

Abstract
The influenza virus hemagglutinin (HA) mediates the first essential step in the viral life cycle, virus entry into target cells. Influenza virus HA is synthesised as a precursor protein in infected cells and requires cleavage by host cell proteases to transit into an active form. Cleavage is essential for influenza virus infectivity and the HA‐processing proteases are attractive targets for therapeutic intervention. It is well established that cleavage by ubiquitously expressed subtilisin‐like proteases is a hallmark of highly pathogenic avian influenza viruses (HPAIV). In contrast, the nature of the proteases responsible for cleavage of HA of human influenza viruses and low pathogenic avian influenza viruses (LPAIV) is not well understood. Recent studies suggest that cleavage of HA of human influenza viruses might be a cell‐associated event and might be facilitated by the type II transmembrane serine proteases (TTSPs) TMPRSS2, TMPRSS4 and human airway trypsin‐like protease (HAT). Here, we will introduce the different concepts established for proteolytic activation of influenza virus HA, with a particular focus on the role of TTSPs, and we will discuss their implications for viral tropism, pathogenicity and antiviral intervention. Copyright © 2010 John Wiley & Sons, Ltd.
INTRODUCTION
Infection with influenza viruses, enveloped viruses with a negative stranded, segmented RNA genome, is a significant source of global morbidity and mortality 1. The ever‐changing nature of influenza viruses allows these agents to continuously evade host immunity and to pose a constant threat to human and animal health 1, 2. The acquisition of subtle alterations (antigenic drift), mainly amino acid changes in the viral surface protein hemagglutinin (HA), is responsible for the yearly occurring influenza epidemics (seasonal influenza), which claim 36 000 lives per year in the Unites States alone 3. The exchange of entire genomic segments (reassortment) between influenza viruses of different subtypes in a co‐infected cell can result in the emergence of new viruses (antigenic shift), which, upon introduction into an immunological naïve population, can initiate a pandemic spread (pandemic influenza) with potentially severe medical and social consequences 1, 4, 5. It is estimated that up to 50 million people, or about 2% of the world's population, succumbed to the influenza virus circulating in 1918, the causative agent of the disastrous Spanish influenza 6, 7. A distantly related virus, which emerged in Mexico in February 2009 8, 9, is responsible for the first influenza pandemic of the 21st century, with 258 698 laboratory confirmed cases by 5 December 2009 (the number of actual cases is expected to be much higher) 10. Albeit infection by the 2009 H1N1 virus usually does not cause severe disease in patients without underlying illness, it is notable that the virus shows augmented pathogenicity in animal models compared to contemporary seasonal influenza viruses 11, 12, raising concerns that viral variants with increased pathogenicity might emerge in the course of the pandemic.
Vaccines and antivirals target the viral surface proteins HA (16 subtypes: H1–H16), neuraminidase (NA, 9 subtypes N1–N9) and the ion channel M2; these proteins normally facilitate entry (HA), release (NA) and viral uncoating (M2) in target cells 13, 14, 15, 16. However, the variability of influenza viruses compromises the antiviral defences directed against the surface proteins. Thus, as a result of antigenic drift, the vaccine needs to be reformulated on an annual basis to provide protection against the viral strains expected to be circulating during the next ‘influenza season’, and seasonal vaccination will not be effective against newly emerged viruses, like the 2009 H1N1 virus. In addition, viruses frequently acquire mutations, which confer resistance in the molecular targets of M2 and NA inhibitors 17. Therefore, novel strategies to combat influenza are required, which should be based on a thorough understanding of viral and host cell factors essential for influenza virus spread and pathogenesis. Proteolytic activation of the influenza virus HA by cellular enzymes is indispensable for the virus to transform into an infectious form 18, and the responsible enzymes are potential targets for intervention. However, the nature of the proteases, which activate human influenza viruses, is unclear. Recently, evidence was reported that type II transmembrane serine proteases (TTSPs) might play a major role 19, 20. Here, we will review our knowledge on proteolytic activation of human influenza viruses, with a particular focus on the role of TTSPs, and we will discuss the implications of different modes of HA activation for viral tropism, pathogenicity and antiviral intervention.
INFLUENZA VIRUS HEMAGGLUTININ: THE VIRAL KEY TO THE HOST CELL
The HA mediates influenza virus binding to host cells and fusion of the viral membrane with the limiting membrane of host cell endosomes, thereby providing the virus with access to the host cell interior. These processes are essential for infection, are tightly regulated and involve the formation of several structural HA intermediates, which have been characterised at the atomic level. Here, we will describe how HA brings about membrane fusion and why HA cleavage is essential for its activity.
Hemagglutinin drives fusion of the viral membrane with the membrane of host cell endosomes
Mature HA is composed of two subunits, the globular surface unit HA1 and the stalk‐like transmembrane unit HA2, which are covalently linked by a disulfide bond (Figure 1) 21, 22. The subunits function differently in the entry process: HA1 facilitates binding to receptors on the host cell surface while HA2 drives membrane fusion. Important receptor determinants for influenza viruses are alpha 2–6 (preferred by human viruses) and alpha 2–3 (preferred by avian viruses) linked sialic acids, which are bound by a pocket located in the membrane distal part of HA1 21, 22. Upon uptake of bound particles, which can proceed via multiple endocytic pathways 23, the low pH environment in endosomal vesicles induces conformational changes in HA2, which facilitate membrane fusion 24. The successful membrane merger requires the concerted action of several functional elements in HA2, including an N‐terminal fusion peptide (FP), the transmembrane domain and a central coiled coil, which is located between the FP and the transmembrane domain 21, 22. The membrane fusion reaction commences with a pH‐induced loop‐to‐helix transition of a sequence located between the FP and the coiled coil. As a consequence of this transition, the hydrophobic FP is propelled towards the target cell and inserts into the cellular membrane 21, 22. Subsequently, the C‐terminal extracellular portion of HA2 folds onto the central coiled coil in an antiparallel fashion, forming a stable post‐fusion structure in which the FP and the membrane anchor of HA are located in close proximity. As a result of this conformational change, the viral and the target cell membrane are pulled into close contact and ultimately fuse 21, 22.
Figure 1.
