

Summary
Transforming growth factor‐β (TGF‐β) signaling pathway is a key network in cell signaling that controls vital processes such as proliferation, differentiation, apoptosis, epithelial‐mesenchymal transition, and migration, thus acting as a double‐edged sword in normal development and diseases, in particular organ fibrosis, vascular disorders, and cancer. Early in tumorigenesis, the pathway exerts anti‐tumor effects through suppressing cell cycle and inducing apoptosis, while during late stages, it functions as a tumor promoter by enhancing tumor invasiveness and metastasis. This signaling pathway can be perturbed by environmental and genetic factors such as microbial interference and mutation, respectively. In this way, the present review describes the modulation of the TGF‐β pathway by oncogenic human viral pathogens and other viruses. The main mechanisms by which viruses interferes with TGF‐β signaling seems to be through (1) the alteration of either TGF‐β protein expression or activation, (2) the modulation of the TGF‐β receptors or SMADs factors (by interfering with their levels and functions), (3) the alteration of none‐SMAD pathways, and (4) indirect interaction with the pathway by the modulation of transcriptional co‐activator/repressor and regulators of the pathway. Given the axial role of this pathway in tumorigenesis, it can be regarded as an attractive target for cancer therapy. Hence, further investigations on this subject may represent molecular targets among either TGF‐β signaling molecules or viral factors for the treatment and management of viral infection consequences such as cancer.
Keywords: cancer, TGF‐β, viruses
ABBREVIATIONS
- EMT
Epithelial‐to‐mesenchymal transition
- MCV
Merkel cell polyomavirus
- TGF‐β
Transforming growth factor‐β
- TGFbRI
TGF‐β receptor I
- LOH
Allelic loss of heterozygosity
- R‐SMADs
Receptor‐regulated SMADs
- I‐SMADs
Inhibitory SMADs
- JNK
Jun N‐terminal kinase
- HCC
Hepatocellular carcinoma
- CLD
Chronic liver disease
- THBS
Thrombospondin
- GRP94
Glucose‐regulated protein 94
- TβRE
TGF‐β‐responsive element
- ECM
Extracellular matrix
- pSMAD3L
Linker‐phosphorylated SMAD3
- PPM1a
Protein phosphatase magnesium dependent 1A
- LMP
Latent membrane protein
- EBNA
EBV nuclear antigens
- KS
Kaposi's sarcoma
- MCD
Multicentric Castleman's disease
- PEL
Primary effusion lymphoma
- Vflip
Viral Fas‐associated death domain IL‐1β‐converting enzyme inhibitory protein
- vCyc
Viral cyclin
- LANA
Latency‐associated nuclear antigen
- ATL
Adult T‐cell leukemia
- HBZ
HTLV‐1 bZIP factor
- SBE
SMAD binding elements
- MMP‐2
Matrix metalloprotease 2
- EVT
Extravillous cytotrophoblast
- HCE
Human corneal epithelial cells
- SARS‐CoV
Severe acute respiratory syndrome‐associated coronavirus
- Plpro
Papain‐like protease
- PAI‐1
Plasminogen activator inhibitor‐1
- TLR‐3
Toll like receptor 3
- AV
Adenovirus
- IFP
Idiopathic pulmonary fibrosis
- Huh‐7
Human hepatocellular carcinoma cells
- EBOV
Ebola virus
- Treg
Regulatory T cells
1. INTRODUCTION
With approximately a million cancer‐caused deaths per year, oncogenic viruses are a major cause of cancer‐related mortality; indeed, approximately 10% to 18% of human malignancies are linked to viruses, globally.1, 2 To date, 7 human viruses, namely, HCV, HBV, HPV, EBV, HTLV‐1, Kaposi's sarcoma‐associated herpes virus (KSHV), and Merkel cell polyomavirus (MCV) have been etiologically involved in the development of human cancers.3 In addition, there are other viruses implicated in human carcinogenesis whose causal roles have yet to be fully confirmed.4 To prevent and manage viral‐mediated cancers appropriately, the molecular events underlying the interactions between both oncogenic viruses and cells should be clearly understood. Transforming growth factor (TGF)‐β signaling pathway is a key signaling network, with a diverse range of pathophysiological activities, playing essential roles in processes such as cell proliferation and differentiation, apoptosis, inflammation, angiogenesis, epithelial‐to‐mesenchymal transition (EMT), and tumorigenesis.5 Accordingly, it is not surprising that its loss of regulation can contribute to a broad range of pathologies such as cancer.6, 7 In this regard, the present review focuses on describing the modulation of this pivotal pathway by either tumor‐caused or tumor‐associated viruses. It first outlines the key aspects of the TGF‐β pathway, before focusing on the existing literature on the subject of viral interference with TGF‐β signaling.
2. THE TGF‐β SIGNALING PATHWAY
TGF‐β signaling plays vital roles in both biological processes and diseases. Regarding tumorigenesis, it exhibits a dual role by demonstrating anti‐tumor effects (through inhibiting the proliferation and inducing apoptosis) and pro‐oncogenic activities (via inducing EMT and tumor metastasis) during early and late stages of oncogenesis, respectively.6, 8 This pathway mediates its functions through TGF‐β cytokines whose binding to type II and type I serine‐threonine kinase receptors (known as TGFbRI and TGFbRII, respectively) results in the activation of 2 different downstream pathways; SMAD‐dependent and SMAD‐independent pathways.9 Indeed, the effects of TGF‐β are often controlled by 3 TGFβ ligands, TGF‐β1, TGF‐β2, and TGF‐β3, secreted as latent protein complexes, required to be activated by proteolytic cleavage before binding to receptors.9 Activated TGF‐β interacts with and activates TGFbRII, leading to the phosphorylation of TGFbRI, whose activation results in signal transduction via recruiting and phosphorylating receptor‐regulated SMADs (R‐SMAD), SMAD2 and SMAD3, which subsequently interact with Co‐SMADs (eg, SMAD4) to form the activated SMADs complex, that thereafter, translocate into the nucleus, wherein it exerts its regulatory effects on the expression of target genes, by recruiting other co‐activator or co‐repressor factors for transcription (eg, p300, CBP) (Figure 1).10, 11 Furthermore, TGFbRIII is another TGF‐β receptor, playing a coreceptor role for TGFbRII.12 SMAD‐dependent cascade, as the major TGF‐β signaling mediator, conveys signals from TGFbRI to the nucleus in a linear pathway, a process blocked by inhibitory SMADs (I‐SMADs); for instance, SMAD7 represses this pathway either via interacting with activated TGFbRI, and hence, preventing R‐SMAD activation or through recruiting SMURF2, a ubiquitin‐ligase, which promotes SMAD2 and TGFbRI decrease and downmodulates SMAD3 function.10, 13, 14 Perturbations of this cascade can lead to tumorigenesis and tumor promotion via either direct or indirect effects on key cellular process.15 For example, SMAD4 and TGFbRII were shown to be often disrupted by mechanisms such as allelic loss of heterozygosity (LOH) and mutation in multiple carcinomas.16 Similarly, disturbed SMAD4 has also been observed in a number of malignancies, representing a tumor suppressor role for this pathway.17 In addition to the SMADs cascade, which plays the central role,10 TGF‐β can also interact with other signaling cascades, those mediated independently of the SMAD factors, such as p38 MAP kinases, c‐Jun N‐terminal kinase (JNK), Ras‐ERK, PI3K‐Akt, and small GTPases (eg, RhoA), in a cell type‐specific manner.18, 19, 20
Figure 1.
