

Abstract
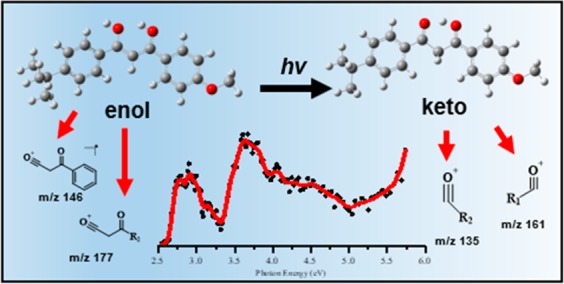
Avobenzone (AB) is a widely used UVA filter known to undergo irreversible photodegradation. Here, we investigate the detailed pathways by which AB photodegrades by applying UV laser-interfaced mass spectrometry to protonated AB ions. Gas-phase infrared multiple-photon dissociation (IRMPD) spectra obtained with the free electron laser for infrared experiments, FELIX, (600–1800 cm–1) are also presented to confirm the geometric structures. The UV gas-phase absorption spectrum (2.5–5 eV) of protonated AB contains bands that correspond to selective excitation of either the enol or diketo forms, allowing us to probe the resulting, tautomer-dependent photochemistry. Numerous photofragments (i.e., photodegradants) are directly identified for the first time, with m/z 135 and 161 dominating, and m/z 146 and 177 also appearing prominently. Analysis of the production spectra of these photofragments reveals that that strong enol to keto photoisomerism is occurring, and that protonation significantly disrupts the stability of the enol (UVA active) tautomer. Close comparison of fragment ion yields with the TD-DFT-calculated absorption spectra give detailed information on the location and identity of the dissociative excited state surfaces, and thus provide new insight into the photodegradation pathways of avobenzone, and photoisomerization of the wider class of β-diketone containing molecules.
1. Introduction
So that the skin can be protected against radiation damage linked to sunlight exposure, considerable effort has been put into the development of effective sunscreens.1−4 Over recent years, a number of advanced laser spectroscopic studies have been conducted on sunscreen molecules under highly controlled conditions, with the aim of improving our fundamental knowledge of the mechanisms by which molecular sunscreens function.1,5−13 Such measurements aim to provide fundamental insights into the properties of the sunscreen molecule free from the complications of the complex environment of a real sunscreen lotion. One group of experiments has focused on “isolated” gas-phase sunscreen molecules to provide information on how UV light absorption varies as a function of molecular structure.11,12,14−16 A second group of studies have used transient absorption spectroscopy to probe relaxation dynamics of two-component mixtures of a single organic sunscreen molecule in a single solvent.1,6−8,10 Experiments are typically complemented by high-level quantum chemical calculations,17−22 which then provide important insights into the mechanism by which the sunscreen molecule operates.
Avobenzone (AB: 4-tert-butyl-4′-methoxydibenzoylmethane; BD-DBM or Parsol 1789) is a widely used UVA filter which is known to suffer from irreversible UV-driven photodegradation.1,23,24 AB is a dibenzoylmethane derivative and a member of a family of molecules known to take multiple isomerization pathways following UV excitation.25−30 Photoexcitation of AB is believed to lead to nonadiabatic population of high-lying S0 vibrational levels, which can lead to isomerization to a less photostable form.31 Previous photolysis experiments performed in hexane have shown that this photodegradation of AB occurs by a Norrish type 1 mechanism.32,33 (The key tautomeric forms of AB involved in this photoisomerization are illustrated in Scheme 1.) In the ground state, AB is mostly found in its chelated enol (CE) form due to the stabilizing intramolecular hydrogen bond, although the diketo (DK) form is also typically present, with the relative tautomeric ratios being strongly solvent dependent. The CE form is the active UVA blocker (λmax = 355 nm), with photoisomerization leading to nonchelate forms, which gradually populate the DK form. The DK tautomer absorbs at higher UV energies (λmax = 265 nm) and has been shown to be responsible for formation of a reactive triplet. Indeed, the instability of the diketo form is well established.32,34−36 Wang et al., for example, demonstrated that AB photodegrades (in the UVA) more quickly as a function of chlorine substitution at the α-carbon (C2) since this promotes the diketo form.37
Scheme 1. Schematic Diagram Illustrating the UVA Active Chelated-Enol (CE) and UVB Active Diketo (DK) Tautomer of Avobenzone. Irradiation of the DK Tautomer Causes Photoinitiated α-Cleavage.

A considerable number of studies have been performed to explore how AB tautomerization, and hence photostability, is affected by the solvent.38−40 These studies prompt questions of whether other local environmental factors can influence the extent of tautomerization. In this work, we directly address one such issue, namely the potential effect of acid conditions on the photochemistry of AB by directly studying the protonated form of AB, i.e. AB·H+, as an isolated system in the gas phase. We use laser-interfaced mass spectrometry (LIMS) to select ions that are then subjected to wavelength-dependent laser photodissociation, allowing us to measure the gas-phase absorption spectrum.41,42 LIMS has recently been shown to be a useful tool for studying isomeric systems.43−47 Applying LIMS to AB·H+ allows us to selectively investigate the photochemistry of both enol and keto tautomers, and to probe the extent to which tautomerization occurs, away from any influences of the bulk-phase environment. Importantly, our experiments also allow us to measure the photodegradants of the individual AB·H+ tautomers. To complement the UV photochemistry, we present infrared multiple-photon dissociation (IRMPD) spectroscopy of AB·H+ to confirm which tautomers are present.
Fundamental studies of the effect of pH on sunscreen molecules to date are sparse.5,41,48,49 Recent measurements from our group have highlighted the impact of protonation and deprotonation on oxybenzone, in terms of the nature and wavelength dependence of its UV photofragmentation pathways.41 A separate study of deprotonated 2-phenylbenzimidazole-5-sulfonic acid provided direct evidence for the production of free radical photoproducts following UVA excitation.42 We note, however, that there is broad interest in the effect of pH in the field of environmental chemistry and degradation of man-made pollutants present in surface water,50−56 so that further work assessing the effect of pH on individual sunscreens is highly desirable.
2. Methods
Gas-phase UV photodissociation experiments were conducted in an AmaZon SL dual funnel electrospray ionization quadrupole ion trap (ESI-QIT) mass spectrometer (Bruker Daltonics Inc., Billerica, MA, USA), which was modified to allow LIMS.41,43,57 Avobenzone (AB) was purchased from Sigma-Aldrich (St. Louis, MO, USA) and HPLC-grade EtOH was purchased from Fisher Scientific, Inc. (Pittsburgh, PA, USA), both used as received. Solutions of AB (1 × 10–4 mol dm–3) in EtOH were electrosprayed using typical instrumental parameters (nebulizing gas pressure of 10.0 psi; injection rate of 0.33 mL/h; drying gas flow rate of 8.0 L/min), and run in positive ion mode at a capillary temperature of 100 °C.
AB·H+ was mass selected and isolated in the ion-trap prior to laser irradiation. UV–vis photons were produced by a 10 Hz Nd:YAG (Surelite, Amplitude Laser Group, San Jose, CA, USA) pumped OPO (Horizon, Amplitude Laser Group) laser, giving ∼0.3 mJ across the range 400–216 nm (3.10–5.80 eV). A 2 nm laser step size was used to record the photodepletion and photofragment spectra of the AB·H+ parent ion. To check for single-photon conditions, the laser power dependence on signal was determined as described previously.42
Photofragmentation experiments were conducted with an ion accumulation time of 20 ms and a fragmentation time of 100 ms, thereby ensuring that each mass-selected ion packet interacted with one laser pulse, minimizing the likelihood of multiphoton events. When fluorescence is negligible,58 the UV excited gaseous ion will fragment upon excited state relaxation, yielding an action absorption spectrum by photodepletion (PD).57,59,60 PD was measured as a function of the scanned wavelength, with the photofragment production (PF) also recorded simultaneously at each wavelength, both of which according to
| 1 |
| 2 |
where IntOFF and IntON are the peak intensities with laser off and on respectively; IntFRAG is the selected fragment intensity with the laser on; λ is the excitation wavelength (nm); and P is the tunable laser pulse energy (mJ). The photodepletion intensities were taken from an average of three runs at each wavelength of the ranges studied. We note that fragment ions with m/z < 50 are not detectable in our mass spectrometer because of the low-mass cutoff of the quadrupole ion trap. The UV photodepletion (absorption) spectra of the major AB photofragments produced upon the photoexcitation of AB·H+ were also recorded using MSn scheme and mass-isolation feature available on the trapControl version 7.2 (Bruker Daltonics Inc.) software.