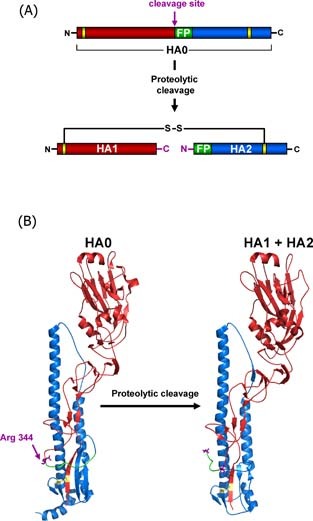
Structural rearrangements associated with influenza virus hemagglutinin cleavage by host cell proteases. The HA precursor HA0 is cleaved into two subunits HA1 (red) and HA2 (blue) by cellular proteases which recognise either a multibasic or monobasic cleavage site located in a loop between HA1 and HA2. Multibasic cleavage sites harbour multiple arginines and/or lysines and are found in the HA‐proteins of HPAIV. Monobasic cleavage sites consist of a single arginine (Arg 344 (purple) in case of the 1918 influenza virus) or lysine and are found in the HA‐proteins of human influenza viruses and LPAIV. Upon proteolytic cleavage, the fusion peptide (green) in HA2 is liberated from HA1 and the HA is primed for activation by low pH, which involves insertion of the fusion peptide into a negatively charged pocket located adjacent to the cleavage site‐bearing loop in HA0. The subunits HA1 and HA2 remain covalently linked by a single disulfide bond (yellow), the newly formed C‐terminus of HA1 and N‐terminus of HA2 are labelled in purple. (A) Schematic representation of HA cleavage. (B) Structural changes associated with cleavage of the 1918 influenza HA (A/South Carolina/1/18 (H1N1), Accession‐No: ADD17229, HA0 (1RD8.pdb), HA1 + HA2 (1RUZ.pdb)
Proteolytic cleavage is essential for hemagglutinin‐driven membrane fusion
The ability of HA to drive membrane fusion critically depends on a ‘priming step’, during which HA transits into a conformation responsive to low pH, the trigger for membrane fusion. The influenza virus HA is initially synthesised as a precursor protein, HA0, in the secretory pathway of infected cells 21, 22. Despite proper glycosylation, folding and trimerisation of HA0 25, 26—intimately connected processes 27—HA0 is unable to mediate membrane fusion. To acquire its membrane fusion potential, HA0 must be cleaved into HA1 and HA2 by host cell proteases 18, 28, 29. Cleavage occurs at a linker sequence connecting the HA1 and HA2 subunits, which is located on a partially surface exposed loop, and proceeds in two steps. First, an endoprotease cleaves HA at the carboxyl terminus of an arginine located at the border between HA1 and HA2 and thereby generates the N‐terminus of mature HA2 28, 29. Subsequently, a carboxypeptidase removes the arginine and thereby creates the C‐terminus of mature HA1 30. Cleavage is associated with a structural change in HA, during which the FP, probably due to electrostatic interactions, inserts into a conserved cavity formed by residues within HA1 and HA2 31. This configuration is essential for fusion, since only the liberated (separated from the C‐terminus of HA1) FP is able to insert into the target cell membrane upon a low pH stimulus. Amino acids within the cavity and the FP are essential for membrane fusion and determine at which pH fusion is triggered 27, 32, suggesting that the cavity and the FP are potential targets for therapeutic intervention 28.
The sequence of the cleavage site in hemagglutinin is an important determinant of pathogenicity of avian influenza viruses
The nature of the linker sequence critically determines access to proteases, which in turn impacts viral tropism and pathogenicity, at least in the context of avian viruses. Thus, for low pathogenic avian influenza viruses (LPAIV) the linker consists of a single arginine (in some cases lysine), which is accessible to only a limited number of trypsin‐like proteases 28, 29. Tissue expression of these proteases is believed to be restricted to the respiratory tract (terrestrial birds) and gastrointestinal tract (water fowl, terrestrial birds). Consequently, viral replication is confined to these compartments. In contrast, the linker sequence in highly pathogenic avian influenza viruses (HPAIV) contains several arginines, with R‐X‐R/K‐R as consensus motif 33, 34, 35. This sequence is surface‐exposed and readily recognised by furin and PC5/6, members of the proprotein convertase family of eukaryotic subtilisin‐like serine endoproteases, which are ubiquitously expressed 36, 37. Therefore, HPAIVs can replicate systemically and cause severe disease. In this context, it needs to be noted that some HPAIVs do not harbour a furin consensus sequence. However, cleavability of these viruses might be increased due to the absence of a carbohydrate side chain, which would otherwise limit recognition by proteases 38, 39, 40, 41, or due to amino acid substitutions close to the cleavage site, which might increase surface exposure of this sequence and thus accessibility to proteases 21.
Considering the clear correlation between cleavability, viral tropism and pathogenicity observed for avian viruses, a similar interdependence could also be expected for human viruses. However, none of the influenza virus subtypes previously pandemic in humans (H1N1, H2N2 and H3N2) contained a multi‐basic cleavage site, including the highly pathogenic 1918 influenza virus 42. The implications of the cleavage site for spread and pathogenicity of human viruses are thus not overt, and can only be elucidated once the HA‐activating proteases have been defined.
Collectively, HA exhibits a particular domain organisation, which reflects the molecular mechanism by which it drives membrane fusion. A similar architecture and membrane fusion mechanism have been identified for other viral glycoproteins, like the HIV envelope protein, which are termed class I fusion proteins 22. Most class I fusion proteins, including influenza virus HA, require cleavage by host cell proteases to transit into a fusion‐competent state. Proteolytic activation of influenza viruses can occur in the Golgi apparatus or at the plasma membrane of infected cells, as well as in the extracellular space and in target cell vesicles, so the nature of the cleavage site and the respective activating proteases have important implications for the biological properties of influenza virus as well as for therapeutic intervention, as discussed below.
PROTEOLYTIC ACTIVATION OF INFLUENZA VIRUS HEMAGGLUTININ: PROTEASES AND CELLULAR COMPARTMENTS
HPAIV are activated by furin in the trans‐Golgi apparatus
Early, groundbreaking studies on the activation of influenza viruses noted that viral infectivity correlated with the status of HA cleavage and that activation of viruses by trypsin correlated with HA cleavage 43, 44, 45. Within these studies it was also observed that some viruses contained cleaved HA and were highly infectious irrespective of the cellular systems used, while HA cleavage and infectivity of others were strongly dependent on the host cell 43, 44, 45. Viruses of the former phenotype were subsequently shown to harbour several arginine and lysine residues at the cleavage site 33, with an R‐X‐R/K‐R consensus sequence being indispensable for efficient cleavage 46. In addition, evidence was obtained that cleavage of HA might occur in the trans‐Golgi network (TGN) 47. Stieneke‐Gröber and colleagues then demonstrated that these viruses are activated by furin (Figure 2) and that peptide derivatives spanning the R‐X‐R/K‐R consensus sequence were able to suppress viral spread 36. Furin, which is mainly expressed in the Golgi apparatus but also found on the cell surface, processes a host of cellular pro‐proteins in the secretory pathway and is indispensable for normal embryonic development 37. Increase of influenza virus pathogenicity in poultry is associated with acquisition of a multibasic cleavage site 1, 33, 48 and this effect is reversed when the cleavage site is mutated 49, underlining that the protease recognition site in HA of avian influenza viruses is an important determinant of pathogenicity. Nevertheless, introduction of a multibasic cleavage site is not necessarily sufficient to immediately transform a LPAIV into a HPAIV 50, suggesting that determinants in HA other than the cleavage site might contribute to viral pathogenicity. Besides influenza virus, several other viruses, including HIV 51, Respiratory Syncytial Virus 52 and Chikungunya virus 53, misuse furin and potentially related proteases (seven pro‐protein convertases were identified in humans: furin, PC2, PC1/3, PC4, PACE4, PC5/6 and PC7) to facilitate their activation by cleavage. In addition, furin activates several bacterial toxins, including the anthrax toxin 54, 55 and Clostridium septicum α‐toxin 56, making furin and related enzymes attractive targets for therapeutic intervention 37. Identification of the pro‐protein convertases with the highest activity for the pathogen protein in question 57 and generation of specific inhibitors are therefore important tasks 58.