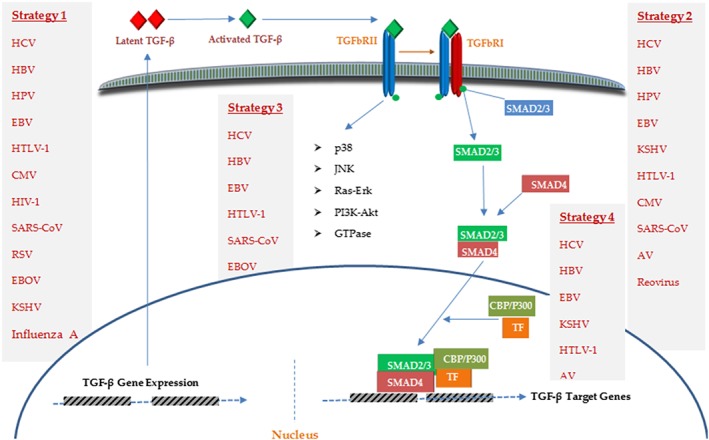
An overview of the TGF‐β signaling pathway
3. TUMOR‐CAUSED VIRUSES AND TGF‐β SIGNALING
3.1. Hepatitis C virus
HCV is a member of the Flaviviridae family with a single stranded RNA, acting as a major cause of liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). In patients with chronic HCV infection, chronic liver disease (CLD) occurs in 50% of the cases leading to 5% to 20% cirrhosis, of which, 1% to 2% result in HCC, a pathology causing for approximately 600 000 deaths per year.21 Dysregulation of TGF‐β signaling has been implicated in the pathogenesis of all stages of the liver diseases,22 a cytokine whose upregulated level has been reported both in liver tissue and serum of patients with chronic HCV infection and HCC.23 Recently, Jee found high levels of expressing TGF‐β in hepatocytes of HCV patients, as well as, in HCV‐infected hepatocytes cultured in vitro, as that of cultured cells was sufficient to activate liver fibrosis‐associated cells, hepatic stellate cells (HSC), a new mechanism underlying liver fibrogenesis.24 Accordingly, the HCV E2 protein was reported as cause of this overproduction, by acting through glucose‐regulated protein 94 (GRP94) mediated NF‐κB activation, which proposed GRP94 as a potential target for preventing HCV‐caused liver cirrhosis. Relevant in this regard, other investigations have shown an upregulated level of TGF‐β either directly by HCV factors, in particular core protein, or via NF‐κB and oxidative/ER stress activation.25, 26, 27, 28, 29 Taniguchi indicated TGF‐β upmodulation at the transcriptional levels by HCV core protein.28 Furthermore, HCV core protein also increases active TGF‐β levels by thrombospondin (THBS), an anti‐angiogenic factor whose function is mediated in part through activating the latent form of TGF‐β, and thus SMAD2/3 phosphorylation, indicating TGF‐β signaling activation.29 Similarly, HCV increases ROS production, which in turn activates the p38 MAPK, JNK, and ERK pathways, leading to NFκB activation that induces TGF‐β production.30 Conversely, HCV NS5A protein was able to block a pivotal transcriptional activator for TGF‐β gene expression, known as AP‐1, through interrupting the Ras‐ERK pathway.24 In addition to overexpression or activation of TGF‐β cytokine, HCV also interferes with downstream TGF‐β signaling components and mediators.31, 32, 33 TGF‐β arrests cell cycle promotion by upmodulation of inhibitory factors such as p21, a key role whose inhibition exerts a fundamental effect on tumor progression.34 Recently, Choi showed HCV NS5A protein acts as a negative modulator, where it interacts with TGFbRI, resulting in inhibition of SMAD2 phosphorylation and SMADs complex formation and then in downmodulation of p21 expression.31 Furthermore, HCV core protein suppresses TGF‐β–induced p21 overexpression in a transcriptional‐dependent manner. In fact, HCV core protein functions through the TGF‐β‐responsive element (TβRE) positioned in the p21 promoter region to repress the p21 promoter by suppression of TGF‐β pathway; a mechanism by which HCV results in cell growth induction.32 In the same way, Cheng showed 2 physical interactions between the HCV core and NS3 proteins with SMAD3 factor, which was shown to suppress the SMAD3‐mediated transcriptional activation of target genes via reducing the SMAD3 binding ability to the SMAD binding elements (SBE) located in the promoter regions.33 Pavio by assessing core variants isolated from both tumoral and non‐tumoral tissues of the same HCC patient reported in contrast to tumor derived variants, which block TGF‐β signaling, no such effects were exerted by non‐tumoral ones.35 Additionally, their findings demonstrated a direct interaction between core and SMAD3, which in turn inhibited the DNA‐binding activity of SMAD3. Hence, it could be concluded that during chronic infection, those viral variants would be selected that provoke cell transformation via enhancing resistance to anti‐proliferative activities of TGF‐β. Battaglia similarly found HCC‐derived HCV core proteins are capable of shifting TGF‐β responses from anti‐tumor to pro‐tumor effects via reducing SMAD3 signaling.36 This duality of function of TGF‐β in tumorigenesis was also further observed in another study, where a TGFbRI‐independent SMAD3 activation by TGF‐β resulted in a shift of TGF‐β activity from tumor‐suppressor to fibrogenic in HCV‐chronically infected patients; in these patients, TGF‐β activated c‐JNK, which in turn phosphorylated SMAD3 into linker‐phosphorylated SMAD3 (pSMAD3L) and led to extracellular matrix (ECM) deposition via upmodulating plasminogen activator inhibitor 1 (PAI‐1), a process, which increased during liver disease course from chronic hepatitis through cirrhosis to HCC.37 Interestingly, HCV may be able to indirectly block TGF‐β mediated‐apoptosis via inducing the PI3K‐Akt survival pathway. This PI3K‐Akt‐mediated survival signal could be directed by NS5A, which is capable of downmodulating PTEN, an inhibitor of the PI3K‐Akt pathway.38 HCV also affects TGF‐β signaling via interacting with SMURF2, a key negative regulator of the TGF‐β pathway. HCV NS3‐4A targets SMURF2, resulting in enhanced SMAD2/3 phosphorylation and then in increased TGF‐β signaling, an event, which is inhibited by SMURF2 overexpression.14 Taken together, these observations show how TGF‐β signaling can be both negatively and positively regulated by HCV to affect the pathogenesis of HCV infection outcomes (Figure 2).
Figure 2.
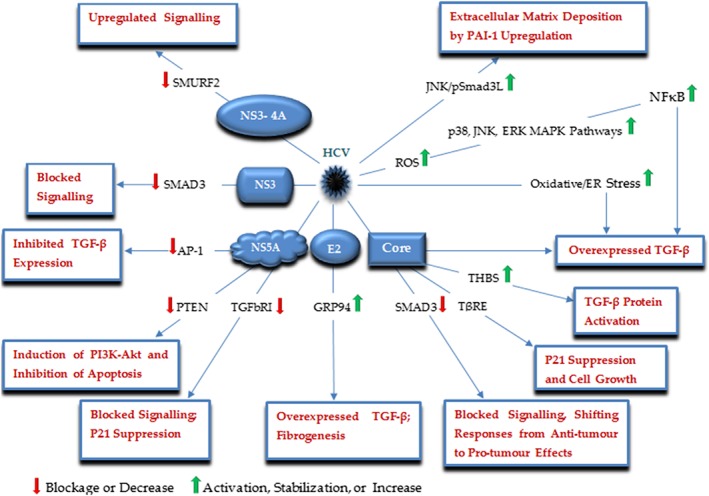
A schematic illustration of the major HCV proteins and corresponding targets, by which HCV modulates the TGF‐β signaling pathway
3.2. Hepatitis B virus
HBV is a DNA virus pertained to the Hepadnaviridae family, known as another major risk factor for cirrhosis and HCC progression.39, 40 HBV chronic infection is currently estimated to influence almost 240 million people, globally, of which approximately 10% have an increased risk of developing cirrhosis and HCC.4, 41 Similar to HCV, HBV also interacts with the TGF‐β pathway, a phenomenon mainly mediated by HBV X protein (HBx), which may exert diverse effects on the pathogenesis of the HBV‐mediated liver pathologies7, 21, 42, 43 (Figure 3). Evidence by Murata further supported the hypothesis that the TGF‐β pathway is directly involved in liver tumorigenesis, where HBx was shown to switch the target of hepatocytic TGF‐β signaling from the TGFbRI/pSmad3C/p21 anti‐tumor pathway to the JNK/pSmad3L/c‐Myc tumor supportive pathway in the early stages of liver carcinogenesis.43 Subsequently, the same result was reported by WU investigating the effect of TGF‐β on HBx‐expressing well‐differentiated HCC cells.42 As a result, the JNK/pSmad3L pathway can be regarded as a potential candidate toward preventing and treating HBV‐mediated HCC. It is thought the upregulated TGF‐β pathway enhances the invasive and metastatic potential of HCC cells, by suppressing E‐cadherin expression and promoting https://www.frontiersin.org/articles/10.3389/fmicb.2012.00247/full (Treg) induction.7, 21, 44 In this respect, Liu suggested a new mechanism underlying the pathogenesis of HBV‐caused HCC; HBx downmodulates protein phosphatase magnesium‐dependent 1A (PPM1a) levels, resulting in overactivation of the TGF‐β signaling pathway.7 In fact, HBx was shown to promote the degradation of PPM1a, a p‐Smad2/3 phosphatase required for terminating TGF‐β signaling, through elevating its ubiquitination. Furthermore, Yoo showed HBx upregulates TGF‐β expression by acting through the Egr transcription factors binding site.45 A study by Lee also demonstrated that HBx directly interacts with SMAD4 leading to the SMAD complex stabilization and enhanced TGF‐β signaling.46 In this way, HBV may affect cancer invasion in HCC, by upmodulating the TGF‐β pathway. Moreover, HBx has also been shown to prevent TGF‐β mediated‐apoptosis via upregulating the PI3‐kinase signaling pathway activity.47 In addition to HBx, HBV transcripts have also been implicated in the pathogenesis of HCC.48 A recent study demonstrated HBV mRNAs could bind to and absorb microRNA 15a/16 to inhibit apoptosis.49 The importance of this interaction was subsequently highlighted, when the authors interestingly found that SMAD7 acts as a target of miR‐15a.48 Thus, HBV can increase the level of SMAD7 and then block TGF‐β signaling and corresponding responses (eg, apoptosis) by downmodulating miR‐15a, an attractive molecular target for therapeutic development.
Figure 3.
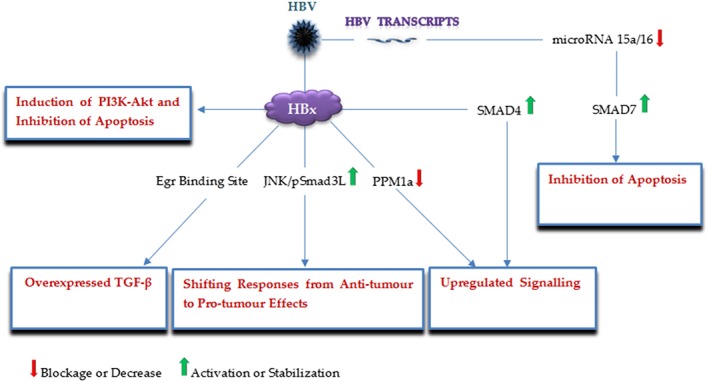
A schematic illustration of the major HBV factors and corresponding targets, by which HBV modulates the TGF‐β signaling pathway
3.3. Human papilloma virus
HPVs are DNA viruses associated with the development of cervical, anal, and head and neck cancers. The majority of cervical cancer is mediated by HPV16 and HPV18 encoding the E6 and E7 oncoproteins.50, 51 There is a close relationship between HPV infection and the TGF‐β pathway in cervical tissues; investigations have reported upmodulated TGF‐β levels in HPV‐positive cervical cancers compared with HPV‐negative ones and have suggested a positive correlation between the expression levels of TGF‐β and the HPV E6/E7 oncogenes, which may be due to trans‐regulatory functions of the E6/E7 oncoproteins, so that, HPV16 E6/E7 have been shown to enhance TGF‐β promoter activity by interacting with an Sp1‐binding site placed in the TGF‐β core promoter.52 However, a prominent aspect of malignant transformation in cervical epithelial cells is the progressive loss of TGF‐β responsiveness.53 In this respect, studies demonstrate a contributing role for HPV in the acquisition of TGF‐β resistance in cervical cancer (Figure 4). HPV16 E7 interacts with SMAD2, SMAD3, and SMAD4 leading to inhibition of SMAD3 binding to its DNA targets and then blocking the pathway.54 HPV16 E5 downregulates the TGF‐β pathway by lowering the expression of TGFbRII, as well as, by reducing SMAD2 phosphorylation and SMAD4 nuclear translocation, which may play a crucial role in the HPV‐induced cervical carcinogenesis.55 Through decreasing the intracellular levels of TIP‐2/GIPC, a PDZ protein involved in the expression of TGFbRIII, HPV18 E6 was shown to render HeLa cells less sensitive to the anti‐growth activities of TGF‐β.56 HPV16 E7 was also found to interfere with the growth‐inhibitory effects of the cyclin‐dependent kinase inhibitors p21CIP1, p27KIP1, and p15INKB, which are induced by TGF‐β.57 Furthermore, Mendoza demonstrated a similar activity for HPV5, a causative agent for skin carcinomas; it was shown that HPV5 E6 could bind to SMAD3 and destabilize the SMAD3/SMAD4 complex.58 Similarly, the cutaneous HPV8 and MmuPV1 (a recently detected HPV) E6 proteins interact with SMAD2/SMAD3 and suppress the TGF‐β pathway.59 These findings show that how may these interactions be important in the development of HPV‐associated tumors, in particular cervical cancer.
