

Abstract
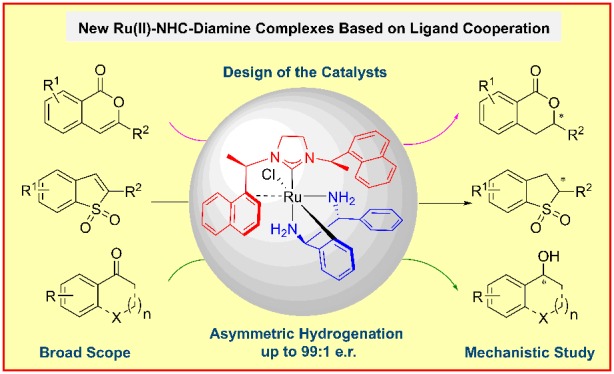
A modular synthesis of Ru(II)-NHC-diamine complexes from readily available chiral N-heterocyclic carbenes (NHCs) and chiral diamines is disclosed for the first time. The well-defined Ru(II)-NHC-diamine complexes show unique structure and coordination chemistry including an unusual tridentate coordination effect of 1,2-diphenylethylenediamine. The isolated air- and moisture-stable Ru(II)-NHC-diamine complexes act as versatile precatalysts for the asymmetric hydrogenation of isocoumarines, benzothiophene 1,1-dioxides, and ketones. Moreover, on the basis of the identification of reaction intermediates by stoichiometric reactions and NMR experiments, together with the DFT calculations, a possible catalytic cycle was proposed.
1. Introduction
The development of new chiral catalysts is key to improving efficiency and selectivity in asymmetric catalysis, even to discovering new modes of catalysis and new reactions.1 Conventionally, the chiral environment of a transition-metal catalyst is optimized by the modification of the structure of a single chiral ligand. However, this strategy is limited to ligand structures that can be readily accessed. Even though much endeavor and time are spent on the ligand optimization, satisfactory results are often out of reach. The utilization of ligand cooperation is an alternative strategy, which has the potential to accelerate the optimization process through a simple mix of two different chiral ligands (Scheme 1a). Remarkably, the introduction of a second, readily available ligand not only adds a new tunable site but also offers a practical way to avoid the tedious synthetic problems caused by the modification of a complicated ligand.2
Scheme 1. Ligand Cooperation Strategy for the Design of New Chiral Catalysts.
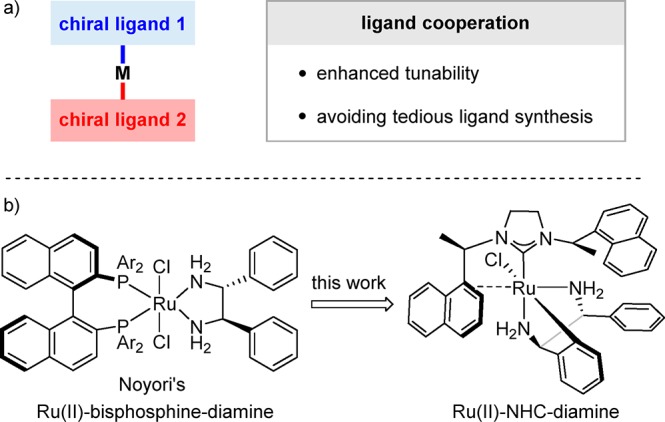
Noyori’s elegant Ru(II)-bisphosphine-diamine catalyst is a representative example utilizing the ligand cooperation concept (Scheme 1b).3 Since then, the cooperation of chiral phosphines and chiral diamines was applied widely in asymmetric catalysis. Nevertheless, novel cooperative systems beyond phosphine-type ligands have been rarely investigated for late transition metals.2,4 Inspired by Noyori’s catalyst and our ongoing interest in N-heterocyclic carbenes (NHCs),5,6 we recently reported a new combination of chiral NHC and chiral diamine ligands for the Ru(II)-catalyzed enantioselective hydrogenation of isocoumarins.7 However, the proposed Ru(II)-NHC-diamine species was only supported by control experiments. Undoubtedly, the isolation and characterization of the Ru(II)-NHC-diamine complexes would not only confirm the previous proposal but also provide us a new platform to design NHC-based catalysts directed by the ligand cooperation concept. Herein, we report the synthesis and structure determination of the chiral ruthenium precatalysts and related complexes (Scheme 1b). Unprecedented procedures for the preparation of air- and moisture-stable Ru(II)-NHC-diamine precatalysts from commercial or other readily available materials were described in this endeavor. Furthermore, the newly established catalysts were successfully applied to asymmetric hydrogenations of several important substrate classes. In the end, detailed mechanistic studies of asymmetric hydrogenation catalyzed by the Ru(II)-NHC-diamine complexes were conducted.
2. Results and Discussion
2.1. Synthesis and Structure of Ru(II)-NHC-Diamine Complexes
First, we developed a stepwise way to install a chiral NHC ligand and a chiral diamine ligand into one ruthenium complex (Figure 1). We hypothesized that silver carbene complexes could serve as carbene transfer agents for the synthesis of the ruthenium complexes as this strategy had previously been used for the synthesis of palladium and gold complexes.8 The NHC silver complex C0 was prepared from imidazolinium chloride (R,R)-INpEt·HCl and Ag2O according to the literature.9 The desired heteroleptic complex C1 was obtained in 56% overall yield following the transmetalation of the NHC from silver complex C0 to the ruthenium precursor and subsequent replacement of benzene with (R,R)-1,2-diphenylethylenediamine (DPEN). The isolated complex C1 is stable to air and moisture and can be stored in an ordinary vial for months. X-ray crystallographic analysis of C1 indicated a distorted octahedral geometry of the ruthenium center with a trans-dichloro geometry. Interestingly, the NHC ligand is acting as a chelate ligand with a dative carbene bond and an additional η2-coordination of the naphthyl ring to ruthenium. This was further verified by the distinct NMR signals at δ 4.99 (1H NMR, HC12), δ 92.0 (13C NMR, C11), and δ 72.7 (13C NMR, C12). The NMR analysis of trans-RuCl2(INpEt)(DPEN) C1 shows that C1 exists as a single conformer in solution (C6D6, CDCl3, or THF-d8).
Figure 1.
