle
4. DISCUSSION
Bovine coronavirus, an important pathogen of calves, is globally responsible for severe economic losses in farming (Azizzadeh et al., 2012; BOK et al., 2015; Johnson & Pendell, 2017). In China, the prevalence of BCoV is still largely unknown. Therefore, in this study, we screened 190 diarrhoea faecal samples from calves, 36 of which were found to be BCoV‐positive, and the positive samples were distributed across 13 of the 14 farms we screened across six provinces in the major dairy cattle production areas of China. The results showed that BCoV is circulating widely in these dairy cattle, a finding that should help with the diagnosis and control of diarrhoea in these animals. Most of the strains from this study are unique in their evolutionary histories based on our analysis of the full‐length S, HE, N and M genes, a finding similar to that for BCoV in Korean (Ko et al., 2006; Park et al., 2010). This may be the result of geographical, environmental and natural selection patterns (Bidokhti et al., 2013; Hasoksuz, Sreevatsan, Cho, Hoet, & Saif, 2002; Martínez et al., 2012).
The BCoV S protein is involved in receptor recognition, host specificity, antigenic diversity and immunogenicity (Fang Li, 2016). Its gene sequences are variable, and mutations in this protein are associated with alterations in viral antigenicity, viral pathogenicity, host range and tissue tropism (Gallagher & Buchmeier, 2001; Peng et al., 2012). In this study, compared with other BCoV S genes, we found that nine out of 13 of our sequences and 13 Chinese BCoV sequences (one strain from cattle and 12 strains from Yaks), which clustered on a large independent branch of the phylogenetic tree, each had an identical aa variant (N1192Y) in the S2 subunit. As a transmembrane protein, the S2 subunit mediates the fusion of viral and cellular membranes (Luo & Weiss, 1998); hence, the biological significance of this variant warrants further investigation. In addition, four out of 13 sequences and 12 sequences from Chinese Yaks were found to have an identical aa variant (E121V) in the S1 receptor‐binding region, compared with the other S genes. The S1 subunit in the N‐terminal of BCoV (aa 1–330) recognizes a sugar receptor (Peng et al., 2012), and aa substitutions in this region can change the receptor‐binding capacity (Li et al., 2005) and host receptor specificity (Sheahan et al., 2008). Two BCoV strains (GenBank accession number MK095177 and MK095178) from Shanxi Province were found to have unique aa substitutions (E1051V, S1076P) in the heptad repeat region. This region is crucial for viral entry and for viral and host cell membrane interactions to occur, and it promotes lipid bilayer fusion and nucleocapsid release into the cytoplasm (Forni et al., 2015). Thus, aa substitutions in this region may affect the interaction between the coiled‐coil structure and the host cell receptor (Martínez et al., 2012). The HE protein has a receptor‐binding function, which also plays a critical role in the infection process of BCoV (Langereis, Zeng, Heesters, Huizinga, & Groot, 2012). The BCoV HE receptor‐binding region comprises six surface loops, among which five are grafted onto the beta‐sandwich core of the lectin domain, named the R1‐loop, R2‐loop, R3‐loop, R4‐loop and the RBS‐hairpin, and another is present on the esterase domain (E‐loop; Langereis et al., 2012). In our study, 10 of the 13 strains were identified as having the same recombination events in the HE gene, located between the esterase and lectin region (Figure 5); this recombination phenomenon may affect receptor‐binding capacity.
The 10 strains in which recombination had occurred in the HE gene, and two other Chinese strains (GenBank accession number KU886219 and MH810163), share the same recombination event that we identified in this study. Moreover, these strains each had an identical aa variant (F181V) in the R2‐loop of the HE gene, while nine of the 10 recombinant strains had another identical aa variant (P, S158A) in the adjacent R1‐loop of the HE gene and this variant differs from all other available BCoV HE sequences in GenBank, and these recombinant strains clustered into a large independent branch in the phylogenetic tree (Figure 3). The 10 recombinant strains were recovered from eight farms in four provinces across a wide geographical distance, with the two furthest provinces being more than 1,000 km apart. Thus, these novel HE recombinant strains have been circulating widely in dairy cattle in China. To the best of our knowledge, this is the first report of a recombination event in the HE gene from BCoV in cattle, a finding that augments current understanding about the evolution of BCoV. In fact, MHV, which is another lineage A member of the betacoronavirus genus, has also reportedly undergone a non‐homologous recombination event in the HE gene (Luytjes et al., 1988). Recombination in the HE gene from Influenza C virus and in toroviruses has also been observed (Groot, 2006; Smits et al., 2005). Recombination in the HE gene may be a strong driving force for generating strains with new genotypes, host spectra and tissue tropisms (Groot, 2006; Luytjes et al., 1988; Smits et al., 2005). Notably, in our study, the HE recombinant and non‐recombinant strains simultaneously existed on the same cattle farm in Liaoning Province.