Figure 4.
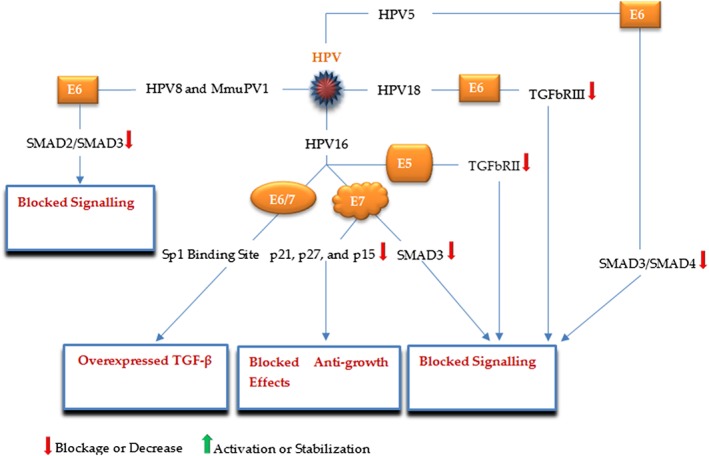
A schematic illustration of the major HPV proteins and corresponding targets, by which HPV modulates the TGF‐β signaling pathway
3.4. Epstein‐Barr virus
EBV is a DNA virus from the Herpesviridae family, involved in Hodgkin's lymphoma, Burkitt's lymphoma (as B‐cell cancres), nasopharyngeal carcinoma, gastric cancer (as epithelial malignancies),60, 61 and autoimmune diseases.62 In these tumors, EBV establishes latent infections, expressing only a subset of viral genes, allowing the virus to affect cellular signaling contributing to oncogenesis.63 EBV may interfere with TGF‐β signaling to promote tumorigenesis (Figure 5). Takanashi reported EBV‐encoded latent membrane protein 1 (LMP‐1) caused loss of TGF‐β sensitivity in rodent fibroblasts.64 Arvanitakis earlier had demonstrated LMP‐1 inhibited TGF‐β‐mediated growth suppression in EBV‐positive B lymphocytes.65 Subsequently, LMP‐1 was indicated to suppress the activation of TGF‐β signaling and TGFβ‐induced growth suppression via an NF‐κB‐dependent mechanism.66 Indeed, LMP‐1 was suggested to activate NF‐κB that in turn and in competition with SMAD proteins interacts with the transcriptional coactivator CBP and p300, those factors essential for SMADs to function as transcriptional effectors. In other words, it seems the LMP‐1‐mediated inhibitory effect on SMAD‐dependent transcription results from inhibition of the transcriptional cofactors involved in SMAD transcriptional function, but not owing to suppression of TGF‐β‐induced SMAD signaling through mechanisms such as affecting the formation of SMAD heteromers or their DNA binding activities; similarly, LMP‐1 suppresses ATF3, a transcriptional repressor induced by SMAD signaling, whose association with SMADs blocks the expression of a growth promoting gene, known as Id1. However, in the absence of this factor, SMAD3 binds to Id1 promoter directly, resulting in Id1 upmodulation.67 In this way, it can be seen how the well‐documented “double‐edged sword” nature of TGF‐β signaling is manipulated by EBV. While, LMP‐1 increases the expression of TGF‐β and SMAD2 phosphorylation but also blocks SMAD‐dependent transcription and TGFβ‐mediated cytostasis, and exerts its effects by using the non‐SMAD arm of TGF‐β signaling; it enhances the secretion of fibronectin through the JNK/SAPK pathway,68 which can contribute to tumor invasiveness. EBV also affects the pathway via EBNA‐1 protein, so that, it decreases SMAD2 levels by enhanced protein turnover.69, 70 Accordingly, EBNA‐1 represses TGF‐β‐induced transcription of βig‐h3 (a TGF‐β‐target gene implicated in cell growth, differentiation, and apoptosis), and PAI‐1 (a classical target of TGF‐β) in carcinoma cells,70 as well as, PTPRK (a functional tumor suppressor in Hodgkin lymphoma cells) leading to the growth and survival of these cancer cells.69 EBV‐encoded BARF1 is another EBV factor by which the virus modulates the TGF‐β pathway enhancing cell proliferation in gastric cancer; in an NFκB‐dependent manner, BARF1 upmodulate miR‐146a‐5p levels that directly targets SMAD4 and reduces its levels in gastric cancer cells.71 LMP‐1 also induces miR‐146a expression that may similarly suppress SMAD4 protein.72 In summary, reports suggest these EBV proteins as promising target for therapeutic interventions, so that a BARF1‐specific mAb was proposed as a novel immunotherapeutic tool toward the management of EBV‐associated malignancies.73
Figure 5.
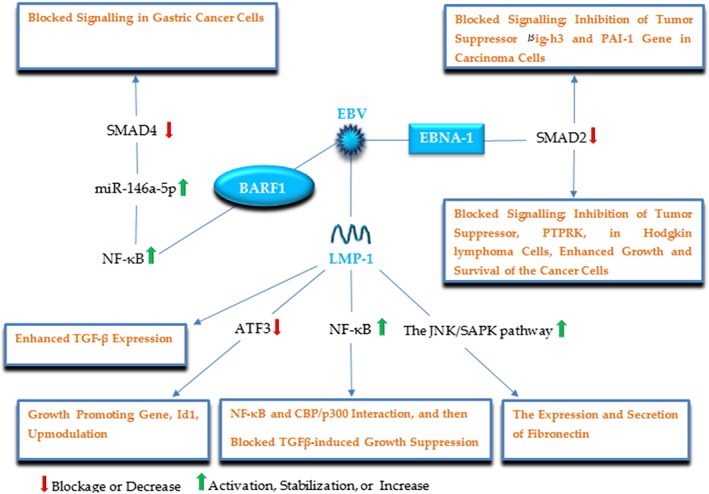
A schematic illustration of the major EBV proteins and corresponding targets, by which EBV modulates the TGF‐β signaling pathway
3.5. Kaposi's sarcoma‐associated herpes virus
KSHV is a Herpesvirus implicated in the onset of Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma (PEL).74 KSHV infection comprises 2 latent and lytic phases, which its transforming properties mainly root from the expression of latent genes such as kaposin, viral micro RNAs, viral Fas‐ associated death domain IL‐1β‐converting enzyme inhibitory protein (vFLIP), viral cyclin (vCyc), and latency‐associated nuclear antigen (LANA), through enhancing the survival and proliferation of the virus‐ infected cancer cells.75 Dysregulation of TGF‐β signaling importantly contributes to cell survival and proliferation in KSHV infection,76 and in this way, investigations have reported an interfering role of KSHV‐encoded products with this pathway (Figure 6).77 TGF‐β signaling has been reported to be suppressed by LANA in PEL via the association of this KSHV‐encoded protein with the promoter region of TGFbRII and subsequently histone methylation and deacetylation and then downregulation of this receptor, contributing to resistance to the antiproliferative effects of TGF‐β.77 Choi was able to show that vFLIP and vCyclin upregulate the expression of the oncogenic miR‐17‐92 cluster that in turn interact with the TGF‐β pathway via downmodulating SMAD2 protein.75 KSHV not only modifies the expression of cellular miRNAs, but also encodes for viral miRNAs to interact with TGF‐β signaling. KSHV‐encoded miR‐K12‐11 targets SMAD5 to block TGFβ signaling, and hence, facilitating cell survival and proliferation. Furthermore, suppression of this viral miRNA eliminates this suppression in B cells infected by KSHV.76 Similarly, KSHV miR‐K10 variants repress the pathway through targeting TGFbRII, so that its expression is adequate to block TGF‐β‐mediated cell apoptosis in KSHV‐infected PEL cells.74 THBS1 has been suggested as another KSHV‐encoded miRNAs target; miR‐K12 targets this factors resulting in repression of TGF‐β signaling.78 In addition to the latent proteins, 2 KSHV lytic products also interfere with TGF‐β signaling; viral interferon regulatory factor 1 (vIRF‐1) was demonstrated to perturb TGF‐β‐induced transcription and growth arrest through direct interaction with both SMAD3 and SMAD4, leading to interrupting the formation of SMAD3‐SMAD4 complex and their DNA binding activity.79 Furthermore, by binding to the transcriptional coactivator CBP and blocking its recruitment into transcription initiation complexes on TGF‐β‐responsive elements, the KSHV lytic protein, K‐bZIP, also inhibits TGF‐β signaling.80
Figure 6.