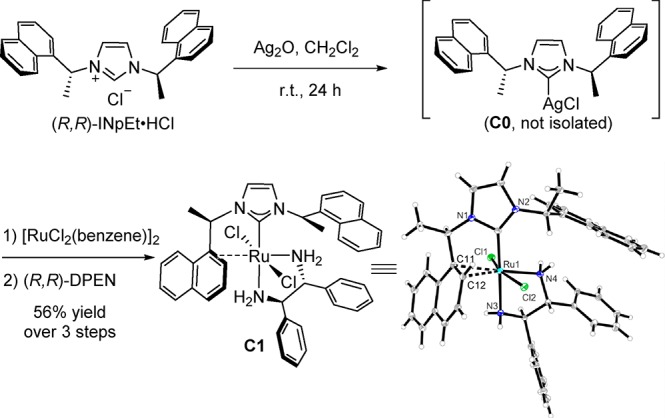
Synthesis and structure of trans-RuCl2(INpEt)(DPEN) C1. Selected bond lengths (Å) and bond angles (deg) of C1: Ru1–C1, 1.986(6); Ru1–N4, 2.135(5); Ru1–N3, 2.187(5); Ru1–C12, 2.218(5); Ru1–C11, 2.239(6); Ru1–Cl2, 2.4265(16); Ru1–Cl1, 2.4266(16); C11–C12 1.418(8); C1–Ru1–N3, 177.6(3); N4–Ru1–N3, 78.46(18); Cl2–Ru1–Cl1, 62.53(5); C12–Ru1–C11, 37.1(2).
Furthermore, we developed a one-pot procedure for the synthesis of Ru(II)-NHC-diamine complexes (Figure 2). The direct isolation of the Ru(II)-NHC-diamine species formed in situ by reacting [Ru(2-methylallyl)2(COD)], NHC precursor (R,R)-SINpEt·HBF4, diamine ligand (R,R)-DPEN, and NaOt-Bu in n-hexane was unsuccessful. We rationalized that the introduction of a chloride ligand might stabilize the ruthenium complex, thus simplifying the purification and isolation. After quenching the reaction with HCl solution (4.0 M in dioxane), ruthenium dichloride C2 and ruthenium monochloride C3 were isolated by flash chromatography on silica gel in 3% yield and 40% yield, respectively. In contrast, the combination of (R,R)-SINpEt·HBF4 and (S,S)-DPEN gave only trace amounts of Ru(II)-NHC-diamine complexes, thus demonstrating a strong matched/mismatched effect. X-ray and NMR analyses revealed that ruthenium complex C2 is structurally similar to complex C1. Unexpectedly, ruthenium monochloride C3 contains an unusual tridentate binding of the diamine through an additional cyclometalation. This metal–carbon bond is formed through C–H activation at the 2-positon of the phenyl ring (13C NMR shift: δ 176.5 in toluene-d8).10 Similar to C1 and C2, the chelating NHC ligand in C3 binds via carbene coordination and η2-naphthyl coordination. Despite the polar carbon–metal bond, complex C3 is not sensitive to air and moisture in the solid state. However, C3 decomposes in solution over time. C3 was also confirmed to remain a single conformer in solution by NMR spectroscopy (in C6D6, toluene-d8, or THF-d8). It is noteworthy that the addition of an excess of HCl (4.0 M in dioxane) to the C3 solution (n-hexane, toluene, or THF) did not convert the monochloride complex to dichloride complex C2. Likewise, treatment of dichloride C2 with a strong base like NaOt-Bu did not yield monochloride C3. According to the same one pot procedure, precatalysts C4, C4a, C5, and C6 were prepared (Table 1).
Figure 2.
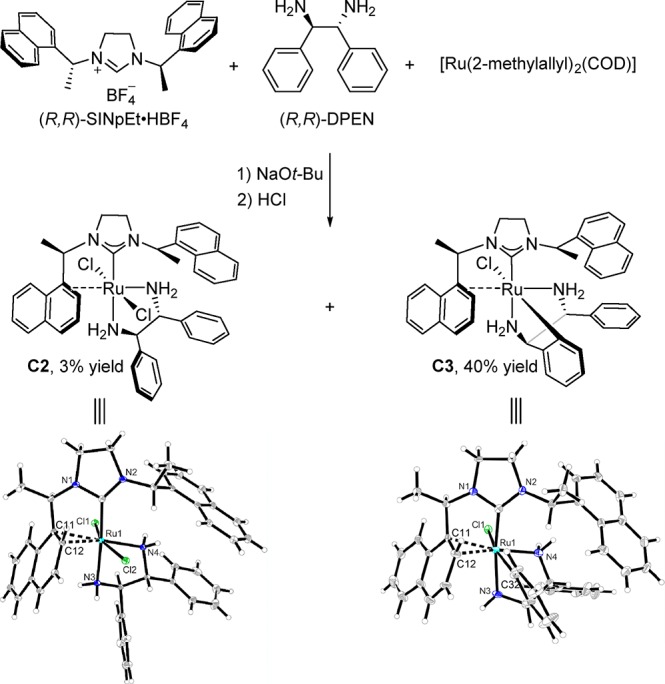
Synthesis and structure of trans-RuCl2(SINpEt)(DPEN) C2 and RuCl(SINpEt)(DPEN) C3. Selected bond lengths (Å) and bond angles (deg) of C2: Ru1–C1, 1.984(3); Ru1–N4, 2.146(3); Ru1–N3, 2.214(2); Ru1–C12, 2.222(3); Ru1–C11, 2.229(2); Ru1–Cl2, 2.4205(7); Ru1–Cl1, 2.4211(7); C11–C12, 1.423(5); C1–Ru1–N4, 103.22(11); C1–Ru1–N3, 172.73(11); N4–Ru1–N3, 78.58(9); C12–Ru1–C11, 37.30(12); Cl2–Ru1–Cl1, 160.63(2). Selected bond lengths (Å) and bond angles (deg) of C3: Ru1–C1, 1.953(9); Ru1–C32, 2.061(9); Ru1–N4, 2.153(7); Ru1–C12, 2.192(8); Ru1–N3, 2.203(7); Ru1–C11, 2.206(7); Ru1–Cl1, 2.549(2); C1–Ru1–C32, 99.3(3); C1–Ru1–N3, 173.8(3); N4–Ru1–N3, 75.5(3); C32–Ru1–Cl1, 160.0(3).
Table 1. Catalyst Test for the Enantioselective Hydrogenation of Isocoumarinsa.