Figure 2.
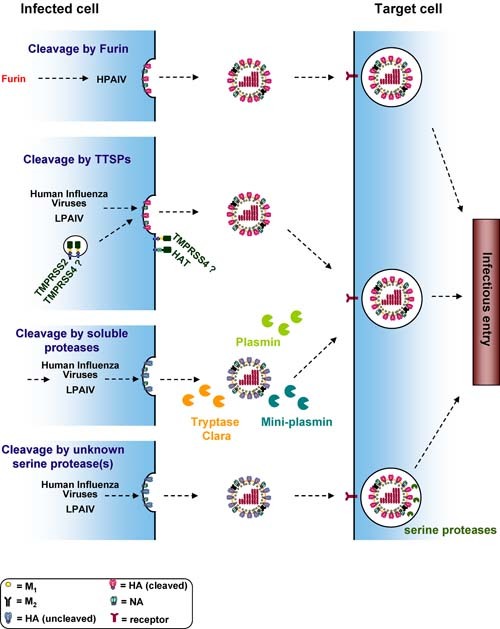
Activation of the influenza virus hemagglutinin in different cellular compartments. The influenza virus HA binds to alpha 2–3 linked (avian viruses) or alpha 2–6 linked (human viruses) sialic acids presented by proteins or lipids on the host cell surface. Upon endocytic uptake of virions, the acidic environment of the endosome triggers HA‐driven fusion of the viral and the endosomal membrane. After fusion the viral ribonucleoproteins are released into the host cell cytoplasm and transported to the nucleus, where viral genomic and messenger RNAs are synthesised. The viral membrane proteins HA, NA and M2 are synthesised in the secretory pathway. The HA proteins of HPAIV are cleaved in the Golgi apparatus/TGN by pro‐protein convertases of the subtilisin family, like furin or PC6, and cleaved HA is incorporated into progeny particles. The HA proteins of human influenza viruses and LPAIV can be cleaved by different proteases and in different cellular locations. HA can be transported to the cell surface, incorporated into virions in an uncleaved form and cleavage can subsequently be mediated by soluble proteases like plasmin in the extracellular space or by unknown serine protease(s) in endosomal vesicles of target cells. Alternatively, HA can be cleaved by the TTSPs TMPRSS2, TMPRSS4 and HAT, and cleavage could either occur en route to the cell membrane (TMPRSS2) or upon insertion into the target cell membrane (HAT). The cellular location where HA is cleaved by TMPRSS4 is at present unclear.
Soluble trypsin‐like proteases can activate influenza viruses in the extracellular space
Activation of human viruses and LPAIV is generally believed to be mediated by soluble proteases, which are secreted by lung epithelial cells. Several such enzymes have been identified, including, among others, tryptase Clara 59, mini‐plasmin 60 and ectopic anionic trypsin I 61 (Figure 2). The role of these proteases in influenza virus infection has recently been reviewed 62, and will only be briefly discussed here. For some of the soluble, HA‐activating proteases distinct expression patterns in the respiratory epithelium have been identified, suggesting that different enzymes might activate influenza viruses in different sections of the respiratory tract 62. In addition, endogenous inhibitors of these enzymes, like secretory leukoprotease inhibitor and pulmonary surfactant, have been identified and were shown to inhibit viral replication in vitro and in experimentally infected rats 62. Although these observations suggest a role of soluble cellular proteases in influenza virus spread in the infected host, direct evidence to support this hypothesis, for instance reduced viral spread in knockout animals, is largely missing.
Soluble bacterial proteases generated by Staphylococcus aureus and Aerococcus viridans strains can also activate influenza viruses 63, 64. Since bacterial superinfection by i.e. Staphylococcus aureus is a frequent complication in influenza 65, it is conceivable that bacterial proteases might promote viral spread and pathogenesis in a substantial fraction of influenza patients. Indeed, co‐infection of mice with a Staphylococcus aureus strain secreting an HA‐activating protease and an influenza virus with a monobasic cleavage site caused severe disease, while infection with bacteria or virus alone did not induce overt symptoms 64. Severe disease was also not observed when bacteria were inoculated, which did not secrete the HA‐activating protease, or when a virus was employed 64, which was not activated by the protease in question, indicating that the HA‐activation by the Staphylococcus aureus protease was the pathogenic mechanism underlying the above described observations. In summary, soluble proteases produced by both host cells and bacteria 63, 64, 66 can activate influenza virus HA. While an important contribution of the former enzymes to influenza virus spread remains to be demonstrated, experimental infection of mice indicates that the latter might contribute to pneumonia development in influenza patients with bacterial superinfection.
Activation of influenza viruses at the plasma membrane by TTSPs
Many studies assessing influenza virus HA cleavage were conducted with permanent cell lines. The establishment of culture systems for primary human lung epithelial cells allowed the field to revisit earlier findings on HA activation in a more relevant experimental model. Zhirnov and colleagues used primary human adenoid epithelial cells (HAEC) to study proteolytic activation of an influenza virus harbouring a monobasic cleavage site 67. HAEC contain differentiated cells, which exhibit ciliary motion, secrete mucins and express markers of Clara cells, and are thus considered an adequate model for the epithelium of the upper respiratory tract 67, 68. Notably, analysis in this system revealed that HA cleavage was largely cell associated, and occurred either during HA transport in the secretory pathway of productively infected cells or during entry of non‐activated viruses into uninfected cells 67. Cleavage was reduced by serine protease inhibitors 67, suggesting that the enzymes responsible for cleavage of influenza viruses in human respiratory epithelium might be cell‐associated serine protease(s).