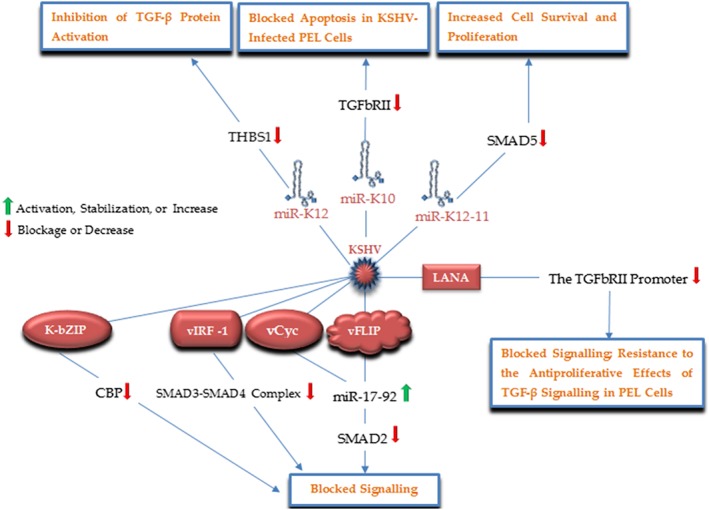
A schematic illustration of the major KSHV factors and corresponding targets, by which KSHV modulates the TGF‐β signaling pathway
3.6. Human T‐cell Lymphotropic virus type 1
HTLV‐1, the causative agent of adult T‐cell leukemia (ATL), has also been involved in TGF‐β signaling interruption (Figure 7).81, 82, 83 Reports indicate the HTLV‐1 Tax protein overexpresses high levels of TGF‐β mRNA and protein in mice models.84 However, Tax was reported to repress the TGF‐β‐mediated transcriptional activation and growth inhibition, via competitive interactions with both SMAD factors and the transcriptional co‐activator p300, and also by preventing the SMADSs complex formation and binding to target sequences.81 Furthermore, Tax inhibits TGF‐β signaling by JNK/c‐Jun activation and then the association of c‐Jun with SMAD3 and suppressing its DNA binding ability, which may significantly affect ATL leukemogenesis.83 Similar to natural Tregs, ATL cells represent a CD4+CD25+ phenotype, and two‐thirds of ATL cases express FoxP3 suggesting that the cancer may root from https://www.frontiersin.org/articles/10.3389/fmicb.2012.00247/full‐infected natural Treg cells; nonetheless, recently Zhao found the HTLV‐1 bZIP factor (HBZ) interacts with SMAD3 and p300, forming a ternary complex that leads to increased association of SMAD3 and p300, enhanced signaling, and thereafter, induced Foxp3 expression in naive T cells, which allows the virus to convert infected T cells into Treg cells. Additionally, they reported that HBZ could overcome the Tax‐induced inhibition.82 Taken together, these interactions proposed that the modulation of TGF‐β signaling may play a critical role in the HTLV‐1‐associated leukemogenesis.
Figure 7.
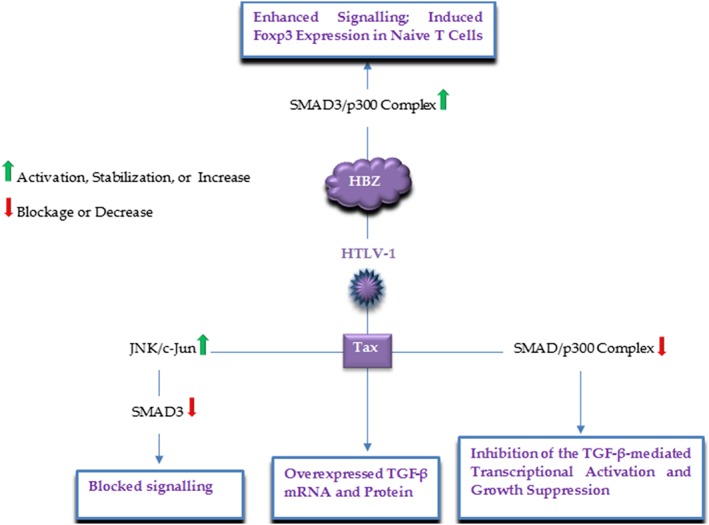
A schematic illustration of the major HTLV‐1 proteins and corresponding targets, by which HTLV‐1 modulates the TGF‐β signaling pathway
4. TUMOR‐ASSOCIATED VIRUSES AND TGF‐β SIGNALING
4.1. Cytomegalovirus
CMV is a Herpesvirus associated with several tumors, such as colon, breast, and prostate cancers, acting as an oncomodulator by which enhance tumor growth through interfering with cell signaling in an established malignancy.63 CMV also associates with poor graft outcome in renal disorders, and can result in miscarriage, stillbirth, and retardation of fetal growth in pregnancy.85, 86 Investigations showed the expression of TGF‐β in a variety of cells and tissues during CMV infection; brain samples of AIDS patients who had CMV encephalitis were indicated to have viral inclusions co‐localized with TGF‐β protein in cells with astrocyte‐specific glial filaments.87 CMV increases in vitro expression of TGF‐β mRNA and protein from infected astrocytes, fibroblasts, and osteosarcoma cells.87, 88, 89 Furthermore, in vitro transient expression of the CMV immediate early 1 and 2 (IE1, IE2) genes also induces TGF‐β expression.88, 90 For instance, the IE2 stimulates TGF‐β expression in human glioma cells, via interacting with the Egr‐1 DNA‐binding protein.90 Additionally, CMV also augments the production of this cellular immune suppressive cytokine in several tumor cell types (eg, glioblastoma, leukemia, and osteosarcoma cells), affecting host immune responses toward these tumor cells.91 CMV DNA and proteins were shown to be correlated with increased TGF‐β and arterial intimal thickening in tubuli and arterial endothelium in CMV infected human kidney allografts compared with uninfected allografts.92 Similarly, enhanced urinary excretion of TGF‐β has also been found in urine samples of CMV‐positive renal transplant patients, which was associated with exacerbated fibrosis in biopsies taken from patients.93 This association of CMV infection with enhanced TGF‐β expression reinforces the possibility that CMV may promote renal allograft rejection through CMV infection‐induced TGF‐β leading to fibrosis inside the allograft; CMV‐infected human renal tubular epithelial cells that were exposed to TGF‐β in vitro, developed EMT, after which the cells also activated extracellular latent form of TGF‐β. This effect may root from the induction of a known activator of latent TGF‐β, matrix metalloprotease 2 (MMP‐2), by IE1 and IE2.85 Moreover, manipulated TGF‐β signaling by CMV may also be involved in defective placental development, and then in pregnancy‐associated complications. In CMV‐infected placental blood vessels, the virus infected endothelial cells induced the expression of TGF‐β (and TGF‐β signaling) and collagen IV through increasing αvβ6 integrin production, proposing that CMV infection of the placenta may modify ECM, enabling the virus to translocate throughout the placenta to infect the fetus.94 Extravillous cytotrophoblast (EVT) invasion into the endometrium is a critical event to remodel the uterine arterioles during normal pregnancy, whose dysfunction results in impaired placental blood flow.95 An in vitro study revealed that CMV may inhibit EVT proliferation and invasion by dysregulating the TGF‐β pathway; so that, the levels of TGF‐β, SMAD2, SMAD3, and SMAD4 mRNA were significantly increased, but those of TGFβRI, TGFβRII, SMAD7, MMP2, and MMP9 were reported to be decreased by HCMV infection.86
HIV‐1, a lentivirus, whose infection is associated with an increased risk of developing cancers of other infectious agents,96 also may affect TGF‐β signaling. Elevated concentrations of active TGF‐β were recorded in patients with HIV‐1 infection.97 Furthermore, HIV‐1 gp160 increases the levels of TGF‐β in human PBMC,98 and also inactivated HIV and gp120 elevate TGF‐β expression, by which enhance HCV replication, and then, exacerbate HCV‐caused liver disease.99
5. OTHER HUMAN VIRUSES
Viral pathogens currently not associated with oncogenesis also modulate the TGF‐β pathway. Severe acute respiratory syndrome (SARS)‐coronavirus (CoV), a RNA virus known to cause outbreak of SARS disease, targets TGF‐β signaling100, 101, 102, 103; SARS‐CoV papain‐like protease (Plpro) stimulates the TGF‐β mRNA and protein production and pro‐fibrotic genes expression by the p38 MAPK and ERK1/2‐mediated pathways in human promonocytes (Figure 8).102 In human lung epithelial cells and mouse models, SARS‐CoV Plpro also upregulated the level of Egr‐1 through the ROS‐induced p38 MAPK and STAT3 activation, leading to the increase in TGF‐β production by direct Egr‐1 interacting with the Egr‐1 binding site located in the TGF‐β promoter region.100 SARS‐CoV PLpro similarly upmodulated p38 MAPK/STAT3‐mediated expression of Type I collagen in vitro and in vivo, mediated by non‐SMAD TGF‐β signaling including STAT6 activation, correlated with upregulated pulmonary pro‐fibrotic responses.101 Furthermore, SARS‐CoV nucleocapsid (N) protein inhibited the formation of SMAD3/4 complex, resulting in blocked TGF‐β‐induced apoptosis in SARS‐CoV‐infected host cells. Meanwhile, N protein also interacted with SMAD3 and increased SMAD3‐p300 complex formation, contributing to promoted plasminogen activator inhibitor‐1 (PAI‐1) levels, and hence, tissue fibrosis,103 suggesting novel therapeutic targets for SARS treatment.
Figure 8.