IR experiments were performed in a modified commercial quadrupole ion-trap mass spectrometer (Bruker, AmaZon Speed ETD).61,62 Ions were generated in an Apollo ESI source. Solutions of AB·H+ (1 × 10–6 mol dm–3) in MeOH were introduced at 180 μL/h flow rates, electrosprayed, and transferred into the trap. The ions of interest were mass-selected and fragmented by IRMPD. IR spectra in the 700–1800 cm–1 region were recorded using the FELIX infrared free electron laser.63 FELIX was set to produce IR radiation in the form of 5–10 μs macropulses of 80–120 mJ per pulse at a 10 Hz repetition rate and with a bandwidth of ∼0.4% of the center frequency. The mass-selected ions were irradiated with one macropulse. Resonant absorption of IR radiation leads to an increase of the internal energy of an ion mediated by intramolecular vibrational redistribution (IVR), which eventually leads to unimolecular dissociation.64 After irradiation, a mass spectrum of the resulting ions in the trap is recorded. At each IR frequency point, six mass spectra were averaged. The dissociation was calculated from the mass spectra by relating the precursor ion and fragment ion intensities (eq 3) and plotted as a function of IR frequency.65
| 3 |
The IRMPD intensity was linearly corrected for frequency-dependent laser pulse energy. Spectra were also recorded at two levels of laser-pulse energy attenuation (factors of 2.00 and 3.16) to prevent excessive depletion of the precursor ions (saturation) and minimize formation of low m/z fragment ions which may be undetected in the ion trap and which would result in underestimated IRMPD intensities.65
Calculations were performed using density functional theory in Gaussian 09.66 All reported structures correspond to true minima, as confirmed by frequency calculations. Conformational molecular dynamics searches implemented by Schrodinger’s MacroModel were conducted with mixed Monte Carlo torsional and low-mode sampling to generate possible conformers which were rapidly energy gradient minimized with the OPLS3e force field.67 Unique structures (RMSD 0.4 Å, <100 kJ/mol) were then energy minimized at the ωB97X-D/def2-SVP and M06-2X/def2-SVP levels of theory. An implicit solvent model was used to calculate the relative energies of conformers upon solvation. Frequency calculations at the B3LYP/def2-TZVPP level were performed to test that optimized structures are true minima and were used to interpret the IRMPD spectra.68
3. Results
3.1. Quantum Chemical Calculations of Gas-Phase and Solution-Phase AB·H+
Scheme 2 illustrates the key tautomeric structures of AB·H+. The atomic numbering system previously used by Kojic et al. is also followed here.27 The carbonyl group is the primary protonation site, although a natural population analysis of the low-energy structures indicates that the excess charge is delocalized (section S1). A conformational molecular dynamics search was conducted on AB·H+, with the proton located on either the O1 or O2 site, followed by DFT optimization. Table 1 displays the tautomer relative energies, in the gas-phase and solution (rotamers are omitted from Table 1 as their electronic spectra are expected to be identical at our experimental resolution). The results presented in Table 1 suggest that solvation has little effect on the relative tautomer energies. We note that for the Kb structure, it was challenging to identify the minimum, which was only observed in the presence of solvent at the ωB97X-D level of theory. The calculated relative energies for the various AB·H+ tautomers lead us to predict that the Ka isomer will dominate, both in the gas phase and in ethanol solution, with the Ea isomer also being present at a much reduced but still significant level.
Scheme 2. Schematic Diagram of the Key Tautomeric Structures of AB·H+ with Atom and Rotation Coordinate Labels. The Ka Ketone and Ea Enol Tautomers Are Protonated at the O2 Atom, the Kb Ketone and the Eb Enol at the O1 Atom.

Table 1. Calculated Relative Energies of AB·H+ and Population Analysis. Values in Parentheses Were Calculated in Ethanol Solvent. NM = No Minima Found.
| relative
energy (kJ mol–1)a |
population (%)b |
|||||
|---|---|---|---|---|---|---|
| structure | ωB97X-D | M06-2X | B3LYP | ωB97X-D | M06-2X | B3LYP |
| Ka | 0 (0) | 0 (0) | 0 (0) | 99 (77) | 75 (83) | 84 (91) |
| Kb | NM (4) | NM (NM) | NM (NM) | NM (23) | NM (NM) | NM (NM) |
| Ea | 16 (17) | 4 (6) | 8 (11) | <1 (<1) | 20 (12) | 11 (6) |
| Eb | 20 (20) | 9 (9) | 12 (13) | 4 (5) | 4 (3) | |
Relative electronic energy values are zero-point energy corrected.
Boltzmann probability distribution calculated at 100 °C from relative energy difference column values.
The lowest-energy isomers, Ka and Kb, are planar across the aromatic and diketone moieties. Suffixes a and b identify tautomer protonation sites as O2 and O1, respectively, and this nomenclature is applied to the enol-like structures as well. The Ea structure represents the protonated enol form of AB·H+, which is nonplanar, with the O1H1 bond (H1/O1/C1/R11) directed +10° out-of-plane and the R1-arene by −30° (O1/C1/R11/R12). The Eb structure is similarly nonplanar with the O2H2 bond (H2/O2/C3/R21) positioned +12° and the R2-arene by −26° (O2/C3/R21/R22). The Ka and Ea tautomer energies vary significantly with different functionals, with ωB97X-D placing Ea substantially higher in energy than when using M06-2X. This disagreement mirrors the previous reported issues in predicting relative energy values for the neutral keto and enol tautomers of dibenzoylmethane systems without anharmonic methods.27,69 Many of these previous papers also identified twisted Z and E rotamer geometries. Our conformational search identified many such twisted forms, but their relative energies lay at much higher energies (>40 and 30 kJ/mol using the ωB97X-D and M06-2X functionals, respectively) so they have been omitted from this work. The high energy of the twisted geometries highlights the electronic stability of the planar, hyperconjugated, AB·H+ ion.
As noted above, the Kb isomer only exists as a geometric minimum when optimized using the ωB97X-D functional and with a solvent present. Figure 1 shows the relaxed coordinate scan between Ka and Kb proton transfer geometries as a function of the O2H bond distance from 1.05 to 1.50 Å (a relaxed energy minimization at the ωB97X-D/def2-SVP level was performed at each scan coordinate). The calculated intermolecular hydrogen bond length is 1.41 and 1.46 Ε in the gas phase and in ethanol, respectively. Our calculations reveal that while a 4 kJ/mol energy difference exists between the proton transfer isomers there is no barrier to this reaction in the absence of solvent. In ethanol, there exists a very small 1 kJ/mol barrier to the shallow potential well. Nonetheless, the Kb isomer exists by way of a motional resonance with Ka due to the in-plane intermolecular hydrogen bond. A symmetric C2/O2/H in-plane bend (section S1) defines the Kb ← Ka proton migration and has a zero-point energy of 0.28 eV.
Figure 1.
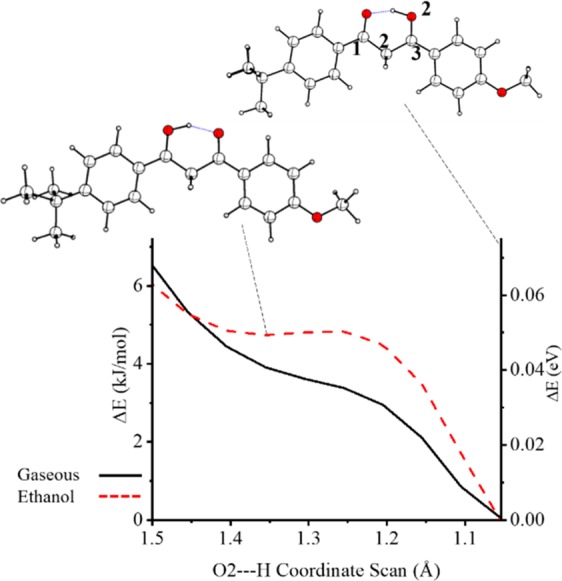
Coordinate energy scans about the O2–H bond, presented with selected molecular structures.
TD-DFT calculations were performed to determine the electronic excitations of the Ka, Ea, and Eb structures. The resulting simulated electronic spectra are presented in Figure 2 for excitation energies between 2.5 and 6.0 eV and reveal that the absorption spectra associated with the low-energy cis structures, Ka and Ea/Eb, are quite different. The same was observed previously in experiments on neutral AB,35,40 and is directly related to the substantial change of electron density within the pseudocyclic moiety that occurs upon keto–enol tautomerization.27 Three calculated spectra are overlaid for each isomer to explore the effect of changing functional. The Ka spectrum shows good agreement between the different dispersive functional treatments, while the PBE0 spectrum is red-shifted by 0.2 eV. The convolved spectral shape of Ka is nearly identical for all functionals chosen here, with each calculation predicting a strong transition followed by a medium strength transition 0.5 eV higher in energy. The Ea spectral profile is also similar for each of the dispersive functionals, with the PBE0 spectrum red-shifted by 0.26 eV. The convolved spectral shape of Ea shows two well-separated transitions, with the most intense being the lower-energy transition. Similar results were obtained for the Eb tautomer, with the spectrum being red-shifted by 0.06 eV compared to Ea. The difference between the most intense Ka and Ea transitions is 0.76 eV.
Figure 2.