| entry | precatalyst | time (h) | yield (%)b | e.r.c |
|---|---|---|---|---|
| 1 | C1 | 24 | trace | ND |
| 2 | C2 | 24 | trace | ND |
| 3 | C3 | 18 | 80 | 98:2 |
| 4 | C4 | 18 | 83 | 98.5:1.5 |
| 5 | C5 | 18 | 80 | 97:3 |
| 6 | C6 | 18 | 78 | 96:4 |
| 7 | C4a | 18 | 65 | 72:28 |
Reactions were carried out with 1a (0.2 mmol), the indicated ruthenium complex (0.004 mmol, 2 mol %), and NaOt-Bu (0.02 mmol, 10 mol %) in n-hexane (4.0 mL) under 50 bar of H2 at 15 °C for the indicated time.
Isolated yields.
Determined by HPLC analysis using a chiral stationary phase.
2.2. Applications of Ru(II)-NHC-Diamine Complexes in the Catalytic Asymmetric Hydrogenation
The isolated ruthenium complexes were then tested as precatalysts for the enantioselective hydrogenation. Asymmetric hydrogenation of 3-substituted isocoumarins is a direct way to form chiral 3-substituted 3,4-dihydroisocoumarins which represent a key motif in a wide range of natural products and biological active molecules.11,12 By use of the isolated precatalysts, the enantioselective hydrogenation of 3-substituted isocoumarines was probed (Table 1). A strong base like NaOt-Bu was determined to be necessary to activate the precatalysts. Only trace amounts of product were obtained in the presence of dichloride ruthenium complex C1 or C2 (Table 1, entries 1 and 2). Notably, RuCl(SINpEt)(DPEN) C3 exhibited a good activity under basic conditions (entry 3), giving the desired product 2a in 80% yield and 98:2 e.r.13 Next, variations on the chiral diamine ligand were investigated. Complex C4 with (1R,2R)-1,2-di-p-tolylethane-1,2-diamine gave slightly better results, delivering the product in 83% yield and 98.5:1.5 e.r. (entries 4). Further para-substituents on the diamine were tested and gave comparable results (entries 5 and 6). However, additional substituents in the ortho position turned out to be unfavorable for the enantioselective hydrogenation of 1a (entry 7). Diamines containing meta-substituents were previously tested through the in situ system, giving lower enantioselectivities and yields.7a We then explored the substrate scope of the reaction using the optimized conditions (Table 1, entry 4) as shown in the Supporting Information (page S36). A broad scope of isocoumarin derivatives was hydrogenated while tolerating differential position and electronic nature of the substituents as well as functional groups like thiophenes or methyl ethers (see Supporting Information for further details).
Next, we explored the enantioselective hydrogenation of benzothiophene 1,1-dioxides. The chiral 2,3-dihydrobenzothiophene 1,1-dioxide framework is a versatile motif in pharmaceutical research and agrochemistry.14 Recently, the Pfaltz group reported a novel way to prepare chiral 2,3-dihydrobenzothiophene 1,1-dioxides by the iridium-catalyzed asymmetric hydrogenation of substituted benzothiophene 1,1-dioxides.15,16 Remarkably, high reactivity and enantioselectivity of the hydrogenation of substituted benzothiophene 1,1-dioxides were obtained with the ruthenium complex C3 (Scheme 2). After optimization of the reaction conditions, 2-methylbenzo[b]thiophene 1,1-dioxide 3a was smoothly reduced under mild conditions using 0.5 mol % of precatalyst C3 and a low H2 pressure (5 bar) furnishing the corresponding product 4a in 99% yield and 97:3 e.r. The absolute configuration of 4a was determined to be S by comparing its optical rotation data to the literature.15 The effect of substituents on the phenyl ring was probed. Electron-poor substrate 3d provided the corresponding product 4d with excellent enantioselectivity and in a good yield, while the electron-rich substrates provided slightly lower e.r. values (4b and 4c). Halogen substituents F, Cl, and Br were well-tolerated under the mild reaction conditions (4e–g). Substrates containing longer alkyl chains and functional groups were hydrogenated with good e.r. values and excellent yields (4h–j). Poor enantioselectivity was observed when 2-phenylbenzothiophene 1,1-dioxide was used, due to the deprotonation of the product’s chiral center under basic conditions.
Scheme 2. Ruthenium-Catalyzed Asymmetric Hydrogenation of Benzothiophene 1,1-Dioxides.

Unless otherwise noted, reactions were conducted with 3 (0.3 mmol), C3 (0.0015 mmol, 0.5 mol %), and NaOt-Bu (0.02 mmol 6.7 mol %) under 5 bar of H2 in toluene (2.0 mL) at 25 °C for 24 h. Isolated yields after column chromatography are reported. The e.r. values were determined by HPLC analysis using a chiral stationary phase.
Using 30 bar of H2 at 0 °C.
The enantioselective hydrogenation of ketones is among the most important asymmetric reactions, producing chiral alcohols that are common core structures in biologically active molecules and natural products.3,17 To our delight, Ru(II)-NHC-diamine complexes can also be applied in this reaction (Scheme 3). Good enantioselectivities were obtained for acetophenones and 1-(thiophen-2-yl)ethan-1-one in the presence of ruthenium complex C4 (6a–c). Moreover, various benzo-fused cyclic ketones underwent the hydrogenation smoothly in the current catalytic system, giving the corresponding chiral alcohols with high e.r. values (6d–j).18
Scheme 3. Enantioselective Hydrogenation of Ketones Catalyzed by Ru(II)-NHC-Diamine Complexes.

Unless otherwise noted, reactions were carried out with 5 (1.0 mmol), C4 (0.0003 mmol, 0.03 mol %), and NaOt-Bu (0.02 mmol, 2.0 mol %) under 5–10 bar of H2 and in i-PrOH (1.0 mL) at 22 °C for 24 h. Isolated yields after column chromatography are reported. The e.r. values were determined by HPLC analysis using a chiral stationary phase.
Using 0.05 mol % of C4.
Using 0.5 mol % of C4.