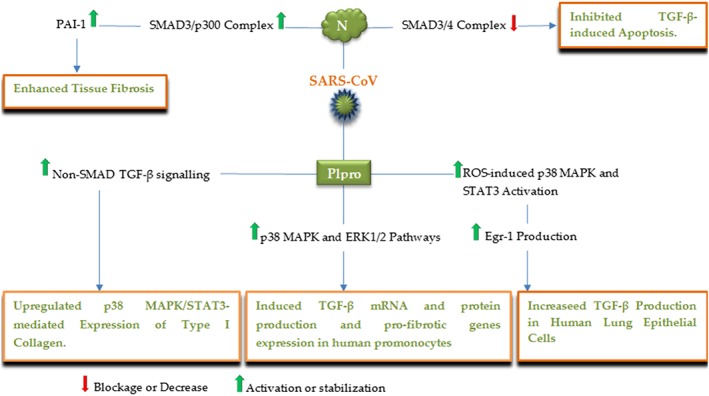
A schematic illustration of the major SARS‐CoV proteins and corresponding targets, by which the virus modulates the TGF‐β signaling pathway
Influenza A is a RNA virus, pathogenic for respiratory tract that was reported to intensify fibrotic lung diseases such as idiopathic pulmonary fibrosis (IPF) through an αvβ6 integrin dependent TGF‐β activity.104 In fact, pulmonary infection with H1N1 virus can result in the activation of SMAD2/3 and also can increase toll like receptor 3 (TLR3) stimulation, which in turn enhances RhoA activity, a novel mechanism contributing to αvβ6 integrin‐mediated TGF‐β activation, resulting in enhanced epithelial apoptosis, collagen deposition, and then pulmonary fibrosis (Figure 9).104 In addition, both influenza virus and bacterial neuraminidases (NA) directly activate latent TGF‐β by removing sialic acid motifs from the latent form in cell free systems.105 Interestingly, through the activation of TGF‐β, influenza A virus NA upregulates host adhesion molecules required for bacterial binding, resulting in elevated bacterial loading in lung, a phenomenon that was inhibited after the suppression of TGF‐β signaling, proposing TGF‐β and cellular adhesins as potential molecular targets for the prevention of postinfluenza bacterial pneumonia.106
Figure 9.
A schematic illustration of the major influenza A proteins and corresponding targets, by which the virus modulates the TGF‐β signaling pathway
Adenovirus (AV), an infectious agent of respiratory tract,107 also influences TGF‐β signaling. Adenovirus E1A reduces the level of TGFβRII mRNA and protein in adenovirus‐infected cells.108 The E1A protein also inhibits TGF‐β‐induced cell growth arrest by its binding to p300 and then preventing TGF‐β‐mediated expression of the cyclin‐dependent kinase inhibitors, p15 and p21.109 Moreover, E1A interacts with SMAD3 and then further represses the formation of SMAD3/p300 complex.110 Human hepatocellular carcinoma cells (Huh‐7) infected with AV were also reported to be insensitive to TGF‐β‐induced apoptosis; the E1A protein and E1B‐19 K were required for this resistance.111
RSV, a common cause of infant bronchiolitis and pneumonia,112, 113 increases TGF‐β production and signaling in human epithelial cells to facilitate its infection. RSV also induces TGF‐β and the TGF‐β dependent SMAD‐2/3 signaling in macrophages.114
Further, infection of human hepatocytes with ebola virus (EBOV), a major cause of hemorrhagic fevers, enhanced TGF‐β secretion, suggested to activate TGF‐β‐regulated signaling pathways, ERK1/2, p38 MAPK, and SMAD3, leading to the induction of an EMT‐like phenotype in infected cells.115 Similarly, reovirus also stimulates TGF‐β signaling in murine model of encephalitis in vivo, by inducing TGFβRI expression, and by activating SMAD3 factor, contributing to neuronal survival.116
6. CONCLUSIONS
TGF‐β signaling is a critical target of viruses during course of tumorigenesis. It also seems to be involved in viral‐induced pulmonary fibrosis, renal diseases, and pregnancy‐associated complications. Viruses utilize various mechanisms to modulate this pathway, which appears to be through (Table 1, Figure 1) (1) modulation of either TGF‐β protein expression or activation, (2) modulation of the TGF‐β receptors or SMADs factors (by interfering with their levels and functions), (3) modulation of none‐SMAD cascades, and (4) indirect interaction by the modulation of transcriptional co‐activator/repressor and regulators of the pathway. Two major TGF‐β‐regulated events that are modulated by most, if not all, viruses are exacerbated cell growth and induction of fibrosis. Hence, as a pivotal pathway modified by viruses in particular in cancer, TGF‐β signaling should be assessed as an appropriate therapeutic target. So that, TGF‐β antibodies, antisense oligonucleotides, and small molecules targeting TGFβRI have demonstrated therapeutic potentials.8
Table 1.
Viral‐mediated alterations in the TGF‐β signaling pathway
| Viral pathogen | Viral Factor | Mechanism/Effect | References |
|---|---|---|---|
| HCV | Core | ‐upregulated TGF‐β expression, both in vivo and in vitro. | 28 |
| ‐suppressed TGF‐β–induced p21 expression by acting through the TGF‐β‐responsive element (TβRE) positioned in the p21 promoter region. | 32 | ||
| ‐inhibited SMAD3‐mediated transcriptional activation via reducing the SMAD3 DNA binding ability. | 35 | ||
| ‐activated TGF‐β protein through the induction of THBS. | 29 | ||
| E2 | ‐overexpressed TGF‐β expression by enhancing GRP94. | 24 | |
| NS5A | ‐downregulated TGF‐β expression via reducing AP‐1. | 24 | |
| ‐repressed TGF‐β mediated‐apoptosis via inducing the PI3K‐Akt survival pathway by PTEN inhibition. | 38 | ||
| ‐blocked TGF‐β signaling and P21 expression through TGFbRI suppression. | 31 | ||
| NS3 | ‐suppressed SMAD3‐induced transcriptional activation via decreasing the SMAD3 DNA binding activity. | 33 | |
| NS3‐4A | ‐increased TGF‐β signaling by the inhibition of SMURF2. | 14 | |
| ‐ | ‐TGF‐β overexpression, mediated by ROS‐induced p38, JNK, ERK MAPK pathways induction leading to NF‐κB activation. | 30 | |
| ‐extracellular matrix (ECM) deposition by JNK/pSmad3L‐mediated PAI‐1 upregulation. | 37 | ||
| ‐upregulated TGF‐β through induced ER stress responses. | 26 | ||
| HBV | HBx | ‐induction of the JNK/pSmad3L/c‐Myc tumor supportive pathway in the early stages of liver carcinogenesis. | 43 |
| ‐upregulated signaling through PPM1a downmodulation. | 7 | ||
| ‐upregulated TGF‐β expression by acting through the Egr transcription factors binding site. | 45 | ||
| ‐stabilized SMADs complex by interacting with SMAD4. | 46 | ||
| ‐impeded TGF‐β induced‐apoptosis via upregulating the PI3‐kinase signaling pathway. | 47 | ||
| HBV transcripts | ‐suppressed TGF‐β induced‐apoptosis through absorbing miR‐15a that in turn targets and increases SMAD7. | 48 | |
| HPV | HPV16 E6 | ‐enhanced TGF‐β expression through the interaction with the Sp1‐binding site located in the TGF‐β promoter. | 52 |
| HPV16 E7 | ‐blocked signaling through the inhibition of SMAD3 DNA binding activity. | 54 | |
| ‐interrupted anti‐growth effects by interfering with the cyclin‐dependent kinase inhibitors p21CIP1, p27KIP1, and p15INKB. | 57 | ||
| ‐increased TGF‐β levels via the interaction with the Sp1‐binding site located in the TGF‐β promoter. | 52 | ||
| HPV16 E5 | ‐blocked signaling through the downregulation of TGFbRII, and by decreasing SMAD2 phosphorylation and SMAD4 nuclear translocation. | 55 | |
| HPV18 E6 | ‐suppressed TGFbRIII expression via targeting TIP‐2/GIPC. | 56 | |
| HPV8 and MmuPV1 E6 | ‐blocked signaling through interacting with SMAD2/SMAD3. | 59 | |
| HPV5 E6 | ‐destabilizing the SMAD3/SMAD4 complex by interacting with SMAD3. | 58 | |
| EBV | LMP‐1 | ‐blocked SMAD/CBP‐p300 complex formation by the induction of NF‐κB that competitively interacts with CBP‐p300. | 66 |
| ‐upmodulated Id1 expression through ATF3 suppression. | 67 | ||
| ‐increased fibronectin expression and secretion by upregulating the JNK/SAPK pathway. | 68 | ||
| ‐enhanced TGF‐β expression and SMAD2 phosphorylation. | 68 | ||
| EBNA‐1 | ‐repression of the tumor suppressor, PTPRK, in Hodgkin lymphoma cells by decreasing SMAD2 levels. | 69 | |
| ‐inhibition of the tumor suppressor, βig‐h3, and PAI‐1 in carcinoma cells via reducing SMAD2 levels. | 70 | ||
| BARF1 | ‐reduced SMAD4 levels, mediated by NF‐κB upregulation, which in turn enhances miR‐146a‐5p to target SMAD4 in gastric cancer. | 71 | |
| KSHV | LANA | ‐downregulated TGFbRII by LANA‐mediated histone methylation and deacetylation of TGFbRII promoter. | 77 |
| vFLIP | ‐downregulated SMAD2 through the induction of oncogenic miR‐17‐92. | 75 | |
| vCyc | ‐downmodulated SMAD2 via the induction of oncogenic miR‐17‐92. | 75 | |
| miR‐K12‐11 | ‐suppressed SMAD5 by targeting SMAD5 mRNAs. | 76 | |
| miR‐K10 | ‐downregulated TGFbRII through targeting TGFbRII mRNAs. | 74 | |
| miR‐K12 | ‐repressed THBS1 by targeting THBS1 mRNAs. | 78 | |
| vIRF‐1 | ‐inhibited SMAD3/SMAD4 complex formation by vIRF‐1 interaction with both SMAD3/4. | 79 | |
| K‐bZIP | ‐suppression of transcription initiation complexes via CBP inhibition by K‐bZIP. | 80 | |
| HTLV‐1 | Tax | ‐overexpressed TGF‐β mRNA and protein in mice model. | 84 |
| ‐inhibited SMAD/p300 complex formation by competitive interactions with both SMAD and p300. | 81 | ||
| ‐suppressed SMAD3 DNA binding activity through JNK/c‐Jun activation and then c‐Jun/SMAD3 interaction. | 83 | ||
| HBZ | ‐increased SMAD/p300 complex formation by HBZ‐mediated ternary complex formation (SMAD3‐HBZ‐p300). | 82 | |
| CMV | IE1 | ‐enhanced TGF‐β expression in vitro. | 88, 89 |
| ‐activation of latent TGF‐β via increasing matrix metalloprotease 2 (MMP‐2). | 85 | ||
| IE2 | ‐promoted TGF‐β expression through interacting with the Egr‐1 DNA‐binding protein in human glioma cells. | 90 | |
| ‐activation of latent TGF‐β by inducing matrix metalloprotease 2 (MMP‐2). | 85 | ||
| ‐ | ‐repressed EVT proliferation and invasion by disrupting the TGF‐β signaling pathway. | 86 | |
| ‐manipulated both TGF‐β protein and signaling in renal transplant patients. | 85, 93 | ||
| ‐increased collagen IV expression in the placenta, as a result of αvβ6 integrin‐mediated TGF‐β protein and signaling activation. | 94 | ||
| HIV‐1 | gp160 | ‐elevated TGF‐β mRNA expression and protein secretion in human PBMC. | 98 |
| gp120 | ‐exacerbate HCV‐caused liver disease by upregulating TGF‐β expression. | 99 | |
| SARS‐CoV | PLpro | ‐promoted TGF‐β mRNA and protein production by p38 MAPK and ERK1/2‐mediated pathways in human promonocytes. | 102 |
| ‐stimulated TGF‐β production by Egr‐1 upmodulation mediated via ROS‐induced p38 MAPK and STAT3 activation. | 100 | ||
| ‐enhanced p38 MAPK/STAT3‐mediated expression of type I collagen in vitro and in vivo, through non‐SMAD TGF‐β signaling. | 101 | ||
| N | ‐suppressed TGF‐β‐induced apoptosis by disrupting SMAD3/SMAD4 complex. | 103 | |
| ‐enhanced PAI‐1‐induced tissue fibrosis via SMAD3/p300 complex promotion. | 103 | ||
| Influenza A | NA | ‐upregulated host adhesion molecules required for bacterial binding, as a result of both TGF‐β protein and signaling activation, leading to postinfluenza bacterial pneumonia. | 106 |
| ‐ | ‐promoted pulmonary fibrosis by αvβ6 integrin‐mediated both TGF‐β protein and signaling activation. | 104 | |
| AV | E1A | ‐blocked signaling by downregulating TGFβRII mRNA and protein. | 108 |
| ‐suppressed TGF‐β‐mediated cell growth arrest by interacting with p300 and SMAD3. | 109 | ||
| RSV | ‐ | ‐promoted TGF‐β production and signaling in human epithelial cells and macrophages. | 114 |
| EBOV | ‐ | ‐enhanced TGF‐β production and signaling, leading to ERK1/2 and p38 MAPK activation, and then an EMT‐like phenotype in infected cells. | 115 |
| Reovirus | ‐ | ‐elevated neuronal survival by increasing TGFβRI expression, and by activating SMAD3. | 116 |
CONFLICT OF INTEREST
None declared.
