

Abstract
The development of multi-resistant strains of plasmodium parasite has become a global problem, therefore, the discovery of new antimalarial agents is the only available solution. In order to improve and propose new compounds with antimalarial activity, the three-dimensional quantitative structure-activity relationship (3D-QSAR) and molecular docking studies were carried on aurone analogues acting as Qo site inhibitors in cytochrome b. The 3D-QSAR model was established in this study based on the Comparative Molecular Field Analysis (CoMFA) and the Comparative Molecular Similarity Indices Analysis (CoMSIA). The good predictability was obtained using the CoMFA model (Q2 = 0.5; R2 = 0.97; = 0.72) and the best CoMSIA model (Q2 = 0.526; R2 = 0.915; 0.765). The predictive capacity of the developed model was evaluated through external validation using a test set compound and an applicability domain technique. In this study, the Steric, electrostatic and hydrogen bond acceptor fields played a key role in antimalarial activity. The results of the molecular docking revealed theoretically the importance of the residues his183 and his82 in the active site of the heme bL, this result was validated by a new assessment method. Based on the previous results, we designed several new potent Cytochrome b inhibitors and their inhibitory activities were predicted by the best model. Furthermore, these new inhibitors were analyzed for their ADMET properties and drug likeness. These results would be of great help in leading optimization for new drug discovery that can solve the problem of multiple drug resistance.
Keywords: Theoretical chemistry, Pharmaceutical chemistry, Azaaurone, Aurone, 3D-QSAR, Molecular docking, ADMET properties, Druglikeness, Malarial
Theoretical chemistry; Pharmaceutical chemistry, Azaaurone; Aurone; 3D-QSAR; Molecular docking; ADMET properties; Druglikeness; Malarial.
1. Introduction
Malaria is one of the most infectious diseases caused mainly by parasites of the genus Plasmodium. Among the five species of parasite (P. knowlesi, P. falciparum, P. vivax, P. malariae, and P. ovale) that affect humans [1, 2], P. falciparum is the most fatal and responsible for the majority (90%) of fatalities [3]. According to statistics from the World Health Organization (WHO) Report 2019 [4], the number of malaria cases is about to be above 228 million globally in 2018 and causing 405000 deaths. Despite intensive efforts to produce a vaccination [5], drugs remain the only available therapeutic alternative [6]. Moreover, all efforts made to overcome the disease of malaria have failed due to the continued multi-resistance of P. falciparum to available drugs [7]. Therefore, it is extremely urgent to discover and develop new therapeutic agents targeting the Plasmodium parasite.
In this regard, several research teams have shown great interest in aurones and their nitrogen analogues of azaaurones, which are very important therapeutic targets against malaria. Aurones (2-arylidenebenzofuran-3(2H)-ones) (Figure 1), and azaaurones (2-araylideneindol-3(2H)ones) (Figure 2), belong to the flavonoid family containing an exocyclic double bond. Aurones have been reported to have important biological activities [8, 9], including anticancer [10, 11], antimicrobial [12], antileishmanial [13], anti-alzheimer [14] antiparasitic [15], antifungal [16] and anti-inflammatory [17] activity. In a particular way, aurones and azaaurones have shown a high antimalarial activity because they act as dual-stage antimalarial agent, i.e. they are capable of inhibiting exoerythrocytic and intraerythrocytic stages [18].
Figure 1.

Structures of aurones.
Figure 2.

Structures of azaaurones.
Aurone and azaaurone have the potential to inhibit the cytochrome bc1 by acting as inhibitors of the mitochondrial respiratory chain of P. falciparum, by blocking the bifurcated electron transfer by inhibiting the oxidation or reduction of quinol which allows the released or captured protons, which are essential in the synthesis of ATP in cellular respiration. The cytochrome b subunit, which contains two types heme b of different redox potential, the site of quinol oxidation (Qo) with heme bL and the site of quinol reduction (Qi) with heme bH [19]. According to the molecular docking study, the aurone and azaaurone act on the Qo site of heme bL to the iron–sulfur protein subunit, blocking the electron transfer at the Qo site by inhibiting the oxidation of quinol to quinone which interrupt the synthesis of ATP in cellular respiration [20, 21].
Since the last decade, the Drug's use design techniques by computer software has provided very excellent results for drug discovery and research process [22, 23], among the effective and useful methods for drug design are the three-dimensional quantitative structure-activity relationship (3D-QSAR), hence to molecular docking and the pharmacokinetic parameters (ADMET). In the purpose to pursue our previous works on antimalarial [24, 25, 26].
In this research, the 3D-QSAR study on 35 aurone derivatives was used to build QSAR model [27], which was generated using Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). Molecular docking studies of these compounds with the protein of cytochrome b were also conducted to better understand the main structural requirements and analyzing the key interactions between ligand and receptor, the promising results in good accordance with experimental results were obtained. Moreover, to evaluate their drug-like ability, each designed compound has been tested by standard computational pharmacokinetic parameters (ADMET) and druglikeness.
2. Material and methods
2.1. Database and biological activity
3D-QSAR models were performed on a set of 35 aurone derivatives were taken from the literature [27]. The biological activities against the P. falciparum were expressed as the effective Concentration IC50 (μM). These values were converted into pIC50 (-Log (IC50) values to construct the 3D-QSAR models (Tables 1 and2). For performing the 3D-QSAR models, we divided a data set containing 35 compounds randomly into a training set (25 compounds) to build the models and a test set (10 compounds) to test the performance of the established models.
Table 1.
Aurone compounds studied and their observed and predicted antimalarial activities.
| Compounds | R | R1 | R2 | IC50 (μM) | pIC50(obs) | pIC50(pred) | Residual |
|---|---|---|---|---|---|---|---|
| 1 | H | OH | OH | 94.5 | 4.024 | 4.041 | -0.017 |
| 2 | H | OMe | OMe | 60.3 | 4.220 | 4.306 | -0.086 |
| 3a | 4′-Me | OH | OH | 63.4 | 4.198 | 4.055 | 0.143 |
| 4 | 2′-Et | OMe | OMe | 21 | 4.678 | 4.541 | 0.137 |
| 5 | 2′-Et | OH | OH | 113.5 | 3.945 | 4.273 | -0.328 |
| 6 | 4′-Et | OH | H | 28 | 4.553 | 4.43 | 0.123 |
| 7a | 4′-tBu | OMe | OMe | 13.3 | 4.876 | 4.794 | 0.082 |
| 8 | 4′-Bu | OMe | OMe | 11.8 | 4.928 | 4.845 | 0.083 |
| 9 | 4′-Br | OMe | OMe | 49.8 | 4.303 | 4.334 | -0.031 |
| 10a | 4′-F | OMe | OMe | 86.7 | 4.062 | 4.392 | -0.33 |
| 11 | 4′-OH | OH | H | 130 | 3.886 | 4.033 | -0.147 |
| 12 | 4′-OMe | OMe | OMe | 11 | 4.959 | 4.697 | 0.262 |
| 13a | 4′-Ph | OMe | OMe | 234 | 3.631 | 4.238 | -0.607 |
| 14 | 4′-Py | OMe | OMe | 85 | 4.071 | 3.95 | 0.121 |
Compounds in the test set.