TMPRSS2, TMPRSS4 and HAT cleave the hemagglutinin of human influenza viruses
A milestone study by Böttcher and colleagues provided evidence that the elusive proteases responsible for cell‐associated cleavage of influenza viruses with a monobasic cleavage site might be members of the TTSP family, TMPRSS2 and human airway trypsin‐like protease (HAT) 19 (Figure 2). Both proteases are expressed in the human lung, and engineered expression of either of the two enzymes in MDCK cells was sufficient to support trypsin‐independent spread of influenza viruses representing the subtypes previously pandemic in humans (H1N1, H2N2 and H3N2) 19. A subsequent study by Wang et al. confirmed cleavage‐activation of HA (subtypes H1, H3 and H5) by TMPRSS2 and HAT 69, and Chaipan et al. showed that TMPRSS2 and a related TTSP, TMPRSS4, activate the HA of the 1918 influenza virus by cleavage 20. In addition, evidence was presented that mosaic serine protease large form (MSPL) and a splice variant thereof, TMPRSS13, can cleave an influenza virus HA‐derived peptide with a particular multibasic cleavage site (M‐R‐N‐V‐P‐Q‐K‐K‐K‐R↓‐G‐L‐F‐G from A/chick/Penn/1370/83 (H5N2)), which is not efficiently recognised by furin 70. Although information on the contribution of TTSPs to influenza virus spread is missing at present, it is conceivable that these enzymes play a major role. We will therefore introduce the relevant TTSPs in more detail.
TTSPs in health and disease
The members of the TTSP family, 17 and 19 of which have been identified in humans and mice, respectively, play important roles in homeostasis and development 71. For instance, mutations in tmprss3 are associated with deafness 72 and matriptase is required for survival, epidermal barrier function, hair follicle development and thymic homeostasis in mice 73. In addition, deregulation of TTSPs is associated with several cancers 74. TTSPs are synthesised as precursor proteins, zymogens, which undergo autoproteolytic activation. Although a disulfide bond links the catalytic domain to the membrane‐inserted remainder of the molecule, release of the protease domain into the extracellular space has been described for several TTSPs, including HAT 75. TTSPs exhibit a distinct domain organisation: The N‐terminal domain of TTSPs is localized in the cytoplasm, and is followed by a hydrophobic transmembrane domain, a stem region and a protease domain, containing a catalytic AA triad composed of H, D and S, which is essential for proteolytic activity. The intracellular domain might interact with components of the cellular cytoskeleton as well as signalling molecules and might be required for correct intracellular trafficking of TTSPs, while the stem region might mediate protein–protein interactions and binding to macromolecules 76. The catalytic domain cleaves cell‐membrane receptors, growth factors, cytokines and components of the extracellular matrix 71, 74. In summary, the TTSP family comprises several enzymes which play an important role in health and disease and has therefore received increasing attention in the recent years.
Domain organisation, tissue expression and cellular localisation of TMPRSS2, TMPRSS4 and HAT
Two of the TTSPs, which cleave influenza virus HA with a monobasic cleavage site, TMPRSS2 and TMPRSS4, exhibit an identical domain organisation (Figure 3). The low‐density lipoprotein receptor domain class A (LDLA) and scavenger receptor cysteine‐rich domain (SR) present in the stem region of these enzymes are not found in the third influenza virus HA‐activating TTSP, HAT, which instead harbours a single sea urchin sperm protein (SEA) domain (Figure 3). The role of these domains in TTSP biology and HA recognition is not well understood. The protease domains of TMPRSS2, TMPRSS4 and HAT exhibit high sequence identity (43–44%), in agreement with their shared specificity for influenza virus HA. However, it is at present unclear if lack of HA cleavage by other TTSPs, for instance TMPRSS3 (S.B., I.G. and S.P., unpublished observations), is due to differences in the protease domain or in the spatial presentation of the protease domain by the stem region.
Figure 3.
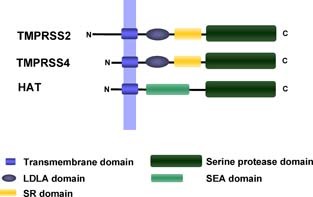
Structural organisation of the hemagglutinin‐activating enzymes TMPRSS2, TMPRSS4 and HAT.
Localisation at the cell surface has been reported for many TTSPs, including TMPRSS2 77, HAT 78 and TMPRSS4 79, suggesting that HA cleavage by these proteases occurs upon insertion of HA into the plasma membrane. However, a recent study indicates that there might be differences in the localisation of the HA cleavage event facilitated by TMPRSS2 and HAT 78. Thus, cleavage by TMPRSS2 but not HAT was resistant to certain protease inhibitors and resistance seemed to correlate with cleavage of HA inside the cell compared to cleavage at the plasma membrane 78 (Figure 2). This finding has implications for inhibitor development (since membrane permeable inhibitors will be required to block TMPRSS2 but not HAT) and for our understanding of HA cleavage in lung epithelial cells. Furthermore, the nature of the cellular compartment(s) where TTSPs mediate HA cleavage deserves further characterisation.
Expression of all known influenza virus‐activating TTSPs in lung tissue has been detected. TMPRSS2 is expressed in an epithelial‐cell‐specific fashion and expression was found in lung 80, 81. However, the nature of the TMPRSS2‐positive lung cells awaits further characterisation. Initial studies with human tracheo‐bronchial epithelium indicate expression in a subset of non‐ciliated cells 82. Expression of HAT protein was detected on ciliated cells in bronchial epithelium but was absent from basal and goblet cells 83. Messenger RNA for TMPRSS4 has been detected in a variety of tissues, but data on protein expression are largely missing 20, 79, 84. Messenger RNA for MSPL has been detected in several tissues, including lung, and TMPRSS13 mRNA seems to be ubiquitously expressed 70, 85. It remains to be determined if TMPRSS2, TMPRSS4 and HAT are co‐expressed with alpha 2–6 and alpha 2–3 linked sialic acids, respectively, and if TTSPs‐positive cells are targeted by influenza virus in the lung of infected individuals.
Elucidating the role of TTSPs in influenza virus infection
The intriguing data on influenza virus HA cleavage by TTSPs in cell culture should stimulate efforts to determine the relevance of these proteins for influenza virus spread in vivo. Expression and knock‐down studies with cultures of human respiratory epithelium should be an important part of these endeavours. It will also be interesting to determine if HA‐activating TTSPs are expressed in cell lines, which support trypsin‐independent influenza virus spread, most prominently Caco‐2 cells 86. Analysis of knockout mice should then allow the contribution of specific TTSPs to influenza virus spread in the infected host to be defined. Finally, cloning and functional analysis of the respective TTSPs from animals susceptible to influenza virus infection, particularly birds (water fowl is the natural reservoir) and swine (believed to favour reassortment due, at least in part, to susceptibility to infection by viruses with different receptor specificities) 1, should provide further insights into the importance of these enzymes for viral spread in and between species.