Mirzaei H, Faghihloo E. Viruses as key modulators of the TGF‐β pathway; a double‐edged sword involved in cancer. Rev Med Virol. 2018;28:e1967 10.1002/rmv.1967
REFERENCES
- 1. Parkin DM. The global health burden of infection‐associated cancers in the year 2002. Int J Cancer. 2006;118(12):3030‐3044. [DOI] [PubMed] [Google Scholar]
- 2. Fiorina L, Ricotti M, Vanoli A, et al. Systematic analysis of human oncogenic viruses in colon cancer revealed EBV latency in lymphoid infiltrates. Infect Agents Cancer. 2014;9(1):18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. White MK, Pagano JS, Khalili K. Viruses and human cancers: a long road of discovery of molecular paradigms. Clin Microbiol Rev. 2014;27(3):463‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zuylen WJ, Rawlinson WD, Ford CE. The Wnt pathway: a key network in cell signalling dysregulated by viruses. Rev Med Virol. 2016;26(5):340‐355. [DOI] [PubMed] [Google Scholar]
- 5. Fabregat I, Fernando J, Mainez J, Sancho P. TGF‐beta signaling in cancer treatment. Curr Pharm Des. 2014;20(17):2934‐2947. [DOI] [PubMed] [Google Scholar]
- 6. Fu S, Zhou R‐r, Li N, Huang Y, Fan X‐G, Hepatitis B. Virus X protein in liver tumor microenvironment. Tumor Biol. 2016;37(12):15371‐15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Y, Xu Y, Ma H, et al. Hepatitis B virus X protein amplifies TGF‐β promotion on HCC motility through down‐regulating PPM1a. Oncotarget. 2016;7(22):33125‐33135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Syed V. TGF‐β signaling in cancer. J Cell Biochem. 2016;117(6):1279‐1287. [DOI] [PubMed] [Google Scholar]
- 9. Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415‐424. [DOI] [PubMed] [Google Scholar]
- 10. Imamura T, Oshima Y, Hikita A. Regulation of TGF‐β family signaling by ubiquitination and deubiquitination. J Biochem. 2013;154(6):481‐489. [DOI] [PubMed] [Google Scholar]
- 11. Budi EH, Duan D, Derynck R. Transforming growth factor‐β receptors and Smads: regulatory complexity and functional versatility. Trends Cell Biol. 2017;27(9):658‐672. [DOI] [PubMed] [Google Scholar]
- 12. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neuzillet C, Tijeras‐Raballand A, Cohen R, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22‐31. [DOI] [PubMed] [Google Scholar]
- 14. Verga‐Gérard A, Porcherot M, Meyniel‐Schicklin L, André P, Lotteau V, Perrin‐Cocon L. Virus/human interactome identifies SMURF2 and the viral protease as critical elements for the control of TGF‐β signaling. FASEB J. 2013;27(10):4027‐4040. [DOI] [PubMed] [Google Scholar]
- 15. Huang JJ, Blobe GC. Dichotomous roles of TGF‐β in human cancer. Biochem Soc Trans. 2016;44(5):1441‐1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levy L, Hill CS. Alterations in components of the TGF‐β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17(1):41‐58. [DOI] [PubMed] [Google Scholar]
- 17. Bornstein S, White R, Malkoski S, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119(11):3408‐3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Papageorgis P. TGFβ signaling in tumor initiation, epithelial‐to‐mesenchymal transition, and metastasis. J Oncol. 2015;2015:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mu Y, Gudey SK, Landström M. Non‐Smad signaling pathways. Cell Tissue Res. 2012;347(1):11‐20. [DOI] [PubMed] [Google Scholar]
- 20. Itoh S, ten Dijke P. Negative regulation of TGF‐β receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19(2):176‐184. [DOI] [PubMed] [Google Scholar]
- 21. Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV‐ and HCV‐associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13(2):123‐135. [DOI] [PubMed] [Google Scholar]
- 22. Fabregat I, Moreno‐Càceres J, Sánchez A, et al. TGF‐β signalling and liver disease. FEBS Journal. 2016;283(12):2219‐2232. [DOI] [PubMed] [Google Scholar]
- 23. Shirai Y, Kawata S, Tamura S, et al. Plasma transforming growth factor‐β1 in patients with hepatocellular carcinoma. Comparison with chronic liver diseases. Cancer. 1994;73(9):2275‐2279. [DOI] [PubMed] [Google Scholar]
- 24. Jee MH, Hong KY, Park JH, et al. New mechanism of hepatic fibrogenesis: hepatitis C virus infection induces transforming growth factor β1 production through glucose‐regulated protein 94. J Virol. 2016;90(6):3044‐3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shin JY, Hur W, Wang JS, et al. HCV core protein promotes liver fibrogenesis via up‐regulation of CTGF with TGF‐[beta] 1. Exp Mol Med. 2005;37(2):138‐145. [DOI] [PubMed] [Google Scholar]
- 26. Chusri P, Kumthip K, Hong J, et al. HCV induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci Rep. 2016;6(1): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin W, Tsai WL, Shao RX, et al. Hepatitis C virus regulates transforming growth factor β1 production through the generation of reactive oxygen species in a nuclear factor κB–dependent manner. Gastroenterology. 2010;138(7):2509‐2518. e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taniguchi H, Kato N, Otsuka M, et al. Hepatitis C virus core protein upregulates transforming growth factor‐β1 transcription. J Med Virol. 2004;72(1):52‐59. [DOI] [PubMed] [Google Scholar]
- 29. Benzoubir N, Lejamtel C, Battaglia S, et al. HCV core‐mediated activation of latent TGF‐β via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J Hepatol. 2013;59(6):1160‐1168. [DOI] [PubMed] [Google Scholar]
- 30. Lin W, Tsai W‐L, Shao R‐X, et al. HCV regulates TGF‐β1 production through the generation of reactive oxygen species in an NFκB‐dependent manner. Gastroenterology. 2010;138(7):2509‐2518.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi S‐H, Hwang SB. Modulation of the transforming growth factor‐β signal transduction pathway by hepatitis C virus nonstructural 5A protein. J Biol Chem. 2006;281(11):7468‐7478. [DOI] [PubMed] [Google Scholar]
- 32. Lee MN, Jung EY, Kwun HJ, et al. Hepatitis C virus core protein represses the p21 promoter through inhibition of a TGF‐β pathway. J Gen Virol. 2002;83(9):2145‐2151. [DOI] [PubMed] [Google Scholar]
- 33. Cheng P‐L, Chang M‐H, Chao C‐H, Lee Y‐HW. Hepatitis C viral proteins interact with Smad3 and differentially regulate TGF‐β/Smad3‐mediated transcriptional activation. Oncogene. 2004;23(47):7821‐38. [DOI] [PubMed] [Google Scholar]
- 34. Bauer J, Sporn JC, Cabral J, Gomez J, Jung B. Effects of activin and TGFβ on p21 in colon cancer. PloS one. 2012;7(6):e39381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pavio N, Battaglia S, Boucreux D, et al. Hepatitis C virus core variants isolated from liver tumor but not from adjacent non‐tumor tissue interact with Smad3 and inhibit the TGF‐β pathway. Oncogene. 2005;24(40):6119‐6132. [DOI] [PubMed] [Google Scholar]
- 36. Battaglia S, Benzoubir N, Nobilet S, et al. Liver cancer‐derived hepatitis C virus core proteins shift TGF‐beta responses from tumor suppression to epithelial‐mesenchymal transition. PloS one. 2009;4(2):e4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsuzaki K, Murata M, Yoshida K, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46(1):48‐57. [DOI] [PubMed] [Google Scholar]
- 38. Cheng D, Zhang L, Yang G, et al. Hepatitis C virus NS5A drives a PTEN‐PI3K/Akt feedback loop to support cell survival. Liver Int. 2015;35(6):1682‐1691. [DOI] [PubMed] [Google Scholar]
- 39. Shin E‐C, Sung PS, Park S‐H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat Rev Immunol. 2016;16(8):509‐523. [DOI] [PubMed] [Google Scholar]
- 40. Li Y‐W, Yang F‐C, Lu H‐Q, Zhang J‐S. Hepatocellular carcinoma and hepatitis B surface protein. World J Gastroenterol. 2016;22(6):1943‐1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tu T, Budzinska MA, Shackel NA, Urban SHBVDNA. Integration: molecular mechanisms and clinical implications. Virus. 2017;9(4):75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu YH, Ai X, Liu FY, Liang HF, Zhang BX, Chen XP. c‐Jun N‐terminal kinase inhibitor favors transforming growth factor‐β to antagonize hepatitis B virus X protein‐induced cell growth promotion in hepatocellular carcinoma. Mol Med Rep. 2016;13(2):1345‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murata M, Matsuzaki K, Yoshida K, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)‐β signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49(4):1203‐1217. [DOI] [PubMed] [Google Scholar]