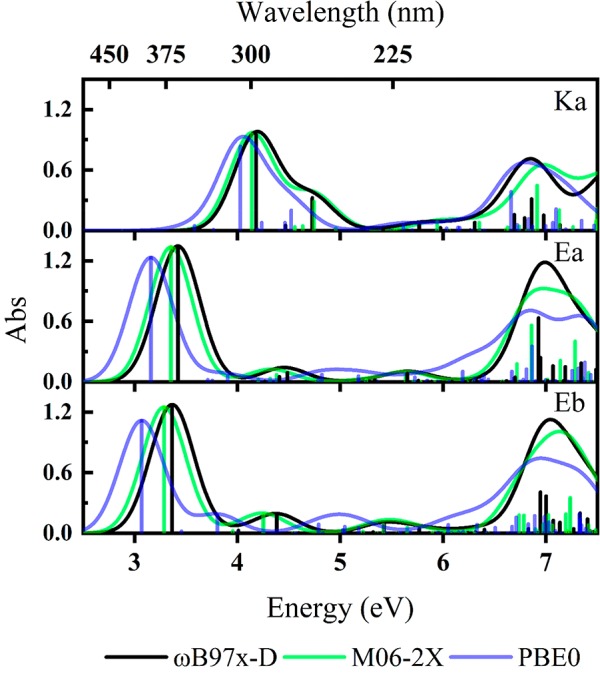
Calculated gas-phase TD-DFT excitation energies and simulated electronic absorption spectra (ωB97X-D, M06-2X, PBE0) for Ka, Ea, and Eb. Oscillator strengths of individual transitions are given as vertical bars, while the full line spectrum is a convolution of the calculated transitions with a Gaussian function (0.5 eV FWHM). Excitation energies are presented unshifted.
3.2. IRMPD Spectroscopy of AB·H+
Figure 3 shows the IRMPD total ion yield spectra of AB·H+ (summed from yield spectra of photofragments at m/z 135 and 161 (major) and m/z 177 (minor) and m/z 255 (minor)), overlaid with the calculated vibrational spectrum of Ka, Ea, and Eb. The Ka species is predicted to have a dominant vibrational band at 1420 cm–1. A strong vibrational feature is evident in the experimental spectra in this region, consistent with Ka being present in the experimental ion ensemble. For the Ea/Eb tautomers, the dominant vibrational band over the scanned spectral region is predicted to occur at 1562–5 cm–1. We assign the experimental feature at 1575 cm–1 to this Ea/Eb tautomer vibration. The experimental spectrum is therefore consistent with both Ka and Ea/Eb tautomers being present in the electrospray ion ensemble, as predicted by the ab initio population analysis reported in section 3.1. The spectral features that occur across the 1100–1350 cm–1 region could be attributed to either keto or enol tautomers and so are not discussed in detail here. A series of composite calculated Ka and Ea infrared spectra (Section S2) support our assignment of the IRMPD spectrum as arising from a combination of Ka and Ea/Eb tautomers.
Figure 3.
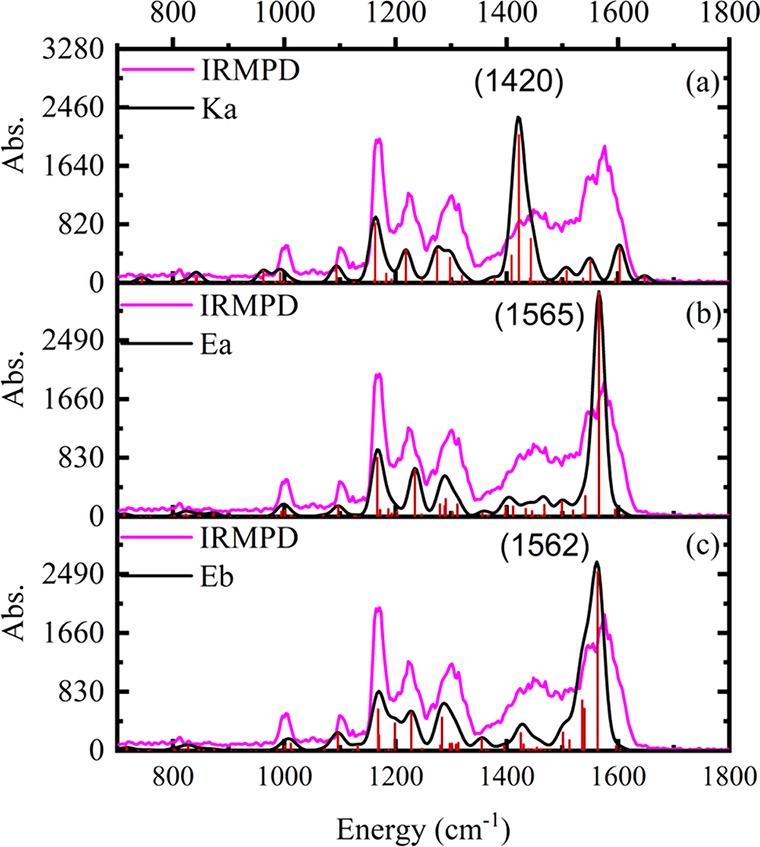
IRMPD spectrum of AB·H+ (in pink) is shown overlaid with simulated spectra (in black) for (a) Ka, (b) Ea, and (c) Eb. The calculated frequencies are scaled by 0.97.
3.3. Gas-Phase UV Absorption Spectrum of AB·H+
Figure 4a displays the gas-phase absorption photodepletion spectra of AB·H+ (m/z 311) over the range 2.5–5.7 eV. There are two distinct bands, labeled I and II, with λmax values at 2.9 and 3.6 eV, respectively. We note that the falling edge of band I contains a minor shoulder which is likely to be an experimental artifact caused by an OPO crystal changeover at that energy. Band II is asymmetric with an extended shoulder to higher energies. The photodepletion spectrum was recorded under single-photon absorption conditions, confirmed by power studies at the λmax of bands I and II (section S3).
Figure 4.
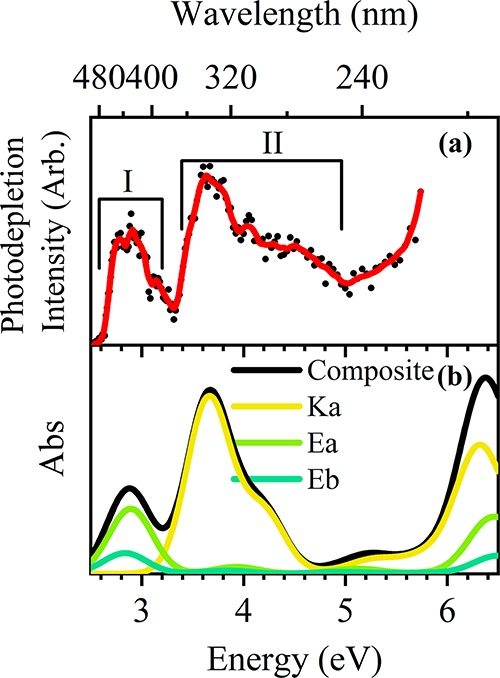
(a) Gas-phase photodepletion spectrum of AB·H+. The solid line is a 5-point adjacent average of the data points. (b) Composite TD-DFT curves for Ka, Ea, Eb at the ωB97X-D/def2-SVP level. The TD-DFT calculated electronic transitions have been red-shifted by 0.53 eV,70 and intensities weighted by the Boltzmann populations.
The main features of the experimental photodepletion spectrum can be well reproduced by the calculated gas-phase absorption spectrum (Figure 4b), which sums the TD-DFT spectra of the Ka, Ea, and Eb isomers (Section 3.1). (The contributions of each isomer are scaled to match the calculated gas-phase Boltzmann populations (Table 1) and the calculated electronic transitions have been red-shifted by 0.53 eV to match the observed experimental band positions).70 This allows confident assignment of band I to the bright S1 transitions of the Ea/Eb pair of isomers, and band II to bright S1 and S3 transitions of the Ka isomer (note that depending on the functional used there are minor contributions from weak transitions within band II and small differences in the ordering of states S1–S4). We note that the increase in photodepletion signal above band II is poorly matched to the minor feature at 5.2 eV of the composite TD-DFT; however, there is a strong feature at 6.4 eV which likely corresponds to this region.
3.4. UV Photofragmentation of AB·H+
Figure 5 displays the photofragment difference mass spectra of AB·H+ photoexcited at the band I and II maxima (2.6 and 3.6 eV, respectively). The dominant photofragments observed are m/z 161 [eq 4a] and m/z 135 [eq 4b], with both being products of McLafferty-like retro-heteroene hydrogen rearrangements adjacent to the a and b protonation centers.71
| 4a |
Figure 5.
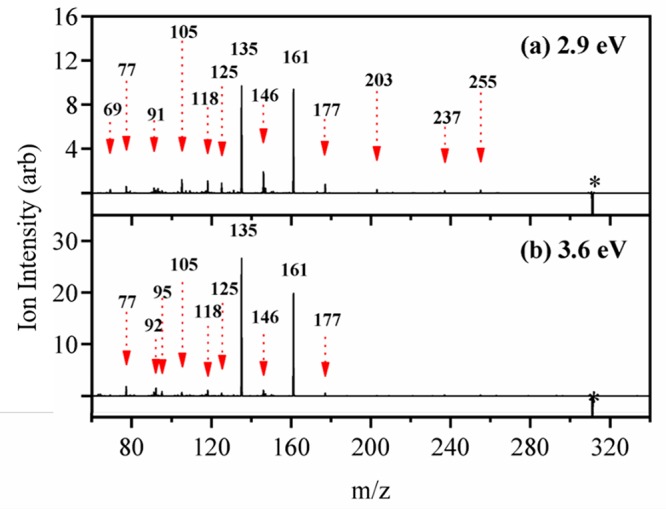
Photofragment difference (laser on–laser off) mass spectrum of AB·H+ excited at (a) 2.9 eV, and (b) 3.6 eV. The asterisk (∗) represents the depleted parent ion signal at m/z 311.