Table 2.
Azaaurone compounds studied and their observed and predicted antimalarial activities.
| Compounds | R | IC50(μM) | pIC50(obs) | pIC50(pred) | Residual |
|---|---|---|---|---|---|
| 15 | 4′-Br | 49.8 | 4.303 | 4.444 | -0.141 |
| 16 | 4′-Cl | 17 | 4.770 | 4.538 | 0.232 |
| 17a | 2′-Cl | 9.9 | 5.004 | 5.086 | -0.082 |
| 18 | 2′,5′-Cl | 8.4 | 5.076 | 5.049 | 0.0267 |
| 19 | 2′-Cl, 6′-F | 9 | 5.046 | 4.937 | 0.109 |
| 20 | 4′-Et | 1 | 6 | 5.841 | 0.159 |
| 21 | 2′-Et | 12.8 | 5.893 | 5.635 | 0.258 |
| 22 | 2′,6′-Me | 9.1 | 5.041 | 5.549 | -0.508 |
| 23a | 2′,4′-Me | 3.6 | 5.444 | 5.515 | -0.071 |
| 24 | 2′,4′,5′-Me | 5.6 | 5.252 | 5.174 | 0.078 |
| 25 | 2′,3′,5′,6′-Me | 8.9 | 5.051 | 5.147 | -0.096 |
| 26 | 4′-i-Pr | 4.4 | 5.356 | 5.383 | -0.027 |
| 27a | 4′-t-Bu | 7.2 | 5.143 | 5.214 | -0.071 |
| 28 | 4′-Bu | 4.1 | 5.387 | 5.381 | 0.006 |
| 29a | 4′-CCH | 13.4 | 4.873 | 5.489 | -0.616 |
| 30 | 2′,4′-OMe | 5 | 5.301 | 5.349 | -0.048 |
| 31a | 2′,4′,6′-OMe | 1.9 | 5.721 | 5.449 | 0.272 |
| 32 | 3′,4′,5′-OMe | 1.9 | 5.721 | 5.81 | -0.0889 |
| 33 | 4′-SMe | 6.7 | 5.174 | 5.204 | -0.03 |
| 34 | 4′-Morpholino | 8.9 | 5.051 | 5.108 | -0.057 |
| 35a | 4′-N (Me)2 | 3.7 | 5.432 | 5.115 | 0.317 |
Compounds in the test set.
2.2. Molecular modeling and alignment
Molecular alignment considered as one of the most sensitive parameters for CoMFA and CoMSIA analysis [28, 29]. In this paper, the ligand-based alignment rule was used by the simple alignment method available in SYBYL-X 2.0 software. All Molecular structures were built with sketch module and minimized under the Tripos force standard field [30] using corresponding Gasteiger–Huckel atomic partial charges on SYBYL-X 2.0 platform [30, 31]. Moreover, 0.005 kcal/(mol Å) was set as convergence criterion of Powell gradient algorithm and maximum of 10,000 iterations to get stable conformation [32].
2.3. Generation of 3D-QSAR models
CoMFA and CoMSIA studies were performed using Sybyl X-2.0 software (Tripos,Inc., USA). We analyzed the CoMFA model using steric and electrostatic fields according to Lennard Jones and Coulomb potentials, the steric and electrostatic energies were calculated using a sp3 hybridized carbon atom with a Van Der Waals radius of 1.52 Å and a net +1.0 charge, with the default value of 30 kcal/mol was set for energy cutoff calculations. The CoMSIA model calculates more physico-chemical descriptors as hydrophobic, hydrogen bond acceptor and hydrogen bond donor fields using the probe atom. The CoMSIA method was executed with the same lattice box as employed in CoMFA method. The column filtering and attenuation factor were set to the default value of 2.0 kcal/mol and 0.3, respectively. In this paper, in order to obtain the best CoMSIA model, 16 different combinations of fields were employed to build the CoMSIA model (see Table 3).
Table 3.
The PLS statistical results of CoMFA and CoMSIA models in different molecular field combinations.