2.3. Mechanistic Studies of the Ru(II)-NHC-Diamine Catalyzed Asymmetric Hydrogenation of Benzothiophene 1,1-Dioxides
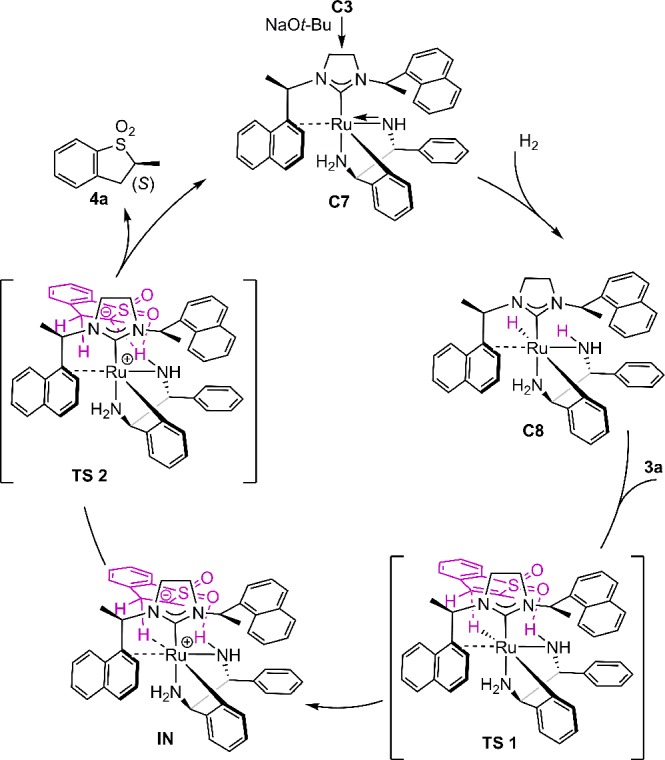
To gain information on the mechanism of the catalytic process, several stoichiometric reactions were conducted followed by in situ NMR measurements (Scheme 4). First, precatalyst C3 underwent a dehydrochlorination reaction upon treatment with NaOt-Bu (1.0 equiv) in toluene-d8 giving the amido complex C7 quantitively. This reaction was accompanied by a strong color change from yellow to dark red and took place in 5 min at room temperature. Removing t-BuOH under reduced pressure enabled the characterization of C7 (Scheme 4, step 1; for detailed information, see Supporting Information). According to NMR analysis, the η2-coordination of the naphthyl ring and the tridentate coordination of DPEN to the ruthenium center persisted at this stage. Complex C7 then reacted instantaneously with H2 (1 bar) in toluene-d8 at room temperature to produce metal hydride species C8 with a typical Ru–H hydride signal in the 1H NMR spectrum (−2.14 ppm) (Scheme 4, step 2). NMR characterization of C8 confirmed that no hydrogenolysis of the ruthenium–carbon occurred (for details, see Suppporting Information). Complex C8 decomposes rapidly in solution (0.14 M in toluene-d8) at room temperature but is stable for days at −78 °C. After release of H2 from the NMR tube, an amount of 2 equiv of 3a was added to C8 in toluene-d8 accompanied by a color change from brown to dark red. 1H resonances associated with complex C7 appeared while the signal corresponding to the Ru–H disappeared (Scheme 4, step 3). Meanwhile, 4a was observed with 97:3 e.r. and roughly 50% NMR yield with respect to the amount of 3a. If the mixture of step 3 was placed under H2 atmosphere again, C8 could be regenerated along with full conversion of 3a. Finally, quenching of complex C7 with an HCl solution (4 M in dioxane) regenerated the air-stable precatalyst C3 (Scheme 4, step 4).
Scheme 4. Stoichiometric Reactions and NMR Experiments.

On the basis of the above experiments, previous mechanistic studies of Ru(II)-diphosphine-diamine catalyzed asymmetric hydrogenations of ketones,19 and DFT calculations as described below (Scheme 6; see Supporting Information for details and unfavorable alternative pathways), we propose a mechanism for ruthenium-catalyzed asymmetric hydrogenation of benzothiophene 1,1-dioxides including the mode of enantioinduction (Scheme 5). The reaction between precatalyst C3 and NaOt-Bu should give the active amido complex C7. We postulate that an unobserved dihydrogen intermediate might be generated when placing complex C7 under an H2 atmosphere. Then, an intramolecular heterolytic splitting of dihydrogen would provide hydride complex C8. When substrate 3a is added to C8, a hydrogen-bond interaction between the amine proton and the oxygen atom of the sulfone group occurs. The resulting intermediate is stabilized by 0.8 kcal/mol relative to the reactants 3a and C8 for the pathway leading to the (S)-product according to our DFT (PBE0/def2-SVP) results (Scheme 6). A possible transition state TS1 is proposed based on the outer-sphere bifunctional catalysis mechanism by Noyori and other groups.19 The nucleophilic Ru–H hydride should attack the β position of the sulfone ring according to the reaction nature of benzothiophene 1,1-dioxide compounds.20 Our calculations confirm the existence of a transition state in which the Ru–H hydride is transferred to the β position of the sulfone ring in the presence of the above-mentioned hydrogen bond (Scheme 6, TS1), resulting in a destabilization of 3.7 kcal/mol. An intrinsic reaction coordinate (IRC) analysis shows, however, that hydrogen and proton transfers proceed in a stepwise manner. The IRC analysis leads to intermediate IN (Scheme 6, IN), in which the proton still resides on the amine group. This intermediate is stabilized by 12.5 kcal/mol compared to TS1. The product complex is reached via a second transition state (Scheme 6, TS2, destabilized by 2.8 kcal/mol compared to IN) in which the proton is transferred. In this second step, the proton of the amine group in the intermediate IN should transfer to the α position of the sulfone group from the Re face of the prochiral center, thus giving rise to the product 4a with S configuration and regenerating amido complex C7, resulting in a final stabilization of 14.2 kcal/mol.
Scheme 6. Calculated Relative Gibbs Free Energies for the Ruthenium-Catalyzed Asymmetric Hydrogenation of Benzothiophene 1,1-Dioxides Leading to the (S)-Product.

All values in kcal/mol.
Scheme 5. Proposed Mechanism for Ruthenium-Catalyzed Asymmetric Hydrogenation of Benzothiophene 1,1-Dioxides.
3. Conclusions
We introduced a practical and general procedure for the synthesis of chiral Ru(II)-NHC-diamine complexes based on the concept of ligand cooperation. The characterized ruthenium dichloride and monochloride complexes show a unique structure with unusual binding of the ligands. The ruthenium monochloride complexes serve as versatile catalysts for the enantioselective hydrogenation of isocoumarines, benzothiophene 1,1-dioxides, and ketones. A possible mechanism was proposed based on the identification of reaction intermediates via stoichiometric experiments, NMR analyses, and a computational study.
Acknowledgments
The authors acknowledge financial support from the European Research Council (ERC Advanced Grant Agreement No. 788558), the Deutsche Forschungsgemeinschaft (Leibniz Award and Grant IRTG 2027), the Alfried Krupp von Bohlen und Halbach Foundation, and Zhejiang University (W.L.). The authors also thank Dr. M. P. Wiesenfeldt, Dr. M. J. James, Dr. J. Li, and Dr. S. Gressies for many helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00985.