In addition to the hydrogen rearrangement products, several minor photofragments are produced across all probed wavelengths. Two of the more prominent minor photofragments are m/z 146 [eq 4c] and m/z 177 [eq 4d].
| 4c |
These photoproducts arise from proton transfer from a carbonyl to a ring group, with subsequent elimination of neutral molecules. The m/z 146 fragment is an unusual odd-electron photofragment, which is not produced in the collision induced dissociation (CID) of AB·H+ (section S4). However, m/z 147 is measured in both CID and UVPD studies, but only nominally by the latter measurement. This is a notable observation since the m/z 146 fragment, and the accompanying free radical(s), are therefore produced from AB·H+ following UVA excitation only. Table 2 presents proposed structural assignments of the major observed AB·H+ photofragment ions, as well as those produced by thermal fragmentation (CID; section S4). Additional minor fragmentation channels are tabulated in section S4.
Table 2. Proposed Structures for the Major Ionic and Associated Neutral Fragments of AB·H+ (m/z 311) Produced during CID and UV Laser Photoexcitation.

Notation: very strong (vs), strong (s), medium (m), and weak (w).
The IRMPD photofragments are identical to the CID fragments listed here and have similar relative intensities.
To determine whether any of the observed photofragments arise from secondary fragmentation, the major photofragment ions m/z 161 and m/z 135 were isolated in the ion trap and then subjected to photoexcitation. The resulting photofragment action spectra are displayed in section S5 and reveal that neither m/z 161 nor m/z 135 directly photofragments to produce any other observed photofragments.
Figure 6 displays a plot of the wavelength-dependent relative ion yields of the key AB·H+ photofragments, providing a concise overview of the photofragment branching ratios. Full photofragment action spectra are included in section S5. At the maxima of band I (2.9 eV) the major fragment ions m/z 161 and m/z 135 appear in equal proportions with each accounting for roughly 32% of the total photofragmentation signal. The onset of band II (3.3 eV) sees an increase in the relative ion intensity of m/z 135, with peaks in production of this ion at 3.4 and 4.3 eV (where it constitutes 50% and 60% of the total photofragmentation, respectively). The relative intensity of the m/z 161 photofragment falls to 25% at these energies. At photon energies above 4.8 eV, the relative ion intensities of the m/z 161 and m/z 135 pair of photofragments are again approximately equal, with the total branching into these fragments being slightly higher than in the band I region.
Figure 6.
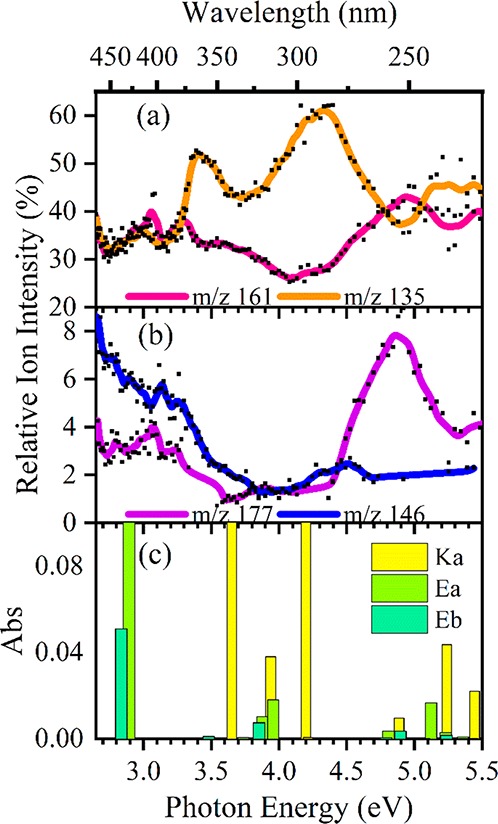
Relative ion yield plots for (a) the m/z 161, 135, (b) m/z 146, 177 photofragments of AB·H+ between 2.6 and 5.5 eV, and (c) the TD-DFT calculated excitation energies for each isomer at the ωB97X-D/def2-SVP level. The curved lines included with the data points are a five-point adjacent average of such data points. Calculated excitation energies have been red-shifted by 0.53 eV.
The m/z 146 fragment is produced most intensely in the low-energy spectral region between 2.7 and 3.6 eV, with its relative intensity decreasing across the band I region from 8% to 3%, and with a small enhancement in production around 4.5 eV. The m/z 177 ion is similar across the band I region, with respect to the m/z 146 spectrum. Between 4.5 and 5.2 eV, the relative ion intensity of m/z 177 increases dramatically, peaking strongly around 4.7 eV. Notably, this peak in production of m/z 177 does not follow the same profile as band II of the photodepletion spectra, but does coincide with a pair of predicted weak Ea and Eb excitations at 4.8 and 4.9 eV (Figure 6c).
Approximately 15–20% parent ion depletion (Ioff–Ion) is not recovered as measurable photofragmentation signal, an effect that can be attributed to production of ion fragments with m/z < 50 which is below the instrumental low mass cutoff. For example, heterolytic dissociation of the R2-bound methoxy group would produce an even-electron ion at m/z 31 along with zwitterionic C19H20O2.
Finally, it is useful to review the photofragments produced compared to those observed via CID. CID, like IRMPD, is equivalent to thermal-induced dissociation and produces so-called statistical fragments.41 For AB·H+, CID produces m/z 161 and m/z 135 as the dominant ionic products (Figure S8), with approximately equal intensity. Notably, these are the major photofragments observed in this study. From the relative ion intensities shown in Figure 6, it is evident that production of the m/z 161 and m/z 135 pair of photofragments is broadly statistical in the regions from 2.7 to 3.2 eV (i.e., the band I region), and above 5 eV (i.e., above band II). However, in the region between 3.7 and 4.7 eV (vis to near-UVA), photofragmentation is clearly nonstatistical with production of the m/z 135 photofragment being photochemically enhanced.42,72−76
4. Discussion
4.1. Mechanisms of Dissociation for the Ka and Ea/Eb Isomers of Protonated Avobenzone
From the calculated TD-DFT spectra presented in section 3.1, excitations associated with the Ka tautomer dominate in the region between 3.7 and 5.0 eV, while Ea/Eb excitations dominate for energies between 2.5 and 3.7 eV. One of the striking features of the ion yield production spectra is that production of the m/z 135 photofragment peaks strongly at 3.6 and 4.3 eV. These energies coincide with the calculated bright transitions for the Ka tautomer (Figure 6c), leading to the conclusion that enhanced production of the m/z 135 photofragment follows excitation of keto tautomer transitions. At the energies where production of m/z 135 is enhanced, the relative ion intensity of m/z 161 decreases, which is expected for a relative ion intensity plot (i.e., if one relative ion intensity increases, another ion intensity must decrease).
Scheme 3 presents the proposed fragmentation pathways for the m/z 161 and m/z 135 fragments, associated with hydrogen rearrangement within the protonated diketo bridge of Ka. Since the odd-electron m/z 162 and m/z 136 photofragments are not observed, we can rule out direct α-cleavage of the Ka and Kb tautomers, as occurs for the corresponding neutral tautomers in solution.32 Instead, a retro-heteroene addition of the γ-hydrogen (charge carrier) to the Ka and Kb tautomers generates the m/z 135 and m/z 161 ions, respectively (Kb is included in this scheme, as it was established (section 3.1) to be vibrationally populated by Ka). The Scheme 3 mechanism resembles the classic McLafferty rearrangement for ionic and radical molecules containing a keto group which undergoes β-cleavage.71,77−80 (For AB·H+, a protonated heteroatom occupies the saturated γ-carbon position typical to McLafferty systems.) Scheme 3 illustrates that the m/z 161 and m/z 135 fragments can be produced in a straightforward mechanism from a keto tautomer, but no similar McLafferty-like rearrangement can be deduced from an enol tautomer (the reduced C2 β-carbon of either Ea/Eb species needs an additional hydrogen for this reaction (as shown) to occur). Thus, Scheme 3 provides further evidence that m/z 161 and m/z 135 derive from a Ka/Kb tautomer.
Scheme 3. Proposed McLafferty-like Rearrangement to Form Products 4b and 4a.

The ion fragments m/z 177 and m/z 146 are photoproducts which can be assigned as being unique to the isomer Ea. The spectral profile traces of m/z 177 and m/z 146 shown in Figure 6b match the computed spectra of Ea/Eb (Figure 6c). Scheme 4a presents a tentative mechanism for the formation of m/z 177 from Ea via a proton transfer from the enol to the nearby ring. Following this transfer a ring-walk and subsequent elimination yields the detected ion fragment. The proton transfer ring-walk mechanism proposed here is akin to those proposed previously for fragmentation patterns of elimination substitution (ortho, meta, para) of aromatic systems.81−83 An analogous mechanism from the Eb isomer (not shown) would yield the m/z 255 ion fragment which was measured with very low intensity and therefore omitted from Figure 6b. At the same time, the observation of the m/z 255 photofragment indicates that Eb is present at low concentration in our ion ensemble. Scheme 4b includes the homogeneous dissociation of the R2-methoxy group followed by a mechanism similar to 4a to yield m/z 146.
