

Abstract
Viruses have caused much mortality and morbidity to humans and pose a serious threat to global public health. The virome with the potential of human infection is still far from complete. Novel viruses have been discovered at an unprecedented pace as the rapid development of viral metagenomics. However, there is still a lack of methodology for rapidly identifying novel viruses with the potential of human infection. This study built several machine learning models to discriminate human‐infecting viruses from other viruses based on the frequency of k‐mers in the viral genomic sequences. The k‐nearest neighbor (KNN) model can predict the human‐infecting viruses with an accuracy of over 90%. The performance of this KNN model built on the short contigs (≥1 kb) is comparable to those built on the viral genomes. We used a reported human blood virome to further validate this KNN model with an accuracy of over 80% based on very short raw reads (150 bp). Our work demonstrates a conceptual and generic protocol for the discovery of novel human‐infecting viruses in viral metagenomics studies.
Keywords: human‐infecting virus, machine learning, viral metagenomics, virome
1. INTRODUCTION
Viruses are the most abundant biological entities on Earth and exist in all habitats of the world (Paez‐Espino et al., 2016). They can infect all kinds of organisms in a range from the bacteria to animals, including humans. Humans are constantly exposed to a vast diversity of viruses, and over two‐thirds of human pathogens belong to viruses (Woolhouse & Gaunt, 2007). The viruses have caused colossal mortality and morbidity to the human society in history, such as the devastating smallpox and Spanish flu outbreaks (Johnson & Mueller, 2002; Riedel, 2005). Despite the continuous progress in the prevention and control of viral disease, recent serial outbreaks caused by the Middle East respiratory syndrome coronaviruses (Breban, Riou, & Fontanet, 2013), avian influenza H7N9 viruses (Gao et al., 2013), Ebola viruses (Maganga et al., 2014) and Zika viruses (Mlakar et al., 2016) indicate that viruses still pose a severe threat to global public health.
To this date, the virome with the potential for human infection is still far from complete. Generally, a new pathogen would not be identified until it caused epidemics or pandemics. Many viruses that may have been introduced into human populations remain undiscovered (Rosenberg, 2015). Traditional diagnostic methods such as polymerase chain reaction, immunological assays and pan‐viral microarrays are inadequate for the quick identification of novel human‐infecting viruses (Corman et al., 2012; Wootton et al., 2011). The rapid development of viral metagenomic sequencing in recent years provides powerful high‐throughput and culture‐independent methods to identify new viruses, which lead to the accumulation of new viruses at an unprecedented pace (Alavandi & Poornima, 2012). The Global Virome Project (GVP), which was proposed and initiated at the beginning of 2018, estimated that there are over 1.67 million yet‐to‐be‐discovered viruses in animal reservoirs, while 631,000–827,000 of these unknown viruses can infect humans (Carroll et al., 2018). Therefore, the development of rapid methods for identifying the potential human‐infecting viruses is in great need.
Two kinds of methods, the sequence alignment‐based and alignment‐free methods, have been developed to predict the viral host. For example, the methods based on k‐mers extracted from viral genomes (Ahlgren, Ren, Lu, Fuhrman, & Sun, 2016; Li & Sun, 2018) or sequence blast have been developed to predict the hosts of the phage (Bolotin, Quinquis, Sorokin, & Ehrlich, 2005; Edwards, McNair, Faust, Raes, & Dutilh, 2015). Some studies also attempted to identify the human virus by using these methods. Xu, Tan, Li, Jiang, and Peng (2017) developed SVM models to predict the hosts of influenza viruses based on word vectors. However, all of these studies focused on one or a few specific types of viruses, such as the coronavirus and influenza virus. These methods are not suitable for the identification of novel human‐infecting viruses from the viral metagenomic sequences. Herein, we employed machine learning models to establish a generic protocol for the rapid identification of potential human‐infecting viruses.
2. MATERIALS AND METHODS
2.1. Virus‐host interactions and viral genomes
The virus‐host relationship and the viral genomic sequences were obtained from the database of Virus‐Host DB (available at https://www.genome.jp/virushostdb/) on 15 July 2018 (Mihara et al., 2016). The viroid, satellites and the viruses with genomic sequence <1 kb were removed. The resulting dataset of 9,428 viruses includes 1,236 viruses infecting humans (defined as human‐infecting viruses) and 8,192 viruses infecting other species.