Hemagglutinin cleavage activation in target cell vesicles
The different modes of cleavage‐activation discussed so far involved proteolytic cleavage of HA in the secretory pathway of productively infected cells and cleavage of HA on progeny particles released into the extracellular space. Evidence from other viruses indicates that proteolytic activation can occur even after attachment of progeny virions to target cells. The envelope proteins of Ebola virus (EBOV) and SARS‐coronavirus are activated by the endosomal cysteine proteases cathepsin B and L 87, 88, and these enzymes constitute potential therapeutic targets. Cathepsins require low pH for their activity, and the previously reported pH‐dependence of EBOV and SARS‐coronavirus entry is due to the requirement for cathepsin activity rather than direct glycoprotein activation by low pH 87, 88. Notably, activation in target cell vesicles has also been described for influenza virus A/WSN/33 89 (Figure 2). This process was observed with MDBK but not CV‐1 cells and was sensitive to serine‐ but not cysteine protease inhibitors 89, suggesting that influenza virus activation in target cells might occur in a cell‐type dependent fashion and is not facilitated by cathepsins. The protease(s) responsible for influenza virus activation in MDBK cells remains to be identified. However, the above described observation by Zhirnov and colleagues that HA activation can occur upon influenza virus uptake into cultures of respiratory epithelium 67 suggests that this mode of HA cleavage might be important in the infected host and thus deserves further investigation.
NEURAMINIDASE PROMOTED CLEAVAGE OF THE HEMAGGLUTININ OF A/WSN/33 AND THE 1918 INFLUENZA VIRUS
Despite the ample possibilities for HA cleavage discussed above, two prominent influenza viruses evolved additional mechanisms, which depend on the presence of the viral NA. One is the frequently studied, neurovirulent 90 laboratory strain A/WSN/33 (H1N1), which was obtained by extensive passaging of the parental virus, WS/33, in different animals. The spread of WSN/33 in infected mice is not limited to brain and lung tissue but is rather systemic 91, suggesting that the virus evolved a mechanism to ensure efficient HA cleavage in a broad spectrum of cell types. Studies dating back to the 1970s demonstrated that (i) WSN/33 was activated by the serum protease plasmin 92, (ii) NA was intimately involved in HA‐cleavage 93 and that (iii) NA 90, specifically adequate glycosylation of NA 94, was required for neurovirulence in mice. Work by Goto and Kawaoka linked these observations by demonstrating that WSN/33 NA recruits serum plasminogen, which upon conversion to plasmin, activates HA 95, 96, 97. One of several amino acid exchanges present in WSN/33 NA but not the parental WS/33 NA was found to be crucial for HA activation: Mutation N146R (N2 numbering), which inactivated a signal for N‐linked glycosylation, conferred plasminogen binding to WS/33 while the reverse exchange abrogated plasminogen recruitment by WSN/33. In addition, the carboxy‐terminal lysine residue 453, which is conserved among NAs of the N1 subtype, was essential for plasminogen recruitment 95, 96, 97, suggesting that certain influenza virus NAs might have the intrinsic capacity to bind plasminogen, which is masked by posttranslational modification of N146 by a glycan side chain.
The recruitment of plasminogen by WSN/33 NA and the resulting expansion of the viral tropism raised the intriguing question, whether a similar mechanism might contribute to spread of other influenza viruses, most importantly the highly pathogenic 1918 influenza virus. Analysis of the 1918 NA revealed the presence of the lysine residue crucial for plasminogen binding but also showed the presence of the glycosylation signal incompatible with plasminogen recruitment by WSN/33 98. Notably, the 1918 virus replicated in the dog kidney cell line MDCK with high efficiency in the absence of trypsin, and trypsin‐independent spread was dependent on the presence of the 1918 NA 99. These results suggest that the 1918 NA also facilitates HA cleavage, at least in some cellular systems (MDCK cells), and might do so by recruiting a protease. A subsequent study demonstrated that WSN/33 NA but not 1918 NA was able to sequester plasminogen and to activate WSN/33 HA 20. Conversely, and unexpectedly, the 1918 HA but not the WSN/33 HA bound plasminogen and the biological significance of the plasminogen capture is at present unclear 20. These results argue against 1918 NA facilitating HA cleavage in a manner directly comparable to WSN/33 NA. It can be speculated, however, that 1918 NA might recruit a factor which promotes plasminogen conversion into plasmin, like annexin II, which was shown to be incorporated into influenza virus particles and to support viral replication by activating plasminogen 100. Regardless of the, at present unclear, mechanism underlying NA‐dependent 1918 HA cleavage, it needs to be noted that this process was not detected in the human hepatoma cell line Huh‐7 or the human kidney‐derived cell line 293T 20, and is thus of uncertain relevance to 1918 virus spread in the human host. Finally, it is noteworthy that the 1918 influenza virus and a seasonal influenza virus replicated in the human lung cell line Calu‐3 with high efficiency in the absence of trypsin 99. Cleavage of the HA proteins by TTSPs, which does not depend on NA, might have been responsible. Expression analysis and knock‐down studies are needed to test this hypothesis.
PROTEOLYTIC CLEAVAGE OF HEMAGGLUTININ AS A TARGET FOR THERAPEUTIC INTERVENTION
As to be expected for a highly variable RNA virus, usage of NA and M2 inhibitors as monotherapy for influenza triggers emergence of resistant viruses, and a substantial proportion of the currently circulating viruses are insensitive against at least one or more of the drugs available. The potential of combination therapy should therefore be explored, and the development of new drugs is imperative. The cellular proteases which activate HA are attractive targets for several reasons. For one, they are essential for viral spread and, as host cell‐encoded factors, of invariable nature. As a consequence of the latter property, viral resistance due to mutation of the drug target can be excluded. The alteration of the cleavage site in response to protease inhibition is conceivable. However, it is important to note that evolution of seasonal influenza viruses with a multibasic cleavage site, which would confer resistance to inhibitors directed against proteases recognising monobasic cleavage sites, has never been observed in humans. Another advantage of protease inhibitors is the mere fact that ample proof of concept is already available: protease inhibitors have been documented to suppress influenza virus spread in cell culture 78, in experimentally infected animals 101, 102 and, most importantly, in humans 102. Major unwanted side effects were not observed in these studies but are of concern when considering clinical development of protease inhibitors for influenza therapy. However, identifying which of the so far described HA‐processing proteases indeed support(s) spread of human influenza virus in infected individuals would allow the generation of highly specific inhibitors, which can be expected to be both safe and effective. The discovery of TTSPs as HA‐activating enzymes combined with the observation that knockout of TMPRSS2 has no phenotype in mice 103, might be the first step in this direction.
