

Abstract
Enterovirus species G (EV‐G) comprises a highly diversity of 20 genotypes that is prevalent in pig populations, with or without diarrhoea. In the present study, a novel EV‐G strain (KOR/KNU‐1811/2018) that resulted from cross‐order recombination was discovered in diagnostic faecal samples from neonatal pigs with diarrhoea that were negative for swine enteric coronaviruses and rotavirus. The recombinant EV‐G genome possessed an exogenous 594‐nucleotide (198‐amino acid) sequence, flanked by two viral 3Cpro cleavage sites at the 5′ and 3′ ends in its 2C/3A junction region. This insertion encoded a predicted protease similar to the porcine torovirus papain‐like cysteine protease (PLCP), which was recently found in the EV‐G1, ‐G2, and ‐G17 genomes. The complete KNU‐1811 genome shared 73.7% nucleotide identity with a prototype EV‐G1 strain, but had 83.9%–86.7% sequence homology with the global EV‐G1‐PLCP strains. Genetic and phylogenetic analyses demonstrated that the Korean recombinant EV‐G's own VP1 and inserted foreign PLCP genes are most closely related independently to contemporary chimeric G1‐PLCP and G17‐PLCP strains respectively. These results implied that the torovirus‐derived PLCP gene might have undergone continuous nucleotide mutations in the respective EV‐G genome following its independent acquisition through naturally occurring recombination. Our results advance the understanding of the genetic evolution of EV‐G driven by infrequent viral recombination events, by which EV‐G populations laterally gain an exotic gene encoding a virulence factor from heterogeneous virus families, thereby causing clinical disease in swine.
Keywords: cross‐order recombination, enterovirus G, horizontal gene transfer, papain‐like cysteine protease, porcine diarrhoea, Torovirus
Enteroviruses (EVs) are small, non‐enveloped viruses of 27 nm in diameter with icosahedral symmetry. They belong to the genus Enterovirus within the family Picornaviridae of the order Picornavirales, which contains 15 species (EV A–L and rhinovirus A–C) (Knowles et al., 2012). The EVs possess an approximately 7.4–7.5‐kb, single‐stranded, positive‐sense RNA genome, flanked by 5′ and 3′ untranslated regions (UTRs) and a 3′ poly(A) tail, which contains a single open reading frame (ORF). Genome translation yields a large polyprotein that is initially cleaved into three precursor protein products, which are further proteolytically matured to generate four structural (VP1–4) and seven nonstructural viral proteins (2Apro, 2B, 2C, 3A, 3B, 3Cpro, and 3Dpol) (Semler & Wimmer, 2002). Enteroviruses species comprise viruses that infect humans (species A–D), cows (species E and F), swine (species G), and nonhuman primates (A, B, D, H, and J) (Knowles et al., 2012). Porcine enteroviruses (PEVs) were originally classified into 13 types (PEV‐1 to PEV‐13). On the basis of genomic analyses, however, the original PEV‐1 to ‐7 and PEV‐11 to ‐13 have been reclassified and assigned to the genus Teschovirus; and PEV‐8, formally named PEV‐A, has been renamed porcine sapelovirus 1 and reclassified into the genus Sapelovirus (Kaku, Sarai, & Murakami, 2001). The remaining PEV‐9 and ‐10, formally belonging to PEV‐B, were reclassified as EV species G (EV‐G), which currently encloses 20 genotypes (Knowles et al., 2012) and has been reported in Europe, North America, and Asia (Anbalagan, Hesse, & Hause, 2014; Boros et al., 2012; Tsuchiaka et al., 2018; Van Dung et al., 2014).