| Q2 | R2 | SEE | F | NOC | Fractions |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ster | Elec | Hyd | Don | Acc | |||||||
| CoMFA | 0.501 | 0.97 | 0.115 | 156.689 | 4 | 0.72 | 0.370 | 0.630 | |||
| CoMSIA/SEA | 0.526 | 0.915 | 0.191 | 53.800 | 4 | 0.765 | 0.283 | 0.433 | 0.284 | ||
| CoMSIA/SEH | 0.485 | 0.882 | 0.219 | 52.278 | 3 | 0.715 | 0.272 | 0.404 | 0.324 | ||
| CoMSIA/SED | 0.483 | 0.882 | 0.219 | 52.458 | 3 | 0.728 | 0.273 | 0.365 | 0.365 | ||
| CoMSIA/SHD | 0.48 | 0.858 | 0.24 | 42.335 | 3 | 0.751 | 0.281 | 0.329 | 0.390 | ||
| CoMSIA/SHA | 0.501 | 0.907 | 0.199 | 48.682 | 4 | 0.738 | 0.299 | 0.378 | 0.323 | ||
| CoMSIA/SDA | 0.506 | 0.859 | 0.239 | 42.666 | 3 | 0.694 | 0.279 | 0.415 | 0.306 | ||
| CoMSIA/EHA | 0.502 | 0.909 | 0.192 | 68.819 | 3 | 0.728 | 0.390 | 0.336 | 0.274 | ||
| CoMSIA/EHD | 0.482 | 0.902 | 0.199 | 64.451 | 3 | 0.709 | 0.357 | 0.300 | 0.343 | ||
| CoMSIA/EDA | 0.507 | 0.895 | 0.207 | 59.665 | 3 | 0.634 | 0.352 | 0.365 | 0.283 | ||
| CoMSIA/HDA | 0.513 | 0.875 | 0.226 | 48.937 | 3 | 0.710 | 0.322 | 0.377 | 0.302 | ||
| CoMSIA/EHDA | 0.52 | 0.899 | 0.203 | 61.985 | 3 | 0.721 | 0.271 | 0.234 | 0.279 | 0.217 | |
| CoMSIA/SHDA | 0.512 | 0.871 | 0.229 | 47.126 | 3 | 0.726 | 0.205 | 0.256 | 0.311 | 0.228 | |
| CoMSIA/SEDA | 0.516 | 0.886 | 0.215 | 54.536 | 3 | 0.697 | 0.203 | 0.288 | 0.298 | 0.212 | |
| CoMSIA/SEHA | 0.525 | 0.944 | 0.158 | 64.397 | 5 | 0.732 | 0.194 | 0.331 | 0.254 | 0.221 | |
| CoMSIA/SEHD | 0.494 | 0.887 | 0.215 | 54.878 | 3 | 0.74 | 0.204 | 0.282 | 0.233 | 0.281 | |
| CoMSIA/SEHDA | 0.518 | 0.889 | 0.212 | 88.840 | 3 | 0.72 | 0.162 | 0.232 | 0.195 | 0.240 | 0.172 |
2.4. Partial least squares (PLS) analysis
The PLS method [33] was performed to evaluate a linear correlation between the CoMFA and CoMSIA descriptors and the biological activity values. In PLS analysis, the leave-one-out (LOO) cross-validation method was used to determine the optimum number of components (NOC) using the highest cross-validation correlation coefficient (Q2) with the lowest standard estimation error (SEE). After determining (NOC), the non-cross-validation methods were then carried out to test the overall significance of the models, which calculated by Statistical parameters: coefficient of determination (R2), the value F (Fischer test) and standard estimation error (SEE). To further assess the robustness and statistical validity of the established models, several external validation strategies were also applied [34].
2.5. External validation of the CoMFA and CoMSIA models
To assess the predictive capabilities of the 3D-QSAR models based on the training set, the biological activities of the external test set of 10 compounds were predicted. These molecules were aligned to the template using the same method described above, and then their predictive ability and the accuracy of the model was measured using the external validation determination coefficient () by the following formula: = (SD-PRESS)/SD, where SD is the sum of the squared deviations between the activity values of the test set and the average activity values of the training set, and PRESS is the of the squared deviations between predicted and experimental activity values of the test set compounds. According to Golbraikh and Tropsha study [35], The following equation introduces an additional statistic for the external validation:
Where is a squared correlation coefficient value between predicted and experimental activity values of the test set.
and is squared correlation coefficient values of predicted versus experimental and experimental versus predicted activity for the test set at zero intercept, respectively.
K and K' are the slope of the plot of predicted versus observed and observed versus predicted activity for the test set at zero intercept, respectively.
Moreover, according to Roy study [36], it is necessary to compute the difference between the values of , and for further validate the predictive ability of the model using the following equation:
2.6. Applicability domain
All QSAR models were developed on a limited number of compounds that do not cover the entire chemical space, the domain of applicability (DA) is the region of the chemical space in which the QSAR model can reliably predict new compounds. Thus, determination of DA is an important tool for the reliable application of QSAR models [37]. In this study, to determine the applicability of the QSAR model, the leverage approach was employed, in which standardized residuals (h) versus leverage values were illustrated for fast and simple graphical detection of outliers (Williams plot). Then, a diagram allows to validate the reliability of the QSAR model if the leverage value (h) is lower than critical leverage value (h∗) [38].
2.7. Molecular docking
The Molecular docking simulation was carried out using AutoDock 4.2.3 software, in order to analyze the interactions mechanism and investigate binding modes to gain insight into the key structural and geometric requirements. In this study, we carried out the molecular docking study of two compounds, compound 13 (the lowest active) and compound 20 (the highest active), with the Qo site of cytochrome bc1 (PDB Id: 3CX5), which is downloaded from the RCSB Protein Data Bank [39]. First, we have removed the ligands and all water molecules from the protein using the Discovery Studio software [40]. The docked conformations and the ligand-protein interactions studies were carried out in AutoDock Tools version1.5.6 [41]. The 3D grid was generated using the AutoGrid algorithm to evaluate ligand-protein interactions energy [42]. The grid maps were created were set to 60 Å in all directions (X,Y,Z axes), with default grid space size 0.375 Å, The center grid box is about (14.903Å, -22.842Å and 30.627Å) by the ligand location in the protein. After completion the docking, the docked conformations of the ligands were analyzed for their binding interactions using 2D and 3Dvisualizations by Discovery Studio software [40].
2.8. Docking validation protocol
The docking results were validated by extracting the co-crystallized ligand of the protein (PDB Id: 3CX5) and re-docking it into the same position. The lowest energy pose obtained on re-docking and the co-crystallized ligand were superimposed, and its root mean square deviation (RMSD) was calculated between these two superimposed ligands. To validate the docking process, The RMSD must be within the reliable range of 2 Å [43, 44].
2.9. In silico pharmacokinetics ADMET and drug likeness prediction
In order to identify potential drug candidates, the ADMET and drug likeness profile was developed for the preliminary estimation of the pharmacokinetic, physicochemical and drug-like parameters in the discovery drug process. In silico study provides a pathway to access pharmacokinetic parameters (Adsorption, Distribution, Metabolism, Excretion and Toxicity; ADMET) [45], its absorption in the human intestine, penetration of the blood-brain barrier and the central nervous system, the metabolism refers to the chemical biotransformation of a drug by the body, total clearance of drugs and the toxicity levels of the molecules. Prediction of the drug likeness of the designed compound was assessed by rule-based filters from Lipinski, Ghose, Veber and Egan, and the synthetic accessibility difficulty scale was 1–10.
3. Results and discussion
3.1. Molecular alignment
The molecular alignment method is the sensitive step used to build a performing 3D-QSAR model. The highest biological activity from the compound 20 was chosen as template molecule for aligned the data set and to serve to visualize the contour maps in 3D-QSAR studies. Figure 3 shows the all 3D molecular structures in training and test sets were aligned on the common core and the template using the alignment technique based on the best-docked conformation of compound 20.
Figure 3.

Superposition and alignment of the 35 studied compounds using molecule 20 as a template.