Experimental details for data acquisition, full quantum chemical study concerning the reaction mechanism, and additional discussion (PDF)
CCDC-1879259 (C1), -1879260 (C2) and -1879261 (C3) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Author Present Address
† W.L.: Department of Chemistry, Zhejiang University, Hangzhou 310027, China.
The authors declare no competing financial interest.
Supplementary Material
References
- Jacobsen E. N., Pfaltz A., Yamamoto H., Eds. Comprehensive Asymmetric Catalysis; Springer: New York, 1999; Vol. I-III, Suppl. I-II. [Google Scholar]
- a Privileged Chiral Ligands and Catalysts; Zhou Q.-L., Ed.; Wiley-VCH: Weinhem, Germany, 2011. [Google Scholar]; b Cooperative Catalysis: Designing Efficient Catalysts for Synthesis; Peters R., Ed.; Wiley-VCH: Weinhem, Germany, 2015; Chapter 3. [Google Scholar]; c Ding K.; Du H.; Yuan Y.; Long J. Combinatorial Chemistry Approach to Chiral Catalyst Engineering and Screening: Rational Design and Serendipity. Chem. - Eur. J. 2004, 10, 2872. 10.1002/chem.200305707. [DOI] [PubMed] [Google Scholar]; d Ding K. Synergistic Effect of Binary Component Ligands in Chiral Catalyst Library Engineering for Enantioselective Reactions. Chem. Commun. 2008, 909. 10.1039/b710668h. [DOI] [PubMed] [Google Scholar]; e Reetz M. T. Combinatorial Transition-Metal Catalysis: Mixing Monodentate Ligands to Control Enantio-, Diastereo-, and Regioselectivity. Angew. Chem., Int. Ed. 2008, 47, 2556. 10.1002/anie.200704327. [DOI] [PubMed] [Google Scholar]; f Piovesana S.; Scarpino Schietroma D. M.; Bella M. Multiple Catalysis with Two Chiral Units: An Additional Dimension for Asymmetric Synthesis. Angew. Chem., Int. Ed. 2011, 50, 6216. 10.1002/anie.201005955. [DOI] [PubMed] [Google Scholar]; g Hong L.; Sun W.; Yang D.; Li G.; Wang R. Additive Effects on Asymmetric Catalysis. Chem. Rev. 2016, 116, 4006. 10.1021/acs.chemrev.5b00676. [DOI] [PubMed] [Google Scholar]
- a Ohkuma T.; Ooka H.; Hashiguchi S.; Ikariya T.; Noyori R. Practical Enantioselective Hydrogenation of Aromatic Ketones. J. Am. Chem. Soc. 1995, 117, 2675. 10.1021/ja00114a043. [DOI] [Google Scholar]; b Ohkuma T.; Ooka H.; Ikariya T.; Noyori R. Preferential Hydrogenation of Aldehydes and Ketones. J. Am. Chem. Soc. 1995, 117, 10417. 10.1021/ja00146a041. [DOI] [Google Scholar]; c Doucet H.; Ohkuma T.; Murata K.; Yokozawa T.; Kozawa M.; Katayama E.; England A. F.; Ikariya T.; Noyori R. trans-[RuCl2(phosphane)(1,2-diamine)] and Chiral trans-[RuCl2(diphosphane)(1,2-diamine)]: Shelf-Stable Precatalysts for the Rapid, Productive, and Stereoselective Hydrogenation of Ketones. Angew. Chem., Int. Ed. 1998, 37, 1703.. [DOI] [PubMed] [Google Scholar]; d Noyori R.; Ohkuma T. Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem., Int. Ed. 2001, 40, 40.. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Han Z.; Ding K. The N-H Functional Group in Organometallic Catalysis. Angew. Chem., Int. Ed. 2013, 52, 4744. 10.1002/anie.201204921. [DOI] [PubMed] [Google Scholar]
- Selected reviews of N-heterocyclic carbene ligands in transition-metal catalysis:; a Herrmann W. A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem., Int. Ed. 2002, 41, 1290.. [DOI] [PubMed] [Google Scholar]; b Perry M. C.; Burgess K. Chiral N-Heterocyclic Carbene-Transition Metal Complexes in Asymmetric Catalysis. Tetrahedron: Asymmetry 2003, 14, 951. 10.1016/S0957-4166(03)00037-5. [DOI] [Google Scholar]; c César V.; Bellemin-Laponnaz S.; Gade L. H. Chiral N-Heterocyclic Carbenes as Stereodirecting Ligands in Asymmetric Catalysis. Chem. Soc. Rev. 2004, 33, 619. 10.1039/B406802P. [DOI] [PubMed] [Google Scholar]; d N-Heterocyclic Carbenes in Synthesis; Nolan S. P., Ed.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]; e Gade L. H.; Bellemin-Laponnaz S. Mixed Oxazoline-Carbenes as Stereodirecting Ligands for Asymmetric Catalysis. Coord. Chem. Rev. 2007, 251, 718. 10.1016/j.ccr.2006.05.015. [DOI] [PubMed] [Google Scholar]; f Dragutan V.; Dragutan I.; Delaude L.; Demonceau A. NHC–Ru Complexes—Friendly Catalytic Tools for Manifold Chemical Transformations. Coord. Chem. Rev. 2007, 251, 765. 10.1016/j.ccr.2006.09.002. [DOI] [Google Scholar]; g Hahn F. E.; Jahnke M. C. Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem., Int. Ed. 2008, 47, 3122. 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]; h Díez-González S.; Marion N.; Nolan S. P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612. 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]; i Gaillard S.; Cazin C. S. J.; Nolan S. P. N-Heterocyclic Carbene Gold(I) and Copper(I) Complexes in C–H Bond Activation. Acc. Chem. Res. 2012, 45, 778. 10.1021/ar200188f. [DOI] [PubMed] [Google Scholar]; j Valente C.; Çalimsiz S.; Hoi K. H.; Mallik D.; Sayah M.; Organ M. G. The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2012, 51, 3314. 