The major hallmark of enteroviral evolution is represented by a heterogeneous group of viruses resulting from high mutation and recombination rates (Lukashev, 2005). In particular, genetic recombination, a key factor affecting EV genomic construction, occurs frequently between members of the same or different species, or infrequently between viruses belonging to two distinct families or orders (Muslin, Joffret, Pelletier, Blondel, & Delpeyroux, 2015). EV‐Gs have continued to undergo intratypic and intertypic recombination events and furthermore, EV‐G infections were reported to be endemic among swine herds in several pig‐raising countries (Tsuchiaka et al., 2018; Van Dung et al., 2014; Vilar et al., 2016). However, despite their genetic diversity and high prevalence, EV‐Gs have been generally relevant to subclinical diseases in their natural host. Recently, novel EV‐G1, ‐G2, and ‐G17 variants were reported in faecal samples from independent porcine diarrhoeic diseases in the United States, Belgium, and Japan. These recombinant viruses identically carry a porcine torovirus (ToV) papain‐like cysteine protease (PLCP) gene in the 2C/3A junction region of their genome, arising from a natural cross‐order recombination between EV‐G (order Picornavirales) and torovirus (order Nidovirales) (Conceição‐Neto et al., 2017; Knutson, Velayudhan, & Marthaler, 2017; Shang, Misra, Hause, & Fang, 2017; Tsuchiaka et al., 2018). Although the pathogenic association of EV‐Gs with enteric diseases has not been determined, acquiring ToV‐PLCP from a recombinant event appears to influence EV‐G pathogenesis by acting as an innate immune antagonist, thus leading to clinical manifestations in pigs (Shang et al., 2017)
Enteric illness is not only an important health problem in piglets but also causes substantial economic losses to the global pork industry. A large diversity of viral and bacterial species is known to cause clinical and subclinical enteritis in pigs. Among these aetiological agents, swine enteric coronaviruses (SECoVs), including transmissible gastroenteritis virus (TGEV), porcine epidemic diarrhoea virus (PEDV), and porcine deltacoronavirus (PDCoV), are considered to be representative enteropathogenic viruses that cause serious diarrhoea outbreaks, accompanied by high neonatal mortality, which pose significant financially threats in most pig‐producing countries. In South Korea, monoinfections of each SECoV or rotavirus, and their co‐infections, are typically responsible for diarrhoeal diseases in piglets, which are routinely diagnosed using a RT‐PCR‐based method in veterinary diagnostic laboratories (Jang, Lee, Kim, & Lee, 2017; Lee & Lee, 2018). In a minority of clinical cases, none of aforementioned viral pathogens could be detected in diarrhoeal samples, suggesting that other viruses are involved in porcine scours. Viral metagenomics studies indicated that kobuviruses, sapoviruses, astroviruses (AstV), and EVs are the most commonly detected porcine viruses in the faecal virome (Sachsenröder et al., 2012; Shan et al., 2011); therefore, genomic detection and sequencing analysis have been applied to detect these stool viruses from clinical diarrhoeic pigs that were negative for SECoVs and rotavirus. In this study, we identified a novel recombinant EV‐G from diarrhoeic piglets that contains a 198‐amino‐acid (aa) insertion encoding a ToV‐PCLP protein and characterized its complete genome to reveal new genetic features.