3.2. CoMFA and CoMSIA studies
Table3 show the statistical parameters of CoMFA and CoMSIA models. For CoMFA analysis, the contributions of the steric and electrostatic fields explain 37% and 63% of the variance, respectively. The cross-validated determination coefficient Q2 fitted by the LOO method in PLS is 0.501, with optimum number of principal components as 4, the non-cross-validated determination coefficient R2 was 0.97 with a reliable SEE of 0.115 and F-test value is 156.689, a high predictive value for the external validation of test set (=0.72).
For CoMSIA analysis, the different combinations of the five fields were used to develop the different CoMSIA models, the results indicate (Table 3) that CoMSIA model based on the Steric, Electrostatic and Hydrogen bond Acceptor fields (SEA) and the Steric, Electrostatic, Hydrophobic, and Hydrogen bond Acceptor fields (SEHA) having the best prediction of biological activity, but the model CoMSIA/SEA (Tables 1, 2 and 3) show the greatest value of external predictive of test set (= 0.765). In this study, we considered the CoMSIA/SEA model as the most appropriate and provided the best statistical keys, the Q2 in our model is 0.526 with the four as optimum number of principal components. The R2 is 0.915 with a reliable SEE of 0.191 and F -test value is 53.8.
Q2 is the Cross-validated determination coefficient, N is the Optimum number of components, R2 is the non-cross-validated determination coefficient. SEE is the standard estimation error. F is the F -test value, NOC is the Optimum number of components, is the external validation determination coefficient, Ster is the steric field, Elec is the electrostatic field, Hyd is the hydrophobic field, Don is the Hydrogen bond donor field and Acc is the Hydrogen bond acceptor field.
Overall, the 3D-QSAR model considered reliable predictive [46], if the values of R2, and Q2 are greater than 0.6, 0.6 and 0.5, respectively. Therefore, the CoMFA and CoMSIA/SEA models indicate a favorable estimation of stability and a good predictive quality, this was confirmed by the prediction ability of external validation. In order to verify these results, we tested the robustness and predictability of the best models. The results of the external validation test computed for the CoMFA and CoMSIA/SEA models are presented in Table 4.
Table 4.
Statistical parameters for the validation of CoMFA and CoMSIA/SEA model.
| Parameter | Validation Criteria | CoMFA | CoMSIA/SEA |
|---|---|---|---|
| 0.501 | 0.526 | ||
| r2 | r2 > 0.6 | 0.718 | 0.765 |
| 0.21 | 0.057 | ||
| k | 0.85 < k < 1.15 | 1.1 | 1.09 |
| 0.02 | 0 | ||
| K′ | 0.85 < k’<1.15 | 0.84 | 0.87 |
| 0.31 | 0.06 | ||
| 0.63 | 0.765 | ||
| 0.37 | 0.582 |
According to the results of Table 4, the CoMSIA/SEA model passed all tests successfully and in perfect agreement with the criteria of the Roy as well as Golbraikh and Tropsha, however, the CoMFA model was unsuccessful in some criteria. The CoMSIA/SEA model showed the better understanding of the activity compared to the CoMFA model, because he has better predictive power for new compound and it respects all validation methods. For this reason, we used the CoMSIA/SEA contour maps to understand the criteria structures for increasing activity so as to discover new active compounds.
3.3. Applicability domain
The William plot for the applicability domain (AD) of the model is shown in Figure 4.
Figure 4.

William plot for the developed CoMSIA/SEA model.
The AD of the CoMSIA/SEA model was defined by leverage analysis expressed as Williams plot (Figure 4). In the William plots, the results indicate that the leverage values of all the compounds in the training and test sets were lower than the warning leverage (h∗ = 0.40) except for compound 20, which was greater than the warning lever effect, this compound belong to the training set. The CoMSIA/SEA model of test sets was predicted correctly because they're void of outliers. Therefore, all compounds tested are in the AD, indicating that their predicted activity values are reliable.
3.4. Graphical interpretation of CoMSIA model
To visualize the information contained in the best 3D-QSAR model, we based on CoMSIA/SEA contour map with the most active compound 20 of the series as a reference. The Steric, Electrostatic and Hydrogen bond acceptor fields of the CoMSIA contour maps are shown in Figure 5.
Figure 5.

(a) Steric, (b) Electrostatic and (c) Hydrogen bond acceptor Contour maps of CoMSIA analysis with 2 Å grid spacing in combination with compound 20.
In the steric field, the green contours (80% contribution) indicate regions where bulky groups enhance the biological activity, while yellow contours (20% contribution) indicate regions where bulky groups decrease activity. In the electrostatic field, the blue contours (80% contribution) indicate where the positive electrostatic favored, whereas red contours (20% contribution) indicated where the negative electrostatic favored regions. Furthermore, the hydrogen-bond acceptor field, the magenta contour maps (contribution 80%) for hydrogen-bond acceptor groups increase activity, while red contours (contribution 20%) indicate the disfavored region.
In the CoMSIA steric contour map, we have observed a large green contour covers at R substitutions in para position for azaaurone compounds, which indicates that bulky substitution group selection is required in this region. That can explain why compounds 20 with the –CH2-CH3 on para position is more active than than the compound 21 with the same substitution on ortho position. Therefore, the R2 bulky substitution at the para position of the benzene ring might have better activity.
In the CoMSIA electrostatic contour maps, we observed a blue contour near at R substitutions in para position for azaaurone compounds, indicating that introducing high electropositive groups or atoms in this position can improve the activity. This can be explained by the fact that compounds 20 with the -CH2-CH3 at para position shows higher activity than compounds 15 and 16 with -Br and -Cl in the same position. Therefore, the compounds 20, 26, 27, 28, 29 and 33 have higher activity. Moreover, we have observed a red contour is near at R substitutions in meta position for azaaurone compounds, indicating that introducing high electronegative groups or atoms can improve the activity. This can be explained by the fact that compounds 18 with the –Cl at ortho and meta position shows higher activity than compounds 17 with –Cl in ortho position only and then adding –CH3 at the meta position in compound 24 has decreased the activity relative to compound 23.
In the hydrogen-bond acceptor contour map, the red contours near R substitution in para position indicated that the substitution the hydrogen-bond acceptor in this position is unfavorable for increasing the activity, this result can explain why compounds 15, 16, 20, 26, 27, 28, 29, 33 and 35 have higher activity.