Scheme 4. Proposed Product Formation from the AB·H+ Enol Tautomers by (a) Proton Transfer Preceding Ring-Walk and Elimination [4d] and by (b) Homolytic Fission of the R2-Methoxy Bond Prior to Proton Transfer Preceding Ring-Walk and Elimination [4c]. Dotted Arrows Indicate the Proposed Reaction Path. *m/z 280 Is Not Detected.

4.2. Implied Photodynamics of Ka and Ea/Eb Conformers
Previous work by Dunkelberger et al.25 on neutral AB in solution measured nonradiative formation of transient hot ground-state intermediates formed upon excitation of the CE form. These transients were found to be accessed by a series of largely rotational coordinate-driven conical intersections (CIs) in less than 1 ps, with the chelation of CE being broken by either out-of-plane OH motion, or C1–C2, C2–C3 centered dihedral (i.e., twisted) rotations. Solvent effects were found to influence the relaxation rates back to the CE equilibria, with the rotated OH form being the most rapid to equilibrate at τ = 1.3 ps, while the twisted forms displayed tens of picoseconds to microsecond–millisecond measured lifetimes. A CI was identified along the intramolecular proton transfer tautomerization coordinate which had a relaxation lifetime of 6 ps. Overall, these observations revealed that the rate of enol to keto tautomerization for the neutral in solution is low.
It is useful to consider how the known photodynamics of the neutral change upon protonation. From the discussion above, it is clear that pathways involving the OH group (i.e., out-of-plane OH rotation and tautomerization) play key roles in the decay dynamics. Protonation is highly likely to disrupt these pathways, since protonation of the carbonyl of the CO of the CE will lead to a decrease in the intramolecular hydrogen-bond strength.
Given that AB·H+ produces the m/z 135 and 161 photofragments across the Ea/Eb excitation wavelengths, and the fact that Ea/Eb structures cannot produce these product ions (section 4.1), our results indicate that enol to keto photoisomerization AB·H+ must be occurring, followed by subsequent degradation into m/z 135 and 161. Even at the peak of Ea/Eb absorption (2.9 eV), 75% of the detected photofragments arise from keto structures. For neutral AB, enol to keto tautomerization has a very low cross-section from the measured rates.25,30,40 If we assume that the photofragment cross-section is directly proportional to the relaxation rates of the initially populated excited states into photoproducts, then our experiment suggests that the cross-section for dissociative excited-state decay observed for AB·H+ is substantially greater than for AB in solution. One explanation of this observation is that protonation lowers the energetic barriers to “twisting” mechanisms that enhance enol to keto photoisomerization. High level computational potential energy scans of transition state geometries would be valuable to clarify this and solution-phase studies on the protonates species are clearly warranted.
This work has shown that the protonation of the β-diketone bridge is responsible for a shift from type I to II photoelimination of AB.31,84 Neutral AB in solution is known to undergo Norris Type I (photoinitiated α-cleavage) dissociation and subsequent recombination.32 This dissociation process produces radical fragments aided, in part, by the long-range repulsive force of an excited triplet spin-state.31 Norris Type II reactions, which are analogous to thermally activated McLafferty rearrangements seen in mass spectrometry, rely on the acidic nature of excited states to access an S0 ← S1/T1 CI by way of a downhill γ-hydrogen potential coordinate, widely known as excited-state intramolecular proton transfer.31,84−86 Whether the photodegradation of Ea/Eb proceeds through an excited triplet state cannot be directly experimentally identified here. However, the observation of the m/z 146 photofragment (a free-radical fragment) is indicative of fragmentation via a triplet state.
Computational work on neutral AB by Kojić et al. showed that under both gaseous and solvated conditions (MeCN), the S1 state of CE has a T1 state of alternate spin parity available energetically within the zero-point-energy window of S1 at the Franck–Condon geometry.27 If T1 is accessed, there is a flat potential about the out-of-plane OH dihedral coordinate which crosses the contorted S0 state. An easily accessible T1 state for Ea/Eb at S1 Franck–Condon geometries may facilitate the rearrangements shown in Scheme 4. Future calculations of the excited state potential energy surfaces would be valuable to provide further insight into the photophysics.
5. Concluding Remarks
In summary, we have investigated the photochemistry of selected enol and keto isomers of protonated avobenzone in the gas phase. IRMPD spectroscopy was employed to confirm the presence of both enolic (Ea/Eb) and diketone (Ka) isomers of AB·H+ in the electrosprayed ion ensemble. The dominant electronic excitations of the enol and keto tautomers of gaseous AB·H+ were found to be well separated, allowing us to determine the tautomer-dependent photofragmentation products. Such measurements would be extremely challenging to perform in solution, due to the solution-phase tautomers displaying overlapping spectral profiles. Analysis of the wavelength-dependent production of the various photofragments revealed that near-UVA photoexcitation provides a route to enol-to-keto tautomerization, consistent with previously proposed mechanisms for photodegradation of avobenzone in the condensed phase. Finally, we observed odd-electron photofragments in low quantum yield from the enol tautomer of AB·H+, which indicates that a previously unidentified photosensitizing pathway is present for this isomer. Prior studies of the neutral AB system have identified triplet excited states for the keto form.
We note that gas-phase photoisomerization has been studied by Bieske and co-workers, using ion mobility to detect photoisomers generated from protonated azobenzones.87−89 Sheps and co-workers have performed an elegant series of experiments on gaseous neutral acetylacetone (a prototypical β-diketone), to probe keto to enol tautomerization using a combination of photoionization techniques.90 However, work presented here provides an important complement to these earlier studies since it demonstrates a method for directly detecting the photoproducts of a photoisomerizing system.
In the context of the current study of AB·H+, it is useful to compare the results to our previous work on protonated oxybenzone, OB·H+. Oxybenzone is another widely used UV sunscreen, and provides an interesting contrast to AB since it is known to exhibit excellent photostability.4,91−94 The OB·H+ contains a pseudocycle, formed by the intramolecular bond between the hydroxy and carbonyl groups. The McLafferty-like elimination reaction responsible for the most intense AB·H+ fragments cannot be produced straightforwardly from the enolic tautomers of AB·H+, or similarly, from OB·H+ due to the pseudocyclic nature of both structures. Moreover, while an enol-to-keto tautomerization in AB·H+ can lead to this elimination reaction, the direct conjugation of the pseudocycle and benzene ring in OB·H+ restricts this reaction pathway. Such comparative measurements are valuable for gaining a broader understanding of the molecular-level performance of different sunscreen molecules, and may be useful in the context of efforts to develop new sunscreen molecules.2,95
Acknowledgments
This work was funded through the Leverhulme Trust Research Project Grant RPG-2017-147. We thank the University of York and the Department of Chemistry for provision of funds for the OPO laser system, and the University of York High Performance Computing service, Viking, and the Research Computing team, for the provision of computational resources. We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. The research leading to this result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.0c01295.