CONCLUSIONS
The cleavage of HA by cellular proteases is essential for virus spread and pathogenesis, so the responsible proteases are attractive targets for intervention. For avian influenza viruses, a correlation between the cleavage site sequence, the nature of the HA‐processing proteases and the degree of viral pathogenicity has been firmly established. For human influenza viruses, a similar correlation does not exist and the paradigm on HA cleavage shifted from cleavage by soluble proteases to cleavage by cell‐associated proteases. Recent studies suggests that members of the TTSP family, specifically TMPRSS2, TMPRSS4 and HAT, mediate cell‐associated HA‐cleavage, and future studies must define the importance of the respective enzymes. Protease inhibitors are effective as treatment for influenza 102, and TTSPs might emerge as prime targets. Finally, inhibitors of TTSPs might be of therapeutic value for conditions others than influenza, since recent evidence suggests that TTSPs are also involved in the proteolytic activation of human metapneumovirus 104 and SARS‐coronavirus 105, and an important role of TTSPs in several cancers is well documented 71, 74.
Abbreviations used
- EBOV
Ebola virus
- FP
fusion peptide
- HA
hemagglutinin
- HAECs
human adenoid epithelial cells
- HAT
human airway trypsin‐like protease
- HPAIV
highly pathogenic avian influenza viruses
- LDLA
low‐density lipoprotein receptor domain class A
- LPAIV
low pathogenic avian influenza viruses
- MSPL
mosaic serine protease large form
- NA
neuraminidase
- SEA
single sea urchin sperm protein
- SR
scavenger receptor cysteine‐rich domain
- TGN
trans‐Golgi network
- TTSPs
type II transmembrane serine proteases
Acknowledgements
The authors thank T.F. Schulz for support and BMBF (01KI 0703 to I.G., S.B. and S.P) for funding.
REFERENCES
- 1. Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine‐origin H1N1 influenza virus. Nature 2009; 459: 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palese P. Influenza: old and new threats. Nat Med 2004; 10: S82–S87. [DOI] [PubMed] [Google Scholar]
- 3. Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. J Am Med Assoc 2003; 289: 179–186. [DOI] [PubMed] [Google Scholar]
- 4. Belshe RB. The origins of pandemic influenza–lessons from the 1918 virus. N Engl J Med 2005; 353: 2209–2211. [DOI] [PubMed] [Google Scholar]
- 5. Parrish CR, Kawaoka Y. The origins of new pandemic viruses: the acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu Rev Microbiol 2005; 59: 553–586. [DOI] [PubMed] [Google Scholar]
- 6. Morens DM, Taubenberger JK. Understanding influenza backward. J Am Med Assoc 2009; 302: 679–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 2007; 195: 1018–1028. [DOI] [PubMed] [Google Scholar]
- 8. Dawood FS, Jain S, Finelli L, et al. Emergence of a novel swine‐origin influenza A (H1N1) virus in humans. N Engl J Med 2009; 360: 2605–2615. [DOI] [PubMed] [Google Scholar]
- 9. Fraser C, Donnelly CA, Cauchemez S, et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 2009; 324: 1557–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. http://www.who.int/csr/disease/swineflu/laboratory18_12_2009/en/index.html WHO, update 79, accessed on 21 Dec 2009.
- 11. Itoh Y, Shinya K, Kiso M, et al. In vitro and in vivo characterization of new swine‐origin H1N1 influenza viruses. Nature 2009; 460: 1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Munster VJ, de Wit E, van den Brand JM, et al. Pathogenesis and transmission of swine‐origin 2009 A(H1N1) influenza virus in ferrets. Science 2009; 325: 481–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellebedy AH, Webby RJ. Influenza vaccines. Vaccine 2009; 27 (Suppl 4): D65–D68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beigel J, Bray M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res 2008; 78: 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. von Itzstein M, Thomson R. Anti‐influenza drugs: the development of sialidase inhibitors. Handb Exp Pharmacol 2009; 111–154. [DOI] [PubMed] [Google Scholar]
- 16. Basler CF. Influenza viruses: basic biology and potential drug targets. Infect Disord Drug Target 2007; 7: 282–293. [DOI] [PubMed] [Google Scholar]
- 17. Poland GA, Jacobson RM, Ovsyannikova IG. Influenza virus resistance to antiviral agents: a plea for rational use. Clin Infect Dis 2009; 48: 1254–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Steinhauer DA. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 1999; 258: 1–20. [DOI] [PubMed] [Google Scholar]
- 19. Böttcher E, Matrosovich T, Beyerle M, et al. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 2006; 80: 9896–9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaipan C, Kobasa D, Bertram S, et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol 2009; 83: 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 2000; 69: 531–569. [DOI] [PubMed] [Google Scholar]
- 22. Harrison SC. Viral membrane fusion. Nat Struct Mol Biol 2008; 15: 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microb Infect 2004; 6: 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skehel JJ, Bayley PM, Brown EB, et al. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus‐mediated membrane fusion. Proc Natl Acad Sci USA 1982; 79: 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Copeland CS, Zimmer KP, Wagner KR, et al. Folding, trimerization, and transport are sequential events in the biogenesis of influenza virus hemagglutinin. Cell 1988; 53: 197–209. [DOI] [PubMed] [Google Scholar]
- 26. Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 1995; 81: 425–433. [DOI] [PubMed] [Google Scholar]
- 27. Daniels RS, Downie JC, Hay AJ, et al. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 1985; 40: 431–439. [DOI] [PubMed] [Google Scholar]
- 28. Garten W, Klenk HD. Understanding influenza virus pathogenicity. Trend Microbiol 1999; 7: 99–100. [DOI] [PubMed] [Google Scholar]
- 29. Klenk HD, Garten W. Host cell proteases controlling virus pathogenicity. Trend Microbiol 1994; 2: 39–43. [DOI] [PubMed] [Google Scholar]
- 30. Garten W, Klenk HD. Characterization of the carboxypeptidase involved in the proteolytic cleavage of the influenza haemagglutinin. J Gen Virol 1983; 64: 2127–2137. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Lee KH, Steinhauer DA, et al. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998; 95: 409–417. [DOI] [PubMed] [Google Scholar]
- 32. Reed ML, Yen HL, DuBois RM, et al. Amino acid residues in the fusion peptide pocket regulate the pH of activation of the H5N1 influenza virus hemagglutinin protein. J Virol 2009; 83: 3568–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bosch FX, Garten W, Klenk HD, et al. Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of Avian influenza viruses. Virology 1981; 113: 725–735. [DOI] [PubMed] [Google Scholar]