3.5. Molecular docking
The molecular docking study was used to obtain information on the structural basis and interaction established of the difference in sensitivity to Qo site inhibitors of the heme bL. The heme bL Binding Sites is built by nonpolar residues (Leu40, Gln43, Gly47, Ala 51, Ala83, Ala86, Phe89, Thr127, Gly131, Tyr132, Val135 Tyr184 and Pro187) with the exception of the iron ligands (His82 and His183) [47]. The interactions mode obtained by molecular docking for compounds 13 and 20 are illustrated in Figure 6.
Figure 6.

2D and 3D docking poses showing interactions of compounds 13 and 20 in the binding sites of cytochrome b. (a) Compound 13:(binding energy -10.33 kcal/mol). (b) Compound 20: (binding energy -10.49 kcal/mol).
In molecular docking study, the compound 20 interaction of azaaurone with the active site of heme bL shows two hydrogen bonds between the nitrogen atoms of the azaaurone and the amino acids His82 and His183 with the distance of 2.66 Ǻ and 2.69Ǻ, respectively. In addition, the Pi-Pi interactions are observed between a benzene with amino acid His180 (5.1 Ǻ) and Tyr54 (4.77 Ǻ). However, the compound 13 of aurone was performed the two Pi-Pi interactions between a benzene and pyrrolidine with amino acids His183 (5.17 Ǻ) and His82 (4.44 Ǻ), respectively.
The most active compound has shown the hydrogen bonds interactions with the amino acids His82 and His183. However, the low active compound has formed the Pi-Pi interactions with the same amino acids. The residues His82 and His183 are ligands of heme bL in the Qo site in the cytochrome b, the hydrogen bonds are much stronger interactions than Pi-Pi bonds [48], because they assure stability to the protein structure and selectivity to protein-ligand interactions as well as the distances of hydrogen bond interactions are much closer to key amino acids than to Pi-Pi bond interactions. Overall, the azaaurone is more active than aurone because the nitrogen atom of azaaurone plays a crucial role in antimalarial activity by inhibiting mitochondrial cytochrome b.
3.6. Docking validation protocol
In order to test the capacity of docking algorithms to predict the conformation of the protein-bound ligand, re-docking of the Co-Crystallized Ligand was applied to validate the accuracy of the docking procedure. Figure 7 clarifies the superimposed view between the docked ligand conformation and the co-crystallized ligand conformation and the RMSD value is 5.885 Ǻ. After the majority of publications, high RMSD value (5.885 Ǻ) would suggest an inaccurate pose prediction. However, several studies have shown that the RMSD parameter suffers from serious problems [49, 50]. On the one hand, the large, almost symmetric molecules almost symmetrical can be exchanged in the binding site during docking, as the case in this study, the RMSD would be at a very high level and would suggest a poor placement [49]. On the other hand, the Small compounds can easily achieve low RMSD even when placed randomly. Moreover, there is no information on the quality of the representation of the complex, the interactions of a ligand with the protein. Several studies [49, 51, 52] have proposed a new benchmark for the quality of docking poses based on visual inspection.
Figure 7.
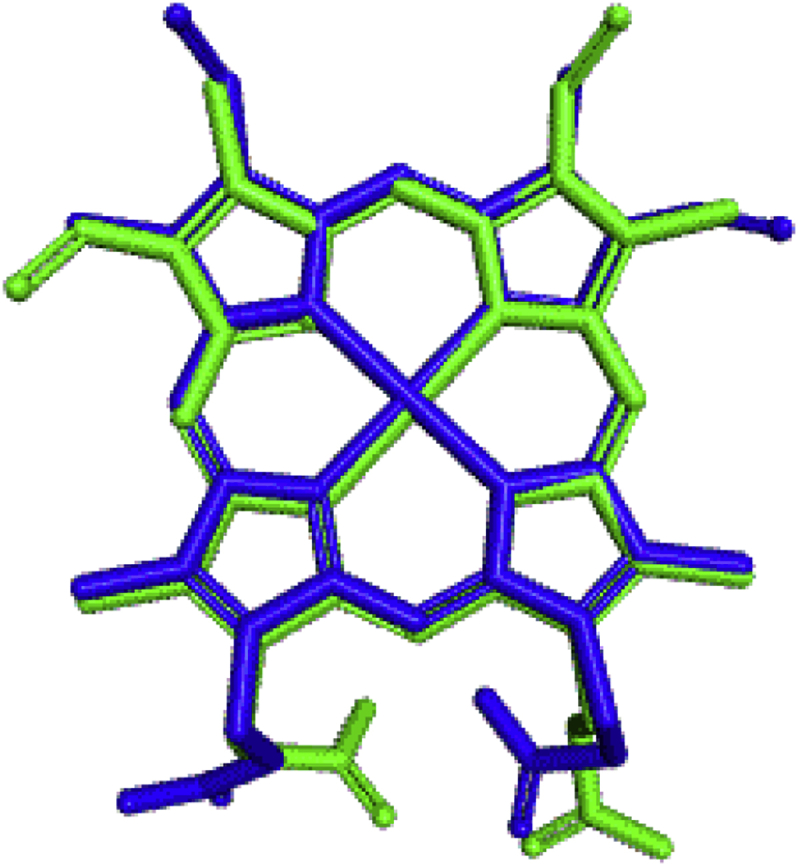
Re-docking pose with the RMSD value of 5.885 Å (Green = Original, blue = Docked).
For visual inspection, Figure 8 presents the 2D visualization of the interactions between a generated docking pose and the experimental ligand conformation. The results of this visual inspection show the same interactions as in the experimental binding mode, as observed in Figure 8. This result confirms that the à alone is not a reliable parameter for the quality of docking poses for docking validation and the use of visual inspection as a new reference is necessary. Thus, the docking process in this study was successfully validated.
Figure 8.

(a) 2D visualization showing interactions of ligand pose prediction result. (b) 2D visualization showing interactions of the crystallographic ligand pose.
3.7. Design of new compounds
The principal aim of this study is the design of new inhibitors of Qo site in cytochrome b, through the recommendations we have extracted from the 3 D-QSAR and molecular docking studies on the structural characteristics of the highest active compound (compound number 20). In this study, four azaaurone derivatives were designed to improve and propose a new antimalarial agent, newly designed compounds were aligned to the database using compound 20 as a template, and we used the CoMSIA/SEA model to predict the activity of the newly designed compounds (Figure 9). The new candidate compounds with antimalarial activity are presented in Table 5.
Figure 9.

Structures of newly designed molecules.
Table 5.