- 44. Yang P, Markowitz GJ, Wang X‐F. The hepatitis B virus‐associated tumor microenvironment in hepatocellular carcinoma. National Sci Rev. 2014;1(3):396‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoo Y, Ueda H, Park K, et al. Regulation of transforming growth factor‐beta 1 expression by the hepatitis B virus (HBV) X transactivator. Role in HBV pathogenesis. J Clin Investig. 1996;97(2):388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee DK, Park SH, Yi Y, et al. The hepatitis B virus encoded oncoprotein pX amplifies TGF‐β family signaling through direct interaction with Smad4: potential mechanism of hepatitis B virus‐induced liver fibrosis. Genes Dev. 2001;15(4):455‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shih W‐L, Kuo M‐L, Chuang S‐E, Cheng A‐L, Doong S‐L, Hepatitis B. Virus X protein inhibits transforming growth factor‐β‐induced apoptosis through the activation of phosphatidylinositol 3‐kinase pathway. J Biol Chem. 2000;275(33):25858‐25864. [DOI] [PubMed] [Google Scholar]
- 48. Liu N, Jiao T, Huang Y, Liu W, Li Z, Ye X. Virus regulates apoptosis and tumorigenesis through the microRNA‐15a‐Smad7‐transforming growth factor beta pathway. J Virol. 2015;89(5):2739‐2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu N, Zhang J, Jiao T, et al. Hepatitis B virus inhibits apoptosis of hepatoma cells by sponging the MicroRNA 15a/16 cluster. J Virol. 2013;87(24):13370‐13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dadashi M, Eslami G, Faghihloo E, et al. Detection of human papilloma virus type 16 in epithelial ovarian tumors samples. Arch Clin Infect Dis. 2017. January;12(1):e39666 [Google Scholar]
- 51. Faghihloo E, Akbari A, Adjaminezhad‐Fard F, Mokhtari‐Azad T. Transcriptional regulation of E‐cadherin and oncoprotein E7 by valproic acid in HPV positive cell lines. Iran J Basic Med Sci. 2016;19(6):601‐607. [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu H, Luo H, Shen Z, Hu X, Sun L, Zhu X. Transforming growth factor‐β1 in carcinogenesis, progression, and therapy in cervical cancer. Tumor Biol. 2016;37(6):7075‐7083. [DOI] [PubMed] [Google Scholar]
- 53. Ki K‐D, Tong S‐Y, Huh C‐Y, Lee J‐M, Lee S‐K, Chi S‐G. Expression and mutational analysis of TGF‐β/Smads signaling in human cervical cancers. J Gynecol Oncol. 2009;20(2):117‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee DK, Kim B‐C, Kim IY, Cho E‐a, Satterwhite DJ, Kim S‐J. The human papilloma virus E7 oncoprotein inhibits transforming growth factor‐β signaling by blocking binding of the Smad complex to its target sequence. J Biol Chem. 2002;277(41):38557‐38564. [DOI] [PubMed] [Google Scholar]
- 55. French D, Belleudi F, Mauro MV, et al. Expression of HPV16 E5 down‐modulates the TGFbeta signaling pathway. Mol Cancer. 2013;12(1):38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Favre‐Bonvin A, Reynaud C, Kretz‐Remy C, Jalinot P. Human papillomavirus type 18 E6 protein binds the cellular PDZ protein TIP‐2/GIPC, which is involved in transforming growth factor β signaling and triggers its degradation by the proteasome. J Virol. 2005;79(7):4229‐4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McLaughlin‐Drubin ME, Münger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384(2):335‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mendoza J‐A, Jacob Y, Cassonnet P, Favre M. Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor β signaling pathway by binding to SMAD3. J Virol. 2006;80(24):12420‐12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meyers JM, Uberoi A, Grace M, Lambert PF, Munger K. Cutaneous HPV8 and MmuPV1 E6 proteins target the NOTCH and TGF‐β tumor suppressors to inhibit differentiation and sustain keratinocyte proliferation. PLoS Pathog. 2017;13(1):e1006171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kempkes B, Robertson ES. Epstein‐Barr virus latency: current and future perspectives. Curr Opin Virol. 2015;14:138‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Faghihloo E, Saremi MR, Mahabadi M, Akbari H, Saberfar E. Prevalence and characteristics of Epstein‐Barr virus‐associated gastric cancer in Iran. Arch Iran Med. 2014;17(11):767‐770. [PubMed] [Google Scholar]
- 62. Mahabadi M, Faghihiloo E, Alishiri GH, Ataee MH, Ataee RA. Detection of Epstein‐Barr virus in synovial fluid of rheumatoid arthritis patients. Electron Physician. 2016;8(3):2181‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mirzaei H, Goudarzi H, Eslami G, Faghihloo E. Role of viruses in gastrointestinal cancer. J Cell Physiol. 2017; [DOI] [PubMed] [Google Scholar]
- 64. Takanashi M, Li J, Shirakata M, Mori S, Hirai K. Tumorigenicity of mouse BALB/c 3T3 fibroblast cells which express Epstein‐Barr virus‐encoded LMP1 and show normal growth phenotypes in vitro is correlated with loss of transforming growth factor‐β1‐mediated growth inhibition. Arch Virol. 1999;144(2):241‐257. [DOI] [PubMed] [Google Scholar]
- 65. Arvanitakis L, Yaseen N, Sharma S. Latent membrane protein‐1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF‐beta 1‐mediated growth inhibition in EBV‐positive B cells. J Immunol. 1995;155(3):1047‐1056. [PubMed] [Google Scholar]
- 66. Mori N, Morishita M, Tsukazaki T, Yamamoto N. Repression of Smad‐dependent transforming growth factor‐β signaling by Epstein‐Barr virus latent membrane protein 1 through nuclear factor‐κB. Int J Cancer. 2003;105(5):661‐668. [DOI] [PubMed] [Google Scholar]
- 67. Lo AK, Dawson CW, Lo KW, Yu Y, Young LS. Upregulation of Id1 by Epstein‐Barr virus‐encoded LMP1 confers resistance to TGFβ‐mediated growth inhibition. Mol Cancer. 2010;9(1):155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morris MA, Dawson CW, Laverick L, et al. The Epstein‐Barr virus encoded LMP1 oncoprotein modulates cell adhesion via regulation of activin A/TGFβ and β1 integrin signalling. Sci Rep. 2016;6(1): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Flavell JR, Baumforth KR, Wood VH, et al. Down‐regulation of the TGF‐beta target gene, PTPRK, by the Epstein‐Barr virus–encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood. 2008;111(1):292‐301. 1 [DOI] [PubMed] [Google Scholar]
- 70. Wood V, O'neil J, Wei W, Stewart S, Dawson C, Young L. Epstein‐Barr virus‐encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGF [beta] signaling pathways. Oncogene. 2007;26(28):4135‐4147. [DOI] [PubMed] [Google Scholar]
- 71. Kim DH, Chang MS, Yoon CJ, et al. Epstein‐Barr virus BARF1‐induced NFκB/miR‐146a/SMAD4 alterations in stomach cancer cells. Oncotarget. 2016;7(50):82213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Motsch N, Pfuhl T, Mrazek J, Barth S, Grässer FA. Epstein‐Barr virus‐encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR‐146a. RNA Biol. 2007;4(3):131‐137. [DOI] [PubMed] [Google Scholar]
- 73. Turrini R, Merlo A, Martorelli D, et al. A BARF1‐specific mAb as a new immunotherapeutic tool for the management of EBV‐related tumors. OncoImmunology. 2017;6(4):e1304338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lei X, Zhu Y, Jones T, Bai Z, Huang Y, Gao S‐JA. Kaposi's sarcoma‐associated herpesvirus microRNA and its variants target the transforming growth factor β pathway to promote cell survival. J Virol. 2012;86(21):11698‐11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi HS, Jain V, Krueger B, et al. Kaposi's sarcoma‐associated herpesvirus (KSHV) induces the oncogenic miR‐17‐92 cluster and down‐regulates TGF‐β signaling. PLoS Pathog. 2015;11(11):e1005255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu Y, Sun R, Lin X, Liang D, Deng Q, Lan K. Kaposi's sarcoma‐associated herpesvirus‐encoded microRNA miR‐K12‐11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J Virol. 2012;86(3):1372‐1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Di Bartolo DL, Cannon M, Liu Y‐F, et al. KSHV LANA inhibits TGF‐β signaling through epigenetic silencing of the TGF‐β type II receptor. Blood. 2008;111(9):4731‐4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Samols MA, Skalsky RL, Maldonado AM, et al. Identification of cellular genes targeted by KSHV‐encoded microRNAs. PLoS Pathog. 2007;3(5):e65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Seo T, Park J, Choe J. Kaposi's sarcoma‐associated herpesvirus viral IFN regulatory factor 1 inhibits transforming growth factor‐β signaling. Cancer Res. 2005;65(5):1738‐1747. [DOI] [PubMed] [Google Scholar]
- 80. Tomita M, Choe J, Tsukazaki T, Mori N. The Kaposi's sarcoma‐associated herpesvirus K‐bZIP protein represses transforming growth factor [beta] signaling through interaction with CREB‐binding protein. Oncogene. 2004;23(50):8272‐8281. [DOI] [PubMed] [Google Scholar]