10.1002/anie.201106131. [DOI] [PubMed] [Google Scholar]; k Wang F.; Liu L.-j.; Wang W.; Li S.; Shi M. Chiral NHC–Metal-Based Asymmetric Catalysis. Coord. Chem. Rev. 2012, 256, 804. 10.1016/j.ccr.2011.11.013. [DOI] [Google Scholar]; l Hopkinson M. N.; Richter C.; Schedler M.; Glorius F. An Overview of N-Heterocyclic Carbenes. Nature 2014, 510, 485. 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]; m Zhao D.; Candish L.; Paul D.; Glorius F. N-Heterocyclic Carbenes in Asymmetric Hydrogenation. ACS Catal. 2016, 6, 5978. 10.1021/acscatal.6b01736. [DOI] [Google Scholar]; n Janssen-Müller D.; Schlepphorst C.; Glorius F. Privileged Chiral N-Heterocyclic Carbene Ligands for Asymmetric Transition-Metal Catalysis. Chem. Soc. Rev. 2017, 46, 4845. 10.1039/C7CS00200A. [DOI] [PubMed] [Google Scholar]
- Our selected studies on carbene ligands:; a Urban S.; Ortega N.; Glorius F. Ligand-Controlled Highly Regioselective and Asymmetric Hydrogenation of Quinoxalines Catalyzed by Ruthenium N-Heterocyclic Carbene Complexes. Angew. Chem., Int. Ed. 2011, 50, 3803. 10.1002/anie.201100008. [DOI] [PubMed] [Google Scholar]; b Urban S.; Beiring B.; Ortega N.; Paul D.; Glorius F. Asymmetric Hydrogenation of Thiophenes and Benzothiophenes. J. Am. Chem. Soc. 2012, 134, 15241. 10.1021/ja306622y. [DOI] [PubMed] [Google Scholar]; c Zhao D.; Beiring B.; Glorius F. Ruthenium-NHC-Catalyzed Asymmetric Hydrogenation of Flavones and Chromones: General Access to Enantiomerically Enriched Flavanones, Flavanols, Chromanones, and Chromanols. Angew. Chem., Int. Ed. 2013, 52, 8454. 10.1002/anie.201302573. [DOI] [PubMed] [Google Scholar]; d Li W.; Schlepphorst C.; Daniliuc C.; Glorius F. Asymmetric Hydrogenation of Vinylthioethers: Access to Optically Active 1,5-Benzothiazepine Derivatives. Angew. Chem., Int. Ed. 2016, 55, 3300. 10.1002/anie.201512032. [DOI] [PubMed] [Google Scholar]; e Li W.; Wollenburg M.; Glorius F. Enantioselective Synthesis of 2-Oxazolidinones by Ruthenium(II)–NHC-Catalysed Asymmetric Hydrogenation of 2-Oxazolones. Chem. Sci. 2018, 9, 6260. 10.1039/C8SC01869C. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Wiesenfeldt M. P.; Nairoukh Z.; Li W.; Glorius F. Hydrogenation of Fluoroarenes: Direct Access to All-cis-(multi)fluorinated Cycloalkanes. Science 2017, 357, 908. 10.1126/science.aao0270. [DOI] [PubMed] [Google Scholar]
- a Li W.; Wiesenfeldt M. P.; Glorius F. Ruthenium–NHC–Diamine Catalyzed Enantioselective Hydrogenation of Isocoumarins. J. Am. Chem. Soc. 2017, 139, 2585. 10.1021/jacs.6b13124. [DOI] [PubMed] [Google Scholar]; For a related achiral NHC/amine ligand system, see the following:; b Filonenko G. A.; Aguila M. J. B.; Schulpen E. N.; van Putten R.; Wiecko J.; Müller C.; Lefort L.; Hensen E. J. M.; Pidko E. A. Bis-N-heterocyclic Carbene Aminopincer Ligands Enable High Activity in Ru-Catalyzed Ester Hydrogenation. J. Am. Chem. Soc. 2015, 137, 7620. 10.1021/jacs.5b04237. [DOI] [PubMed] [Google Scholar]
- McGuinness D. S.; Cavell K. J. Donor-Functionalized Heterocyclic Carbene Complexes of Palladium(II): Efficient Catalysts for C-C Coupling Reactions. Organometallics 2000, 19, 741. 10.1021/om990776c. [DOI] [Google Scholar]
- a Wang H. M. J.; Lin I. J. B. Facile Synthesis of Silver(I)-Carbene Complexes. Useful Carbene Transfer Agents. Organometallics 1998, 17, 972. 10.1021/om9709704. [DOI] [Google Scholar]; b Pytkowicz J.; Roland S.; Mangeney P. Synthesis of Chiral Silver(I) Diaminocarbene Complexes From (R,R)-4,5-di-tert-butylimidazoline. J. Organomet. Chem. 2001, 631, 157. 10.1016/S0022-328X(01)01013-0. [DOI] [Google Scholar]; c Winn C. L.; Guillen F.; Pytkowicz J.; Roland S.; Mangeney P.; Alexakis A. Enantioselective Copper Catalysed 1,4-Conjugate Addition Reactions Using Chiral N-Heterocyclic Carbenes. J. Organomet. Chem. 2005, 690, 5672. 10.1016/j.jorganchem.2005.07.024. [DOI] [Google Scholar]
- This cyclometalation might be formed via a 2-methylallyl-assisted concerted-metalated-deprotonation (CMD) C–H bond activation. For related works involving cyclometalation of diamine with ruthenium, see the following:; a Koike T.; Ikariya T. Reaction of 16-Electron Ruthenium and Iridium Amide Complexes with Acidic Alcohols: Intramolecular C-H Bond Activation and the Isolation of Cyclometalated Complexes. Organometallics 2005, 24, 724. 10.1021/om049205x. [DOI] [Google Scholar]; b Sortais J.-B.; Pannetier N.; Holuigue A.; Barloy L.; Sirlin C.; Pfeffer M.; Kyritsakas N. Cyclometalation of Primary Benzyl Amines by Ruthenium(II), Rhodium(III), and Iridium(III) Complexes. Organometallics 2007, 26, 1856. 10.1021/om060973t. [DOI] [Google Scholar]; c Matsumura K.; Arai N.; Hori K.; Saito T.; Sayo N.; Ohkuma T. Chiral Ruthenabicyclic Complexes: Precatalysts for Rapid, Enantioselective, and Wide-Scope Hydrogenation of Ketones. J. Am. Chem. Soc. 2011, 133, 10696. 10.1021/ja202296w. [DOI] [PubMed] [Google Scholar]; d Matsunami A.; Kuwata S.; Kayaki Y. A Bifunctional Iridium Catalyst Modified for Persistent Hydrogen Generation from Formic Acid: Understanding Deactivation via Cyclometalation of a 1,2-Diphenylethylenediamine Motif. ACS Catal. 2017, 7, 4479. 10.1021/acscatal.7b01068. [DOI] [Google Scholar]