Molecular docking scoring and predicted pIC50 based on CoMSIA/SEA model for the newly designed compounds.
| compounds | CoMSIA/SEA model |
Docking score |
|
|---|---|---|---|
| pIC50(pred) | IC50(pred) | (-Log ki) | |
| T1 | 5.429 | 3.72 | 8.126 |
| T2 | 5.425 | 3.76 | 6.407 |
| T3 | 5.409 | 3.9 | 7.558 |
| T4 | 5.407 | 3.92 | 6.961 |
3.8. ADMET prediction and druglikeness
All newly designed molecules predicted by the CoMSIA/SEA model have almost the same activities. Therefore, to ensure that the designed molecules are the viable drugs, we used the pharmacokinetic parameters ADMET and druglikeness. The pkCSM online tool [53] was employed to predict in silico ADMET properties (Table 6), and druglikeness properties were further predicted using the SwissADME online tool [54] (Table 7).
Table 6.
In silico ADMET prediction of new designed compounds.
| Compounds | Absorption |
Distribution |
Metabolism |
Excretion |
Toxicity |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intestinal absorption (human) |
VDss (human) |
BBB permeability |
CNS permeability |
Substrate |
Inhibitor |
Total Clearance |
AMES toxicity |
||||||
| CYP | |||||||||||||
| 2D6 |
3A4 |
1A2 |
2C19 |
2C9 |
2D6 |
3A4 |
|||||||
| Numeric (% Absorbed) |
Numeric (Log L/kg) |
Numeric (Log BB) | Numeric (Log PS) | Categorical (Yes/No) | Numeric (Log ml/min/kg) |
Categorical (Yes/No) | |||||||
| T1 | 93.864 | 0.205 | 0.041 | -0.568 | No | Yes | No | Yes | Yes | No | Yes | 0.366 | No |
| T2 | 96.318 | 0.172 | -0.708 | -2.933 | No | Yes | Yes | Yes | Yes | No | Yes | 0.501 | No |
| T3 | 94.608 | 0.535 | 0.024 | -1.935 | No | Yes | Yes | Yes | Yes | No | Yes | 0.498 | No |
| T4 | 95.671 | 0.563 | 0.111 | -1.715 | No | Yes | Yes | Yes | Yes | No | Yes | 0.56 | No |
Table 7.
Drug likeness prediction of the compound T1, T2, T3 and T4 basing on lipinski, Ghose, veber and Egan, and their synthetic accessibility.
| Lipinski | Ghose | Veber | Egan | Synthetic accessibility | |
|---|---|---|---|---|---|
| T1 | Yes | Yes | Yes | Yes | 3.41 |
| T2 | Yes | Yes | Yes | Yes | 3.31 |
| T3 | Yes | Yes | Yes | Yes | 3.63 |
| T4 | Yes | No | Yes | Yes | 3.9 |
Absorbance value below 30% signifies poor absorbance, all compounds displaying a value above 90%, which reveals a good absorbance in the human intestine. Volume of distribution (VDss) is considered high if the value is greater than 0.45. In addition, blood brain barrier (BBB) and central nervous system (CNS) permeability standard values (>0.3 to < -1 Log BB and > -2 to < -3 LogPS), respectively. For a given compound a LogBB < -1 are poor distributed to brain, while LogBB >0.3 are potential to cross BBB and LogPS > -2 considered to penetrate the CNS, while LogPS < -3 are difficult to move in the CNS [55]. Therefore, the compounds T3 and T4 have the best significant potential to cross the barriers.
The enzymatic metabolism indicate the chemical biotransformation of a drug in the body, which plays an important role in conversion of drug compounds. In the body, drugs produce several enzymatic metabolites, that play a major role in catalysing the reaction with Different drug concentration [56]. It is required to consider their metabolism of drugs, which may have different physicochemical and pharmacological properties. The cytochrome P450 (CYP450) plays a major role in drug metabolism because the major liver enzyme system involved in phase 1 metabolism (oxidation) as the case of our study. Thus far, 57 CYP genes from 17 families have been identified in human, but only CYP1, CYP2, CYP3 and CYP4 families are involved in drug metabolism, with CYP (1A2, 2C9, 2C19, 2D6 and 3A4) being responsible for the biotransformation of greater than 90% of drugs undergoing phase I metabolism [56, 57]. However, among these families, CYP3A4 is the most important inhibition in this study [58]. All new designed compounds were found to be the substrate and the inhibitor of CYP3A4.
Clearance is a constant that describes the relationship between drug concentration in the body and the rate of elimination of the drug. Therefore, All new compounds designed show a somewhat high value, but still acceptable in persistence of the drug in the body. Moreover, it is necessary to examine whether if the predicted compounds are non-toxic because this plays a critical role in the selection of drugs. Fortunately, all the compounds we designed are non-toxic. Overall, the new molecules designed T3 and T4 with good pharmacokinetic properties.
The compounds designed were evaluated for their synthetic accessibility, the Synthetic accessibility values for all compounds designed is about 3, therefore, they are easy to synthetic. In addition, the new compounds T1, T2 and T3 respect all drug likeness rules.
4. Conclusion
In the aim of producing new antimalarial drug candidates, the 3D-QSAR and molecular docking study was used to explore the structural determinants and specific binding modes of 35 aurone analogues acting as Qo site inhibitors at the cytochrome b. The excellent predictive power of CoMSIA/SEA model observed for a test set of compounds indicates a significant statistical quality and the significance was confirmed by the external validation methods, This shows that this model can be successfully used to predict the activity of new molecules designed.
The molecular docking study explained how aurone analogues can inhibit cytochrome b by acting at the Qo site by intractions with the amino acids His183 and His82 which are ligands of heme bL, these residues play a decisive role for antimalarial activity by blocking electron transfer from the respiratory chain of P. falciparum. In the docking validation protocol, RMSD is not a good parameter in the case of large, nearly symmetrical molecules. however, visual inspection is the perfect solution. in this study, visual inspection showed very convincing results for the validation of molecular docking.
The powerful approaches such as 3D-QSAR study using CoMSIA contour map results and molecular docking analysis led to design of four new compounds (T1-4) that can be useful for further design of novel cytochrome b inhibitors. The combination of ADMET prediction and Druglikeness has shown promising results in silico, because the new designed molecules have improved kinetic properties and it respect all druglikeness rules as well as an interesting result in terms of biological activity. The results of this study could represent good drug candidates for the treatment of malaria.
Declarations
Author contribution statement
Hanine Hadni: Conceived and designed the analysis; Analyzed and interpreted the data; Wrote the paper.