- 34. Perdue ML, Garcia M, Senne D, et al. Virulence‐associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res 1997; 49: 173–186. [DOI] [PubMed] [Google Scholar]
- 35. Webster RG, Rott R. Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell 1987; 50: 665–666. [DOI] [PubMed] [Google Scholar]
- 36. Stieneke‐Grober A, Vey M, Angliker H, et al. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin‐like endoprotease. EMBO J 1992; 11: 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 2002; 3: 753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ohuchi M, Orlich M, Ohuchi R, et al. Mutations at the cleavage site of the hemagglutinin after the pathogenicity of influenza virus A/chick/Penn/83 (H5N2). Virology 1989; 168: 274–280. [DOI] [PubMed] [Google Scholar]
- 39. Ohuchi R, Ohuchi M, Garten W, et al. Human influenza virus hemagglutinin with high sensitivity to proteolytic activation. J Virol 1991; 65: 3530–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawaoka Y, Naeve CW, Webster RG. Is virulence of H5N2 influenza viruses in chickens associated with loss of carbohydrate from the hemagglutinin? Virology 1984; 139: 303–316. [DOI] [PubMed] [Google Scholar]
- 41. Deshpande KL, Fried VA, Ando M, et al. Glycosylation affects cleavage of an H5N2 influenza virus hemagglutinin and regulates virulence. Proc Natl Acad Sci USA 1987; 84: 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reid AH, Fanning TG, Hultin JV, et al. Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci USA 1999; 96: 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klenk HD, Rott R, Orlich M, et al. Activation of influenza A viruses by trypsin treatment. Virology 1975; 68: 426–439. [DOI] [PubMed] [Google Scholar]
- 44. Klenk HD, Rott R, Orlich M. Further studies on the activation of influenza virus by proteolytic cleavage of the haemagglutinin. J Gen Virol 1977; 36: 151–161. [DOI] [PubMed] [Google Scholar]
- 45. Lazarowitz SG, Choppin PW. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 1975; 68: 440–454. [DOI] [PubMed] [Google Scholar]
- 46. Vey M, Orlich M, Adler S, et al. Hemagglutinin activation of pathogenic avian influenza viruses of serotype H7 requires the protease recognition motif R‐X‐K/R‐R. Virology 1992; 188: 408–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Curtis I, Simons K. Isolation of exocytic carrier vesicles from BHK cells. Cell 1989; 58: 719–727. [DOI] [PubMed] [Google Scholar]
- 48. Ito T, Goto H, Yamamoto E, et al. Generation of a highly pathogenic avian influenza A virus from an avirulent field isolate by passaging in chickens. J Virol 2001; 75: 4439–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horimoto T, Kawaoka Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J Virol 1994; 68: 3120–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stech O, Veits J, Weber S, et al. Acquisition of a polybasic hemagglutinin cleavage site by a low‐pathogenic avian influenza virus is not sufficient for immediate transformation into a highly pathogenic strain. J Virol 2009; 83: 5864–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hallenberger S, Bosch V, Angliker H, et al. Inhibition of furin‐mediated cleavage activation of HIV‐1 glycoprotein gp160. Nature 1992; 360: 358–361. [DOI] [PubMed] [Google Scholar]
- 52. Bolt G, Pedersen LO, Birkeslund HH. Cleavage of the respiratory syncytial virus fusion protein is required for its surface expression: role of furin. Virus Res 2000; 68: 25–33. [DOI] [PubMed] [Google Scholar]
- 53. Ozden S, Lucas‐Hourani M, Ceccaldi PE, et al. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J Biol Chem 2008; 283 21899–21908. [DOI] [PubMed] [Google Scholar]
- 54. Klimpel KR, Molloy SS, Thomas G, et al. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci USA 1992; 89: 10277–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Molloy SS, Bresnahan PA, Leppla SH, et al. Human furin is a calcium‐dependent serine endoprotease that recognizes the sequence Arg‐X‐X‐Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem 1992; 267: 16396–16402. [PubMed] [Google Scholar]
- 56. Gordon VM, Benz R, Fujii K, et al. Clostridium septicum alpha‐toxin is proteolytically activated by furin. Infect Immun 1997; 65: 4130–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Remacle AG, Shiryaev SA, Oh ES, et al. Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. J Biol Chem 2008; 283: 20897–20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shiryaev SA, Remacle AG, Ratnikov BI, et al. Targeting host cell furin proprotein convertases as a therapeutic strategy against bacterial toxins and viral pathogens. J Biol Chem 2007; 282: 20847–20853. [DOI] [PubMed] [Google Scholar]
- 59. Kido H, Yokogoshi Y, Sakai K, et al. Isolation and characterization of a novel trypsin‐like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem 1992; 267: 13573–13579. [PubMed] [Google Scholar]
- 60. Murakami M, Towatari T, Ohuchi M, et al. Mini‐plasmin found in the epithelial cells of bronchioles triggers infection by broad‐spectrum influenza A viruses and Sendai virus. Eur J Biochem 2001; 268: 2847–2855. [DOI] [PubMed] [Google Scholar]
- 61. Towatari T, Ide M, Ohba K, et al. Identification of ectopic anionic trypsin I in rat lungs potentiating pneumotropic virus infectivity and increased enzyme level after virus infection. Eur J Biochem 2002; 269: 2613–2621. [DOI] [PubMed] [Google Scholar]
- 62. Kido H, Okumura Y, Yamada H, et al. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharm Des 2007; 13: 405–414. [DOI] [PubMed] [Google Scholar]
- 63. Tashiro M, Ciborowski P, Reinacher M, et al. Synergistic role of staphylococcal proteases in the induction of influenza virus pathogenicity. Virology 1987; 157: 421–430. [DOI] [PubMed] [Google Scholar]
- 64. Tashiro M, Ciborowski P, Klenk HD, et al. Role of Staphylococcus protease in the development of influenza pneumonia. Nature 1987; 325: 536–537. [DOI] [PubMed] [Google Scholar]
- 65. Kuiken T, Taubenberger JK. Pathology of human influenza revisited. Vaccine 2008; 26 (Suppl 4): D59–D66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Scheiblauer H, Reinacher M, Tashiro M, et al. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis 1992; 166: 783–791. [DOI] [PubMed] [Google Scholar]
- 67. Zhirnov OP, Ikizler MR, Wright PF. Cleavage of influenza a virus hemagglutinin in human respiratory epithelium is cell associated and sensitive to exogenous antiproteases. J Virol 2002; 76: 8682–8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Endo Y, Carroll KN, Ikizler MR, et al. Growth of influenza A virus in primary, differentiated epithelial cells derived from adenoids. J Virol 1996; 70: 2055–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang W, Butler EN, Veguilla V, et al. Establishment of retroviral pseudotypes with influenza hemagglutinins from H1, H3, and H5 subtypes for sensitive and specific detection of neutralizing antibodies. J Virol Method 2008; 153: 111–119. [DOI] [PubMed] [Google Scholar]