Interestingly, the reported decrease in the HE receptor‐binding capacity of HCoV‐OC43 betacoronaviruses was thought to be caused by an accumulation of aa substitutions (T114N, R177P, E178Q, F247L) in the receptor‐binding region during the course of evolution, and that aa substitutions (D220Y, H) in the adjacent R3‐loop can reduce the HE receptor‐binding capacity of BCoV (Bakkers et al., 2017). The most common betacoronavirus HE receptor is 9‐O‐acetylated sialic acid (9‐O‐Ac‐Sia; Matrosovich, Herrler, & Klenk, 2013). However, the MHV‐S receptor is 4‐O‐Ac‐Sia, not 9‐O‐Ac‐Sia, and this change may be caused by aa substitutions and/or insertions in the receptor‐binding region of the HE lectin domain, as compared with the Mebus BCoV prototype strain (Langereis et al., 2012). The HE protein lectin domain recognizes the sugar receptor, and the R2‐loop in the lectin domain (residues 176–185) plays an important role in ligand binding; aa substitutions in this region may reduce receptor‐binding activity (Bakkers et al., 2017). In the present study, all the strains with recombination in HE share an identical aa variant (F181V) in the R2‐loop of the HE gene. Furthermore, nine of the 10 strains have another identical aa variant (P, S158A) in the adjacent R1‐loop of their HE genes, which is an identical situation to that seen with the HCoV‐OC43 (Bakkers et al., 2017). Thus, further investigation of the significance of the receptor‐binding capacity caused by aa substitutions in the receptor‐binding region of the HE recombinant strains is warranted.
Notable, monoclonal antibodies against the BCoV HE protein efficiently neutralized BCoV infectivity in vitro (Deregt & Babiuk, 1987) and protected the intestinal epithelium of cattle from virus infection in vivo (Deregt et al., 1989), indicating that the HE protein of BCoV may also play a significant role in the induction of protective effect on BCoV infection besides S protein. In this study, the high prevalence of the HE recombinant strain was found in China, which has a significant implication on BCoV vaccine development subsequently in China. It is also interesting to further investigate the protective effect of the vaccine developed by BCoV prototype strain on the HE recombinant strain of BCoV. In this study, we made efforts to isolate the recombinant strain using HRT‐8 cell. Unfortunately, we failed. It may be caused by repeated freeze‐thaw of samples when they were used for detecting other enterovirus before the detection of BCoV. Now, we are monitoring the outbreak of diarrhoea in calves and plan to re‐collect diarrhoeic faecal samples to isolate the virus, in order to further investigate biological characteristics of the HE recombinant strain.
In conclusion, the results of our study have shown that BCoVs are circulating widely in dairy calves in China and that most of these strains have unique evolutionary pattern based on our phylogenetic analysis of the complete S, HE, N and M genes. Recombination events between the esterase and lectin domain of HE were identified as occurring at remarkably high frequencies, and these recombinant strains are widely prevalent in dairy cattle in China. As far as we are aware, this is the first description of a recombination event in the HE gene of BCoV, and our findings will enhance current understanding about the genetic evolution of BCoV.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
ETHICAL APPROVAL
This study did not involve animal experiments besides the faecal sampling of diarrhoea dairy calves that visited farm for clinical treatment.
ACKNOWLEDGEMENT
This work was funded by the 13th Five‐Year Plan National Science and Technology Support Program (grant number 2016YFD0500907), the Innovation team for animal epidemic diseases prevention and control on Qinghai–Tibet Plateau, State Ethnic Affairs Commission (grant number 13TD0057) and the Innovative Research Project of Graduate Students in Southwest University for Nationalities (grant number CX2017SZ058). We thank Sandra Cheesman, PhD, from Liwen Bianji, Edanz Group China, for proofreading the English grammar of drafts of this manuscript.
keha A, Xue L, Yan S, Yue H, Tang C. Prevalence of a novel bovine coronavirus strain with a recombinant hemagglutinin/esterase gene in dairy calves in China. Transbound Emerg Dis. 2019;66:1971–1981. 10.1111/tbed.13228
Funding information
The 13th Five‐Year Plan National Science and Technology Support Program (grant number 2016YFD0500907), the Innovation team for animal epidemic diseases prevention and control on Qinghai–Tibet Plateau, State Ethnic Affairs Commission (grant number 13TD0057) and the Innovative Research Project of Graduate Students in Southwest University for Nationalities (grant number CX2017SZ058).
REFERENCES
- Azizzadeh, M. , Shooroki, H. F. , Kamalabadi, A. S. , & Stevenson, M. A. (2012). Factors affecting calf mortality in Iranian holstein dairy herds. Preventive Veterinary Medicine, 104(3–4), 335–340. 10.1016/j.prevetmed.2011.12.007 [DOI] [PubMed] [Google Scholar]
- Bakkers, M. J. G. , Lang, Y. , Feitsma, L. J. , Hulswit, R. J. G. , Poot, S. A. H. D. , Vliet, A. L. W. V. , & Huizinga, E. G. (2017). Betacoronavirus adaptation to humans involved progress loss of hemagglutinin‐esterase lectin activity. Cell Host & Microbe, 21(3), 356 10.1016/j.chom.2017.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidokhti, M. R. , Tråvén, M. , Krishna, N. K. , Munir, M. , Belák, S. , Alenius, S. , & …Cortey, M. (2013). Evolutionary dynamics of bovine coronaviruses: Natural selection pattern of the spike gene implies adaptive evolution of the strains. Journal of General Virology, 94(9), 2036–2049. 10.1099/vir.0.054940-0 [DOI] [PubMed] [Google Scholar]
- Bok, M. , Miño, S. , Rodriguez, D. , Badaracco, A. , Nuñes, I. , Souza, S. P. , … Parreño, V. (2015). Molecular and antigenic characterization of bovine coronavirus circulating in argentinean cattle during 1994–2010. Veterinary Microbiology, 181(3‐4), 221–229. 10.1016/j.vetmic.2015.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, N. , Li, S. , Zhou, R. , Zhu, M. , He, S. , Ye, M. , & Zhu, J. (2017). Two novel porcine epidemic diarrhea virus (PEDV) recombinants from a natural recombinant and distinct subtypes of PEDV variants. Virus Research, 242, 90–95. 10.1016/j.virusres2017.09.013 [DOI] [PubMed] [Google Scholar]
- Deregt, D. , & Babiuk, L. A. (1987). Monoclonal antibodies to bovine coronavirus: chacteristics and topographical mapping of neutralizing epitopes on the E2 and E3 glycoproteins. Virology, 161(2), 0–420. 10.1016/0042-6822(87)90134-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregt, D. , Gifford, G. A. , Ijaz, M. K. , Watts, T. C. , Gilchrist, J. E. , Haines, D. M. , & Babiuk, L. A. (1989). Monoclonal antibodies to bovine coronavirus glycoproteins E2 and E3: Demonstration of in vivo virus‐neutralizing activity. Journal of General Virology, 70(4), 993–998. 10.1099/0022-1317-70-4-993 [DOI] [PubMed] [Google Scholar]
- Forni, D. , Filippi, G. , Cagliani, R. , De Gioia, L. , Pozzoli, U. , Al‐Daghri, N. , … Sironi, M. (2015). The heptad repeat region is a major selection target in MERS‐CoV and related corona‐ viruses. Scientific Reports, 5, 14480 10.1038/srep14480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, T. M. , & Buchmeier, M. J. (2001). Coronavirus spike proteins in viral entry and pathogenesis. Virology, 279(2), 371–374. 10.1006/viro.2000.0757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gélinas, A. M. , Boutin, M. , Sasseville, A. M. , & Dea, S. (2001). Bovine coronaviruses associated with enteric and respiratory diseases in Canadian dairy cattle display different reactivities to anti‐HE monoclonal antibodies and distinct amino acid changes in their HE, S and ns4.9 protein. Virus Research, 76(1), 43–57. 10.1016/S0168-1702(01)00243-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot, R. J. D. (2006). Structure, function and evolution of the hemagglutinin‐esterase proteins of corona‐ and toroviruses. Glycoconjugate Journal, 23(1–2), 59–72. 10.1007/s10719-006-5438-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasoksuz, M. , Sreevatsan, S. , Cho, K. O. , Hoet, A. E. , & Saif, L. J. (2002). Molecular analysis of the S1 subunit of the spike glycoprotein of respiratory and enteric bovine coronavirus isolates. Virus Research, 84(1), 101–109. 10.1016/S0168-1702(02)00004-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh, A. A. P. M. , Smeenk, I. , Horzinek, M. C. , Rottier, P. J. M. , & Groot, R. J. D. (1998). Feline coronavirus type ii strains 79–1683 and 79–1146 originate from a double recombination between feline coronavirus, type I and canine coronavirus. Journal of Virology, 72(5), 4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, K. R. , Ye, R. , Goebel, S. J. , Jayaraman, P. , & Masters, P. S. (2010). An interaction between the nucleocapsid protein and a component of the replicase‐transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. Journal of Virology, 84(19), 1027 10.1128/JVI.01287-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, K. K. , & Pendell, D. L. (2017). Market impacts of reducing the prevalence of bovine respiratory disease in United States beef cattle feedlots. Frontiers in Veterinary Science, 4, 189 10.3389/fvets.2017.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienzle, T. E. , Abraham, S. , Hogue, B. G. , & Brian, D. A. (1990). Structure and orientation of expressed bovine coronavirus hemagglutinin‐esterase protein. Journal of Virology, 64(4), 1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin, N. , Miszczak, F. , Lin, W. , Gouilh, M. , Vabret, A. , & Consortium, E. (2015). Genomic analysis of 15 human coronaviruses OC43 (HCoV‐OC43s) circulating in france from 2001 to 2013 reveals a high intra‐specific diversity with new recombinant genotypes. Viruses, 7(5), 2358–2377. 10.3390/v7052358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, C. K. , Kang, M. I. , Lim, G. K. , Kim, G. Y. , Yoon, S. S. , Park, J. T. , & Cho, K. O. (2006). Molecular characterization of HE, M, and E genes of winter dysentery bovine coronavirus circulated in korea during 2002–2003. Virus Genes, 32(2), 129–136. 10.1007/s11262-005-6867-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, M. M. C. , & Cavanagh, D. (1997). The molecular biology of coronaviruses. Advances in Virus Research, 48(48), 1–100. 10.1016/S0168-1702(96)01421-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langereis, M. A. , Zeng, Q. , Heesters, B. , Huizinga, E. G. , & Groot, R. J. D. (2012). The murine coronavirus hemagglutinin‐esterase receptor‐binding site: A major shift in ligand specificity through modest changes in architecture. PLoS Path, 8(1), e1002492 10.1371/journal.ppat.1002492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S. K. , Lee, P. , Tsang, A. K. , Yip, C. C. , Tse, H. , Lee, R. A. , & Yuen, K. Y. (2011). Molecular epidemiology of human coronavirus oc43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. Journal of Virology, 85(21), 11325 10.1128/JVI.05512-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S. K. P. , Li, K. S. M. , Huang, Y. , Shek, C.‐T. , Tse, H. , Wang, M. , … Yuen, K.‐Y. (2010). Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome‐related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self‐limiting infection that allows recombination events. Journal of Virology, 84(6), 2808 10.1128/JVI.02219-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. (2016). Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology, 3(1), 1954–1964. 10.1146/annurev-virology-110615-042301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Zhang, C. , Sui, J. , Kuhn, J. H. , Moore, M. J. , Luo, S. , … Farzan, M. (2005). Receptor and viral determinants of SARA‐coronavirus adaptation to human ACE 2. Embo Journal, 24(8), 1634–1643. 10.1038/sj.emboj.7600640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Z. , & Weiss, S. R. (1998). Roles in cell‐to‐cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology, 244(2), 483–494. 10.1006/viro.1998.9121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luytjes, W. , Bredenbeek, P. J. , Noten, A. F. , Horzinek, M. C. , & Spaan, W. J. (1988). Sequence of mouse hepatitis virus A59 mRNA 2: Indications for RNA recombination between coronaviruses and influenza c virus. Virology, 166(2), 415–422. 10.1016/0042-6822(88)90512-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Murrell, B. , Golden, M. , Khoosal, A. , & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus. Evolution, 1(1), vev003 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez, N. , Brandão, P. E. , de Souza, S. P. , Barrera, M. , Santana, N. , de Arce, H. D. , … Pérez, L. J. (2012). Molecular and phylogenetic analysis of bovine coronavirus based on the spike glycoprotein gene. Infection Genetics & Evolution, 12(8), 1870–1878. 10.1016/j.meegid.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrosovich, M. , Herrler, G. , & Klenk, H. D. (2013). Sialic acid receptors of viruses. Topics in Current Chemistry, 367, 1–28. 10.1007/128_2013_466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery, V. D. , Graham, R. L. , & Baric, R. S. (2017). Jumping species—a mechanism for coronavirus persistence and survival. Current Opinion in Virology, 23, 1–7. 10.1016/j.coviro.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami, S. , Kuroda, Y. , Terada, Y. , Yonemitsu, K. , Van Nguyen, D. , Kuwata, R. , … Maeda, K. (2016). Detection of novel ferret coronaviruses and evidence of recombination among ferret coronaviruses. Virus Genes, 52(6), 1–5. 10.1007/s11262-016-1365-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostra, M. , Haan, C. A. M. D. , Groot, R. J. D. , & Rottier, A. P. J. M. (2006). Glycosylation of the severe acute respiratory syndrome coronavirus triple‐spanning membrane proteins 3A and M. Journal of Virology, 80(5), 2326 10.1128/JVI.80.5.2326-2336.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S.‐J. , Jeong, C. , Yoon, S.‐S. , Choy, H. E. , Saif, L. J. , Park, S.‐H. , … Cho, K.‐O. (2006). Detection and characterization of bovine coronaviruses in fecal specimens of adult cattle with diarrhea during the warmer seasons. Journal of Clinical Microbiology, 44(9), 3178–3188. 10.1128/JCM.02667-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. J. , Lim, G. K. , Park, S. I. , Kim, H. H. , Koh, H. B. , & Cho, K. O. (2010). Detection and molecular characterization of calf diarrhoea bovine coronaviruses circulating in South Korea during 2004–2005. Zoonoses & Public Health, 54(6–7), 223–230. 10.1111/j.1863-2378.2007.01045.x [DOI] [PubMed] [Google Scholar]
- Peng, G. , Xu, L. , Lin, Y. L. , Chen, L. , Pasquarella, J. R. , Holmes, K. V. , & Li, F. (2012). Crystal structure of bovine coronavirus spike protein lectin domain. Journal of Biological Chemistry, 287(50), 41931 10.1074/jbc.M112.418210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze, B. , Wahn, K. , Klenk, H. D. , & Herrler, G. (1991). Isolated he‐protein from hemagglutinating encephalomyelitis virus and bovine coronavirus has receptor– destroying and receptor‐binding activity. Virology, 180(1), 221‐228. 10.1016/0042-6822(91)90026-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan, T. , Rockx, B. , Donaldson, E. , Sims, A. , Pickles, R. , Corti, D. , & Baric, R. (2008). Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. Journal of Virology, 82(5), 2274–2285. 10.1128/JVI.02041-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits, S. L. , Gerwig, G. J. , van Vliet, A. L. , Lissenberg, A. , Briza, P. , Kamerling, J. P. , & de Groot, R. J. (2005). Nidovirus sialate‐o‐acetylesterases: Evolution and substrate specificity of coronaviral and toroviral receptor‐destroying enzymes. The Journal of Biological Chemistry, 280(8), 6933–6941. 10.1074/jbc.M409683200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Liu, D. I. , Shi, W. , Lu, R. , Wang, W. , Zhao, Y. , … Tan, W. (2015). Origin and possible genetic recombination of the middle east respiratory syndrome coronavirus from the first imported case in china: Phylogenetics and coalescence analysis. Molecular Biology and Physiology, 6(5), 01280–1315. 10.1128/mBio.01280-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau, S. K. , Huang, Y. , & Yuen, K. Y. (2009). Coronavirus diversity, phylogeny and interspecies jumping. Experimental Biology & Medicine, 234(10), 1117–1127. 10.3181/0903-MR-94 [DOI] [PubMed] [Google Scholar]
- Yoo, D. , & Deregt, D. (2001). A single amino acid change within antigenic domain ii of the spike protein of bovine coronavirus confers resistance to virus neutralization. Clinical & Diagnostic Laboratory Immunology, 8(2), 297 10.1128/CDLI.8.2.297-302.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]