Menana Elhallaoui: Conceived and designed the analysis; Contributed analysis tools or data.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
References
- 1.Murray M.C., Perkins M.E. Chapter 15. Chemotherapy of malaria. Annu. Rep. Med. Chem. 1996;31:141–150. [Google Scholar]
- 2.Singh B., Sung L.K., Matusop A., Radhakrishnan A., Shamsul S.S.G., Cox-Singh J., Thomas A., Conway D.J. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–1024. doi: 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- 3.Sondo P., Derra K., Lefevre T., Diallo-Nakanabo S., Tarnagda Z., Zampa O., Kazienga A., Valea I., Sorgho H., Ouedraogo J.B., Guiguemde T.R., Tinto H. Genetically diverse Plasmodium falciparum infections, within-host competition and symptomatic malaria in humans. Sci. Rep. 2019;9:1–9. doi: 10.1038/s41598-018-36493-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World malaria report 2019. https://www.who.int/news-room/feature-stories/detail/world-malaria-report-2019 (n.d.)
- 5.Ouattara A., Laurens M.B. Vaccines against malaria. Clin. Infect. Dis. 2015;60:930–936. doi: 10.1093/cid/ciu954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlitzer M. Malaria chemotherapeutics Part I: history of antimalarial drug development, currently used therapeutics, and drugs in clinical development. ChemMedChem. 2007;2:944–986. doi: 10.1002/cmdc.200600240. [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty A. Emerging drug resistance in Plasmodium falciparum: a review of well-characterized drug targets for novel antimalarial chemotherapy. Asian Pacific J. Trop. Dis. 2016;6:581–588. [Google Scholar]
- 8.Hassan G.S., Georgey H.H., George R.F., Mohamed E.R. Aurones and furoaurones: biological activities and synthesis. Bull. Fac. Pharm. Cairo Univ. 2018;56:121–127. [Google Scholar]
- 9.Boumendjel A. Aurones: a subclass of flavones with promising biological potential. Curr. Med. Chem. 2005;10:2621–2630. doi: 10.2174/0929867033456468. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence N.J., Rennison D., McGown A.T., Hadfield J.A. The total synthesis of an aurone isolated from Uvaria hamiltonii: aurones and flavones as anticancer agents. Bioorg. Med. Chem. Lett. 2003;13:3759–3763. doi: 10.1016/j.bmcl.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Alsayari A., Bin Muhsinah A., Hassan M.Z., Ahsan M.J., Alshehri J.A., Begum N., Aurone A biologically attractive scaffold as anticancer agent. Eur. J. Med. Chem. 2019:417–431. doi: 10.1016/j.ejmech.2019.01.078. [DOI] [PubMed] [Google Scholar]
- 12.Jardosh H.H., Patel M.P. Antimicrobial and antioxidant evaluation of new quinolone based aurone analogs. Arab. J. Chem. 2017;10:S3781–S3791. [Google Scholar]
- 13.Roussaki M., Costa Lima S., Kypreou A.-M., Kefalas P., Cordeiro da Silva A., Detsi A. Aurones: a promising heterocyclic scaffold for the development of potent antileishmanial agents. Int. J. Med. Chem. 2012;2012:1–8. doi: 10.1155/2012/196921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheng R., Xu Y., Hu C., Zhang J., Lin X., Li J., Yang B., He Q., Hu Y. Design, synthesis and AChE inhibitory activity of indanone and aurone derivatives. Eur. J. Med. Chem. 2009;44:7–17. doi: 10.1016/j.ejmech.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Kayser O., Kiderlen A.F., Folkens U., Kolodziej H. In vitro leishmanicidal activity of aurones. Planta Med. 1999;65:316–319. doi: 10.1055/s-1999-13993. [DOI] [PubMed] [Google Scholar]
- 16.Sutton C.L., Taylor Z.E., Farone M.B., Handy S.T. Antifungal activity of substituted aurones. Bioorg. Med. Chem. Lett. 2017;27:901–903. doi: 10.1016/j.bmcl.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Bandgar B.P., Patil S.A., Korbad B.L., Biradar S.C., Nile S.N., Khobragade C.N. Synthesis and biological evaluation of a novel series of 2,2-bisaminomethylated aurone analogues as anti-inflammatory and antimicrobial agents. Eur. J. Med. Chem. 2010;45:3223–3227. doi: 10.1016/j.ejmech.2010.03.045. [DOI] [PubMed] [Google Scholar]
- 18.Carrasco M.P., Machado M., Gonçalves L., Sharma M., Gut J., Lukens A.K., Wirth D.F., André V., Duarte M.T., Guedes R.C., dos Santos D.J.V.A., Rosenthal P.J., Mazitschek R., Prudêncio M., Moreira R. Probing the azaaurone scaffold against the hepatic and erythrocytic stages of malaria parasites. ChemMedChem. 2016;11:2194–2204. doi: 10.1002/cmdc.201600327. [DOI] [PubMed] [Google Scholar]
- 19.Fisher N., Meunier B. Molecular basis of resistance to cytochrome bc1 inhibitors. FEMS Yeast Res. 2008;8:183–192. doi: 10.1111/j.1567-1364.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 20.Kao W.-C., Hunte C. The molecular evolution of the Qo motif. Genome Biol. Evol. 2014;6:1894–1910. doi: 10.1093/gbe/evu147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brasseur G., Lemesle-Meunier D., Reinaud F., Meunier B. QO site deficiency can be compensated by extragenic mutations in the hinge region of the iron-sulfur protein in the bc1 complex of Saccharomyces cerevisiae. J. Biol. Chem. 2004;279:24203–24211. doi: 10.1074/jbc.M311576200. [DOI] [PubMed] [Google Scholar]
- 22.Sarvagalla S., Syed S.B., Coumar M.S. Silico Drug Des. Elsevier; 2019. An overview of computational methods, tools, servers, and databases for drug repurposing; pp. 743–780. [Google Scholar]
- 23.Roy K., Kar S., Das R.N. Underst. Basics QSAR Appl. Pharm. Sci. Risk Assess. Elsevier; 2015. SAR and QSAR in drug discovery and chemical design—some examples; pp. 427–453. [Google Scholar]