- 70. Kido H, Okumura Y, Takahashi A, et al. Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J Mol Genet Med 2008; 3: 167–175. [PMC free article] [PubMed] [Google Scholar]
- 71. Choi SY, Bertram S, Glowacka I, et al. Type II transmembrane serine proteases in cancer and viral infections. Trends Mol Med 2009; 15: 303–312. [DOI] [PubMed] [Google Scholar]
- 72. Scott HS, Kudoh J, Wattenhofer M, et al. Insertion of beta‐satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet 2001; 27: 59–63. [DOI] [PubMed] [Google Scholar]
- 73. List K, Haudenschild CC, Szabo R, et al. Matriptase/MT‐SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 2002; 21: 3765–3779. [DOI] [PubMed] [Google Scholar]
- 74. Szabo R, Bugge TH. Type II transmembrane serine proteases in development and disease. Int J Biochem Cell Biol 2008; 40: 1297–1316. [DOI] [PubMed] [Google Scholar]
- 75. Yasuoka S, Ohnishi T, Kawano S, et al. Purification, characterization, and localization of a novel trypsin‐like protease found in the human airway. Am J Respir Cell Mol Biol 1997; 16: 300–308. [DOI] [PubMed] [Google Scholar]
- 76. Hooper JD, Clements JA, Quigley JP, et al. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J Biol Chem 2001; 276: 857–860. [DOI] [PubMed] [Google Scholar]
- 77. Jacquinet E, Rao NV, Rao GV, et al. Cloning, genomic organization, chromosomal assignment and expression of a novel mosaic serine proteinase: epitheliasin. FEBS Lett 2000; 468: 93–100. [DOI] [PubMed] [Google Scholar]
- 78. Böttcher E, Freuer C, Steinmetzer T, et al. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 2009; 27: 6324–6329. [DOI] [PubMed] [Google Scholar]
- 79. Wallrapp C, Hahnel S, Muller‐Pillasch F, et al. A novel transmembrane serine protease (TMPRSS3) overexpressed in pancreatic cancer. Cancer Res 2000; 60: 2602–2606. [PubMed] [Google Scholar]
- 80. Jacquinet E, Rao NV, Rao GV, et al. Cloning and characterization of the cDNA and gene for human epitheliasin. Eur J Biochem 2001; 268: 2687–2699. [DOI] [PubMed] [Google Scholar]
- 81. Lucas JM, True L, Hawley S, et al. The androgen‐regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J Pathol 2008; 215: 118–125. [DOI] [PubMed] [Google Scholar]
- 82. Garten W, Matrosovich M, Matrosovich T, et al. Cleavage of influenza virus hemagglutinin by host cell proteases. International Congress Series 2004; 1263: 218–221. [Google Scholar]
- 83. Takahashi M, Sano T, Yamaoka K, et al. Localization of human airway trypsin‐like protease in the airway: an immunohistochemical study. Histochem Cell Biol 2001; 115: 181–187. [DOI] [PubMed] [Google Scholar]
- 84. Jung H, Lee KP, Park SJ, et al. TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial‐mesenchymal transition. Oncogene 2008; 27: 2635–2647. [DOI] [PubMed] [Google Scholar]
- 85. Kim DR, Sharmin S, Inoue M, et al. Cloning and expression of novel mosaic serine proteases with and without a transmembrane domain from human lung. Biochim Biophys Acta 2001; 1518: 204–209. [DOI] [PubMed] [Google Scholar]
- 86. Zhirnov O, Klenk HD. Human influenza A viruses are proteolytically activated and do not induce apoptosis in CACO‐2 cells. Virology 2003; 313: 198–212. [DOI] [PubMed] [Google Scholar]
- 87. Chandran K, Sullivan NJ, Felbor U, et al. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005; 308: 1643–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Simmons G, Gosalia DN, Rennekamp AJ, et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA 2005; 102: 11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Boycott R, Klenk HD, Ohuchi M. Cell tropism of influenza virus mediated by hemagglutinin activation at the stage of virus entry. Virology 1994; 203: 313–319. [DOI] [PubMed] [Google Scholar]
- 90. Sugiura A, Ueda M. Neurovirulence of influenza virus in mice. I. Neurovirulence of recombinants between virulent and avirulent virus strains. Virology 1980; 101: 440–449. [DOI] [PubMed] [Google Scholar]
- 91. Castrucci MR, Kawaoka Y. Biologic importance of neuraminidase stalk length in influenza A virus. J Virol 1993; 67: 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lazarowitz SG, Goldberg AR, Choppin PW. Proteolytic cleavage by plasmin of the HA polypeptide of influenza virus: host cell activation of serum plasminogen. Virology 1973; 56: 172–180. [DOI] [PubMed] [Google Scholar]
- 93. Schulman JL, Palese P. Virulence factors of influenza A viruses: WSN virus neuraminidase required for plaque production in MDBK cells. J Virol 1977; 24: 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Li S, Schulman J, Itamura S, et al. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J Virol 1993; 67: 6667–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Goto H, Kawaoka Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci USA 1998; 95: 10224–10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Goto H, Wells K, Takada A, et al. Plasminogen‐binding activity of neuraminidase determines the pathogenicity of influenza A virus. J Virol 2001; 75: 9297–9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Taubenberger JK. Influenza virus hemagglutinin cleavage into HA1, HA2: no laughing matter. Proc Natl Acad Sci USA 1998; 95: 9713–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Reid AH, Fanning TG, Janczewski TA, et al. Characterization of the 1918 “Spanish” influenza virus neuraminidase gene. Proc Natl Acad Sci USA 2000; 97: 6785–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tumpey TM, Basler CF, Aguilar PV, et al. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 2005; 310: 77–80. [DOI] [PubMed] [Google Scholar]
- 100. Lebouder F, Morello E, Rimmelzwaan GF, et al. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J Virol 2008; 82: 6820–6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ovcharenko AV, Zhirnov OP. Aprotinin aerosol treatment of influenza and paramyxovirus bronchopneumonia of mice. Antiviral Res 1994; 23: 107–118. [DOI] [PubMed] [Google Scholar]
- 102. Zhirnov OP, Ovcharenko AV, Bukrinskaia AG, et al. Antiviral and therapeutic action of protease inhibitors in viral infections: experimental and clinical observations. Vopr Virusol 1984; 29: 491–497. [PubMed] [Google Scholar]
- 103. Kim TS, Heinlein C, Hackman RC, et al. Phenotypic analysis of mice lacking the Tmprss2‐encoded protease. Mol Cell Biol 2006; 26: 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shirogane Y, Takeda M, Iwasaki M, et al. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J Virol 2008; 82: 8942–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kam YW, Okumura Y, Kido H, et al. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One 2009; 4: e7870. [DOI] [PMC free article] [PubMed] [Google Scholar]