- a Van der Merwe K. J.; Steyn P. S.; Fourie L.; Scott D. B.; Theron J. J. Ochratoxin A, a Toxic Metabolite Produced by Aspergillus Ochraceus Wilh. Nature 1965, 205, 1112. 10.1038/2051112a0. [DOI] [PubMed] [Google Scholar]; b Malir F.; Ostry V.; Pfohl-Leszkowicz A.; Malir J.; Toman J. Ochratoxin A: 50 Years of Research. Toxins 2016, 8, 191. 10.3390/toxins8070191. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sun H.; Ho C. L.; Ding F.; Soehano I.; Liu X. W.; Liang Z. X. Synthesis of (R)-Mellein by a Partially Reducing Iterative Polyketide Synthase. J. Am. Chem. Soc. 2012, 134, 11924. 10.1021/ja304905e. [DOI] [PubMed] [Google Scholar]
- a Islam M. S.; Ishigami K.; Watanabe H. Synthesis of (−)-Mellein, (+)-Ramulosin, and Related Natural Products. Tetrahedron 2007, 63, 1074. 10.1016/j.tet.2006.11.068. [DOI] [Google Scholar]; b Chakraborty T. K.; Chattopadhyay A. K. Total Synthesis of Cruentaren B. J. Org. Chem. 2008, 73, 3578. 10.1021/jo800181n. [DOI] [PubMed] [Google Scholar]; c Habel A.; Boland W. Efficient and Flexible Synthesis of Chiral γ- and δ-Lactones. Org. Biomol. Chem. 2008, 6, 1601. 10.1039/b801514g. [DOI] [PubMed] [Google Scholar]; d Feng Y.; Jiang X.; De Brabander J. K. Studies toward the Unique Pederin Family Member Psymberin: Full Structure Elucidation, Two Alternative Total Syntheses, and Analogs. J. Am. Chem. Soc. 2012, 134, 17083. 10.1021/ja3057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Over-reduction of the product converting the lactone into the diol was observed, which lowered the yield and slightly enhanced the enantioselectivity.
- a Rempfler H.; Edmunds A.; De Mesmaeker A.; Seckinger K. Patent WO 9909023, 1999. (Novartis AG).; b Saitou M.; Sekiguchi H.; Ogawa S. Patent WO 2000069853, 2000. (Idemitsu Kosan Co., Ltd.).; c Edmunds A.; Mghlebach M.; Stoller A.; Loiseleur O.; Buchholz A.; Hueter O. F.; Bigot A.; Hall R. G.; Emery D.; Jung P. J. M.; Lu L.; Wu Y.; Chen R. Patent WO 2015000715, 2015. (Syngenta AG).; d Wehn P.; Yang H. Patent US 20160362390, 2016. (Peloton Therapeutics, Inc.).
- Tosatti P.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of Benzo[b]thiophene 1,1-Dioxides. Angew. Chem., Int. Ed. 2017, 56, 4579. 10.1002/anie.201701409. [DOI] [PubMed] [Google Scholar]
- For a related asymmetric hydrogenation of unsaturated sulfones, see the following:; a Zhou T.; Peters B.; Maldonado M. F.; Govender T.; Andersson P. G. Enantioselective Synthesis of Chiral Sulfones by Ir-Catalyzed Asymmetric Hydrogenation: A Facile Approach to the Preparation of Chiral Allylic and Homoallylic Compounds. J. Am. Chem. Soc. 2012, 134, 13592. 10.1021/ja306731u. [DOI] [PubMed] [Google Scholar]; b Peters B. K.; Zhou T.; Rujirawanich J.; Cadu A.; Singh T.; Rabten W.; Kerdphon S.; Andersson P. G. An Enantioselective Approach to the Preparation of Chiral Sulfones by Ir-Catalyzed Asymmetric Hydrogenation. J. Am. Chem. Soc. 2014, 136, 16557. 10.1021/ja5079877. [DOI] [PubMed] [Google Scholar]; c Shi L.; Wei B.; Yin X.; Xue P.; Lv H.; Zhang X. Rh-Catalyzed Asymmetric Hydrogenation of α-Substituted Vinyl Sulfones: An Efficient Approach to Chiral Sulfones. Org. Lett. 2017, 19, 1024. 10.1021/acs.orglett.6b03845. [DOI] [PubMed] [Google Scholar]; d Meng K.; Xia J.; Wang Y.; Zhang X.; Yang G.; Zhang W. Ir/BiphPHOX-Catalyzed Asymmetric Hydrogenation of 3-Substituted 2,5-Dihydropyrroles and 2,5-Dihydrothiophene 1,1-Dioxides. Org. Chem. Front. 2017, 4, 1601. 10.1039/C7QO00248C. [DOI] [Google Scholar]; e Long J.; Shi L.; Li X.; Lv H.; Zhang X. Rhodium-Catalyzed Highly Regio- and Enantioselective Hydrogenation of Tetrasubstituted Allenyl Sulfones: An Efficient Access to Chiral Allylic Sulfones. Angew. Chem., Int. Ed. 2018, 57, 13248. 10.1002/anie.201804891. [DOI] [PubMed] [Google Scholar]; f Yan Q.; Xiao G.; Wang Y.; Zi G.; Zhang Z.; Hou G. Highly Efficient Enantioselective Synthesis of Chiral Sulfones by Rh-Catalyzed Asymmetric Hydrogenation. J. Am. Chem. Soc. 2019, 141, 1749. 10.1021/jacs.8b12657. [DOI] [PubMed] [Google Scholar]; g Liu G.; Zhang H.; Huang Y.; Han Z.; Liu G.; Liu Y.; Dong X.-Q.; Zhang X. Efficient synthesis of chiral 2,3-dihydro-benzo[b]thiophene 1,1-dioxides via Rh-catalyzed hydrogenation. Chem. Sci. 2019, 10, 2507. 10.1039/C8SC05397A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews, see the following:; a Chen Q.-A.; Ye Z.-S.; Duan Y.; Zhou Y.