- 24.Hadni H., Mazigh M., Charif E., Bouayad A., Elhallaoui M. Molecular modeling of antimalarial agents by 3D-QSAR study and molecular docking of two hybrids 4-Aminoquinoline-1,3,5-triazine and 4-Aminoquinoline-oxalamide derivatives with the receptor protein in its both wild and mutant types. Biochem. Res. Int. 2018;2018:1–15. doi: 10.1155/2018/8639173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hadni H., Mazigh M., Elhallaoui M. QSAR and Molecular Docking Studies of 4-anilinoquinoline- Triazine Hybrids as Pf-DHFR Inhibitors. 2019;8:84–93. doi: 10.1016/j.heliyon.2019.e02357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hadni H., Elhallaoui M. Molecular docking and QSAR studies for modeling the antimalarial activity of hybrids 4-anilinoquinoline-triazines derivatives with the wild-type and mutant receptor pf-DHFR. Heliyon. 2019;5 doi: 10.1016/j.heliyon.2019.e02357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Souard F., Okombi S., Beney C., Chevalley S., Valentin A., Boumendjel A. 1-Azaaurones derived from the naturally occurring aurones as potential antimalarial drugs. Bioorg. Med. Chem. 2010;18:5724–5731. doi: 10.1016/j.bmc.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Cramer R.D., Patterson D.E., Bunce J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 29.Klebe G., Abraham U., Mietzner T. Molecular similarity Indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 30.Clark M., Cramer R.D., Van Opdenbosch N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989;10:982–1012. [Google Scholar]
- 31.Tsai K.C., Chen Y.C., Hsiao N.W., Wang C.L., Lin C.L., Lee Y.C., Li M., Wang B. A comparison of different electrostatic potentials on prediction accuracy in CoMFA and CoMSIA studies. Eur. J. Med. Chem. 2010;45:1544–1551. doi: 10.1016/j.ejmech.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 32.Powell M.J.D. Restart procedures for the conjugate gradient method. Math. Program. 1977;12:241–254. [Google Scholar]
- 33.Wold S., Ruhe A., Wold H., Dunn W.J., III The collinearity problem in linear regression. The partial least squares (PLS) approach to generalized inverses. SIAM J. Sci. Stat. Comput. 1984;5:735–743. [Google Scholar]
- 34.Tropsha A., Gramatica P., Gombar V. The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003;22:69–77. [Google Scholar]
- 35.Golbraikh A., Tropsha A. Beware of q2! J. Mol. Graph. Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 36.Roy K. On some aspects of validation of predictive quantitative structure-activity relationship models. Expet Opin. Drug Discov. 2007;2:1567–1577. doi: 10.1517/17460441.2.12.1567. [DOI] [PubMed] [Google Scholar]
- 37.Roy K., Kar S., Ambure P. On a simple approach for determining applicability domain of QSAR models. Chemometr. Intell. Lab. Syst. 2015;145:22–29. [Google Scholar]
- 38.Netzeva T.I., Worth A.P., Aldenberg T., Benigni R., Cronin M.T.D., Gramatica P., Jaworska J.S., Kahn S., Klopman G., Marchant C.A., Myatt G., Nikolova-Jeliazkova N., Patlewicz G.Y., Perkins R., Roberts D.W., Schultz T.W., Stanton D.T., van de Sandt J.J.M., Tong W., Veith G., Yang C. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. Altern. Lab. Anim. 2005;33:155–173. doi: 10.1177/026119290503300209. [DOI] [PubMed] [Google Scholar]
- 39.Kouranov A., Xie L., de la Cruz J., Chen L., Westbrook J., Bourne P.E., Berman H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006;34:D302–D305. doi: 10.1093/nar/gkj120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studio D. 2008. Discovery Studio Life Science Modeling and Simulations. [Google Scholar]
- 41.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 43.Onodera K., Satou K., Hirota H. Evaluations of molecular docking programs for virtual screening. J. Chem. Inf. Model. 2007;47:1609–1618. doi: 10.1021/ci7000378. [DOI] [PubMed] [Google Scholar]
- 44.Warren G.L., Andrews C.W., Capelli A.-M., Clarke B., LaLonde J., Lambert M.H., Lindvall M., Nevins N., Semus S.F., Senger S., Tedesco G., Wall I.D., Woolven J.M., Peishoff C.E., Head M.S. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 45.Ferreira L.L.G., Andricopulo A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today. 2019;24:1157–1165. doi: 10.1016/j.drudis.2019.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Golbraikh A., Tropsha A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers. 2000;5:231–243. doi: 10.1023/a:1021372108686. [DOI] [PubMed] [Google Scholar]
- 47.Hunte C., Koepke J., Lange C., Roßmanith T., Michel H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure. 2000;8:669–684. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 48.Morozov A.V., Kortemme T. Potential functions for hydrogen bonds in protein structure prediction and design. Adv. Protein Chem. 2005;72:1–38. doi: 10.1016/S0065-3233(05)72001-5. [DOI] [PubMed] [Google Scholar]
- 49.Kroemer R.T., Vulpetti A., McDonald J.J., Rohrer D.C., Trosset J.-Y., Giordanetto F., Cotesta S., McMartin C., Kihlén M., Stouten P.F.W. Assessment of docking Poses: interactions-based accuracy classification (IBAC) versus crystal structure deviations. J. Chem. Inf. Comput. Sci. 2004;44:871–881. doi: 10.1021/ci049970m. [DOI] [PubMed] [Google Scholar]
- 50.Kirchmair J., Markt P., Distinto S., Wolber G., Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection - what can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 51.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 52.Kontoyianni M., McClellan L.M., Sokol G.S. Evaluation of docking performance: comparative data on docking algorithms. J. Med. Chem. 2004;47:558–565. doi: 10.1021/jm0302997. [DOI] [PubMed] [Google Scholar]
- 53.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7 doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark D.E. In silico prediction of blood–brain barrier permeation. Drug Discov. Today. 2003;8:927–933. doi: 10.1016/s1359-6446(03)02827-7. [DOI] [PubMed] [Google Scholar]
- 56.Kok-Yong S., Lawrence L. Basic Pharmacokinet. Concepts Some Clin. Appl. InTech; 2015. Drug distribution and drug elimination. [Google Scholar]
- 57.Šrejber M., Navrátilová V., Paloncýová M., Bazgier V., Berka K., Anzenbacher P., Otyepka M. Membrane-attached mammalian cytochromes P450: an overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J. Inorg. Biochem. 2018;183:117–136. doi: 10.1016/j.jinorgbio.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Thapar M.M., Karolinska University Press . Malarone®; 2004. Pharmacokinetics and Dynamics of Atovaquone and Proguanil. [Google Scholar]