Additional DFT and TD-DFT calculations; composite AB·H+ IR Ka and Ea spectra; photodepletion laser power dependence; Collision induced dissociation measurements on AB·H+; additional photofragmentation measurements; possible heterolytic cleavage products of AB·H+ isomers (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Baker L. A.; Marchetti B.; Karsili T. N. V.; Stavros V. G.; Ashfold M. N. R. Photoprotection: Extending lessons learned from studying natural sunscreens to the design of artificial sunscreen constituents. Chem. Soc. Rev. 2017, 46, 3770–3791. 10.1039/C7CS00102A. [DOI] [PubMed] [Google Scholar]
- Losantos R.; Funes-Ardoiz I.; Aguilera J.; Herrera-Ceballos E.; Garcia-Iriepa C.; Campos P. J.; Sampedro D. Rational design and synthesis of efficient sunscreens to boost the solar protection factor. Angew. Chem., Int. Ed. 2017, 56, 2632–2635. 10.1002/anie.201611627. [DOI] [PubMed] [Google Scholar]
- Rodrigues N. D.; Staniforth M.; Stavros V. G. Photophysics of sunscreen molecules in the gas phase: A stepwise approach towards understanding and developing next-generation sunscreens. Proc. R. Soc. London, Ser. A 2016, 472, 20160677. 10.1098/rspa.2016.0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forestier S. Rationale for sunscreen development. J. Am. Acad. Dermatol. 2008, 58, S133–138. 10.1016/j.jaad.2007.05.047. [DOI] [PubMed] [Google Scholar]
- Ignasiak M. T.; Houee-Levin C.; Kciuk G.; Marciniak B.; Pedzinski T. A reevaluation of the photolytic properties of 2-hydroxybenzophenone-based UV sunscreens: Are chemical sunscreens inoffensive?. ChemPhysChem 2015, 16, 628–633. 10.1002/cphc.201402703. [DOI] [PubMed] [Google Scholar]
- Baker L. A.; Horbury M. D.; Greenough S. E.; Ashfold M. N.; Stavros V. G. Broadband ultrafast photoprotection by oxybenzone across the UVB and UVC spectral regions. Photochem. Photobiol. Sci. 2015, 14, 1814–1820. 10.1039/C5PP00217F. [DOI] [PubMed] [Google Scholar]
- Baker L. A.; Horbury M. D.; Greenough S. E.; Coulter P. M.; Karsili T. N.; Roberts G. M.; Orr-Ewing A. J.; Ashfold M. N.; Stavros V. G. Probing the ultrafast energy dissipation mechanism of the sunscreen oxybenzone after UVA irradiation. J. Phys. Chem. Lett. 2015, 6, 1363–1368. 10.1021/acs.jpclett.5b00417. [DOI] [PubMed] [Google Scholar]
- Baker L. A.; Staniforth M.; Flourat A. L.; Allais F.; Stavros V. G. Gas-solution phase transient absorption study of the plant sunscreen derivative methyl sinapate. ChemPhotoChem. 2018, 2, 743–748. 10.1002/cptc.201800060. [DOI] [Google Scholar]
- Luo J.; Liu Y.; Yang S.; Flourat A. L.; Allais F.; Han K. Ultrafast barrierless photoisomerization and strong ultraviolet absorption of photoproducts in plant sunscreens. J. Phys. Chem. Lett. 2017, 8, 1025–1030. 10.1021/acs.jpclett.7b00083. [DOI] [PubMed] [Google Scholar]
- Peperstraete Y.; Staniforth M.; Baker L. A.; Rodrigues N. D.; Cole-Filipiak N. C.; Quan W. D.; Stavros V. G. Bottom-up excited state dynamics of two cinnamate-based sunscreen filter molecules. Phys. Chem. Chem. Phys. 2016, 18, 28140–28149. 10.1039/C6CP05205C. [DOI] [PubMed] [Google Scholar]
- Tan E. M.; Hilbers M.; Buma W. J. Excited-state dynamics of isolated and microsolvated cinnamate-based UV-B sunscreens. J. Phys. Chem. Lett. 2014, 5, 2464–2468. 10.1021/jz501140b. [DOI] [PubMed] [Google Scholar]
- Dean J. C.; Kusaka R.; Walsh P. S.; Allais F.; Zwier T. S. Plant sunscreens in the UV-B: Ultraviolet spectroscopy of jet-cooled sinapoyl malate, sinapic acid, and sinapate ester derivatives. J. Am. Chem. Soc. 2014, 136, 14780–14795. 10.1021/ja5059026. [DOI] [PubMed] [Google Scholar]
- Domingos S. R.; Schnell M. Wet sunscreens in the gas phase: Structures of isolated and microsolvated oxybenzone. J. Phys. Chem. Lett. 2018, 9, 4963–4968. 10.1021/acs.jpclett.8b02029. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y.; Yamamoto K.; Aoki J.; Ikeda T.; Inokuchi Y.; Ehara M.; Ebata T. Experimental and theoretical study on the excited-state dynamics of ortho-, meta-, and para-methoxy methylcinnamate. J. Chem. Phys. 2014, 141, 244313. 10.1063/1.4904268. [DOI] [PubMed] [Google Scholar]
- Rodrigues N. D.; Staniforth M.; Young J. D.; Peperstraete Y.; Cole-Filipiak N. C.; Gord J. R.; Walsh P. S.; Hewett D. M.; Zwier T. S.; Stavros V. G. Towards elucidating the photochemistry of the sunscreen filter ethyl ferulate using time-resolved gas-phase spectroscopy. Faraday Discuss. 2016, 194, 709–729. 10.1039/C6FD00079G. [DOI] [PubMed] [Google Scholar]
- Rodrigo C. P.; James W. H. 3rd; Zwier T. S. Single-conformation ultraviolet and infrared spectra of jet-cooled monolignols: P-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol. J. Am. Chem. Soc. 2011, 133, 2632–2641. 10.1021/ja109218j. [DOI] [PubMed] [Google Scholar]
- Fang Y. G.; Li C. X.; Chang X. P.; Cui G. Photophysics of a UV-B filter 4-methylbenzylidene camphor: Intersystem crossing plays an important role. ChemPhysChem 2018, 19, 744–752. 10.1002/cphc.201701230. [DOI] [PubMed] [Google Scholar]
- Li C. X.; Guo W. W.; Xie B. B.; Cui G. Photodynamics of oxybenzone sunscreen: Nonadiabatic dynamics simulations. J. Chem. Phys. 2016, 145, 074308. 10.1063/1.4961261. [DOI] [PubMed] [Google Scholar]
- Assis Oliveira L. B.; Fonseca T. L.; Costa Cabral B. J.; Coutinho K.; Canuto S. Hydration effects on the electronic properties of eumelanin building blocks. J. Chem. Phys. 2016, 145, 084501. 10.1063/1.4961147. [DOI] [PubMed] [Google Scholar]
- Marchetti B.; Karsili T. N. Theoretical insights into the photo-protective mechanisms of natural biological sunscreens: Building blocks of eumelanin and pheomelanin. Phys. Chem. Chem. Phys. 2016, 18, 3644–3658. 10.1039/C5CP06767G. [DOI] [PubMed] [Google Scholar]
- Chang X. P.; Li C. X.; Xie B. B.; Cui G. Photoprotection mechanism of p-methoxy methylcinnamate: A CASPT2 study. J. Phys. Chem. A 2015, 119, 11488–11497. 10.1021/acs.jpca.5b08434. [DOI] [PubMed] [Google Scholar]
- Karsili T. N.; Marchetti B.; Ashfold M. N.; Domcke W. Ab initio study of potential ultrafast internal conversion routes in oxybenzone, caffeic acid, and ferulic acid: Implications for sunscreens. J. Phys. Chem. A 2014, 118, 11999–12010. 10.1021/jp507282d. [DOI] [PubMed] [Google Scholar]
- Gonzalez H.; Tarras-Wahlberg N.; Stromdahl B.; Juzeniene A.; Moan J.; Larko O.; Rosen A.; Wennberg A. M. Photostability of commercial sunscreens upon sun exposure and irradiation by ultraviolet lamps. BMC Dermatol. 2007, 7, 1. 10.1186/1471-5945-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson K. M.; Narayanan S.; Nichols V. M.; Bardeen C. J. Photochemical degradation of the UV filter octyl methoxycinnamate in solution and in aggregates. Photochem. Photobiol. Sci. 2015, 14, 1607–1616. 10.1039/C5PP00074B. [DOI] [PubMed] [Google Scholar]
- Dunkelberger A. D.; Kieda R. D.; Marsh B. M.; Crim F. F. Picosecond dynamics of avobenzone in solution. J. Phys. Chem. A 2015, 119, 6155–6161. 10.1021/acs.jpca.5b01641. [DOI] [PubMed] [Google Scholar]
- Gacoin P. Studies of the triplet state of carbonyl compounds. I. Phosphorescence of β-diketones. J. Chem. Phys. 1972, 57, 1418–1425. 10.1063/1.1678420. [DOI] [Google Scholar]
- Kojic M.; Petkovic M.; Etinski M. A new insight into the photochemistry of avobenzone in gas phase and acetonitrile from ab initio calculations. Phys. Chem. Chem. Phys. 2016, 18, 22168–22178. 10.1039/C6CP03533G. [DOI] [PubMed] [Google Scholar]
- Verma P. K.; Koch F.; Steinbacher A.; Nuernberger P.; Brixner T. Ultrafast UV-induced photoisomerization of intramolecularly H-bonded symmetric β-diketones. J. Am. Chem. Soc. 2014, 136, 14981–14989. 10.1021/ja508059p. [DOI] [PubMed] [Google Scholar]
- Verma P. K.; Steinbacher A.; Koch F.; Nuernberger P.; Brixner T. Monitoring ultrafast intramolecular proton transfer processes in an unsymmetric β-diketone. Phys. Chem. Chem. Phys. 2015, 17, 8459–8466. 10.1039/C4CP05811A. [DOI] [PubMed] [Google Scholar]
- Tobita S.; Ohba J.; Nakagawa K.; Shizuka H. Recovery mechanism of the reaction intermediate produced by photoinduced cleavage of the intramolecular hydrogen bond of dibenzoylmethane. J. Photochem. Photobiol., A 1995, 92, 61–67. 10.1016/1010-6030(95)04158-X. [DOI] [Google Scholar]