- 81. Lee DK, Kim B‐C, Brady JN, Jeang K‐T, Kim S‐J, Human T. Cell lymphotropic virus type 1 tax inhibits transforming growth factor‐β signaling by blocking the association of Smad proteins with Smad‐binding element. J Biol Chem. 2002;277(37):33766‐33775. [DOI] [PubMed] [Google Scholar]
- 82. Zhao T, Satou Y, Sugata K, et al. HTLV‐1 bZIP factor enhances TGF‐β signaling through p300 coactivator. Blood. 2011;118(7):1865‐1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Arnulf B, Villemain A, Nicot C, et al. Human T‐cell lymphotropic virus oncoprotein tax represses TGF‐β1 signaling in human T cells via c‐Jun activation: a potential mechanism of HTLV‐I leukemogenesis. Blood. 2002;100(12):4129‐4138. [DOI] [PubMed] [Google Scholar]
- 84. Kim SJ, Winokur TS, Lee H, et al. Overexpression of transforming growth factor‐beta in transgenic mice carrying the human T‐cell lymphotropic virus type I tax gene. Mol Cell Biol. 1991;11(10):5222‐5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shimamura M, Murphy‐Ullrich JE, Britt WJ. Human cytomegalovirus induces TGF‐β1 activation in renal tubular epithelial cells after epithelial‐to‐mesenchymal transition. PLoS Pathog. 2010;6(11):e1001170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Liu T, Zheng X, Li Q, et al. Role of human cytomegalovirus in the proliferation and invasion of extravillous cytotrophoblasts isolated from early placentae. Int J Clin Exp Med. 2015;8(10):17248 [PMC free article] [PubMed] [Google Scholar]
- 87. Kossmann T, Morganti‐Kossmann MC, Orenstein JM, Britt WJ, Wahl SM, Smith PD. Cytomegalovirus production by infected astrocytes correlates with transforming growth factor‐β release. J Infect Dis. 2003;187(4):534‐541. [DOI] [PubMed] [Google Scholar]
- 88. Michelson S, Alcami J, Kim S, et al. Human cytomegalovirus infection induces transcription and secretion of transforming growth factor beta 1. J Virol. 1994;68(9):5730‐5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kwon YJ, Kim DJ, Kim JH, Park CG, Cha CY, Hwang ES. Human cytomegalovirus (HCMV) infection in osteosarcoma cell line suppresses GM‐CSF production by induction of TGF‐β. Microbiol Immunol. 2004;48(3):195‐199. [DOI] [PubMed] [Google Scholar]
- 90. Yoo YD, Chiou C‐J, Choi KS, et al. The IE2 regulatory protein of human cytomegalovirus induces expression of the human transforming growth factor beta1 gene through an Egr‐1 binding site. J Virol. 1996;70(10):7062‐7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Michaelis M, Doerr HW, Cinatl J. The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia. 2009;11(1):1‐9 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Helanterä I, Loginov R, Koskinen P, Törnroth T, Grönhagen‐Riska C, Lautenschlager I. Persistent cytomegalovirus infection is associated with increased expression of TGF‐β1, PDGF‐AA and ICAM‐1 and arterial intimal thickening in kidney allografts. Nephrology Dialysis Transplantation. 2005;20(4):790‐796. [DOI] [PubMed] [Google Scholar]
- 93. Helanterä I, Teppo A‐M, Koskinen P, Törnroth T, Grönhagen‐Riska C, Lautenschlager I. Increased urinary excretion of transforming growth factor‐β 1 in renal transplant recipients during cytomegalovirus infection. Transpl Immunol. 2006;15(3):217‐221. [DOI] [PubMed] [Google Scholar]
- 94. Tabata T, Kawakatsu H, Maidji E, et al. Induction of an epithelial integrin αvβ6 in human cytomegalovirus‐infected endothelial cells leads to activation of transforming growth factor‐β1 and increased collagen production. Am J Pathol. 2008;172(4):1127‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhou Y, Yuge A, Rajah AM, Unek G, Rinaudo PF, Maltepe E. LIMK1 regulates human trophoblast invasion/differentiation and is down‐regulated in preeclampsia. Am J Pathol. 2014;184(12):3321‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Borges ÁH, Silverberg MJ, Wentworth D, et al. Predicting risk of cancer during HIV infection: the role of inflammatory and coagulation biomarkers. AIDS (London, England). 2013;27(9):1433‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Maina EK, Abana C, Bukusi E, Sedegah M, Lartey M, Ampofo W. Plasma concentrations of transforming growth factor beta 1 in non‐progressive HIV‐1 infection correlates with markers of disease progression. Cytokine. 2016;81:109‐116. [DOI] [PubMed] [Google Scholar]
- 98. Hu R, Oyaizu N, Than S, Kalyanaraman VS, Wang X‐P, Pahwa S. HIV‐1 gp160 induces transforming growth factor‐β production in human PBMC. Clin Immunol Immunopathol. 1996;80(3):283‐289. [DOI] [PubMed] [Google Scholar]
- 99. Lin W, Weinberg EM, Tai AW, et al. HIV increases HCV replication in a TGF‐β1–dependent manner. Gastroenterology. 2008;134(3):803‐811. [DOI] [PubMed] [Google Scholar]
- 100. Li S‐W, Wang C‐Y, Jou Y‐J, et al. SARS coronavirus papain‐like protease induces Egr‐1‐dependent up‐regulation of TGF‐β1 via ROS/p38 MAPK/STAT3 pathway. Sci Rep. 2016;6(1):25754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wang C‐Y, C‐Y L, Li S‐W, et al. SARS coronavirus papain‐like protease up‐regulates the collagen expression through non‐Samd TGF‐β1 signaling. Virus Res. 2017;235:58‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li SW, Yang TC, Wan L, et al. Correlation between TGF‐β1 expression and proteomic profiling induced by severe acute respiratory syndrome coronavirus papain‐like protease. Proteomics. 2012;12(21):3193‐3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhao X, Nicholls JM, Chen Y‐G. Severe acute respiratory syndrome‐associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor‐β signaling. J Biol Chem. 2008;283(6):3272‐3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jolly L, Stavrou A, Vanderstoken G, et al. Influenza promotes collagen deposition via αvβ6 integrin‐mediated transforming growth factor β activation. J Biol Chem. 2014;289(51):35246‐35263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Carlson CM, Turpin EA, Moser LA, et al. Transforming growth factor‐β: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010;6(10):e1001136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li N, Ren A, Wang X, et al. Influenza viral neuraminidase primes bacterial coinfection through TGF‐β–mediated expression of host cell receptors. Proc Natl Acad Sci. 2015;112(1):238‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Scott MK, Chommanard C, Lu X, et al. Human adenovirus associated with severe respiratory infection, Oregon, USA, 2013–2014. Emerg Infect Dis. 2016;22(6):1044‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Tarakanova VL, Wold WS. Transforming growth factor β1 receptor II is downregulated by E1A in adenovirus‐infected cells. J Virol. 2003;77(17):9324‐9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Datto MB, PP‐c H, Kowalik TF, Yingling J, Wang X‐F. The viral oncoprotein E1A blocks transforming growth factor beta‐mediated induction of p21/WAF1/Cip1 and p15/INK4B. Mol Cell Biol. 1997;17(4):2030‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nishihara A, Hanai J‐i, Imamura T, Miyazono K, Kawabata M. E1A inhibits transforming growth factor‐β signaling through binding to Smad proteins. J Biol Chem. 1999;274(40):28716‐28723. [DOI] [PubMed] [Google Scholar]
- 111. Tarakanova VL, Wold WS. Adenovirus E1A and E1B‐19K proteins protect human hepatoma cells from transforming growth factor β1‐induced apoptosis. Virus Res. 2010;147(1):67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Faghihloo E, Rezaie F, Salimi V, et al. Molecular epidemiology of human respiratory syncytial virus in Iran. Acta Virol. 2011;55(1):81‐83. [DOI] [PubMed] [Google Scholar]
- 113. Salimi V, Ramezani A, Mirzaei H, et al. Evaluation of the expression level of 12/15 lipoxygenase and the related inflammatory factors (CCL5, CCL3) in respiratory syncytial virus infection in mice model. Microb Pathog. 2017. Aug;109:209‐213. [DOI] [PubMed] [Google Scholar]
- 114. Pokharel SM, Shil NK, Bose S. Autophagy, TGF‐β, and SMAD‐2/3 signaling regulates interferon‐β response in respiratory syncytial virus infected macrophages. Front Cell Infect Microbiol. 2016;6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kindrachuk J, Wahl‐Jensen V, Safronetz D, et al. Ebola virus modulates transforming growth factor β signaling and cellular markers of mesenchyme‐like transition in hepatocytes. J Virol. 2014;88(17):9877‐9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Beckham JD, Tuttle K, Tyler KL. Reovirus activates transforming growth factor β and bone morphogenetic protein signaling pathways in the central nervous system that contribute to neuronal survival following infection. J Virol. 2009;83(10):5035‐5045. [DOI] [PMC free article] [PubMed] [Google Scholar]