-G. Homogeneous Palladium-Catalyzed Asymmetric Hydrogenation. Chem. Soc. Rev. 2013, 42, 497. 10.1039/C2CS35333D. [DOI] [PubMed] [Google Scholar]; b Xie J.-H.; Bao D.-H.; Zhou Q.-L. Recent Advances in the Development of Chiral Metal Catalysts for the Asymmetric Hydrogenation of Ketones. Synthesis 2015, 47, 460. 10.1055/s-0034-1378939. [DOI] [Google Scholar]; c Ayad T.; Phansavath P.; Ratovelomanana-Vidal V. Transition-Metal-Catalyzed Asymmetric Hydrogenation and Transfer Hydrogenation: Sustainable Chemistry to Access Bioactive Molecules. Chem. Rec. 2016, 16, 2754. 10.1002/tcr.201600100. [DOI] [PubMed] [Google Scholar]; d Ohkuma T.; Arai N. Advancement in Catalytic Asymmetric Hydrogenation of Ketones and Imines, and Development of Asymmetric Isomerization of Allylic Alcohols. Chem. Rec. 2016, 16, 2801. 10.1002/tcr.201600101. [DOI] [PubMed] [Google Scholar]; e Meemken F.; Baiker A. Recent Progress in Heterogeneous Asymmetric Hydrogenation of C=O and C=C Bonds on Supported Noble Metal Catalysts. Chem. Rev. 2017, 117, 11522. 10.1021/acs.chemrev.7b00272. [DOI] [PubMed] [Google Scholar]; f Xie X.; Lu B.; Li W.; Zhang Z. Coordination Determined Chemo- and Enantioselectivities in Asymmetric Hydrogenation of Multi-Functionalized Ketones. Coord. Chem. Rev. 2018, 355, 39. 10.1016/j.ccr.2017.07.013. [DOI] [Google Scholar]
- For selected examples of the asymmetric hydrogenation of benzo-fused cyclic ketones, see the following:; a Ohkuma T.; Hattori T.; Ooka H.; Inoue T.; Noyori R. BINAP/1,4-Diamine-Ruthenium(II) Complexes for Efficient Asymmetric Hydrogenation of 1-Tetralones and Analogues. Org. Lett. 2004, 6, 2681. 10.1021/ol049157c. [DOI] [PubMed] [Google Scholar]; b Matsumura K.; Arai N.; Hori K.; Saito T.; Sayo N.; Ohkuma T. Chiral Ruthenabicyclic Complexes: Precatalysts for Rapid, Enantioselective, and Wide-Scope Hydrogenation of Ketones. J. Am. Chem. Soc. 2011, 133, 10696. 10.1021/ja202296w. [DOI] [PubMed] [Google Scholar]; c Li Y.; Zhou Y.; Shi Q.; Ding K.; Noyori R.; Sandoval C. A. An Efficient Diphosphine/Hybrid-Amine Combination for Ruthenium(II)-Catalyzed Asymmetric Hydrogenation of Aryl Ketones. Adv. Synth. Catal. 2011, 353, 495. 10.1002/adsc.201000577. [DOI] [Google Scholar]; d Xie J.-B.; Xie J.-H.; Liu X.-Y.; Zhang Q.-Q.; Zhou Q.-L. Chiral Iridium Spiro Aminophosphine Complexes: Asymmetric Hydrogenation of Simple Ketones, Structure, and Plausible Mechanism. Chem. - Asian J. 2011, 6, 899. 10.1002/asia.201000716. [DOI] [PubMed] [Google Scholar]; e Rodríguez S.; Qu B.; Fandrick K. R.; Buono F.; Haddad N.; Xu Y.; Herbage M. A.; Zeng X.; Ma S.; Grinberg N.; Lee H.; Han Z. S.; Yee N. K.; Senanayake C. H. Amine-Tunable Ruthenium Catalysts for Asymmetric Reduction of Ketones. Adv. Synth. Catal. 2014, 356, 301. 10.1002/adsc.201300727. [DOI] [Google Scholar]; f Yin C.; Dong X.-Q.; Zhang X. Iridium/f-Amphol-catalyzed Efficient Asymmetric Hydrogenation of Benzo-fused Cyclic Ketones. Adv. Synth. Catal. 2018, 360, 4319. 10.1002/adsc.201800839. [DOI] [Google Scholar]
- For selected examples, see the following:; a Hartmann R.; Chen P. Noyori’s Hydrogenation Catalyst Needs a Lewis Acid Cocatalyst for High Activity. Angew. Chem., Int. Ed. 2001, 40, 3581.. [DOI] [PubMed] [Google Scholar]; b Abdur-Rashid K.; Faatz M.; Lough A. J.; Morris R. H. Catalytic Cycle for the Asymmetric Hydrogenation of Prochiral Ketones to Chiral Alcohols: Direct Hydride and Proton Transfer from Chiral Catalysts trans-Ru(H)2(diphosphine)(diamine) to Ketones and Direct Addition of Dihydrogen to the Resulting Hydridoamido Complexes. J. Am. Chem. Soc. 2001, 123, 7473. 10.1021/ja015902u. [DOI] [PubMed] [Google Scholar]; c Abdur-Rashid K.; Clapham S. E.; Hadzovic A.; Harvey J. N.; Lough A. J.; Morris R. H. Mechanism of the Hydrogenation of Ketones Catalyzed by trans-Dihydrido(diamine)ruthenium(II) Complexes. J. Am. Chem. Soc. 2002, 124, 15104. 10.1021/ja016817p. [DOI] [PubMed] [Google Scholar]; d Sandoval C. A.; Ohkuma T.; Muñiz K.; Noyori R. Mechanism of Asymmetric Hydrogenation of Ketones Catalyzed by BINAP/1,2-Diamine-Ruthenium(II) Complexes. J. Am. Chem. Soc. 2003, 125, 13490. 10.1021/ja030272c. [DOI] [PubMed] [Google Scholar]; e Dub P. A.; Gordon J. C. The Mechanism of Enantioselective Ketone Reduction with Noyori and Noyori–Ikariya Bifunctional Catalysts. Dalton Trans. 2016, 45, 6756. 10.1039/C6DT00476H. [DOI] [PubMed] [Google Scholar]; f Dub P. A.; Scott B. L.; Gordon J. C. Why Does Alkylation of the N–H Functionality within M/NH Bifunctional Noyori-Type Catalysts Lead to Turnover?. J. Am. Chem. Soc. 2017, 139, 1245. 10.1021/jacs.6b11666. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; McKellin W. H. Benzothiophene Chemistry. IV. Some Addition Reactions of Benzothiophene-1-dioxide. J. Am. Chem. Soc. 1950, 72, 1985. 10.1021/ja01161a030. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.