

Abstract
Increased evidence of porcine deltacoronavirus (PDCoV) causing diarrhoea in pigs has been reported in several countries worldwide. The virus has currently evolved into three separated groups including US, China and Southeast Asia (SEA) groups. In Vietnam, PDCoV was first reported in 2015. Based on phylogenetic analyses of spike, membrane and nucleocapsid genes, it is suggested that Vietnam PDCoV is chimeric virus. In the present study, we retrospectively investigated the presence of PDCoV in Vietnam and the full‐length genomes of six PDCoV isolates identified in 2014–2016 were further characterized. The results demonstrated that Vietnam PDCoV was first detected as early as 2014. All six Vietnam PDCoV are in the SEA group and further divided into two separated subgroups including SEA‐1 and SEA‐2. Vietnam PDCoV in SEA‐2 was closely related to Thai and Lao PDCoV. Recombination analysis demonstrated that three isolates in SEA‐1 were a chimeric virus of which P12_14_VN_0814, the first Vietnam isolate, and US PDCoV isolates were major and minor parents, respectively. The recombination was further evaluated by phylogenetic construction based on 3 recombinant fragments. The first and third fragments, closely related to P12_14_VN_0814, were associated with ORF1a/1b and N genes, respectively. The second fragment, associated with S, E, and M genes, was closely related to US PDCoV isolates. High antigenic and hydrophobic variations were detected in S1 protein. Three‐day‐old pigs challenged with the chimeric virus displayed clinical diseases and villus atrophy. In conclusion, Vietnam PDCoV is genetically diverse influenced by an external introduction from neighbouring countries. The chimeric Vietnam PDCoV can induce a disease similar to Thai PDCoV.
Keywords: chimeric virus, full‐length genome, Porcine deltacoronavirus, Vietnam, viral infectivity
1. INTRODUCTION
Porcine deltacoronavirus (PDCoV) is an enveloped, positive‐sense, single‐stranded RNA virus belonged to the genus Deltacoronavirus, family Coronaviridae, order Nidovirales (Woo, Huang, Lau, & Yuen, 2010). PDCoV is the causative agent of a newly emerged enteric disease in pigs characterized by watery diarrhoea, dehydration and death with 40%–80% mortality following an outbreak (Jung, Hu, & Saif, 2016). PDCoV infects enterocytes of the small intestine leading to villous atrophy and malabsorption (Hu et al., 2015; Jung et al., 2015). Clinical signs caused by PDCoV resemble those with two viruses in the genus Alphacoronavirus, family Coronaviridae, including porcine epidemic diarrhoea virus (PEDV) and transmissible gastroenteritis virus (TGEV) (Jung et al., 2015), but severity of clinical disease is lesser and piglet mortality is lower. PEDV and TGEV infect pigs of all ages and cause 100% morbidity, with 80%–100% mortality rates in piglets. The higher severity of PEDV and TGEV infection in pigs indicates that the two viruses are more suitable for infecting pigs. On the other hand, PDCoV may not be perfectly adapted to pigs, and the virus may be evolving towards better ability to infect a specific host. Recently, an experimental study in calves suggested that calves are susceptible to infection by PDCoV but not PEDV (Jung, Hu, & Saif, 2017).
The full‐length genome of PDCoV is approximately 25 kb in length and contains seven essential genes, including open reading frame 1a/1b (ORF1a/1b), spike (S), envelop (E), membrane (M), and nucleocapsid (N), and specific accessory genes including non‐structural protein 6 (Nsp6) and Nsp7, flanked by a 5′‐ and 3′‐untranslated region (UTR) (Woo et al., 2012). ORF1a and ORF1b, occupying two‐thirds of the genome, encode 2 overlapping viral replicase/transcriptase polyproteins, 1a and 1b, which are cleaved into 15 Nsps, namely Nsp2‐16 (Zhang & Yoo, 2016). The S gene encodes S protein which consists of two domains called S1 and S2 domain. The S1 domain plays an important role in binding to specific host cell receptors and contains a neutralizing epitope. The S2 domain functions in membrane fusion (Shang et al., 2018). The E and M genes are transmembrane proteins associated with viral envelope formation and virus release (Woo et al., 2010). On the other hand, the functions of the N gene are associated with viral RNA replication and pathogenesis (Lee & Lee, 2015).
PDCoV was first detected in Hong Kong (HKU15‐44 and HKU15‐155 isolates) in 2012 (Woo et al., 2012). Although it was first detected, there was no association with clinical disease. The first evidence of PDCoV causing a disease was first evident in Ohio, USA in 2014. Soon after the first emergence in Ohio, USA, the virus was then reported in 18 states (Jung et al., 2016). The rapid widespread of PDCoV was evident when the virus was subsequently reported in China, South Korea, Thailand, Laos, and Vietnam in 2015 (Le et al., 2018; Lee & Lee, 2014; Madapong et al., 2016; Marthaler, Jiang, Collins, & Rossow, 2014; Saeng‐Chuto, Lorsirigool, et al., 2017; Song et al., 2015; Wang, Byrum, & Zhang, 2014a, 2014b). Although the first detection of PDCoV in those countries was in 2014–2015, the retrospective investigation of intestinal samples demonstrated that the presence of PDCoV in China, USA and Thailand was as early as 2004, 2013 and 2013, respectively (Saeng‐Chuto, Stott, et al., 2017; Sinha, Gauger, Zhang, Yoon, & Harmon, 2015).
Presently, PDCoV has evolved into three separated groups, including US, China and Southeast Asia (SEA). PDCoV isolates from Thailand and Laos were clustered in a novel PDCoV group, SEA. The SEA group is genetically distinct from US and China PDCoV groups (Madapong et al., 2016; Saeng‐Chuto, Lorsirigool, et al., 2017; Saeng‐Chuto, Stott, et al., 2017). In Vietnam, PDCoV was first detected in 2015 (Saeng‐Chuto, Lorsirigool, et al., 2017). Genetic analyses based on S, M, and N genes demonstrated that Vietnam PDCoV isolates were genetically different from the SEA group (Saeng‐Chuto, Lorsirigool, et al., 2017). The phylogenetic analyses based on S and M genes demonstrated that Vietnam PDCoV is clustered in the SEA group. However, phylogenetic analysis based on the N gene demonstrated contradictory results, since Vietnam PDCoV was clustered in the US group. The findings implied that Vietnam PDCoV could potentially be a recombinant virus. Whether or not this genotype of PDCoV isolates predominantly existing in Vietnam remains not known. We therefore conducted a retrospective study investigating the presence of PDCoV in intestinal samples collected from pigs less than 5 days old experiencing severe diarrhoea, in Vietnam from 2011 to 2016. The full‐length genome sequences of the detected Vietnam PDCoV were further characterized on the basis of heterogenicity, and recombination analyses together with virus infectivity in cell culture and pathogenicity in piglets were performed.