- Marchetti B.; Karsili T. N. V.; Ashfold M. N. R. Exploring norrish type I and type II reactions: An ab initio mechanistic study highlighting singlet-state mediated chemistry. Phys. Chem. Chem. Phys. 2019, 21, 14418–14428. 10.1039/C8CP07292B. [DOI] [PubMed] [Google Scholar]
- Schwack W.; Rudolph T. Photochemistry of dibenzoyl methane UVA filters Part 1. J. Photochem. Photobiol., B 1995, 28, 229–234. 10.1016/1011-1344(95)07118-L. [DOI] [Google Scholar]
- Roscher N. M.; Lindemann M. K. O.; Bin Kong S.; Cho C. G.; Jiang P. Photodecomposition of several compounds commonly used as sunscreen agents. J. Photochem. Photobiol., A 1994, 80, 417–421. 10.1016/1010-6030(94)01043-9. [DOI] [Google Scholar]
- Cantrell A.; McGarvey D. J. Photochemical studies of 4-tert-butyl-4′-methoxydibenzoylmethane (BM-DBM). J. Photochem. Photobiol., B 2001, 64, 117–122. 10.1016/S1011-1344(01)00226-3. [DOI] [PubMed] [Google Scholar]
- Paris C.; Lhiaubet-Vallet V.; Jimenez O.; Trullas C.; Miranda M. A. A blocked diketo form of avobenzone: Photostability, photosensitizing properties and triplet quenching by a triazine-derived UVB-filter. Photochem. Photobiol. 2009, 85, 178–184. 10.1111/j.1751-1097.2008.00414.x. [DOI] [PubMed] [Google Scholar]
- Lhiaubet-Vallet V.; Marin M.; Jimenez O.; Gorchs O.; Trullas C.; Miranda M. A. Filter-filter interactions. Photostabilization, triplet quenching and reactivity with singlet oxygen. Photochem. Photobiol. Sci. 2010, 9, 552–558. 10.1039/b9pp00158a. [DOI] [PubMed] [Google Scholar]
- Wang C.; Bavcon Kralj M.; Kosmrlj B.; Yao J.; Kosenina S.; Polyakova O. V.; Artaev V. B.; Lebedev A. T.; Trebse P. Stability and removal of selected avobenzone’s chlorination products. Chemosphere 2017, 182, 238–244. 10.1016/j.chemosphere.2017.04.125. [DOI] [PubMed] [Google Scholar]
- Mturi G. J.; Martincigh B. S. Photostability of the sunscreening agent 4-tert-butyl-4′-methoxydibenzoylmethane (avobenzone) in solvents of different polarity and proticity. J. Photochem. Photobiol., A 2008, 200, 410–420. 10.1016/j.jphotochem.2008.09.007. [DOI] [Google Scholar]
- Vallejo J. J.; Mesa M.; Gallardo C. Evaluation of the avobenzone photostability in solvents used in cosmetic formulations. Vitae-Revista De La Facultad De Quimica Farmaceutica 2011, 18, 63–71. [Google Scholar]
- Yamaji M.; Kida M. Photothermal tautomerization of a uv sunscreen (4-tert-butyl-4’-methoxydibenzoylmethane) in acetonitrile studied by steady-state and laser flash photolysis. J. Phys. Chem. A 2013, 117, 1946–1951. 10.1021/jp312774e. [DOI] [PubMed] [Google Scholar]
- Wong N. G. K.; Berenbeim J. A.; Hawkridge M.; Matthews E.; Dessent C. E. H. Mapping the intrinsic absorption properties and photodegradation pathways of the protonated and deprotonated forms of the sunscreen oxybenzone. Phys. Chem. Chem. Phys. 2019, 21, 14311–14321. 10.1039/C8CP06794E. [DOI] [PubMed] [Google Scholar]
- Wong N. G. K.; Berenbeim J. A.; Dessent C. E. H. Direct observation of photochemical free radical production from the sunscreen 2-phenylbenzimidazole-5-sulfonic acid via laser-interfaced mass spectrometry. ChemPhotoChem. 2019, 3, 1231–1237. 10.1002/cptc.201900149. [DOI] [Google Scholar]
- Matthews E.; Dessent C. E. H. Experiment and theory confirm that UV laser photodissociation spectroscopy can distinguish protomers formed via electrospray. Phys. Chem. Chem. Phys. 2017, 19, 17434–17440. 10.1039/C7CP02817B. [DOI] [PubMed] [Google Scholar]
- Matthews E.; Cercola R.; Dessent C. E. H. Protomer-dependent electronic spectroscopy and photochemistry of the model flavin chromophore alloxazine. Molecules 2018, 23, 2036. 10.3390/molecules23082036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E.; Dessent C. E. Locating the proton in nicotinamide protomers via low-resolution UV action spectroscopy of electrosprayed solutions. J. Phys. Chem. A 2016, 120, 9209–9216. 10.1021/acs.jpca.6b10433. [DOI] [PubMed] [Google Scholar]
- Kiefer H. V.; Gruber E.; Langeland J.; Kusochek P. A.; Bochenkova A. V.; Andersen L. H. Intrinsic photoisomerization dynamics of protonated schiff-base retinal. Nat. Commun. 2019, 10, 1210. 10.1038/s41467-019-09225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K.; Chandrasekaran V.; Heber O.; Iron M. A.; Rappaport M. L.; Zajfman D. Ultraslow isomerization in photoexcited gas-phase carbon cluster C10–. Nat. Commun. 2018, 9, 912. 10.1038/s41467-018-03197-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Qiao X.; Zhou C.; Zhang Y. N.; Fu Z.; Chen J. Photochemical transformation of sunscreen agent benzophenone-3 and its metabolite in surface freshwater and seawater. Chemosphere 2016, 153, 494–499. 10.1016/j.chemosphere.2016.03.080. [DOI] [PubMed] [Google Scholar]
- De Laurentiis E.; Minella M.; Sarakha M.; Marrese A.; Minero C.; Mailhot G.; Brigante M.; Vione D. Photochemical processes involving the UV absorber benzophenone-4 (2-hydroxy-4-methoxybenzophenone-5-sulphonic acid) in aqueous solution: Reaction pathways and implications for surface waters. Water Res. 2013, 47, 5943–5953. 10.1016/j.watres.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Tovar-Sanchez A.; Sanchez-Quiles D.; Basterretxea G.; Benede J. L.; Chisvert A.; Salvador A.; Moreno-Garrido I.; Blasco J. Sunscreen products as emerging pollutants to coastal waters. PLoS One 2013, 8, e65451 10.1371/journal.pone.0065451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z.; Spinney R.; Ke R.; Yang Z.; Xiao R. Effect of pH on the sonochemical degradation of organic pollutants. Environ. Chem. Lett. 2016, 14, 163–182. 10.1007/s10311-016-0557-3. [DOI] [Google Scholar]
- Thuyet D. Q.; Watanabe H.; Ok J. Effect of pH on the degradation of imidacloprid and fipronil in paddy water. J. Pestic. Sci. 2013, 38, 223–227. 10.1584/jpestics.D12-080. [DOI] [Google Scholar]
- Loftin K. A.; Adams C. D.; Meyer M. T.; Surampalli R. Effects of ionic strength, temperature, and pH on degradation of selected antibiotics. J. Environ. Qual. 2008, 37, 378–386. 10.2134/jeq2007.0230. [DOI] [PubMed] [Google Scholar]
- Barman B. N.; Preston H. G. The effects of pH on the degradation of isothiazolone biocides. Tribol. Int. 1992, 25, 281–287. 10.1016/0301-679X(92)90065-U. [DOI] [Google Scholar]
- Zhang G.; Wang Q.; Zhang W.; Li T.; Yuan Y.; Wang P. Effects of organic acids and initial solution pH on photocatalytic degradation of bisphenol a (BPA) in a photo-fenton-like process using goethite (alpha-feooh). Photochem. .Photobiol. Sci. 2016, 15, 1046–1053. 10.1039/C6PP00051G. [DOI] [PubMed] [Google Scholar]
- Burbano A. A.; Dionysiou D. D.; Suidan M. T.; Richardson T. L. Oxidation kinetics and effect of pH on the degradation of MTBE with fenton reagent. Water Res. 2005, 39, 107–118. 10.1016/j.watres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Matthews E.; Sen A.; Yoshikawa N.; Bergstrom E.; Dessent C. E. UV laser photoactivation of hexachloroplatinate bound to individual nucleobases in vacuo as molecular level probes of a model photopharmaceutical. Phys. Chem. Chem. Phys. 2016, 18, 15143–15152. 10.1039/C6CP01676F. [DOI] [PubMed] [Google Scholar]
- Wellman S. M.; Jockusch R. A. Moving in on the action: An experimental comparison of fluorescence excitation and photodissociation action spectroscopy. J. Phys. Chem. A 2015, 119, 6333–6338. 10.1021/acs.jpca.5b04835. [DOI] [PubMed] [Google Scholar]
- Sen A.; Luxford T. F.; Yoshikawa N.; Dessent C. E. Solvent evaporation versus proton transfer in nucleobase-pt(cn)(4,6)(2)(−) dianion clusters: A collisional excitation and electronic laser photodissociation spectroscopy study. Phys. Chem. Chem. Phys. 2014, 16, 15490–15500. 10.1039/c4cp00989d. [DOI] [PubMed] [Google Scholar]
- Antoine R.; Dugourd P. Visible and ultraviolet spectroscopy of gas phase protein ions. Phys. Chem. Chem. Phys. 2011, 13, 16494–16509. 10.1039/c1cp21531k. [DOI] [PubMed] [Google Scholar]