2.2. Machine learning models
The machine learning models of k‐nearest neighbor (KNN) (k = 1), support vector machine (SVM) (using the linear kernel function), Gaussian Naive Bayes classifier (GNBC), random forest (RF) and logistic regression (LR) were built with the default parameters using the package “scikit‐learn” (version 0.20.2) (Pedregosa et al., 2011) in Python (version 3.6.2). Because the number of human‐infecting viruses was much smaller than that of other viruses, the “BalanceBaggingClassifier” (Barandiaran, 1998; Breiman, 1996) and “BalanceRandomForest” (Chen, Liaw, & Breiman, 2004) in the package of “imbalanced‐learn” (version 0.4.3) in Python were used to deal with the imbalance of the number of viruses in the modelling with the parameter of “n_estimators” set to be 10.
Ten‐fold cross‐validations were used to evaluate the predictive performances of the machine learning models through the “StratifiedKFold” in the package “scikit‐learn” in Python. The predictive performances of the machine learning models were evaluated by the area under the receiver operating characteristics (ROC) curve (AUC), the accuracy, recall rate, specificity and predictive precision.
2.3. Validation of the KNN model by a reported human blood DNA virome study
A total of 14,242,328 viral reads and 396 viral contigs were obtained from Moustafa's study (Moustafa et al., 2017). Each read or contig was queried against the viral sequences from databases of NCBI RefSeq and Virus‐Host DB by blastn and blastx, and the viruses with the best blast hit from the human‐infecting viruses were considered as human‐infecting viruses.
2.4. Statistical analysis
All the statistical analysis was conducted in R (version 3.5.0).
2.5. Code and data availability
All the data and codes used in this study are publicly available at https://github.com/Fzhang1992/human-infecting_virus_finder
3. RESULTS
3.1. Taxonomy distribution of viruses
A total of 9,428 viruses were used in the study. Among them, 1,236 viruses have been reported to infect humans (human‐infecting viruses), including 1,043 viruses exclusively infecting humans and 193 viruses infecting humans and other species. The human‐infecting viruses covered all groups of viruses in the Baltimore classification (Figure 1a). Over 60% of the human‐infecting viruses were single‐stranded RNA (ssRNA) viruses (Figure S1), which also had the highest proportion of human‐infecting viruses (28%). The second largest component (25%) of the human‐infecting viruses was the double‐stranded DNA (dsDNA) virus (Figure S1). On the level of family, the human‐infecting viruses were from 30 viral families. Among them, the families of Caliciviridae (ssRNA), Picornaviridae (ssRNA) and Papillomaviridae (dsDNA) were the three most abundant ones, which accounted for nearly 60% of the human‐infecting viruses (Figure 1b).
Figure 1.
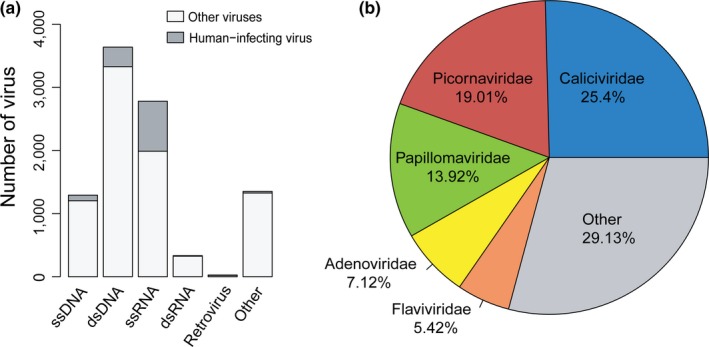
The taxonomy distribution of viruses used in this study. (a) The taxonomy distribution of the human‐infecting viruses and other viruses in the Baltimore classification. (b) The taxonomy distribution of the human‐infecting viruses based on viral families [Colour figure can be viewed at http://wileyonlinelibrary.com]
Besides the human‐infecting viruses, there were 8,192 viruses infecting species other than human, including the archaea, bacteria, fungi, plant, animal and so on. They covered all groups of viruses in the Baltimore classification (Figure 1a) and came from 91 viral families.
3.2. Machine learning models for identifying the human‐infecting viruses based on k‐mer frequencies in the genome
Five machine learning models were built based on k‐mer frequencies in the viral genome to discriminate the human‐infecting viruses from other viruses. These models include KNN, RF, GNBC, SVM and LR. K‐mers containing one to six nucleotides were used in the models to investigate the effect of the k‐mer length on the model performance (Table S1). Figure 2 shows that the AUCs of GNBC, SVM and LR models increased as the increase of the k‐mer length from one to six, while the AUCs of KNN and RF peaked at k‐mer length of four and three, respectively. The AUCs of KNN and RF were visibly higher than those of other models (GNBC, SVM and LR) at all k‐mer length, suggesting that the KNN and RF outperformed other models in discriminating the human‐infecting viruses from other viruses. The KNN model achieved the best performance when the k‐mer was four nucleotides. The model had the optimal overall performance with an accuracy of 0.90 and an AUC of 0.92 and a strong ability to capture the human‐infecting virus with a recall rate of 0.94 (Table 1).
Figure 2.
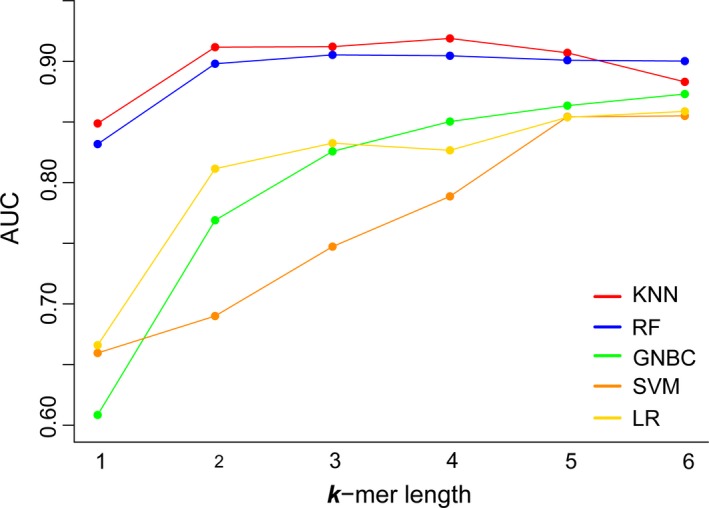
The AUCs of machine learning models with k‐mer lengths in a range from one to six [Colour figure can be viewed at http://wileyonlinelibrary.com]
Table 1.
The optimal performance of machine learning models
| KNN | RF | GNBC | SVM | LR | |
|---|---|---|---|---|---|
| K‐mer length | 4 | 3 | 6 | 6 | 6 |
| Accuracy | 0.90 | 0.91 | 0.84 | 0.84 | 0.85 |
| Recall rate | 0.94 | 0.91 | 0.91 | 0.88 | 0.87 |
| Specificity | 0.90 | 0.91 | 0.83 | 0.83 | 0.85 |
| Precision | 0.58 | 0.59 | 0.45 | 0.45 | 0.46 |
| AUC | 0.92 | 0.91 | 0.87 | 0.86 | 0.86 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.3. Identification of the human‐infecting viruses based on contigs of various lengths
In the metagenomics studies, contigs of varying lengths in a range from several hundred to several thousand nucleotides were assembled. Rapid determination of the human‐infecting viruses from metagenomic sequences is essential for early warnings of newly emerging viruses. Viral genomes were split into non‐overlapping contigs of 500, 1,000, 3,000, 5,000 and 10,000 nucleotides to mimic the metagenomic sequences. The numbers of contigs of various lengths in the human‐infecting viruses and other viruses are listed in Table S2.
Since the KNN model performed best in discriminating the human‐infecting viruses from other viruses based on viral genomes, it was used to identify the human‐infecting virus based on contigs. The KNN models with k‐mers of different size were built on the contigs of given lengths (Table S3). As shown in Figure 3, for all contigs of varying lengths, the AUCs of the KNN models increased as the increase of k‐mer sizes from one to four. The models achieved the best performance when k‐mer length was four nucleotides. The longer the contig was, the better the models performed in identifying the human‐infecting viruses (Figure 3 and Table 2). The model built on the contig of 10,000 nucleotides had an AUC of 0.96 and a recall rate of 0.99, suggesting the model could capture most human‐infecting viruses. While on the contig of 500 nucleotides, the model only had an AUC of 0.86 and a recall rate of 0.88. Typically, for the models built on the contigs of 1,000 nucleotides or longer, they performed similarly to those built on the viral genome.
Figure 3.
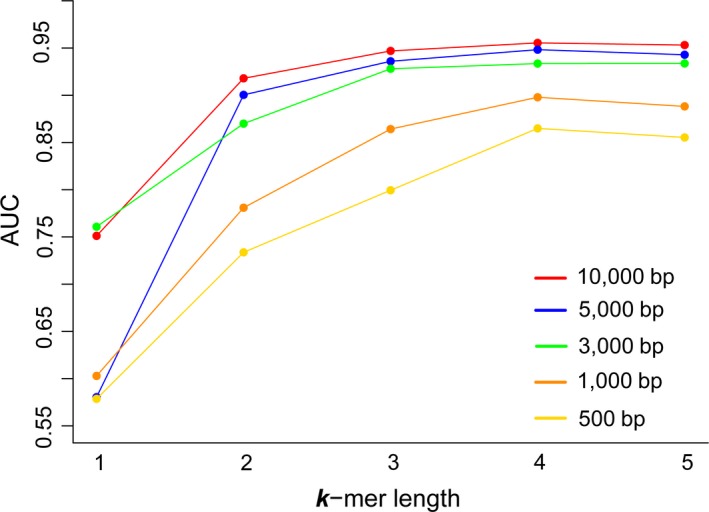
The AUCs of the KNN models with k‐mer lengths from one to five built on contigs of various lengths [Colour figure can be viewed at http://wileyonlinelibrary.com]
Table 2.
The optimal performance of the KNN models with k‐mers of four nucleotides built on contigs of various lengths
| 500 bp | 1,000 bp | 3,000 bp | 5,000 bp | 10,000 bp | |
|---|---|---|---|---|---|
| Accuracy | 0.85 | 0.87 | 0.91 | 0.92 | 0.92 |
| Recall rate | 0.88 | 0.93 | 0.96 | 0.98 | 0.99 |
| Specificity | 0.85 | 0.86 | 0.90 | 0.92 | 0.92 |
| Precision | 0.25 | 0.28 | 0.35 | 0.38 | 0.32 |
| AUC | 0.86 | 0.90 | 0.93 | 0.95 | 0.96 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.4. Validation of the KNN model by a reported human blood DNA virome study
The KNN model with k‐mers of four nucleotides was further validated through a human blood DNA virome study reported by Moustafa et al. (2017). In this study, 19 human viruses were identified from whole‐genome sequencing of blood from 8,240 individuals without any infectious disease. A total of 14,242,328 viral reads of 150 bp, including 14,010,527 reads from the human‐infecting viruses and 231,801 reads from other viruses, were obtained in the study. The KNN model correctly predicted 81% of the reads from the human‐infecting viruses (Table 3). Besides the raw viral reads, a total of 396 viral contigs varying from less than 1,000 bp to more than 10,000 bp were assembled in the study, including 309 human‐infecting viral contigs and 87 other viral contigs. The KNN model correctly predicted 87% of the human‐infecting viral contigs. Also, the proportion of correct predictions of the human‐infecting viral contigs increased from 0.84 to 1 as the contig lengths increased from several hundred nucleotides to more than 10,000 nucleotides.
Table 3.
Predictive performance of the KNN model in identifying the human‐infecting viruses from a reported human blood DNA virome study. a Raw read in the study. 1 kb, 1,000 bp
| Length of read/contig (L) | Number of reads/contigs | Proportion of correct predictions | Overall predictive accuracy | ||
|---|---|---|---|---|---|
| Human‐infecting | Other | Human‐infecting | Other | ||
| L = 150 bpa | 14,010,527 | 231,801 | 0.81 | 0.84 | 0.82 |
| L < 1 kb | 222 | 75 | 0.84 | 0.15 | 0.67 |
| 1 kb ≤ L < 3 kb | 61 | 9 | 0.93 | 0 | 0.81 |
| 3 kb ≤ L < 5 kb | 13 | 3 | 0.92 | 0 | 0.75 |
| 5 kb ≤ L < 10 kb | 10 | 0 | 1 | 0 | 1 |
| L ≥ 10 kb | 3 | 0 | 1 | 0 | 1 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
4. DISCUSSION
Identification of human‐infecting viruses is critical for early warnings of newly emerging viruses. The rapid accumulation of viral metagenomic sequences presents us with unique opportunities to identify more and more novel viruses. Unfortunately, there is currently an unmet challenge to rapidly identify the potential human‐infecting viruses. As far as we know, this study for the first time attempted to discriminate the human‐infecting viruses from non‐human‐infecting viruses in the perspective of virome. The KNN models predicted the human‐infecting viruses with high accuracy and sensitivity. Even if the KNN models were built on the contigs as short as 1 kb, they performed comparably to those built on the viral genomes in the aspect of the AUC and the recall rate (Tables 1 and 2). The KNN model was further validated by a reported human blood virome study. It correctly predicted the human‐infecting viruses with an accuracy of over 80% based on the raw reads as short as 150 bp (Table 3). The performance of our KNN model on the blood virome study suggested that the model can be used in metagenomics for identifying the potential human‐infecting viruses.
The human‐infecting viruses used here have different ability to infect humans. Some of them are human‐specific that have been circulating in human populations for a long time, such as the human papillomavirus (Baseman & Koutsky, 2005) and yellow fever virus (Akondy et al., 2009); while some are zoonotic viruses that infect humans rarely, such as the avian influenza virus and coronavirus (García‐Sastre & Schmolke, 2014; Shipley et al., 2019). As shown in numerous studies, the zoonotic viruses are very likely to cause epidemics or even pandemics after adaptive evolution. The most recent example is Zika viruses. It rarely caused wide‐spread epidemics in humans in the 20th century, even in highly enzootic areas, (Weaver et al., 2016). However, a few mutations in the viral genome facilitated its rapid spread in human populations (Campos, Bandeira, & Sardi, 2015; Duffy et al., 2009; Liu et al., 2017). The virus has caused epidemics in 84 countries and has infected more than one million people since 2014 (European CDC, 2016; de Oliveira Garcia, 2019). Taken together, it is difficult to distinguish the viruses with the varying ability of infecting humans. Therefore, the viruses with the various ability of infecting humans were considered equally in the modelling.
There are some limitations to this study. Firstly, the human‐infecting viruses used here is far from complete when compared to those estimated by the GVP. However, the viruses used in this study covered 30 families in all groups of the Baltimore classification, which were similar to those estimated by the GVP (Carroll et al., 2018). Besides, the machine learning models had excellent performances in discriminating the human‐infecting viruses from other viruses. They could help much in discovering novel human‐infecting viruses. Secondly, the number of human‐infecting viruses was much less than that of other viruses. Such a large imbalance may hinder accurate modelling. Here, the under‐sampling method was used to deal with the imbalance problem so that the KNN model achieved excellent overall performances and high sensitivity in identifying the human‐infecting viruses. Thirdly, the model was not robust to the contamination of human sequences in the viral genomes. It would be better to remove such contamination from the viral sequences before using the model.
In conclusion, this study built novel computational models to predict the human‐infecting viruses in the perspective of virome. The high accuracy and sensitivity of the KNN model built on the viral contigs of various lengths suggest that the model can be used to identify the human‐infecting viruses from the viral metagenomic sequences. This work provides an effective strategy for the identification of novel human‐infecting viruses in metagenomics studies.
CONFLICT OF INTEREST
The authors have declared that no competing interests exist.
ETHICAL APPROVAL
Not applicable because no human or animal samples were collected in this study.
Supporting information
ACKNOWLEDGEMENTS
The authors would like to thank Prof. Heping Zheng in Hunan University for helping improve the language of the manuscript. This work was supported by the National Key Plan for Scientific Research and Development of China (2016YFD0500300), Hunan Provincial Natural Science Foundation of China (2018JJ3039), Changsha Science and Technology Bureau (kq1801040), the National Natural Science Foundation of China (31500126 and 31671371) and the Chinese Academy of Medical Sciences (2016‐I2M‐1‐005).
Zhang Z, Cai Z, Tan Z, et al. Rapid identification of human‐infecting viruses. Transbound Emerg Dis. 2019;66:2517–2522. 10.1111/tbed.13314
Zheng Zhang and Zena Cai contributed equally to this work.
REFERENCES
- Ahlgren, N. A. , Ren, J. , Lu, Y. Y. , Fuhrman, J. A. , & Sun, F. (2016). Alignment‐free oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically‐derived viral sequences. Nucleic Acids Research, 45, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akondy, R. S. , Monson, N. D. , Miller, J. D. , Edupuganti, S. , Teuwen, D. , Wu, H. , … Del Rio, C. (2009). The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. The Journal of Immunology, 183, 7919–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavandi, S. , & Poornima, M. (2012). Viral metagenomics: A tool for virus discovery and diversity in aquaculture. Indian Journal of Virology, 23, 88–98. 10.1007/s13337-012-0075-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barandiaran, I. (1998). The random subspace method for constructing decision forests. IEEE Transactions on Pattern Analysis and Machine Intelligence, 20(8), 832–844. [Google Scholar]
- Baseman, J. G. , & Koutsky, L. A. (2005). The epidemiology of human papillomavirus infections. Journal of Clinical Virology, 32, 16–24. 10.1016/j.jcv.2004.12.008 [DOI] [PubMed] [Google Scholar]
- Bolotin, A. , Quinquis, B. , Sorokin, A. , & Ehrlich, S. D. (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology, 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- Breban, R. , Riou, J. , & Fontanet, A. (2013). Interhuman transmissibility of Middle East respiratory syndrome coronavirus: Estimation of pandemic risk. The Lancet, 382, 694–699. 10.1016/S0140-6736(13)61492-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman, L. (1996). Bagging predictors. Machine Learning, 24, 123–140. 10.1007/BF00058655 [DOI] [Google Scholar]
- Campos, G. S. , Bandeira, A. C. , & Sardi, S. I. (2015). Zika virus outbreak, Bahia, Brazil. Emerging Infectious Diseases, 21, 1885 10.3201/eid2110.150847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, D. , Daszak, P. , Wolfe, N. D. , Gao, G. F. , Morel, C. M. , Morzaria, S. , … Mazet, J. A. (2018). The global virome project. Science, 359, 872–874. 10.1126/science.aap7463 [DOI] [PubMed] [Google Scholar]
- Chen, C. , Liaw, A. , & Breiman, L. (2004). Using random forest to learn imbalanced data (Vol. 110, pp. 1–12). Berkeley, CA: University of California. [Google Scholar]
- Corman, V. V. , Eckerle, I. , Bleicker, T. , Zaki, A. , Landt, O. , Eschbach‐Bludau, M. M. , … Bestebroer, T. (2012). Detection of a novel human coronavirus by real‐time reverse‐transcription polymerase chain reaction. Eurosurveillance, 17(39), 20285 10.2807/ese.17.39.20285-en [DOI] [PubMed] [Google Scholar]
- de Oliveira Garcia, M. H. (2019). Zika: The continuing threat. Bulletin of the World Health Organization, 97, 6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy, M. R. , Chen, T.‐H. , Hancock, W. T. , Powers, A. M. , Kool, J. L. , Lanciotti, R. S. , … Hayes, E. B. (2009). Zika virus outbreak on Yap Island, federated states of Micronesia. New England Journal of Medicine, 360, 2536–2543. 10.1056/NEJMoa0805715 [DOI] [PubMed] [Google Scholar]
- Edwards, R. A. , McNair, K. , Faust, K. , Raes, J. , & Dutilh, B. E. (2015). Computational approaches to predict bacteriophage–host relationships. FEMS Microbiology Reviews, 40, 258–272. 10.1093/femsre/fuv048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European CDC (2016). Zika virus epidemic in the Americas: Potential association with microcephaly and Guillain‐Barré syndrome (first update). Stockholm, Sweden: ECDC. [Google Scholar]
- Gao, R. , Cao, B. , Hu, Y. , Feng, Z. , Wang, D. , Hu, W. , … Shu, Y. (2013). Human infection with a novel avian‐origin influenza A (H7N9) virus. New England Journal of Medicine, 368, 1888–1897. 10.1056/NEJMoa1304459 [DOI] [PubMed] [Google Scholar]
- García‐Sastre, A. , & Schmolke, M. (2014). Avian influenza A H10N8—A virus on the verge? The Lancet, 383, 676–677. 10.1016/S0140-6736(14)60163-X [DOI] [PubMed] [Google Scholar]
- Johnson, N. P. , & Mueller, J. (2002). Updating the accounts: Global mortality of the 1918–1920 “Spanish” influenza pandemic. Bulletin of the History of Medicine, 76, 105–115. [DOI] [PubMed] [Google Scholar]
- Li, H. , & Sun, F. (2018). Comparative studies of alignment, alignment‐free and SVM based approaches for predicting the hosts of viruses based on viral sequences. Scientific Reports, 8, 10032 10.1038/s41598-018-28308-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Liu, J. , Du, S. , Shan, C. , Nie, K. , Zhang, R. , … Cheng, G. (2017). Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature, 545, 482 10.1038/nature22365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maganga, G. D. , Kapetshi, J. , Berthet, N. , Kebela Ilunga, B. , Kabange, F. , Mbala Kingebeni, P. , … Leroy, E. M. (2014). Ebola virus disease in the Democratic Republic of Congo. New England Journal of Medicine, 371, 2083–2091. 10.1056/NEJMoa1411099 [DOI] [PubMed] [Google Scholar]
- Mihara, T. , Nishimura, Y. , Shimizu, Y. , Nishiyama, H. , Yoshikawa, G. , Uehara, H. , … Ogata, H. (2016). Linking virus genomes with host taxonomy. Viruses, 8, 66 10.3390/v8030066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar, J. , Korva, M. , Tul, N. , Popović, M. , Poljšak‐Prijatelj, M. , Mraz, J. , … Avšič Županc, T. (2016). Zika virus associated with microcephaly. New England Journal of Medicine, 374, 951–958. 10.1056/NEJMoa1600651 [DOI] [PubMed] [Google Scholar]
- Moustafa, A. , Xie, C. , Kirkness, E. , Biggs, W. , Wong, E. , Turpaz, Y. , … Telenti, A. (2017). The blood DNA virome in 8,000 humans. PLoS Path, 13, e1006292 10.1371/journal.ppat.1006292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez‐Espino, D. , Eloe‐Fadrosh, E. A. , Pavlopoulos, G. A. , Thomas, A. D. , Huntemann, M. , Mikhailova, N. , … Kyrpides, N. C. (2016). Uncovering Earth's virome. Nature, 536, 425 10.1038/nature19094 [DOI] [PubMed] [Google Scholar]
- Pedregosa, F. , Varoquaux, G. , Gramfort, A. , Michel, V. , Thirion, B. , Grisel, O. , … Dubourg, V. (2011). Scikit‐learn: Machine learning in Python. Journal of Machine Learning Research, 12, 2825–2830. [Google Scholar]
- Riedel, S. (2005). Edward Jenner and the history of smallpox and vaccination. Paper presented at: Baylor University Medical Center Proceedings (Taylor & Francis; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, R. (2015). Detecting the emergence of novel, zoonotic viruses pathogenic to humans. Cellular and Molecular Life Sciences, 72, 1115–1125. 10.1007/s00018-014-1785-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley, R. , Wright, E. , Selden, D. , Wu, G. , Aegerter, J. , Fooks, A. R. , & Banyard, A. C. (2019). Bats and viruses: Emergence of novel lyssaviruses and association of bats with viral zoonoses in the EU. Tropical Medicine and Infectious Disease, 4, 31 10.3390/tropicalmed4010031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, S. C. , Costa, F. , Garcia‐Blanco, M. A. , Ko, A. I. , Ribeiro, G. S. , Saade, G. , … Vasilakis, N. (2016). Zika virus: History, emergence, biology, and prospects for control. Antiviral Research, 130, 69–80. 10.1016/j.antiviral.2016.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse, M. , & Gaunt, E. (2007). Ecological origins of novel human pathogens. Critical Reviews in Microbiology, 33, 231–242. 10.1080/10408410701647560 [DOI] [PubMed] [Google Scholar]
- Wootton, S. C. , Kim, D. S. , Kondoh, Y. , Chen, E. , Lee, J. S. , Song, J. W. , … Collard, H. R. (2011). Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine, 183, 1698–1702. 10.1164/rccm.201010-1752OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, B. , Tan, Z. , Li, K. , Jiang, T. , & Peng, Y. (2017). Predicting the host of influenza viruses based on the word vector. PeerJ, 5, e3579 10.7717/peerj.3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data and codes used in this study are publicly available at https://github.com/Fzhang1992/human-infecting_virus_finder