2. MATERIALS AND METHODS
2.1. Samples and viruses
One hundred and eight intestinal samples collected from 49 swineherds with clinical diarrhoea outbreaks in different regions in Vietnam (Table 1) were collected for viral pathogens causing diarrhoea during 2011–2016. All intestinal samples were individually minced using an Axygen™ Tissue Grinder (Thermo Fisher Scientific) and resuspended in 1X phosphate‐buffered saline (PBS; 0.1 M, pH 7.2). The suspensions were centrifuged at 10,000 × g for 10 min at 4°C, and the supernatant filtered through a 0.45‐µm nylon membrane (Corning Inc.). Virus was isolated, and plaque purification was performed as described in Data S1.
Table 1.
Intestinal sample number that was collected from different regions in Vietnam in 2011–2016
| Year | Regions | No. farms | Total samples | +ve PDCoV | +ve PEDV | +ve TGEV |
|---|---|---|---|---|---|---|
| 2016 | Middle | 2 | 6 | 0 | 5 | 0 |
| South | 2 | 4 | 3 | 4 | 0 | |
| 2015 | South | 14 | 29 | 6 | 15 | 0 |
| 2014 | South | 21 | 47 | 2 | 43 | 0 |
| 2013 | South | 3 | 9 | 0 | 7 | 0 |
| 2012 | South | 3 | 6 | 0 | 6 | 0 |
| 2011 | South | 4 | 7 | 0 | 7 | 0 |
| Total | 49 | 108 | 11 | 87 | 0 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
2.2. Virus detection and full‐length genome sequencing
Viral RNA was extracted from the supernatant using Nucleospin Viral RNA Extraction Kit (Macherey‐Nagel Inc.) and converted to cDNA using M‐MuLV Reverse Transcriptase (New England Biolabs Inc.). All samples were tested with the specific primer for S gene of PEDV and specific primer for N gene of TGEV as in previous studies (Lee & Lee, 2014; Park et al., 2007; Song et al., 2015; Temeeyasen et al., 2014; Wang, Byrum, & Zhang, 2014a; Woo et al., 2010). PDCoV was detected by PCR using specific primer for M and N genes as previously described (Wang, Byrum, & Zhang, 2014b). The PCR products were electrophoresed at 100 V for 30 min on a 1% agarose gel, and the gel was stained with RedSafe™ nucleic acid staining solution (iNtRON Biotechnology Inc.) and examined under a UV light. The PCR products were purified using Nucleospin Gel and PCR Clean‐up kit (Macherey‐Nagel Inc.), and sequencing was performed by First BASE Laboratories Inc. PDCoV‐positive samples were subjected to whole‐genome sequencing using 26 primers (Table S1). All specific PCR products were purified using Nucleospin Gel and PCR Clean‐up kit (Macherey‐Nagel Inc.), and sequencing was done by First BASE Laboratories Inc., using an ABI Prism 3730XL DNA sequencer.
2.3. Phylogenetic and genetic analyses
To determine the genetic relationship between the six Vietnam PDCoV isolates and other PDCoV isolates, the nucleotide (nt) and amino acid (aa) sequences were aligned using the CLUSTALW program (Thompson, Higgins, & Gibson, 1994), together with 84 other PDCoV isolates available in GenBank (Table S2). The nt and aa identities were determined using the sequence identity matrix function implemented in BioEdit software (Hall, 1999). The phylogenetic tree based on the full‐length genome, open reading frame (ORF) 1a/1b, SEMN and S, M and N genes was separately built using the maximum likelihood (ML) method with 1,000 bootstrap replicates, and substitution models were selected by the best‐fit substitution model function implemented in MEGA software version 6.0 (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013). TN93 + G+I model was used in phylogenetic trees based on full‐length genome and SEMN gene construction. TN93 + G model was used in phylogenetic trees based on ORF1a/1b and S genes construction. K2 + G and K2 + G+I models were used in the construction of phylogenetic trees based on M and N genes, respectively.
2.4. Recombination analysis and phylogenetic reconstruction based on recombination fragments
Recombination of six Vietnam PDCoV isolates was further analysed together with the 84 reference PDCoV isolates using automated RDP, GENECONV, BOOTSCAN, MaxChi, CHIMAERA and SISCAN methods implemented in RDP4 software (Martin, Murrell, Golden, Khoosal, & Muhire, 2015). Major and minor parents and breakpoints also were detected, and recombination fragments were generated with the RDP4 software (Martin et al., 2015).
Phylogenetic trees based on recombinant fragments were separately constructed using the ML method with 1,000 bootstrap replicates, and substitution models were designated by the best‐fit substitution model function implemented in MEGA software version 6.0 (Tamura et al., 2013). TN93 + G+I model was used in phylogenetic tree based on all recombination fragments construction.
2.5. Sliding window, antigenicity and hydrophobicity analyses
The sliding window, hydrophobicity and antigenicity were evaluated among the chimeric Vietnam PDCoV isolates in this study, and suspected major and minor parents of the chimeric Vietnam PDCoV, respectively (Figure 2).
The nt sequence variation sites were predicted using dnaSP version 6 with 100 bp and a step size of 25 bp (Rozas et al., 2017). Antigenicity and hydrophobicity were also analysed using IEDB Analysis Resource (http://tools.immuneepitope.org/main) with the Kolaskar and Tongaonkar method and ExPASy‐ProtScale (http://web.expasy.org/protscale) with the Kyte–Doolittle method, respectively.
2.6. In vitro infection of recombinant Vietnam PDCoV
Viral growth curves of the three chimeric Vietnam PDCoV isolates and NT1_1215 isolate were compared. Each isolate was performed in quadruplicate. The NT1_1215 isolate is a Thai PDCoV isolated from a swineherd in Thailand experiencing a diarrhoea outbreak with piglet mortality at approximately 40%. The full‐length genome is deposited in GenBank with accession number KX361345.
The PDCoV‐positive samples were subjected to virus isolation using swine testicular (ST) cells (ATCC CRL‐1746). The viral isolation was approved by Institutional Biosafety Committee of Chulalongkorn University (CU‐IBC) (IBC 1831062). In brief, ST cells were seeded in T25 flasks (Corning, NY, USA) containing Advanced Minimum Essential Medium (adv. MEM) supplemented with 10% heat‐inactivated foetal bovine serum (FBS), 1% HEPES, 1% antibiotic‐antimycotic, 1% L‐glutamine (Gibco) and 0.0001% pancreatin from porcine pancreas (Sigma‐Aldrich) for each viral inoculation. After the cells reached 80% confluency, the cells were inoculated with each virus at a titre of 103 TCID50/ml. The infected cells were incubated at 37°C and 5% CO2. The viruses were harvested at different time points (0, 12, 24, 36, 48, 60 and 72 hr post‐inoculation (hpi.)) and stored at −80°C.
Viral titre at each time point was determined. ST cells were cultured in 96‐well plates (Corning, NY, USA) containing supplemented adv. MEM. Cells were inoculated with each virus at each time point at 10‐fold serial dilutions (100–107). Cytopathic effect was determined daily, and the growth curves were plotted.