- van Outersterp R. E.; Martens J.; Berden G.; Steill J. D.; Oomens J.; Rijs A. M. Structural characterization of nucleotide 5′-triphosphates by infrared ion spectroscopy and theoretical studies. Phys. Chem. Chem. Phys. 2018, 20, 28319–28330. 10.1039/C8CP03314E. [DOI] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Infrared ion spectroscopy in a modified quadrupole ion trap mass spectrometer at the felix free electron laser laboratory. Rev. Sci. Instrum. 2016, 87, 103108. 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Oepts D.; van der Meer A. F. G.; van Amersfoort P. W. The free-electron-laser user facility felix. Infrared Phys. Technol. 1995, 36, 297–308. 10.1016/1350-4495(94)00074-U. [DOI] [Google Scholar]
- Rijs A. M.; Oomens J. Ir spectroscopic techniques to study isolated biomolecules. Top. Curr. Chem. 2014, 364, 1–42. 10.1007/128_2014_621. [DOI] [PubMed] [Google Scholar]
- Berden G.; Derksen M.; Houthuijs K. J.; Martens J.; Oomens J. An automatic variable laser attenuator for IRMPD spectroscopy and analysis of power-dependence in fragmentation spectra. Int. J. Mass Spectrom. 2019, 443, 1–8. 10.1016/j.ijms.2019.05.013. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A., et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Schrödinger Macromodel, release 2019–4; Schrödinger, LLC: New York, NY, 2019. [Google Scholar]
- Rodrigues-Oliveira A. F.; Ribeiro F. W. M; Cervi G.; Correra T. C. Evaluation of common theoretical methods for predicting infrared multiphotonic dissociation vibrational spectra of intramolecular hydrogen-bonded ions. ACS Omega 2018, 3, 9075–9085. 10.1021/acsomega.8b00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petković M.; Etinski M. Intramolecular oho bonding in dibenzoylmethane: Symmetry and spectral manifestations. RSC Adv. 2014, 4, 38517–38526. 10.1039/C4RA05586A. [DOI] [Google Scholar]
- Laurent A. D.; Jacquemin D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. 10.1002/qua.24438. [DOI] [Google Scholar]
- Demarque D. P.; Crotti A. E.; Vessecchi R.; Lopes J. L.; Lopes N. P. Fragmentation reactions using electrospray ionization mass spectrometry: An important tool for the structural elucidation and characterization of synthetic and natural products. Nat. Prod. Rep. 2016, 33, 432–455. 10.1039/C5NP00073D. [DOI] [PubMed] [Google Scholar]
- Debiossac M.; Schatti J.; Kriegleder M.; Geyer P.; Shayeghi A.; Mayor M.; Arndt M.; Kohler V. Tailored photocleavable peptides: Fragmentation and neutralization pathways in high vacuum. Phys. Chem. Chem. Phys. 2018, 20, 11412–11417. 10.1039/C8CP01058G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire G.; Dedonder-Lardeux C.; Jouvet C.; Desfrancois C.; Fayeton J. A. Ultrafast excited state dynamics in protonated GWG and GYG tripeptides. Phys. Chem. Chem. Phys. 2007, 9, 78–82. 10.1039/B613585D. [DOI] [PubMed] [Google Scholar]
- Spezia R.; Martinez-Nunez E.; Vazquez S.; Hase W. L. Theoretical and computational studies of non-equilibrium and non-statistical dynamics in the gas phase, in the condensed phase and at interfaces. Philos. Trans. R. Soc., A 2017, 375, 375. 10.1098/rsta.2017.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.; Skinnerup Byskov C.; Nielsen S. B. Energy flow in peptides after UV photoexcitation of backbone linkages. Phys. Chem. Chem. Phys. 2017, 19, 19640–19645. 10.1039/C7CP01768E. [DOI] [PubMed] [Google Scholar]
- Dessent C.; Fairlamb I.; Lynam J.; Hammarback A.; Wong N.; Garand E.; Scherman S.; Fischer K.; Cercola R.. Direct measurement of the visible to UV photodissociation processes for the photocorm tryptocorm. ChemRxiv 2019, https://doi.org/10.26434/chemrxiv.11369628.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbering N. M. The mclafferty rearrangement: A personal recollection. J. Am. Soc. Mass Spectrom. 2004, 15, 956–958. 10.1016/j.jasms.2004.04.025. [DOI] [PubMed] [Google Scholar]
- Dunbar R. C.; Klein R. Spectroscopy of radical cations. The mclafferty rearrangement product in fragmentation of n-butylbenzene and 2-phenylethanol ions. J. Am. Chem. Soc. 1977, 99, 3744–3746. 10.1021/ja00453a037. [DOI] [Google Scholar]
- Laulhe S.; Bogdanov B.; Johannes L. M.; Gutierrez O.; Harrison J. G.; Tantillo D. J.; Zhang X.; Nantz M. H. Fragmentation of oxime and silyl oxime ether odd-electron positive ions by the mclafferty rearrangement: New insights on structural factors that promote α,β fragmentation. J. Mass Spectrom. 2012, 47, 676–686. 10.1002/jms.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vijlder T.; Valkenborg D.; Lemiere F.; Romijn E. P.; Laukens K.; Cuyckens F. A tutorial in small molecule identification via electrospray ionization-mass spectrometry: The practical art of structural elucidation. Mass Spectrom. Rev. 2018, 37, 607–629. 10.1002/mas.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filges U.; Grützmacher H.-F. Fragmentations of protonated acetophenones via intermediate ion–molecule complexes. Org. Mass Spectrom. 1987, 22, 444–450. 10.1002/oms.1210220711. [DOI] [Google Scholar]
- Grützmacher H.-F., Fragmentation in mass spectrometry. In Encyclopedia of spectroscopy and spectrometry; Lindon J. C., Ed.; Elsevier: Oxford, 1999; pp 637–648. [Google Scholar]
- Kuck D. From fragmentation to construction--from void to massive: Fascination with organic mass spectrometry and the synthesis of novel three-dimensional polycyclic aromatic hydrocarbons. Chem. Rec 2015, 15, 1075–1109. 10.1002/tcr.201500023. [DOI] [PubMed] [Google Scholar]
- De Feyter S.; Diau E. W.; Zewail A. H. Femtosecond dynamics of norrish type-II reactions: Nonconcerted hydrogen-transfer and diradical intermediacy. Angew. Chem., Int. Ed. 2000, 39, 260–263. . [DOI] [PubMed] [Google Scholar]
- Douhal A.; Lahmani F.; Zewail A. H. Proton-transfer reaction dynamics. Chem. Phys. 1996, 207, 477–498. 10.1016/0301-0104(96)00067-5. [DOI] [Google Scholar]
- Wagner P. J. Type II photoelimination and photocyclization of ketones. Acc. Chem. Res. 1971, 4, 168–177. 10.1021/ar50041a002. [DOI] [Google Scholar]
- Bull J. N.; Scholz M. S.; Carrascosa E.; Bieske E. J. From e to z and back again: Reversible photoisomerisation of an isolated charge-tagged azobenzene. Phys. Chem. Chem. Phys. 2018, 20, 509–513. 10.1039/C7CP07278C. [DOI] [PubMed] [Google Scholar]
- Bull J. N.; Carrascosa E.; Scholz M. S.; Coughlan N. J. A.; Bieske E. J. Online measurement of photoisomerisation efficiency in solution using ion mobility mass spectrometry. Analyst 2017, 142, 2100–2103. 10.1039/C7AN00398F. [DOI] [PubMed] [Google Scholar]
- Scholz M. S.; Bull J. N.; Coughlan N. J. A.; Carrascosa E.; Adamson B. D.; Bieske E. J. Photoisomerization of protonated azobenzenes in the gas phase. J. Phys. Chem. A 2017, 121, 6413–6419. 10.1021/acs.jpca.7b05902. [DOI] [PubMed] [Google Scholar]
- Antonov I.; Voronova K.; Chen M. W.; Sztaray B.; Hemberger P.; Bodi A.; Osborn D. L.; Sheps L. To boldly look where no one has looked before: Identifying the primary photoproducts of acetylacetone. J. Phys. Chem. A 2019, 123, 5472–5490. 10.1021/acs.jpca.9b04640. [DOI] [PubMed] [Google Scholar]
- Abid A. R.; Marciniak B.; Pędziński T.; Shahid M. Photo-stability and photo-sensitizing characterization of selected sunscreens’ ingredients. J. Photochem. Photobiol., A 2017, 332, 241–250. 10.1016/j.jphotochem.2016.08.036. [DOI] [Google Scholar]
- Tarras-Wahlberg N.; Stenhagen G.; Larko O.; Rosen A.; Wennberg A. M.; Wennerstrom O. Changes in ultraviolet absorption of sunscreens after ultraviolet irradiation. J. Invest. Dermatol. 1999, 113, 547–553. 10.1046/j.1523-1747.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- Liu Y.-S.; Ying G.-G.; Shareef A.; Kookana R. S. Photostability of the uv filter benzophenone-3 and its effect on the photodegradation of benzotriazole in water. Environ. Chem. 2011, 8, 581. 10.1071/EN11068. [DOI] [Google Scholar]
- Serpone N.; Salinaro A.; Emeline A. V.; Horikoshi S.; Hidaka H.; Zhao J. An in vitro systematic spectroscopic examination of the photostabilities of a random set of commercial sunscreen lotions and their chemical UVB/UVA active agents. Photochem. Photobiol. Sci. 2002, 1, 970–981. 10.1039/b206338g. [DOI] [PubMed] [Google Scholar]
- Pandika M. Looking to nature for new sunscreens. ACS Cent. Sci. 2018, 4, 788–790. 10.1021/acscentsci.8b00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.