Three‐ to four‐week‐old piglets on a commercial farrow‐to‐finish farm located in Chungbuk Province experienced an outbreak of post‐weaning diarrhoea. Four faecal samples (KNU‐1811) from diarrhoeic weaned pigs were submitted to our laboratory in 7 April 2018 and were initially tested for SECoVs and rotavirus using RT‐PCR, which did not detect these enteric viral pathogens. Further RT‐PCR assays, followed by sequencing and BLAST searching, were conducted to identify two pig scours‐associated viruses, AstV and EV‐G, using virus‐specific primers (Conceição‐Neto et al., 2017; Lee, Jang, & Lee, 2015). Using this conventional approach, we were able to find the presence of a chimeric EV‐G possessing an approximately 600‐nucleotide (nt) insertion within the 2C/3A region from all four porcine diarrhoeic samples. Subsequently, ~1,200 nucleotides of sequence from the inserted regions flanked by the EV‐G 2C and 3A genes of the KNU‐1811 isolates from the same farm were determined by the traditional Sanger sequencing method. Initial nucleotide sequencing indicated that all of the KNU‐1811 strains were almost genetically identical to each other, with 99.6%−99.8% sequence similarity and identified an exogenous 594‐nt‐long sequence at the 2C/3A junction of the enterovirus genome (Figure 1a). The unique 594‐nt insertion sequence (encoding a deduced protein of 198 amino acids) was queried against the GenBank nt sequence database and found to be homologous to the PLCP sequence variants that were identified recently in recombinant EV‐G strains from the US, Belgium, and Japan (Conceição‐Neto et al., 2017; Knutson et al., 2017; Shang et al., 2017; Tsuchiaka et al., 2018). The length of the inserted PLCP sequence varied among strains from different geographical locations (194/223‐aa, 212‐aa, and 211/217‐aa for the US, Belgium, and Japanese strains respectively) (Figure 1b). The PLCP sequence of Korean EV‐G only shared 75.0%−89.3% aa sequence identity with other recombinant EV‐G1, ‐G2, and ‐G17 strains, exhibiting the highest identity with the US EV‐G17 strain 08/NC_USA/2015 (Shang et al., 2017). The viral 3Cpro protease trims the EV‐G precursor polyprotein posttranslationally into the 2C and 3A proteins at a well‐defined cleavage sequence (ALFQ↓GPPT) (Blom, Hansen, Brunak, & Blaas, 1996). Furthermore, a previous study using reverse genetics revealed that the US EV‐G17‐PLCP strain functionally produces the exogenous PLCP gene in virus‐infected cells, demonstrating its own ALFQ↓GPPV and AEFQ↓GPPT sequences as the putative cleavage sites (Shang et al., 2017). Considering the sequence similarity of the putative cleavage sites including GPPT−ALFQ, GPPA−ALFQ, and GPPE−ALPQ among global strains (Conceição‐Neto et al., 2017; Knutson et al., 2017; Tsuchiaka et al., 2018), therefore, the cleavage of the inserted PLCP gene appears to be guaranteed using each corresponding cleavage sequence. Consistently, the recombinant ToV‐PLCP of the EV‐G KNU‐1811 strain is bordered by two analogous predicted 3Cpro cleavage sites, ALFQ↓GPPV and AVFQ↓GPPT, at its N and C termini, respectively, indicating its proteolytic processing by 3Cpro into a functional product (Figure 1a). Subsequent phylogenetic analysis based on the PLCP genes of EV‐Gs and nidoviruses revealed that the foreign PLCP gene of KNU‐1811 is most closely related to that of the US EV‐G17 strain, forming a well‐supported cluster with PLCPs of other EV‐Gs, but is only distantly related to those of porcine, bovine, and equine toroviruses, showing lower sequence identities (41.0%−51.3% in the aa sequence) (Supporting Information Figure S1a).
Figure 1.
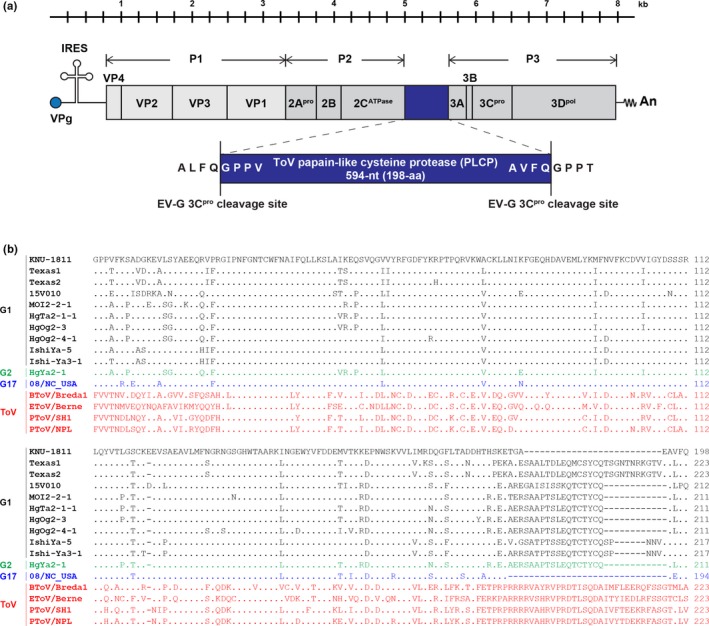