2.7. In vivo infection of recombinant Vietnam PDCoV
Eighteen 3‐day‐old healthy piglets were randomly divided into three groups: (i) mock control group (n = 6), (ii) chimeric Vietnam PDCoV (P29_15_VN_1215) isolate‐infected group (n = 6), and (iii) NT1_1215 isolate‐infected group (n = 6). Each piglet was inoculated with 5 ml of each virus at a titre of 103 TCID50/ml. Clinical signs were observed daily. Three piglets per group were euthanized at 3 and 5 days post‐inoculation (dpi.), and five segments of the small intestine, including duodenum, proximal jejunum, middle jejunum, distal jejunum and ileum, were collected and fixed in 10% formalin. Haematoxylin and eosin (H&E) staining was performed, and villus/crypt ratio was calculated. Comparison between each group was performed with one‐way analysis of variance, followed by Tukey's multiple comparison test implemented in GraphPad Prism 7 (GraphPad Software Inc.). p < .05 was considered significant.
3. RESULTS
3.1. PDCoV detection
One hundred and eight intestinal samples collected from pigs with diarrhoea were assayed for the presence of three viral pathogens including PEDV, TGEV and PDCoV using PCR assays (Table 1). Of 108 intestinal samples tested, none were positive for TGEV. Eleven (10.19%) and 87 (80.56%) samples were positive for PDCoV and PEDV, respectively. All PDCoV‐positive samples were also positive for PEDV. PDCoV was detected in Vietnam as early as 2014, but the most detection was mainly in 2015–2016. Of 11 samples, only six samples including P12_14_VN_0814, P29_15_VN_1215, P30_15_VN_1215, P1_16_VN_0116, P19_16_VN_0416, and P20_16_VN_0416 isolates were able to characterize their full‐length genome sequences and further genetic analyses.
3.2. Full‐length genome characterization
The full‐length genome sequence of the six Vietnam PDCoV isolates, recognized as P12_14_VN_0814, P29_15_VN_1215, P30_15_VN_1215, P1_16_VN_0116, P19_16_VN_0416 and P20_16_VN_0416, were deposited in the GenBank database under accession numbers MH700628, KX998969, MH118333, MH118331, MH118332 and MH700629, respectively. The six isolates were varied in size, varying from 25,405–25,409 nt. The major difference is the deletion in nucleotides in ORF1a/1b region. The full‐length genome sequence of P12_14_VN_0814 isolate was 25,406 nt in length excluding poly(A) tail. P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 isolates were 3 nt longer in which their full‐length genome sequences were 25,409 nt in length excluding poly(A) tail. The full‐length genome sequence of P19_16_VN_0416 and P20_16_VN_0416 isolates were 25,405 nt in length excluding poly(A) tail. All isolates were characterized by the same gene order of the 5′‐UTR, ORF1a/1b, S, E, M, Nsp6, N, Nsp7 and 3′‐UTR. The length of each region is presented in Table S3.
Pairwise nt and aa sequence identity values of each gene between the six Vietnam PDCoV isolates together with two previously reported Vietnam PDCoV isolates, HaNoi6/2015 and Binh21/2015 isolates, and other reference PDCoV isolates are presented in Table S4. Based on the full‐length genome analysis, the P12_14_VN_0814, P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 isolates were closely related to previously reported HaNoi6/2015 and Binh21/2015 isolates (98.7%–100% nt) than Thai and Lao PDCoV isolates (98.1%–98.4% nt), US PDCoV isolates (97.5%–98.1% nt), Japan PDCoV isolates (97.2%–98.1% nt), South Korean PDCoV isolates (97.2%–98.0% nt) and China PDCoV isolates (97.1%–98.3% nt). In contrast, P19_16_VN_0416 and P20_16_VN_0416 isolates were more closely related to the Thai and Lao PDCoV isolates (99.1%–99.8% nt identity) than the HaNoi6/2015 and Binh21/2015 isolates (98.3%–98.4% nt), US PDCoV isolate (97.2%–97.5% nt), China PDCoV isolates (96.9%–97.7% nt), Japan PDCoV isolates (96.9%–97.3% nt) and South Korean PDCoV isolates (96.9%–97.3% nt).
The ORF1a/1b gene of all six Vietnam PDCoV isolates owns aa deletion and insertion in their genomes compared to other PDCoV isolates. The deletions of two (401LK402) and three (758PVG760) aa were observed at positions 401 to 402 and 758 to 760, respectively. Interestingly, these 5 aa deletions in the six Vietnam PDCoV isolates were similar to other PDCoV isolates from SEA countries. The insertion of one aa (51N) at position 51 of the S gene was found in all six Vietnam PDCoV isolates similar to US PDCoV isolates. Based on the E, M and N genes, all six Vietnam PDCoV isolates did not show any aa deletion or insertion compared to other PDCoV isolates.
3.3. Phylogenetic analysis
Phylogenetic trees constructed based on the full‐length genome and ORF1a/1b demonstrated that the six Vietnam PDCoV isolates were grouped in the SEA group together with Thai, Lao, and two previously reported Vietnam PDCoV isolates based on full‐length genome and ORF1a/1b gene sequences. The SEA group was further evolved into two subgroups including subgroup SEA‐1 and SEA‐2 (Figure 1a,b). P19_16_VN_0416 and P20_16_VN_0416 together with Thai and Lao PDCoV isolates were clustered in the subgroup SEA‐1. In contrast, P12_14_VN_0814, P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 were clustered in subgroup SEA‐2 (Figure 1a,b). Interestingly, PDCoV isolates in the subgroup SEA‐2 are from Vietnam only.
Figure 1.
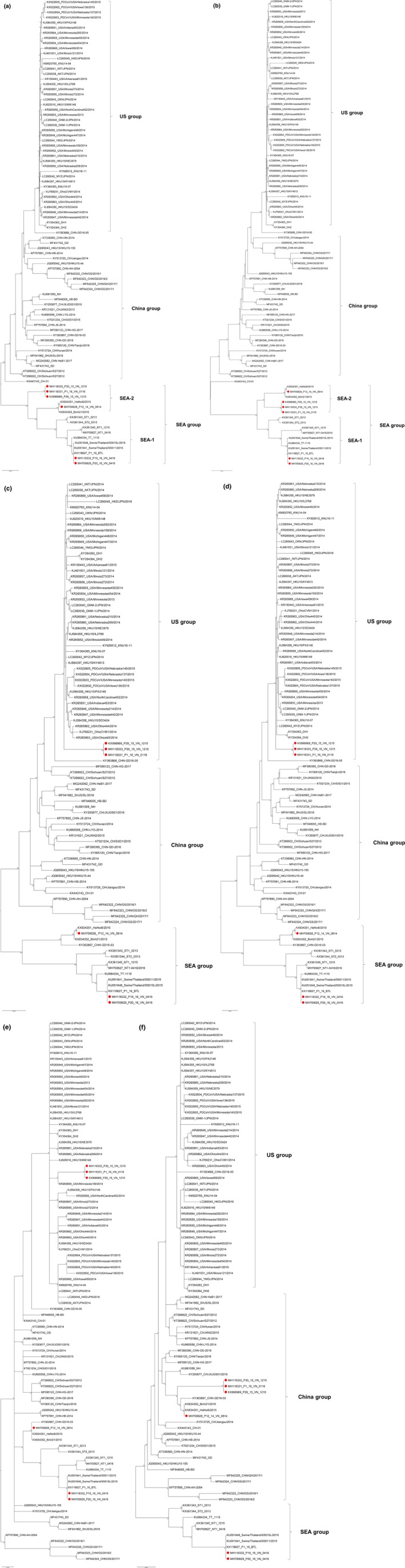
Phylogenetic trees based on nucleotide sequences of full‐length genome (a), ORF1a/1b (b), SEMN (c), S (d), M (e) and N (f) genes. The phylogenetic trees were constructed using the maximum likelihood (ML) method with 1,000 bootstrap replicates implemented in MEGA software version 6.0. Red dots represent the PDCoV isolates from Vietnam in this study. All phylogenetic trees, except M gene, consisted of 3 groups namely US, China, and Southeast Asia (SEA) groups. SEA group was divided into 2 subgroups namely SEA‐1 and SEA‐2 subgroups [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
Phylogenetic trees constructed based on SEMN and S genes demonstrated that P19_16_VN_0416 and P20_16_VN_0416, and P12_14_VN_0814 were clustered together with the SEA group, subgroup SEA‐1 and SEA‐2, respectively (Figure 1c,d). However, three of the six Vietnam PDCoV isolates, including P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116, were clustered together with the US group (Figure 1c,d).
The phylogenetic tree based on N gene demonstrated that P12_14_VN_0814, P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 were clustered together with the China group, differing from P19_16_VN_0416 and P20_16_VN_0416, which were clustered in the SEA group (Figure 1f).
3.4. Recombination analysis and phylogenetic reconstruction based on recombination fragments
Three recombination fragments were generated. The first and second fragments are at nt 1–19,453 and 23,511–25,426 (second fragment) in which are locations of ORF1a/1b and N genes, respectively. The third fragment is at nt 19,454–23,512 in which is located at S, E and M gene region (Figure 2). Recombination analysis suggested that three Vietnam PDCoV isolates including P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 isolates were recombination of the P12_14_VN_0814 isolate (major parent) at the first and third fragments, and USA/Minnesota159/2014 isolate (minor parent) at the second fragment (Figure 2). The phylogenetic trees based on the first and third fragments demonstrated that these three chimeric Vietnam PDCoV isolates were closely related to the P12_14_VN_0814, Hanoi/2015 and Binh21/2015 isolates (Figure 3a,c). However, the phylogenetic trees based on the second fragment suggested that that the three chimeric Vietnam PDCoV isolates were closely related to US PDCoV isolates (Figure 3b).
Figure 2.
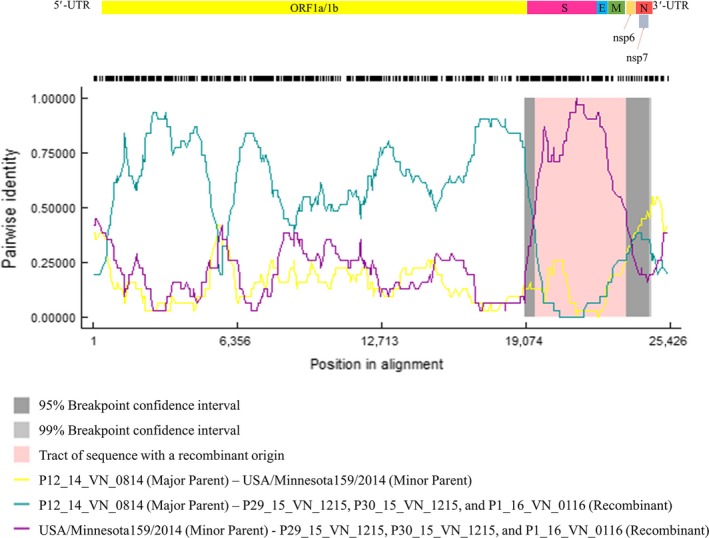
Recombination plot between P12_VN_14_0814, USA/Minnesota159/2014 and three Vietnam PDCoV isolates: P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 isolates as the major parent, minor parent and recombinant, respectively [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
Figure 3.
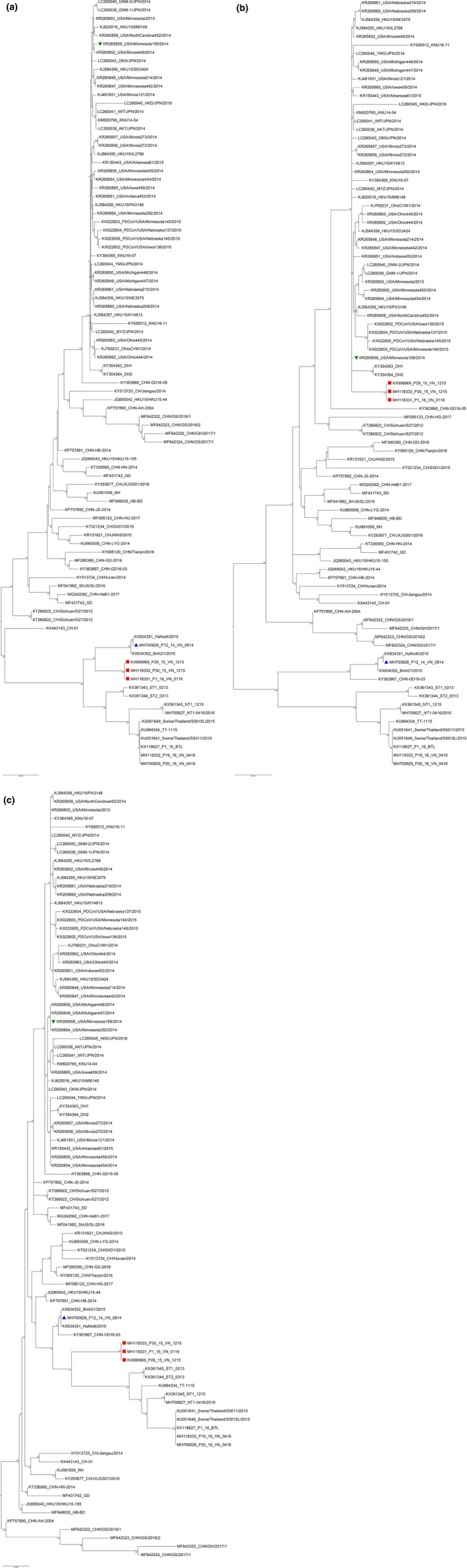
Reconstruction of phylogenetic trees based on the nucleotide sequence of three recombinant fragments, including first (a), second (b) and third (c) fragments. The phylogenetic trees were constructed using the maximum likelihood (ML) method with 1,000 bootstrap replicates implemented in MEGA software version 6.0. Red dots represent the PDCoV isolates from Vietnam in this study. Blue triangle represents P12_VN_14_0814 isolate (major parent). Green triangle represents USA/Minnesota159/2014 isolate (minor parent) [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
3.5. Sliding window, hydrophobicity and antigenicity analyses
The full‐length genome sequences of the five isolates including P29_15_VN_1215, P30_15_VN_1215, P1_16_VN_0116, P12_14_VN_0814 (major parent) and USA/Minnesota159/2014 (minor parent) were used in the sliding window analysis. Five sites, designated R1, R2, R3, R4 and R5, demonstrated high degree of genetic variation between all five isolates. Three sites including R1 (nt position 1,326–1,425), R2 (nt position 2,101–2,225) and R3 (nt position 5,601–5,775) were located within ORF1a/1b regions. Two sites including R4 (nt position 20,101–20,200) and R5 (nt position 25,001–25,100) were located within the S region, and between the N and 3′‐UTR regions (Figure 4).
Figure 4.
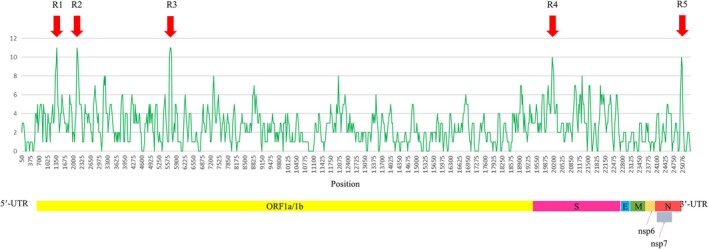
Differences in full‐length genome sequence between five isolates, including P29_15_VN_1215, P30_15_VN_1215, P1_16_VN_0116, P12_VN_14_0814 (major parent) and USA/Minnesota159/2014 (minor parent), were analysed by sliding window method. The red arrows show high variation regions [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
The aa of the S gene encoding the structural protein associated with binding and neutralizing epitopes were subjected for further antigenicity and hydrophobicity analyses. Six segments at positions aa 7–15, 36–44, 116–124, 134–139, 346–352 and 546–554 demonstrated a difference in hydrophobicity and antigenicity indices between the five isolates (Figure 5).
Figure 5.
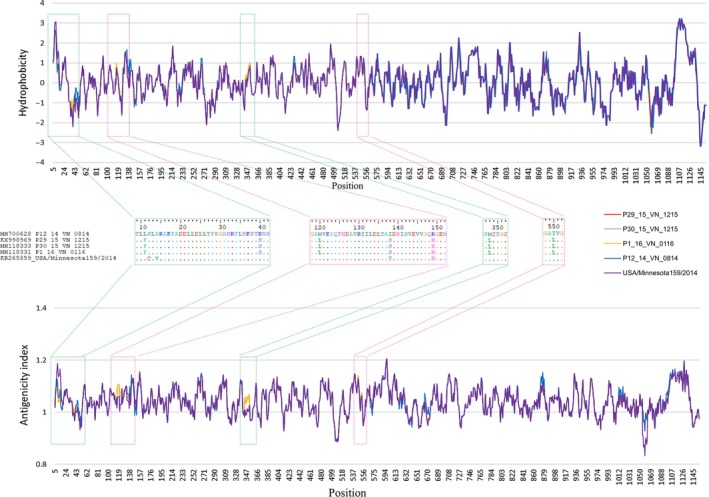
Hydrophobicity and antigenicity differences were found in S protein between the five isolates studied: P29_15_VN_1215, P30_15_VN_1215, P1_16_VN_0116, P12_VN_14_0814 (major parent) and USA/Minnesota159/2014 (minor parent) [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
3.6. In vitro infection with recombinant Vietnam PDCoV
Viral growth curves of the chimeric Vietnam PDCoV (P29_15_VN_1215) and Thai (NT1_1215) isolates in ST cells were compared. The two isolates had a similar pattern of viral growth curves, but virus titres were different (Figure 6). Cytopathic effect, characterized by cell rounding and clumping, with both PDCoV isolates were first observed at 12 hpi with a virus titre of 101 TCID50/ml (mean log10 TCID50/ml ± SD; 1 ± 0). At 24 hpi, the titre of P29_15_VN_1215 remained at 101 TCID50/ml (mean log10 TCID50/ml ± SD; 1 ± 0), while the titre of NT1_1215 increased to 102 TCID50/ml (mean log10 TCID50/ml ± SD; 2 ± 0). The titres of both PDCoV isolates slowly rose from 24 to 48 hpi. At 36 and 48 hpi, the titres of P29_15_VN_1215 were 102 (mean log10 TCID50/ml ± SD; 2 ± 0.25) and 104 TCID50/ml (mean log10 TCID50/ml ± SD; 4 ± 0.25), respectively, which were lower than those of NT1_1215 at 103 (mean log10 TCID50/ml ± SD; 3 ± 0.25) and 105 TCID50/ml (mean log10 TCID50/ml ± SD; 5 ± 0.25), respectively. The titres of both PDCoV isolates rapidly declined to 101 TCID50/ml at 60 hpi (mean log10 TCID50/ml ± SD; 1 ± 0.25). However, at 72 hpi., the titre of NT1_1215 increased to 102 TCID50/ml (mean log10 TCID50/ml ± SD; 2 ± 0), but the titre of P29_15_VN_1215 remained at 101 TCID50/ml (mean log10 TCID50/ml ± SD; 1 ± 0).
Figure 6.
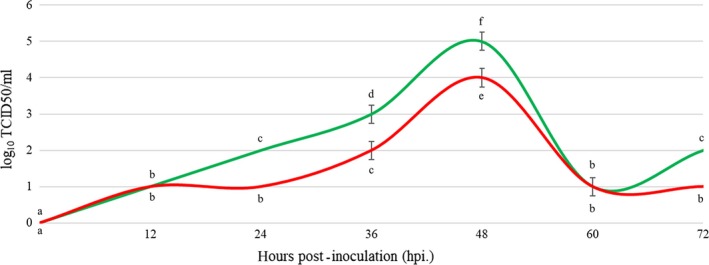
Viral growth curves of the Thai PDCoV (NT1_1215) (green line) and the chimeric Vietnam PDCoV (P29_15_VN_1215) (red line) isolates. Statistical significance between groups annotated by same lower‐case letters. p‐value < 0.05 [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
3.7. In vivo infection with recombinant Vietnam PDCoV
Pigs in the mock control group of which orally inoculated with culture media displayed no clinical disease throughout the study. In contrast, pigs orally inoculated with either Vietnam PDCoV isolates (P29_15_VN_1215) or Thai PDCoV isolate (NT1_1215 isolate) displayed similar level of clinical diseases associated with PDCoV including severe diarrhoea, vomiting, dehydration, weakness and lethargy. Clinical diseases were observed in both Vietnam and Thai PDCoV infected groups at 1 dpi. However, no piglets died before euthanasia.
At 3 and 5 dpi, 3 piglets in each group were necropsied and five parts of small intestine including duodenum, proximal jejunum, middle jejunum, distal jejunum and ileum were collected for further histopathological examination. H&E staining was performed. All H&E‐stained small intestine segments of piglets infected with P29_15_VN_1215 and NT1_1215 showed villous atrophy compared to that of the mock control group (G1) (Figure 7).
Figure 7.
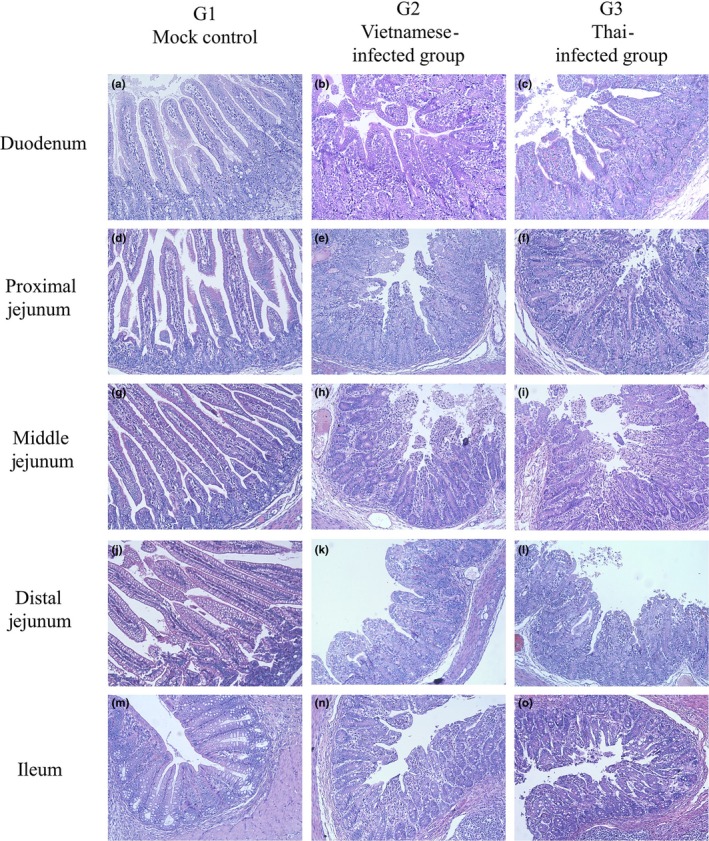
Haematoxylin and eosin (H&E) staining of five segments of small intestine, including duodenum (a–c), proximal jejunum (d–f), middle jejunum (g–i), distal jejunum (j–l) and ileum (m–o). Villous atrophy was observed in all small intestine segments of piglets infected with the chimeric Vietnam PDCoV (P29_15_VN_1215) (b, e, h, k and n) or the Thai PDCoV (NT1_1215) (c, f, i, l and o) isolates, compared to mock control group (a, d, g, j and m). Statistical significance between groups annotated by asterisk. p‐value < 0.05 [Colour figure can be viewed at http://www.wileyonlinelibrary.com/]
Villus/crypt (V/C) ratios of piglets in each group were determined (Figure 8). At 3 dpi, the V/C ratio in the three jejunum segments of piglets infected with both PDCoV isolates was significantly lower compared to that of the mock control group (p < .05) (Figure 8b–d). At 5 dpi, the V/C ratio in middle and distal jejunum segments of piglets infected with both PDCoV isolates was significantly lower than that in piglets in the mock control group (p < .05) (Figure 8c,d). Although no significant difference was observed, the V/C ratio in all parts part in both infected groups trend to reduced when compared between 3 and 5 dpi within the groups (Figure 8a,c).
Figure 8.
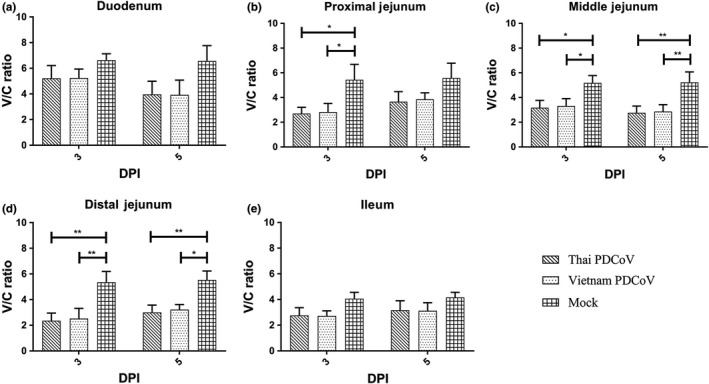
Villus to crypt (V/C) ratio of five segments of small intestine, including duodenum (a), proximal jejunum (b), middle jejunum (c), distal jejunum (d) and ileum (e)
4. DISCUSSION
In Vietnam, the first detection of PDCoV was reported in 2015 in the southern region of Vietnam (Saeng‐Chuto, Lorsirigool, et al., 2017). Genetic analyses based on S, M and N genes discovered contradictory results suggesting that Vietnam PDCoV could be a recombinant virus. However, the recombination of the virus has not been confirmed and the full‐length genome was not attempt in the previous study. We therefore conducted a retrospective study investigating the presence of PDCoV in Vietnam, genetic diversity and characterize the full‐length genome of detected PDCoV. The isolated chimeric virus was further evaluated for its pathogenicity.
In the present study, PDCoV was first detected in southern region of Vietnam as early as 2014. The full‐length genome sequences of six Vietnam PDCoV isolates were characterized. Phylogenetic analysis based on the full‐length genome demonstrated that Vietnam PDCoV was genetically diverse. The six Vietnam PDCoV isolates were grouped in the SEA cluster together with the Vietnam (Hanoi/2015 and Binh21/2015), Thai and Lao PDCoV isolates which all have an amino acid deletion at positions 401 to 402 and 758 to 760 located in the ORF1a gene. However, the six Vietnam PDCoV isolates were separated into two sub‐clusters recognized as SEA‐1 and SEA‐2. Two of six isolates were clustered in SEA‐1 together with Thai and Lao PDCoV isolates while other four isolates were clustered in SEA‐2 together with Hanoi/2015 and Binh21/2015 isolates.
Due to a separated origin, the findings from the present study suggest there were at least 2 possible external introductions of PDCoV into Vietnam. As we mentioned earlier, the Vietnam PDCoV isolated in 2016 (P19_16_VN_0416 and P20_16_VN_0416) shares high degree of genetic similarities and was clustered together with Thai and Lao PDCoV in subgroup SEA‐1, the virus isolates might be introduced from these neighbouring countries. Secondly, the external introduction of US‐related PDCoV was also observed. This introduction seemed to influence the evolution of Vietnam PDCoV as evidenced by chimeric PDCoV in subgroup SEA‐2. The Vietnam PDCoV isolates detected in 2014 (P12_VN_14_0814) were clustered together with other Vietnam PDCoV isolates, HaNoi6/2015 and Binh21/2015, in subgroup SEA‐2. The HaNoi6/2014 isolate was detected in Ha Noi province in the northern region of Vietnam since October 2015 and considered as recombinant virus between Thai and China isolates (Le et al., 2018). This study shows the P12_VN_14_0814 isolate shares 100% similarities with the HaNoi6/2014 isolate which means the P12_VN_14_0814 isolate could be also recombinant virus between Thai and China isolates. Interestingly, three of other four isolates in subgroup SEA‐2 were different. The P29_15_VN_1215, P30_15_VN_1215 and P1_16_VN_0116 isolates were confirmed to be chimeric virus which were a recombination between Vietnam PDCoV (P12_VN_14_0814), the major parent, and US PDCoV (USA/Minnesota159/2014), the minor parent. As previous reports, the recombination in RNA virus occurs when a host cell is infected with at least two viral genomes at the same time and affects host cell tropism, immune evasion, virulence and pathogenesis (Martin et al., 2011; Simon‐Loriere & Holmes, 2011). The findings in this study suggested that there are at least four different genome sequences of PDCoV currently circulating in swine farms in Vietnam. The virus may be trying to evolve, adapt to pigs, emerge and cause a disease. It is remained to be seen whether each of four different genome sequence groups would influence the genetic evolution of each other and become a dominant group, or continuously recombine and evolve in the population, resulting in the emergence of a novel viral isolate.
To analyse the nt variation between chimeric Vietnam PDCoV isolates and their parents, the sliding window method was used. The results showed that high nt variation was detected in five sites of the full‐length genomes. Most nt variations occurred in the ORF1a/1b gene. One of the five sites was found in the S gene which encodes the S protein. The hydrophobicity and antigenicity variations of the S protein were then analysed. Hydrophobicity is associated with protein–protein interaction (e.g. ligand of virus‐host receptor binding) and can be changed by replacement with hydrophobic amino acids (Bosch, Zee, Haan, & Rottier, 2003; Tekewe, Connors, Middelberg, & Lua, 2016), and change in antigenicity can affect immune response (Group et al., 2012; He, Li, Heck, Lustigman, & Jiang, 2006). Differences in hydrophobicity and antigenicity were detected in six parts in the S1 domain (aa 1–552) among the five isolates. The S1 domain plays an important role in host receptor binding and contains a neutralizing epitope (Shang et al., 2018; Woo et al., 2010). These results might support the proposition that the chimeric Vietnam PDCoV isolates are evolving towards improved host infection and immune response evasion.
To investigate the infectivity of the chimeric Vietnam PDCoV (P29_15_VN_1215) isolate compared to the Thai isolate (NT1_1215), both virus isolates were propagated in ST cells. The viral growth curves were developed and compared to each other. The results showed that the growth curve of chimeric Vietnam PDCoV isolate (P29_15_VN_1215) in ST cells was relatively lower compared to the Thai isolate at 12–60 hpi. This finding suggested that the chimeric Vietnam PDCoV isolate (P29_15_VN_1215) had less infectivity in ST cells than the Thai isolate.
To demonstrated whether or not this chimeric Vietnam PDCoV (P29_15_VN_1215) isolate can induce clinical disease, the virus was orally inoculated in piglets. Clinical diseases were compared to piglets that were orally inoculated with the Thai isolate. The results demonstrated that both PDCoV isolates can induce clinical diseases and pathogenicity in piglets. V/C ratio of piglets infected with chimeric Vietnam PDCoV was reduced but not significant difference compared to those of piglets infected with Thai PDCoV. The PDCoV infectivity results are in line with other studies. In 2015, two PDCoV isolates, OH‐FD22 and OH‐FD100, were also shown to cause disease. The infected gnotobiotic pigs showed severe diarrhoea, vomiting and severe atrophic enteritis (Jung et al., 2015). Moreover, in 2018, piglets were challenged with PDCoV KNU16‐07 isolate and showed similar clinical signs, namely diarrhoea, vomiting and viral enteritis (Jang et al., 2018). The findings in our study confirmed that the chimeric Vietnam PDCoV infection could induce small intestinal enteritis.
In conclusion, Vietnam PDCoV is genetically diverse influencing from the external introduction. Vietnam PDCoV is further evolved into 2 separated subgroups including SEA‐1 and SEA‐2. Vietnam PDCoV in SEA‐2 was closely related to Thai and Lao PDCoV. Recombination analysis demonstrated that isolates in SEA‐1 were a chimeric virus. P12_14_VN_0814, the first Vietnam isolate, and US PDCoV isolates were major and minor parents, respectively. The chimeric Vietnam PDCoV could induce clinical diseases.
CONFLICT OF INTEREST
The authors of this work have none connection with other institutions that unsuitable or bias in this work.
ETHICAL APPROVAL
All utilizable international, national, and/or institutional guidelines for concern and usage of animals were followed. The animal challenge study was approved by Institutional Animal Care and Use Committee of Chulalongkorn University (CU‐IACUC) (1931018).
Supporting information
ACKNOWLEDGEMENTS
This project was sponsored by Agricultural Research Development Agency (Public organization) (Grant number PRP6005020990), National Research Council of Thailand and Research and Researcher for Industry (Grant number PHD59I0094) for funding this research. Moreover, the partial funding was supported by Special Task Force for Activating Research (STAR), swine viral evolution and vaccine research (SVEVR), Chulalongkorn University.
Saeng‐chuto K, Jermsutjarit P, Stott CJ, Vui DT, Tantituvanont A, Nilubol D. Retrospective study, full‐length genome characterization and evaluation of viral infectivity and pathogenicity of chimeric porcine deltacoronavirus detected in Vietnam. Transbound Emerg Dis. 2020;67:183–198. 10.1111/tbed.13339
REFERENCES
- Bosch, B. J. , van der Zee, R. , de Haan, C. A. M. , & Rottier, P. J. M. (2003). The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. Journal of Virology, 77, 8801–8811. 10.1128/JVI.77.16.8801-8811.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- He, Y. , Li, J. , Heck, S. , Lustigman, S. , & Jiang, S. (2006). Antigenic and immunogenic characterization of recombinant baculovirus‐expressed severe acute respiratory syndrome coronavirus spike protein: Implication for vaccine design. Journal of Virology, 80, 5757–5767. 10.1128/JVI.00083-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. , Jung, K. , Vlasova, A. N. , Chepngeno, J. , Lu, Z. , Wang, Q. , & Saif, L. J. (2015). Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. Journal of Clinical Microbiology, 53, 1537–1548. 10.1128/JCM.00031-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, G. , Kim, S. H. , Lee, Y. J. , Kim, S. , Lee, D. S. , Lee, K. K. , & Lee, C. (2018). Isolation and characterization of a Korean porcine deltacoronavirus strain KNU16‐07. Journal of Veterinary Science, 19, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , Eyerly, B. , Lu, Z. , Chepngeno, J. , & Saif, L. J. (2015). Pathogenicity of 2 porcine deltacoronavirus strains in gnotobiotic pigs. Emerging Infectious Diseases, 21, 650–654. 10.3201/eid2104.141859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , & Saif, L. J. (2016). Porcine deltacoronavirus infection: Etiology, cell culture for virus isolation and propagation, molecular epidemiology and pathogenesis. Virus Research, 226, 50–59. 10.1016/j.virusres.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , & Saif, L. J. (2017). Calves are susceptible to infection with the newly emerged porcine deltacoronavirus, but not with the swine enteric alphacoronavirus, porcine epidemic diarrhea virus. Archives of Virology, 162, 2357–2362. 10.1007/s00705-017-3351-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, V. P. , Song, S. , An, B. H. , Park, G. N. , Pham, N. T. , Le, D. Q. , … An, D. J. (2018). A novel strain of porcine deltacoronavirus in Vietnam. Archives of Virology, 163, 203–207. 10.1007/s00705-017-3594-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2014). Complete genome characterization of Korean Porcine Deltacoronavirus Strain KOR/KNU14‐04/2014. Genome Announcements, 2, e01191–e1214. 10.1128/genomeA.01191-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2015). Functional characterization and proteomic analysis of the nucleocapsid protein of porcine deltacoronavirus. Virus Research, 208, 136–145. 10.1016/j.virusres.2015.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madapong, A. , Saeng‐Chuto, K. , Lorsirigool, A. , Temeeyasen, G. , Srijangwad, A. , Tripipat, T. , … Nilubol, D. (2016). Complete genome sequence of porcine deltacoronavirus isolated in Thailand in 2015. Genome Announcements, 4, e00408–e416. 10.1128/genomeA.00408-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Jiang, Y. , Collins, J. , & Rossow, K. (2014). Complete genome sequence of strain SDCV/USA/Illinois121/2014, a porcine deltacoronavirus from the United States. Genome Announcements, 2, e00218–e314. 10.1128/genomeA.00218-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Biagini, P. , Lefeuvre, P. , Golden, M. , Roumagnac, P. , & Varsani, A. (2011). Recombination in eukaryotic single stranded DNA viruses. Viruses, 3, 1699–1738. 10.3390/v3091699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Murrell, B. , Golden, M. , Khoosal, A. , & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1, vev003 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. J. , Moon, H. J. , Yang, J. S. , Lee, C. S. , Song, D. S. , Kang, B. K. , & Park, B. K. (2007). Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus Genes, 35, 321–332. 10.1007/s11262-007-0096-x [DOI] [PubMed] [Google Scholar]
- Rozas, J. , Ferrer‐Mata, A. , Sanchez‐DelBarrio, J. C. , Guirao‐Rico, S. , Librado, P. , Ramos‐Onsins, S. E. , & Sanchez‐Gracia, A. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34, 3299–3302. 10.1093/molbev/msx248 [DOI] [PubMed] [Google Scholar]
- Saeng‐Chuto, K. , Lorsirigool, A. , Temeeyasen, G. , Vui, D. T. , Stott, C. J. , Madapong, A. , … Nilubol, D. (2017). Different lineage of porcine deltacoronavirus in Thailand, Vietnam and Lao PDR in 2015. Transboundary and Emerging Diseases, 64, 3–10. 10.1111/tbed.12585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeng‐Chuto, K. , Stott, C. J. , Wegner, M. , Senasuthum, R. , Tantituvanont, A. , & Nilubol, D. (2017). Retrospective investigation and evolutionary analysis of a novel porcine deltacoronavirus strain detected in Thailand from 2008 to 2015. Archives of Virology, 162, 2103–2108. 10.1007/s00705-017-3331-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Zheng, Y. , Yang, Y. , Liu, C. , Geng, Q. B. , Tai, W. B. , … Li, F. (2018). Cryo‐electron microscopy structure of porcine deltacoronavirus spike protein in the Prefusion State. Journal of Virology, 92, JVI01556‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon‐Loriere, E. , & Holmes, E. C. (2011). Why do RNA viruses recombine? Nature Reviews Microbiology, 9, 617–626. 10.1038/nrmicro2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha, A. , Gauger, P. , Zhang, J. , Yoon, K. J. , & Harmon, K. (2015). PCR‐based retrospective evaluation of diagnostic samples for emergence of porcine deltacoronavirus in US swine. Veterinary Microbiology, 179, 296–298. 10.1016/j.vetmic.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , Zhou, X. , Peng, Q. , Chen, Y. , Zhang, F. , Huang, T. , … Tang, Y. (2015). Newly emerged porcine deltacoronavirus associated with diarrhoea in swine in China: Identification, prevalence and full‐length genome sequence analysis. Transboundary and Emerging Diseases, 62, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekewe, A. , Connors, N. K. , Middelberg, A. P. , & Lua, L. H. (2016). Design strategies to address the effect of hydrophobic epitope on stability and in vitro assembly of modular virus‐like particle. Protein Science, 25, 1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temeeyasen, G. , Srijangwad, A. , Tripipat, T. , Tipsombatboon, P. , Piriyapongsa, J. , Phoolcharoen, W. , … Nilubol, D. (2014). Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infection, Genetics and Evolution, 21, 205–213. 10.1016/j.meegid.2013.11.001 [DOI] [PubMed] [Google Scholar]
- Thompson, J. D. , Higgins, D. G. , & Gibson, T. J. (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Research, 22, 4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014a). Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerging Infectious Diseases, 20, 1227–1230. 10.3201/eid2007.140296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014b). Porcine coronavirus HKU15 detected in 9 US states, 2014. Emerging Infectious Diseases, 20, 1594–1595. 10.3201/eid2009.140756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Writing Group , Ampofo, W. K. , Baylor, N. , Cobey, S. , Cox, N. J. , Daves, S. , … Zhang, W. (2012). Improving influenza vaccine virus selection: Report of a WHO informal consultation held at WHO headquarters, Geneva, Switzerland, 14–16 June 2010. Influenza and Other Respiratory Viruses, 6, 142–152 e141–145.21819547 [Google Scholar]
- Woo, P. C. , Huang, Y. , Lau, S. K. , & Yuen, K. Y. (2010). Coronavirus genomics and bioinformatics analysis. Viruses, 2, 1804–1820. 10.3390/v2081803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau, S. K. , Lam, C. S. , Lau, C. C. , Tsang, A. K. , Lau, J. H. , … Yuen, K. Y. (2012). Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. Journal of Virology, 86, 3995–4008. 10.1128/JVI.06540-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , & Yoo, D. (2016). Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling. Virus Research, 226, 128–141. 10.1016/j.virusres.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials