

Abstract
A new highly virulent swine acute diarrhoea syndrome coronavirus (SADS‐CoV) emerged in Guangdong province in 2017 followed by fatal diarrhoea that involved the death of 24,693 piglets. And yet from May 2017 to January 2019, there were no new SADS cases arising in pig herds in Guangdong. In this study, we reported the recent diarrhoea outbreak of SADS‐CoV in Southern China on February 2019. Intestinal samples collected from diarrhoeal piglets were detected for common swine virus and confirmed that SADS‐CoV was responsible for the diarrhoea case. Meanwhile, serological investigation of sows’ sera implied that SADS‐CoV has existed in the farm and PEDV antibody may not directly contribute to the amplification of SADS‐CoV. Homology and phylogenetic analysis of the whole genome showed that the re‐emerging SADS‐CoV strain shared high sequence identities with existing SADS‐CoV strains and all strains clustered together in Alpha coronavirus. All in all, the report herein emphasized the re‐emerging of SADS‐CoV and highlights continuous monitoring for this virus.
Keywords: phylogenetic analysis, re‐emerging, SADS‐CoV, Southern China
1. INTRODUCTION
Swine acute diarrhoea syndrome (SADS) was first identified in South China in January 2017 as a disease causing high mortality in neonatal piglets (Gong et al., 2017; Pan et al., 2017; Zhou et al., 2018). It is quickly demonstrated that the causative agent of SADS is a novel HKU2‐related coronavirus, swine acute diarrhoea syndrome coronavirus (SADS‐CoV) which originated from the same genus of horseshoe bats (Rhinolophus) as severe acute respiratory syndrome coronavirus (SARS‐CoV), but the mechanism of transmission of bat‐originated coronavirus in pigs needs further illumination (Cui, Li, & Shi, 2019; Zhou et al., 2018). Based on the analysis of genomes, SADS‐CoV is classified into the genus Alphacoronavirus within the subfamily Orthocoronavirinae of the family Coronaviridae (Gong et al., 2017; Pan et al., 2017; Zhou et al., 2018). So far, besides porcine epidemic diarrhoea virus (PEDV), transmissible gastroenteritis virus (TGEV) and porcine deltacoronavirus (PDCoV), SADS‐CoV is the fourth porcine enteric coronavirus that circulated in pigs in China (Dong et al., 2015; Xuan et al., 1984). A retrospective investigation showed that SADS‐CoV emerged in China at least since August 2016 (Zhou et al., 2019). Li et al. (2018) reported one SADS‐CoV strain that was isolated from fecal and small intestinal samples collected from Fujian province and suggested that SADS‐CoV might have spread from Guangdong to Fujian province.
In the first outbreak of SADS‐CoV occurring between January and May 2017, in order to reduce mortality in suckling pigs, the minced intestines from acutely infected piglets were filtered, centrifuged and inactivated before feeding back to pregnant sows to allow maternal antibodies to be obtained in neonatal piglets. Hence, after May 2017, the epidemic was controlled. And with a period of quiescence, SADS‐CoV seemed to have disappeared from pig herds in South China. While on February 2019, there was a large‐scale outbreak of approximately 2000 piglets dying of diarrhoea in one farm, which was near the origin of the SADS pandemic in 2017. The extensive aetiological investigation for common swine viruses suggested that the causative agent of this recent diarrhoea outbreak was SADS‐CoV. In this study, we compared the infections of piglets with the original and re‐emerging SADS‐CoV strains by clinical signs, epidemiological investigation, serological investigation and phylogenetic analysis. The results highlight the significance of SADS‐CoV and the disease it causes.
At the beginning of the latest outbreak on February 2019, infected suckling piglets developed acute diarrhoea, vomiting and died after 2–6 days of the diarrhoea, which were consistent with the clinical signs reported in SAD‐CoV outbreaking farms (Zhou et al., 2018). Two weeks after the onset, mortality did not increase due to the effective inactivated SADS‐CoV vaccine which was propagated in vitro (data unpublished). Clinical small intestine samples were collected rapidly from piglets, and intestinal samples were homogenized, centrifuged and filtered for virus isolate. Viral nucleic acid was extracted using TRIzol reagent (Invitrogen, USA) according to the manufacturer's instruction. The potential swine viral pathogens were examined, including SADS‐CoV, PEDV, PDCoV, porcine rotavirus (RV), TGEV, reovirus (REO), senecavirus A (SVA), foot and mouth disease virus (FMDV), porcine pseudorabies virus (PRV), porcine circovirus 2 (PCV2), porcine circovirus 3 (PCV3), porcine reproductive and respiratory syndrome virus (PRRSV), porcine parainfluenza virus 1 (PPIV1), swine influenza virus (SIV), atypical porcine pestivirus (APPV), haemophilus parasuis (HPS), hog cholera (HC), porcine parvovirus (PPV), porcine bocavirus (PBoV), porcine sapelovirus (PSV) and porcine kobuvirus (PKV) by PCR or RT‐PCR following the established protocol of Key Laboratory of Animal Health Aquaculture and Environmental Control. The extensive aetiological investigations for common swine viruses showed that only SADS‐CoV was detected positively, which suggested that the causative agent of the most recent diarrhoea outbreak on February was SADS‐CoV.
Furthermore, a total of 18 blood samples were simultaneously collected from healthy sows for serological investigation. Sera samples were obtained after coagulation and centrifugation (2,000 g, 10 min) and stored at −20°C until ELISA implemented. The PEDV antibody production was detected by means of commercial ELISA test kit (ZhenRui, China), of which, sample S/P ratios with ≥0.4 were considered positive and <0.4 were considered negative. And for SADS‐CoV, ELISA was conducted following the modified procedures established in Key Laboratory of Animal Health Aquaculture and Environmental Control (P/N ≥ 2.1 considered positive). The absorbance at 450 nm wavelength was measured (Multiskan™ FC, Thermo Fisher). The results of ELISA assay revealed that all sera (100%) were negative for the PEDV and 13 of 18 (72.2%) were positive for SADS‐CoV (Figure 1). In our previously clinical investigations, PEDV had caused prior outbreaks at most farms with SADS‐CoV outbreaks (Zhou et al., 2018). It is possible that the presence of PEDV antibody may have somehow facilitated the amplification of the SADS‐CoV. However, our results showed that PEDV antibodies were undetectable in all SADS‐CoV antibody‐positive sera samples, suggesting that it is SADS‐CoV that caused the lethal infection in pigs on February 2019. Collectively, the re‐emergence of SADS‐CoV in pig herds in South China was confirmed by ELISA test and the association between PEDV and SADS‐CoV infection requires further validation.
Figure 1.
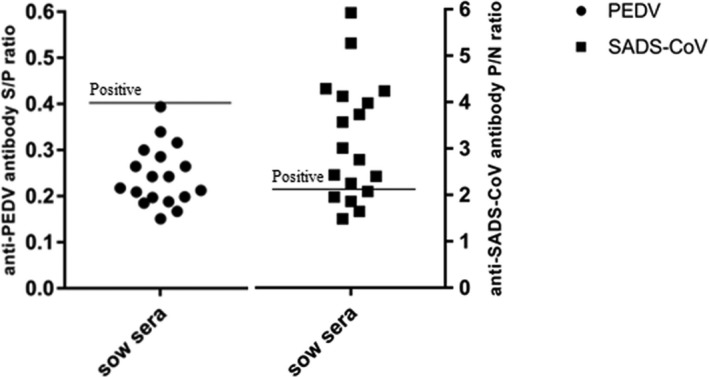
The results of ELISA test, n = 18 sows sera from outbreak farm. Detection of PEDV antibodies, sample S/P ratios ≥ 0.4 were considered positive. Detection of SADS‐CoV antibodies, P/N ratios ≥ 2.1 were considered positive
Subsequently, one SADS‐CoV genome was amplified as previously described (Zhou et al., 2019). The novel SADS‐CoV strain was named as SADS‐CoV/CN/GDLX/2019 (accession number: MK651076). Sequence alignment and phylogenetic analysis were performed with Lasergene coresuite 10 (DNASTAR lnc.). Phylogenetic tree was constructed using the neighbour‐joining method with MEGA 7.0 software (Kumar, Stecher, & Tamura, 2016). Bootstrap analysis was carried out on 1,000 replicate data sets (Felsenstein, 1985). For the whole genomes, sequence alignment results revealed that the nucleotide identities between SADS‐CoV/CN/GDLX/2019 and previously reported SADS‐CoV strains ranged from 99.3% to 100%. Moreover, the phylogenetic tree indicated that the novel SADS‐CoV complete genome clustered together with other reported SADS‐CoV strains and located in the Alpha coronavirus branch (Figure 2). Phylogenetic analysis based on S genes was also conducted, and the result showed that all SADS‐CoV, bat HKU2 and BtRf‐AlphaCoV strains were separated from other AlphaCoVs and clustered within BetaCoVs (Figure 3), which is consistent with previous studies (Pan et al., 2017; Xu et al., 2019; Zhou et al., 2019). Notably, the S gene of SADS‐CoV/CN/GDLX/2019 had high identities of 99.2%–99.9% with other SADS‐CoV strains detected in Guangdong and lowest identities of 97.5% with SADS‐CoV‐CH‐FJWT‐2018 reported from Fujian. Additionally, the comparative analysis of amino acids encoded by S gene revealed that the S protein of the novel strain had a two‐amino‐acid mutation (F306 and I882) compared with that of other SADS‐CoV strains. It is well known that the S protein of coronavirus play an important role in cellular entry and determining the immunogenicity (Li, Frank, He, Peter, & Berend‐Jan, 2016). In view of the lowest identities of Spike gene between the strains from Guangdong and Fujian, continuous monitoring of SADS‐CoV genetic evolution is important for better understanding its epidemiology and establishing efficient strategies against its further circulating in pigs in China.
Figure 2.
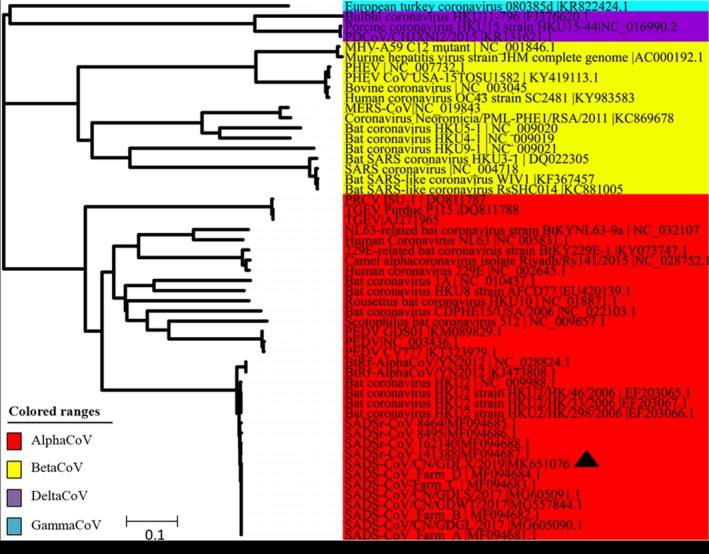
Phylogenetic analysis of the complete genome of SADS‐CoV and reference coronavirus species. The tree was constructed using MEGA 7.0 software with neighbour‐joining methods and 1,000 replicate sets on bootstrap analysis [Colour figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
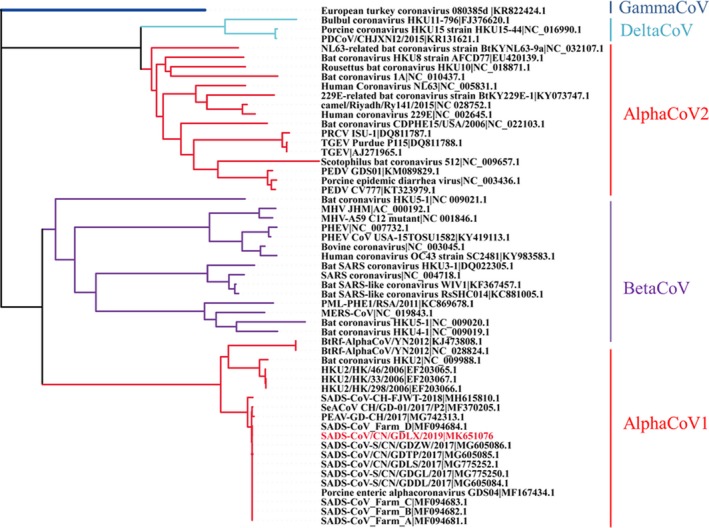
Phylogenetic analysis of the S gene of SADS‐CoV and reference coronavirus species. The tree was constructed using MEGA 7.0 software with neighbour‐joining methods and 1,000 replicate sets on bootstrap analysis [Colour figure can be viewed at http://wileyonlinelibrary.com]
Here, we reported a new case of acute diarrhoea outbreak from a swine farm in Guangdong province on February 2019. Pathogenic and serologic analysis determined that SADS‐CoV re‐emerged in Guangdong and was responsible for the recent acute diarrhoea outbreak. Phylogenetic analysis based on whole genomes showed that SADS‐CoV/CN/GDLX/2019 shared high nucleotide identities with previously reported SADS‐CoV strains. Taken together, our study highlighted the significant threat of SADS‐CoV and the potential outbreak of SADS epidemic in following months in Guangdong. Therefore, the persistent surveillance and vaccination countermeasures will be necessary to control the spread of SADS‐CoV.
CONFLICT OF INTEREST
The authors declare no conflict of interests with any organization.
ACKNOWLEDGEMENTS
This work was supported by the National Key Research and Development Program of China (No. 2016YFD0501304). The Research and Extension of Major Animal Epidemic Prevention and Control Technologies in the Strategic Project of Rural Revitalization of Guangdong Agricultural Department of China (Building Modern Agricultural System) (2018–2020) and the Science and Technology Program of Guangzhou City of China (No. 201904010433).
Zhou L, Li QN, Su JN, et al. The re‐emerging of SADS‐CoV infection in pig herds in Southern China. Transbound Emerg Dis. 2019;66:2180–2183. 10.1111/tbed.13270
REFERENCES
- Cui, J. , Li, F. , & Shi, Z. (2019). Origin and evolution of pathogenic coronavirus. Nat Rew Microbiol, 17, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, N. , Fang, L. , Zeng, S. , Sun, Q. , Chen, H. , & Xiao, S. (2015). Porcine Deltacoronavirus in mainland China. Emerging Infectious Diseases, 21, 2254–2255. 10.3201/eid2112.150283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution, 39, 783–791. [DOI] [PubMed] [Google Scholar]
- Gong, L. , Li, J. , Zhou, Q. , Xu, Z. , Chen, L. , Zhang, Y. , … Cao, Y. (2017). A new bat‐HKU–like coronavirus in swine, China, 2017. Emerging Infectious Diseases, 23, 1607–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Li, H. , Bi, Z. , Gu, J. , Gong, W. , Luo, S. , … Tang, Y. (2018). Complete genome sequence of a novel swine acute diarrhea syndrome coronavirus, CH/FJWT/2018, isolated in fujian, china, in 2018. Microbiol Resour Announc, 7(22), e01259–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Frank, J. , He, Q. , Peter, J. , & Berend‐Jan, B. (2016). Cellular entry of the porcine epidemic diarrhea virus. Virus Research, 226, 117–127. 10.1016/j.virusres.2016.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y. , Tian, X. , Qin, P. , Wang, B. , Zhao, P. , Yang, Y.‐L. , … Huang, Y.‐W. (2017). Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Veterinary Microbiology, 211, 15–21. 10.1016/j.vetmic.2017.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Zhang, Y. , Gong, L. , Huang, L. , Lin, Y. , Qin, J. , … Cao, Y. (2019). Isolation and characterization of a highly pathogenic strain of Porcine enteric alphacoronavirus causing watery diarrhoea and high mortality in newborn piglets. Transboundary and Emerging Diseases, 66, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan, H. , Xing, D. , Wang, D. , Zhu, W. , Zhao, F. , Gong, H. , & Fei, S. (1984). Study on the culture of porcine epidemic diarrhea virus adapted to fetal porcine intestine primary cell monolayer. Chin. J. Vet. Sci., 4, 202–208. [Google Scholar]
- Zhou, L. , Sun, Y. , Lan, T. , Wu, R. , Chen, J. , Wu, Z. , … Ma, J. (2019). Retrospective detection and phylogenetic analysis of swine acute diarrhoea syndrome coronavirus in pigs in southern China. Transboundary and Emerging Diseases, 66, 687–695. 10.1111/tbed.13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Fan, H. , Lan, T. , Yang, X.‐L. , Shi, W.‐F. , Zhang, W. , … Ma, J.‐Y. (2018). Fatal swine acute diarrhoea syndrome caused by an HKU2‐related coronavirus of bat origin. Nature, 556, 255–258. 10.1038/s41586-018-0010-9 [DOI] [PMC free article] [PubMed] [Google Scholar]