Schematic diagram of the genome organization of KOR/KNU‐1811/2018/G1‐PLCP. (a) The Korean KNU‐1811 genome contains a single open reading frame (ORF) flanked by a long 5′ untranslated region (UTR) (813‐nt) and a short 3′ UTR (71‐nt), followed by a poly (A) tail (An). The stem‐loop secondary structure on the 5′ UTR represents an internal ribosome entry site (IRES). The torovirus (ToV) papain‐like cysteine protease (PLCP) gene is presented as a blue box that is inserted at the 2C/3A cleavage junction. The 5′‐ and 3′‐boundary sequences of enterovirus (EV‐G) 3C protease (3Cpro) cleavage sites are shown in an enlarged blue box. Vertical lines indicate the polyprotein processing sites creating precursor polyproteins P1, P2, and P3 by 3Cpro. (b) Multiple alignment of the amino acid sequences of the PLCP regions of the recombinant EV‐G and ToV strains [Colour figure can be viewed at http://wileyonlinelibrary.com]
The full‐length genomic sequence of a representative Korean EV‐G isolate KNU‐1811 was determined using the Sanger method to investigate its genetic relationship to previously reported strains from other countries. To accomplish this, oligonucleotide primers were synthesized based on published known sequences and newly amplified KNU‐1811 sequences. Three overlapping cDNA fragments encompassing the entire viral genome were amplified using RT‐PCR with gene‐specific primer sets, individually cloned into a pGEM‐T Easy Vector System (Promega, Madison, WI), and sequenced in both directions using two commercial vector‐specific T7 and SP6 primers and KNU‐1811‐specific primers. The 5′ and 3′ ends of the KNU‐1811 genome were determined by rapid amplification of cDNA ends (RACE), as described previously (Lee & Lee, 2013). The KNU‐1811 strain sequence was deposited in the GenBank database under accession number MH663501. The complete genome of EV‐G KOR/KNU‐1811/2018 was 7,985‐nt in length, excluding the 3′ poly(A) tail. A single long ORF of 7,101‐nt encoding a 2,366‐aa polyprotein precursor was flanked by 813‐nt 5′ and 74‐nt 3′ UTRs. The P1, P2, and P3 regions of EV‐G KOR/KNU‐1811/2018 comprised 2,505‐nt (835‐aa), 1,734‐nt (578‐aa), and 2,268‐nt (755‐aa) respectively. The complete genome and VP1 sequences of KNU‐1811 shared 73.7% nt and 85.1% aa identities, respectively, with those of the prototype EV‐G strain PEV9/UKG/410/73. Compared with other chimeric EV‐G1, ‐G2, and ‐G17 strains, KNU‐1811 showed relatively high sequence homology (83.9%–86.7% nt and 93.6%–94.6% aa at the genome and VP1 gene levels respectively) with the US, Belgium, and Japanese EV‐G1 strains, but exhibited lower (79.9%–81.2% nt and 61.4%–62.5% aa) identities at the genome and VP1 levels with the Japanese EV‐G2 and US EV‐G17 strains respectively. The per cent nt and aa similarity between the KNU‐1811 and prototypic or recombinant EV‐G strains are summarized in Supporting Information Table S1.
EV‐G classification is solely based on sequence identities of the VP1 gene; therefore, phylogenetic analysis was carried out using the EV‐G VP1 sequences, including those determined in the present study and those available from GenBank (Figure 2a). The Korean KNU‐1811 strain clustered within the G1 genotype, along with other recombinant EV‐G1 strains. Similarly, a phylogenetic tree based on the EV‐G complete genome sequences revealed that the Korean G1‐PLCP strain clustered together with global G1‐PLCP strains (Figure 2b). However, all G1‐PLCP strains, including KNU‐1811, were phylogenetically distant from the authentic G1 strains, but appeared to share a common ancestor within the large clade containing the EV‐G genotypes G3, 9, and 10. Furthermore, phylogenetic analysis excluding the PLCP sequence in all chimeric strains resulted in the same tree topology (Supporting Information Figure S1b). These data indicated that the insertion has occurred through viral recombination and substantial nucleotide variations among related EV‐G genotypes have arisen throughout the entire genome.
Figure 2.

Phylogenetic analysis based on nucleotide sequences of the VP1 genes (a) and full‐length genomes (b) of EV‐G strains. Multiple sequence alignments were created using the ClustalX 2.0 program, and the phylogenetic trees were constructed from the aligned nucleotide sequences using the neighbour‐joining method. Numbers at each branch represent bootstrap values greater than 50% of 1,000 replicates. Hosts of origin, geographical origins, names of the strains, years of isolation, genotypes, and GenBank accession numbers are shown. The genotypes are indicated on the right‐hand side. A solid circle denotes the Korean recombinant EV‐G1‐PLCP strain identified in this study; solid diamonds indicate global EV‐G PLCP strains reported in previous studies. Scale bars indicate nucleotide substitutions per site
In conclusion, we identified a recombinant virus characterized by recombining a foreign porcine ToV‐PLCP gene into the 2C/3A junction region of the EV‐G1 genome from post‐weaning diarrhoea cases in pigs. To the best our knowledge, this is the first documentation of EV‐G recombinant (designated EVG/Porcine/KOR/KNU‐1811/2018/G1‐PLCP) in a Korean pig population. Similar to recent identifications reported in the US, Belgium, and Japan, the current study provided another example of horizontal viral gene transfer, termed cross‐order recombination, between enterovirus and porcine torovirus from two separate orders, Picornavirales and Nidovirales respectively. This rare event seems to occur more frequently in EV‐G1 than in EV‐G2 and ‐G17 genotypes, and might contribute significantly to viral evolution. Sequence and phylogenetic analyses revealed that the Korean recombinant strain is classified into the G1 genotype and is most closely related to other EV‐G1‐PLCP strains, based on the VP1 sequence. However, the inserted PLCP gene of KNU‐1811 is more closely related to that of the US G17‐PLCP strain (89.3% homology), rather than G1‐PLCP strains (75.0%−80.1% homology). More intriguingly, the sizes of the recombinant EV‐G PLCP sequences and their junction sequences are heterogeneous among individual strains, suggesting independent evolution in respective geographical areas. These findings might indicate that the recombination occurred independently in South Korea, and subsequently, the exogenous PLCP sequence has undergone substantial genetic drift (e.g., non‐silent point mutations) after its random insertion into the corresponding EV‐G genome through genetic shift (e.g., viral recombination). However, we cannot exclude that the recombinant virus was introduced from the US or other countries by import of breeding pigs or other unknown sources, and the genetic evolution independently proceed thereafter. Despite EV‐Gs not being associated with clinical illness, all recombinant EV‐G‐PLCP strains were confirmed from diarrhoeic samples, indicating them as potential causative agents of porcine diarrhoeal diseases. Furthermore, a growing body of evidence suggests that the lateral acquisition of the PLCP gene might represent a novel host immune evasion strategy of EV‐Gs to establish their pathogenic potential under certain circumstances. Therefore, future work should to aim to investigate the prevalence of authentic and recombinant EV‐G infections, and their genetic diversity, including inter‐/intra‐genotype and inter‐family recombinations among Korean swine herds. Our study support new insights into EV‐G evolutionary processes, in which EV‐Gs horizontally obtain a virus‐encoded anti‐immune constituent associated with virulence from completely unrelated viruses through recombination, thereby naturally arming the EV‐G population with an acquired gene necessary for viral fitness and long‐term survival in their natural host.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
ACKNOWLEDGEMENTS
We gratefully thank Chang Won Im for providing clinical samples. This research was supported by Bio‐industry Technology Development Program through the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (iPET) funded by the Ministry of Agriculture, Food and Rural Affairs (315021–04).
Lee S, Lee C. First detection of novel enterovirus G recombining a torovirus papain‐like protease gene associated with diarrhoea in swine in South Korea. Transbound Emerg Dis. 2019;66:1023–1028. 10.1111/tbed.13073
REFERENCES
- Anbalagan, S. , Hesse, R. A. , & Hause, B. M. (2014). First identification and characterization of porcine enterovirus G in the United States. PLoS One, 9, e97517 10.1371/journal.pone.0097517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom, N. , Hansen, J. , Brunak, S. , & Blaas, D. (1996). Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Science, 5, 2203–2216. 10.1002/pro.5560051107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros, Á. , Nemes, C. , Pankovics, P. , Bíró, H. , Kapusinszky, B. , Delwart, E. , & Reuter, G. (2012). Characterization of a novel porcine enterovirus in wild boars in Hungary. Archives of Virology, 157, 981–986. 10.1007/s00705-012-1255-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conceição‐Neto, N. , Theuns, S. , Cui, T. , Zeller, M. , Yinda, C. K. , Christiaens, I. ,… Matthijnssens, J. (2017). Identification of an enterovirus recombinant with a torovirus‐like gene insertion during a diarrhea outbreak in fattening pigs. Virus . Evolution, 3, vex024 10.1093/ve/vex024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, G. , Lee, K. K. , Kim, S. H. , & Lee, C. (2017). Prevalence, complete genome sequencing and phylogenetic analysis of porcine deltacoronavirus in South Korea, 2014–2016. Transboundary and Emerging Diseases, 64, 1364–1370. 10.1111/tbed.12690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku, Y. , Sarai, A. , & Murakami, Y. (2001). Genetic reclassification of porcine enteroviruses. Journal of General Virology, 82, 417–424. 10.1099/0022-1317-82-2-417 [DOI] [PubMed] [Google Scholar]
- Knowles, N. J. , Hovi, T. , Hyypiä, T. , King, A. M. Q. , Lindberg, A. M. , Pallansch, M. A. , & Zell, R. (2012). Picornaviridae In King A. M. Q., Adams M. J., Carstens E. B., & Lefkowitz E. J. (Eds.), Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. London: Elsevier Academic Press. [Google Scholar]
- Knutson, T. P. , Velayudhan, B. T. , & Marthaler, D. G. (2017). A porcine enterovirus G associated with enteric disease contains a novel papain‐like cysteine protease. Journal of General Virology, 98, 1305–1310. 10.1099/jgv.0.000799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Jang, G. , & Lee, C. (2015). Complete genome sequence of a porcine astrovirus from South Korea. Archives of Virology, 160, 1819–1821. 10.1007/s00705-015-2436-9 [DOI] [PubMed] [Google Scholar]
- Lee, Y. N. , & Lee, C. (2013). Complete genome sequence of a novel porcine parainfluenza virus 5 isolate in Korea. Archives of Virology, 158, 1765–1772. 10.1007/s00705-013-1770-z [DOI] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2018). Genomic and antigenic characterization of porcine epidemic diarrhoea virus strains isolated from South Korea, 2017. Transboundary and Emerging Diseases, 65, 949–956. 10.1111/tbed.12904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev, A. N. (2005). Role of recombination in evolution of enteroviruses. Reviews in Medical Virology, 15, 157–167. 10.1002/rmv.457 [DOI] [PubMed] [Google Scholar]
- Muslin, C. , Joffret, M. L. , Pelletier, I. , Blondel, B. , & Delpeyroux, F. (2015). Evolution and emergence of enteroviruses through intra‐ and inter‐species recombination: Plasticity and phenotypic impact of modular genetic exchanges in the 5′ untranslated region. PLoS Pathogens, 11, e1005266 10.1371/journal.ppat.1005266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachsenröder, J. , Twardziok, S. , Hammerl, J. A. , Janczyk, P. , Wrede, P. , Hertwig, S. , & Johne, R. (2012). Simultaneous identification of DNA and RNA viruses present in pig faeces using process‐controlled deep sequencing. PLoS One, 7, e34631 10.1371/journal.pone.0034631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semler, B. L. , & Wimmer, E. (2002). Molecular biology of picornaviruses. Washington, D.C.: ASM Press; 10.1128/9781555817916 [DOI] [Google Scholar]
- Shan, T. , Li, L. , Simmonds, P. , Wang, C. , Moeser, A. , & Delwart, E. (2011). The fecal virome of pigs on a high‐density farm. Journal of Virology, 85, 11697–11708. 10.1128/jvi.05217-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, P. , Misra, S. , Hause, B. , & Fang, Y. (2017). A naturally occurring recombinant enterovirus expresses a torovirus deubiquitinase. Journal of Virology, 91, e00450–17. 10.1128/JVI.00450-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiaka, S. , Naoi, Y. , Imai, R. , Masuda, T. , Ito, M. , Akagami, M. ,… Nagai, M. (2018). Genetic diversity and recombination of enterovirus G strains in Japanese pigs: High prevalence of strains carrying a papain‐like cysteine protease sequence in the enterovirus G population. PLoS One, 13, e0190819 10.1371/journal.pone.0190819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dung, N. , Anh, P. H. , Van Cuong, N. , Hoa, N. T. , Carrique‐Mas, J. , Hien, V. B. ,… Simmonds, P. (2014). Prevalence, genetic diversity and recombination of species G enteroviruses infecting pigs in Vietnam. Journal of General Virology, 95, 549–556. 10.1099/vir.0.061978-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar, M. J. , Peralta, B. , García‐Bocanegra, I. , Simon‐Grifé, M. , Bensaid, A. , Casal, J. , & . Pina‐Pedrero, S. (2016). Distribution and genetic characterization of enterovirus G and sapelovirus A in six Spanish swine herds. Virus Research, 215, 42–49. 10.1016/j.virusres.2016.01.019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials