

Summary
Enteroviruses are common human pathogens, and infections are particularly frequent in children. Severe infections can lead to a variety of diseases, including poliomyelitis, aseptic meningitis, myocarditis and neonatal sepsis. Enterovirus infections have also been implicated in asthmatic exacerbations and type 1 diabetes. The large disease spectrum of the closely related enteroviruses may be partially, but not fully, explained by differences in tissue tropism. The molecular mechanisms by which enteroviruses cause disease are poorly understood, but there is increasing evidence that the two enteroviral proteases, 2Apro and 3Cpro, are important mediators of pathology. These proteases perform the post‐translational proteolytic processing of the viral polyprotein, but they also cleave several host‐cell proteins in order to promote the production of new virus particles, as well as to evade the cellular antiviral immune responses. Enterovirus‐associated processing of cellular proteins may also contribute to pathology, as elegantly demonstrated by the 2Apro‐mediated cleavage of dystrophin in cardiomyocytes contributing to Coxsackievirus‐induced cardiomyopathy. It is likely that improved tools to identify targets for these proteases will reveal additional host protein substrates that can be linked to specific enterovirus‐associated diseases. Here, we discuss the function of the enteroviral proteases in the virus replication cycle and review the current knowledge regarding how these proteases modulate the infected cell in order to favour virus replication, including ways to avoid detection by the immune system. We also highlight new possibilities for the identification of protease‐specific cellular targets and thereby a way to discover novel mechanisms contributing to disease. Copyright © 2016 John Wiley & Sons, Ltd.
Abbreviations used
- 2Apro
enteroviral protease 2A, 3Cpro, enteroviral protease 3C
- HFMD
hand‐foot‐and‐mouth disease
- PRR
pattern recognition receptor; VP, viral capsid protein
- IgSF
immonoglobulin superfamily
- ICAM‐1
intracellular adhesion molecule
- CAR
coxsackievirus–adenovirus receptor
- PVR
poliovirus receptor
- DAF
decay accelerating factor
- LDL‐R
low‐density lipoprotein receptor
- PTB
polypyrimidine tract‐binding protein
- 3Dpol
enteroviral RNA‐dependent RNA polymerase
- 3B
uridylylated‐VPg; EV, extracellular vesicle
- EV71
enterovirus 71; EV68, enterovirus 68
- CVB
coxsackievirus; SRF, serum response factor
- miRNA
micro RNA
- HRV
human rhinovirus
- eIF4G
eukaryotic translation initiation factor 4 gamma 1
- IRES
internal ribosome entry site
- CRB
cAMP response element‐binding protein
- Oct‐1
octamer binding transcription factor 1
- NLS
nuclear localization signal
- NPC
nuclear pore complex
- SRp20
cellular splicing factor
- PCBP
cellular RNA‐binding protein poly(rC)‐binding protein
- dsRNA
double stranded RNA
- IFIH1
interferon induced with helicase C domain 1
- TLR
toll‐like receptor
- RIG‐I
retinoic acid‐inducible gene I
- MAVS
mitochondrial antiviral‐signalling protein 1
- SCARB2
scavenger receptor B2
- TRIF
TIR‐domain‐containing adapter‐inducing interferon‐B
- ISG
interferon‐stimulated gene
- G3BP1
Ras GTPase‐activating protein‐binding protein
Introduction
Enterovirus infections are among the most common types of virus infections in humans. The majority of infections are subclinical, but occasionally, they cause diseases such as the common cold, hand‐foot‐and‐mouth disease (HFMD), myocarditis meningitis, otitis media, neonatal sepsis, pancreatitis, poliomyelitis and sinusitis 1, 2. In addition, enterovirus infections have been associated with inflammatory diseases, such as type 1 diabetes, asthma and allergies 1, 3. Our understanding of the complex processes leading to these different disorders is limited, and a better knowledge of how these viruses interact with the host is essential for the discovery of disease‐causing mechanisms and the identification of targets for the development of therapeutic measures. All enteroviruses encode two proteases, 2A (2Apro) and 3C (3Cpro), which are essential for the cleavage of the viral polyprotein into structural‐ and non‐structural proteins. These proteases can also cleave host‐cell proteins, and cellular targets already identified include transcription factors, proteins controlling nuclear import/export, mitochondria‐associated proteins, pattern recognition receptors (PRRs) and other proteins, many of which are involved in the activation of the host immune response 4, 5, 6, 7, 8, 9, 10, 11, 12, 13. The cleavage of host‐cell proteins may contribute to pathology 14, 15, 16, and a better insight into the target specificities of the enteroviral proteases, coupled with information on how protein cleavages affect the biological functions of the cell, is likely to reveal novel disease mechanisms as well as identify ways to treat and prevent enterovirus‐mediated diseases.
Classification and Structure of Enteroviruses
The molecular characteristics, such as the nature of replication, morphology and physiochemical properties of the virion define the genus Enterovirus. The genus belongs to the family of Picornaviridae, under the order of Picornavirales, and the genus is divided into twelve species: Enterovirus A–H, J and Rhinovirus A–C 17.
The enterovirus virion contains a single positive strand RNA genome with a length of around 7.5 kb. The genome is densely packed into an icosahedral capsid, which is composed of 60 copies of four separate viral capsid proteins (VP1–VP4). Upon infection, the capsid undergoes structural changes, causing the release of the viral genome into the cytoplasm where it undergoes translation by the translation machinery of the host. The enterovirus genome encodes a single open reading frame, resulting in translation of all viral proteins as a single polyprotein.
The Enterovirus Life Cycle
Enterovirus receptors and virus entry
Enteroviruses use several types of cell‐surface molecules for binding and initiating their entry into cells (Figure 1). The majority of the known enterovirus receptors belong to the immunoglobulin superfamily (IgSF) 18, and more specifically, the type I transmembrane glycoproteins. They include the intracellular adhesion molecule (ICAM‐1) 19, the coxsackievirus–adenovirus receptor (CAR) 20 and the poliovirus receptor (PVR) 21. Non‐IgSF type receptors include decay accelerating factor (DAF), the low‐density lipoprotein receptor (LDL‐R), scavenger receptor B2 (SCARB2) and integrins 22, 23, 24, 25, 26.
Figure 1.

Proposed model of the enterovirus replication cycle. (1) Entry. After attachment to host‐cell surface receptors virus is internalized and uncoated, leading to the release of viral RNA into the cytoplasm. (2) Translation. Viral polyprotein is translated and then processed by the 2Apro and 3Cpro proteases. Host‐cell translation is also perturbed as a component of the translation machinery (eIF4G) cleaved by 2Apro. (3) Immune evasion. Host‐cell immune response is blunted by proteolysis mediated by viral proteases 2Apro and 3Cpro as intracellular receptors (MDA5/RIG‐1), and proteins relaying innate signalling (IPS‐1) are targeted, blocking the production of interferons and cytokines. (4) Replication. Viral proteins, in orchestration with host‐cell factors, replicate the viral RNA at membrane‐associated replication sites. (5) Release. Enteroviral positive‐stranded RNA genomes are encapsidated by the viral structural proteins, and the new viral progeny are released either by cell lysis or in extracellular vesicles.
The tissue and cell distribution of virus receptors is an important determinant for virus tropism. Polioviruses primarily infect human gastrointestinal lymphoid tissues, such as tonsils and Peyer's patches expressing the PVR 27, 28. If the virus spreads to the circulation and, thereafter, to the central nervous system, neuronal cells expressing PVR can become infected, resulting in muscle weakness and paralysis.
Through their attachment to the cell‐surface receptors, enteroviruses gain access into the cell via endocytotic pathways. Routes of entry depend on the species of the virus and the cell type. The caveolae‐ 29 and the clathrin‐dependent pathways 30, as well as other internalization routes 31, have been described as possible entry mechanisms. The presence of a receptor on the cell surface is, however, not the only determinant for cellular permissiveness. The virus may enter the cell but fail to replicate if, for example, there is a lack of endogenous cellular proteins required for viral propagation. An example of such an endogenous protein is the polypyrimidine tract‐binding protein (PTB) 32. Alternatively, the receptor‐expressing cell can enter an antiviral state and thereby, may not be permissive to infection (reviewed in 33). Therefore, the dependence on various cellular factors makes host susceptibility and permissiveness to infection a multifaceted and complex phenomenon.
Enterovirus translation and replication
After endocytosis, the virus particle undergoes structural changes, resulting in the uncoating of the viral genome and engagement of the capsid proteins with the endosomal membrane, presumably via the VP1 N‐terminus. This allows the delivery of viral RNA with a 5′‐linked VPg protein 34 and a 3′‐polyadenylated tract 35 into the cytosol, where it is translated by the host ribosomes into the viral polyprotein.
The polyprotein encoded by a single open reading frame is divided into three regions, P1–P3 (Figure 1). The P1 region contains four structural proteins (VP1–VP4), whereas the P2 and P3 regions together contain seven non‐structural proteins (2A–2C and 3A–3D), which are required in the different stages of the viruses' replication cycle. The proteolytic processing of the polyprotein into separate proteins is already initiated during translation by the viral proteases 2Apro and 3Cpro 36, 37, 38.
The P1 region, encoding the structural proteins of the capsid, is the first one to be translated, followed by the P2 region, which contains three non‐structural proteins (2A, 2B and 2C). During the translation of the P2 region, as 2Apro is translated first, 2Apro makes an in cis cleavage, separating itself and the P2 region from the P1 region before the full polyprotein has been translated. Translation continues through the P3 region, and this region includes the second protease, 3Cpro, which is responsible for eight out of the 10 cleavages of the viral polyprotein. The cleavage carried out by the two proteases give rise to all of the non‐structural proteins, with several precursor proteins, and three structural proteins: VP1, VP3 and VP0. VP0 is further cleaved into VP2 and VP4 by an unknown mechanism 39, which may entail an RNA‐mediated autocatalytic reaction during the encapsidation process 40.
Viral replication takes place in the proximity of membranous vesicles, derived partly from the endoplasmic reticulum 41. The positive strand RNA is transcribed by the virally encoded polymerase 3Dpol into a complementary negative strand RNA. The RNA synthesis is primed by uridylylated‐VPg (3B), which is associated with the replication complex and recruited to the 3′ end of the negative strand viral genome to initiate RNA synthesis 42. The negative strand RNA then serves as a template for the transcription of the positive strand RNA genome. Multiple positive‐strand RNAs can be synthesized from a single negative‐strand template, making positive‐sense RNA abundant and directly available for translation, synthesis of additional negative‐sense RNA and encapsidation 43 (Figure 1).
Encapsidation and virus release
The accumulation of newly synthesized viral RNA and structural proteins leads to packaging of the viral genome into the capsids, thus forming new viral progeny 44. Surprisingly, very little is known about the encapsidation process, but some studies have indicated that the process of virus assembly is coupled to RNA synthesis 45 on the surface of cytoplasmic membranes 46.
The classical view of enterovirus release is that it occurs by cell lysis. Intriguingly, new observations challenge this model as virus‐containing extracellular vesicles shed by the host cells could potentially disseminate the infection 47, 48. Persistent enterovirus infections without evident cytopathic effect in tissues and cell models have also been reported 49, 50, 51, supporting this recently described nonlytic model of virus release.
Enterovirus‐Mediated Diseases
The most well‐known enteroviral disease is poliomyelitis, which is caused by three different poliovirus serotypes. Poliomyelitis has been virtually eradicated in developed countries, but recently, two other enteroviruses, enterovirus 71 (EV71) and enterovirus 68 (EV68), have been demonstrated to cause an acute flaccid paralysis resembling poliomyelitis 52, 53, 54, 55. Moreover, EV71 and coxsackievirus A6, A10 and A16 can cause HFMD 56. Other enteroviruses, coxsackieviruses (CVBs) in particular, have been associated with acute myocarditis and the later development of dilated cardiomyopathy 14, 15, 57, 58.
Diseases related to enterovirus infections may result either from an acute infection or only appear after the acute phase is over. This indicates that there may be different mechanisms contributing to tissue pathology. Acute infections are typically associated with local inflammation (e.g. the common cold, otitis, pancreatitis and hepatitis) and are cleared relatively rapidly by the immune system. In contrast, conditions like dilated cardiomyopathy and post‐polio syndrome are more likely to result from infections that have not been completely cleared and have entered a persistent infection phase.
Although poliomyelitis caused by poliovirus is the most studied enterovirus‐associated disease, surprisingly little is known about the disease mechanisms 59. Even less is known on how most other enteroviruses cause disease (e.g. EV71 and EV68). An exception, however, is CVB‐induced myocarditis and the subsequent development of chronic dilated myopathy, the latter a severe condition that usually leads to heart failure 15, 58. During the acute phase of the infection, the virus‐encoded protease 2Apro cleaves the cellular protein dystrophin, which leads to sarcolemmal disruption and reduction in myocyte contractility 14, 57. In their recent publication, Matthew et al. postulated a more detailed molecular mechanism for the damage caused by the infection, namely that the C‐terminal 2Apro cleavage product is retained in the sarcoglycan complex. This in turn decouples actin from the sarcolemma and subsequently prevents the recovery of the full‐length dystrophin at the sarcolemmal membrane 16.
A further contribution to impaired cardiac function is the 2Apro‐mediated cleavage of the transcription factor serum response factor (SRF) 60. SRF is normally highly expressed in heart muscle cells and contributes to the regulation and expression of heart tissue‐specific genes, including contractile and regulatory proteins as well as miRNAs controlling specific heart cell functions 61. The 2Apro breaks the transactivation domain of SRF and thereby diminishes the expression of genes regulated by this transcription factor 60.
Coxsackieviruses have been shown to cause persistent infection of the heart both in animal models 62, 63 and humans 64. Characteristic of other persistent CVB infections, they also contain deletions of varying size in their 5′ end 64. The persistent infection may lead to a chronic immune response and also possibly autoimmune responses as exemplified by antibody responses to cardiac antigens such as cardiac myosin and troponin I 15. The chronic inflammation is likely to contribute further to cardiac dysfunction.
The Enterovirus‐Encoded Proteases 2Apro and 3Cpro
Structural features of enterovirus proteases
The enterovirus proteases 2Apro and 3Cpro are multifunctional cysteine proteases, belonging to the chymotrypsin‐related endopeptidase protease family 65 (MEROPS 2Apro: C03.020 and 3Cpro: C03.011). When comparing 2Apro to 3Cpro, a primary sequence alignment of the consensus sequences shows only ~20% identity, even though the two proteases have strikingly similar tertiary structures (Figure 2). Among the different species of enteroviruses, the proteases share approximately 50–75% sequence identity, the rhinoviruses being the most divergent group with around 35–55% identity with the other species (Figure 3). The amino acid residues of the catalytic triad are fully conserved throughout the Enterovirus genus. In addition, the amino acid residues surrounding the catalytic residues are more conserved when compared to the rest of the protein, which is indicative of similarities in the mechanisms involving sequence specificity and cleavage among the enteroviral proteases.
Figure 2.

Sequence alignments of 2Apro and 3Cpro, their topological structure presentations and 3‐dimensional tertiary structures. Orange and cyan colourings are used for 2Apro, and red and blue colourings are used for 3Cpro. Panels (a) and (b) show the primary amino acid sequence alignment of 2Apro and 3Cpro within the Enterovirus family. The residues of the catalytic triad, as well as the ion‐binding residues, are highlighted with arrows underneath the sequences. The secondary structure elements are shown above the alignments (cylinder = alpha‐helical structure; arrow = beta‐sheet structure; turns in purple; 3/10 helices in pink). Secondary structure assignment was made using DSSP 66. Panels (c) and (d) show a topological schematic of the proteases. The same visual secondary structure representations are used as in panels (a) and (b). Panel (e) shows a cartoon representation of EV71 2Apro (PDBID: 4FVB). The side chains of the amino acids of the catalytic triad are shown as sticks. Similarly to panel (e), panel (f) shows a cartoon representation of CVB3 3Cpro (PDBID: 2VB0). Panels (g) and (h) show surface representations of the proteases, with their active sites highlighted with yellow. In comparison, the active site of 2Apro is more confined and restricted by surrounding structures than the active site of 3Cpro.
Figure 3.
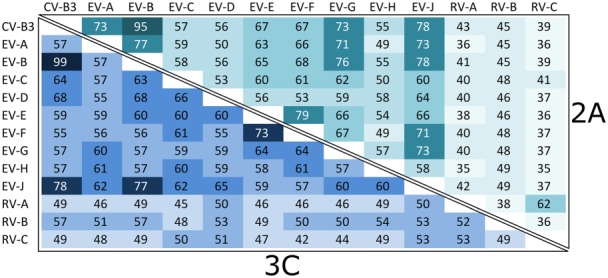
Primary amino acid sequence percentage identity matrix of the enteroviral proteases. The top‐right half of the matrix shows sequence identities between the different enteroviral species and coxsackievirus B3 for 2Apro, and the lower‐left half for 3Cpro. The average sequence conservation between the different species is 53% for 2Apro and 56% for 3Cpro. The rhinoviruses show the most sequence divergence with around 35–50% percentage identities for 2Apro and 45–55% for 3Cpro.
The tertiary structures of both of the proteases are composed of two separate domains. In the case of 2Apro, the two domains include a six‐stranded antiparallel β‐sheet barrel and a β‐sheet pile packed on its side (Figure 2). The tertiary structure of 3Cpro is a combination of two twisted β‐barrels, which are packed perpendicular to each other. In both proteases, these two domains participate in the formation and positioning of the catalytic triad. The catalytic triad is composed of histidine, aspartic acid and cysteine in the case of 2Apro, and histidine, glutamic acid and cysteine in the case of 3Cpro. The cysteine in the catalytic triad acts as a nucleophile in the proteolytic reaction in both proteases. Characteristic for both proteases are also the conserved ion‐binding motifs that are located on the opposite side from the catalytically active site. For 2Apro, a zinc ion is located in one end of the barrel, bound by three cysteines and one histidine residue. For 3Cpro, a chlorine ion is bound to an Asp‐Ile‐Arg stretch residing in the loop connecting the two barrels.
Both monomeric and dimeric quaternary structure forms have been reported for 2Apro. Liebig et al. found that HRV2 2Apro showed a dimeric state in gel filtration analysis, while CVB4 2Apro was found to be monomeric 67. In another study, 2Apro from HRV14 was found to be monomeric by gel filtration analysis 68. In a study by Cai et al., EV71 2Apro was found to form a disulphide‐linked dimer with a negligible monomer–monomer interface in crystal structure, but the oligomeric state in solution could not be shown 69. Mu et al. crystallized EV71 2Apro and found a monomer in the asymmetric unit 70. In another recent study of CVA16 2Apro, both dimeric and hexameric quaternary assemblies in the solution and in crystal were reported 71. The hexameric form was found to dissociate to dimers with an addition of DTT, which could indicate that the hexamer is not present in the reducing intracellular environment. Both dimers and hexamers, separated by size exclusion chromatography, exhibited equally efficient proteolytic activity.
It is most likely that the quaternary structure of 3Cpro is monomeric because it lacks a third domain, whose importance has been shown for dimerization in related coronavirus proteases 72, 73. This is in contrast to what has been observed when solving the crystal structure, in which 3Cpro proteases assembled as dimers. For example, 3Cpro from EV68 and EV93 showed a dimeric assembly in crystal structures. On the contrary, they were found to be monomeric in gel filtration and DLS experiments 74, 75. Therefore, the dimers observed in crystals are not likely to represent the biologically relevant forms.
Sequence specificity of enteroviral proteases
The sequence specificity, and specifically the sequences that the 2Apro and 3Cpro proteases are able to cleave (or not), has not been established or studied comprehensively. To date, the amino acid residues P4, P2, P1, P1′ and P2′ are recognized as being important determinants for the sequence specificity of enteroviral proteases (Figure 4) 65, 76. For the substrate recognition of 2Apro, the most important residue is P1′, which is exclusively a glycine. Following P1′ in order of importance are P2, occupied mainly by threonine and asparagine; P2′, occupied by proline, alanine and phenylalanine; and P4, occupied most frequently by leucine or threonine. For 3Cpro, the residues P1 and P1′ show the least amount of variance in the substrate sequence. The preferred residues for these positions are glutamine or glutamate for P1, and glycine, asparagine or serine for P1′. In addition, the most common residue is alanine in position P4 and proline in position P2′. The most obvious feature for determining the substrate specificity of both 2Apro and 3Cpro is the strong conservation of the glycine residue in position P1′ 76, and the present understanding of which residues are important in the other positions may be revised as new information becomes available (refer to the ‘Methods to Identify New Cellular Substrates for Enteroviral Proteases’ and ‘Cleavage Predictions Using in Silico Analysis Techniques, Bioinformatics’ sections in the succeeding texts).
Figure 4.
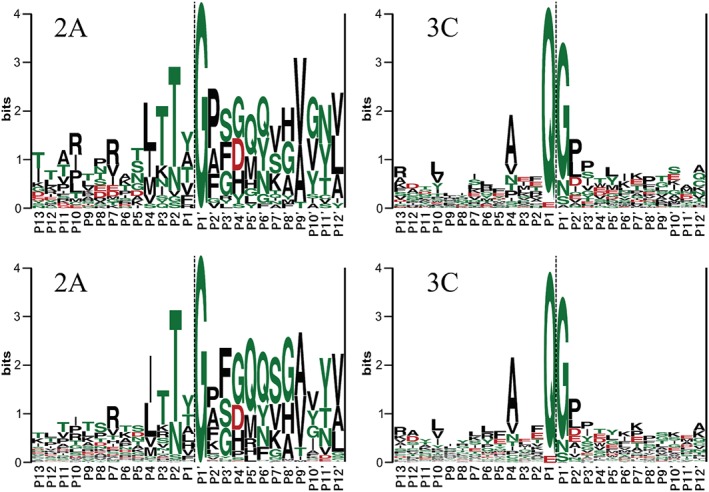
The substrate sequence LOGOs of the enteroviral proteases published by Blom et al. 1996 76 (upper panels) (reprinted with permission from John Wiley and Sons) and new logos based on a larger substrate pool (lower panels; Nurminen et al. manuscript) show the most conserved positions of the substrate sequences around the cleavage site. Hydrophilic residues are shown in green colour, and hydrophobic residues are shown in black colour. Negatively charged residues are coloured red. The lower panel logos were created using all currently available enteroviral polyprotein sequences in the Uniprot database 77. Duplicate sequences were removed to avoid bias towards sequences with multiple entries. The logos were generated using WebLogo 78. Left panels: The most important recognition sites for 2Apro in order of lowest variability are at locations P1′, P2, P2′, P4 and P3. The relatively low variability to the right of P2′ can be a result of the sequence being a functional part of 2Apro itself, as the protease cleaves its own N‐terminal end free from the polyprotein by in cis cleavage. Right panels: The most important recognition sites for 3Cpro in order of lowest variability are at locations P1, P1′, P4 and P2′.
Protease inhibitors as antiviral compounds
As the protease‐dependent processing of the enteroviral polyprotein is indispensable for virus replication, the viral proteases have been recognized as potential targets for antiviral intervention 79, 80. Of the two proteases, 3Cpro in particular, has been considered a compelling target, as the polyprotein has several cleavage sites specific for the protease. Many of the inhibitors that have been developed and studied are small molecule peptide mimetics that target the active site of the proteases, but other small molecular compounds have also been described 81. Structural conservation and the commonly shared proteolytic mechanism seen between different viral proteases make it possible to develop inhibitors that have an antiviral activity towards many species in the Enterovirus genus and furthermore, occasional activity towards more distantly related viruses. Such inhibitor candidates include pyrazole compounds that target 3Cpro from different enteroviruses as well as coronavirus protease homologues of 3Cpro 79, microcyclic inhibitors against enterovirus 3Cpro and noro‐ and SARS‐coronavirus 3Cpro homologues 82. Additionally, a lycorine derivative, 1‐acetyllycorine, has been shown to inhibit EV71 2Apro by stabilizing a special conformation of its zinc finger motive. Similarly, it can furthermore act on the homologous zinc finger of Hepatitis C virus NS3 protease 81. The rhinovirus 3Cpro inhibitor rupintrivir 83 is also active against noroviruses 84.
To date, of all the compounds studied, only rupintrivir and its analogue AG7404 (or compound 1) 85 have progressed to clinical trials 85, 86, 87. Their development as therapeutics for rhinovirus infection has since stalled, possibly a result of their limited activity in clinical trials 88, 89. Recently, rupintrivir has, however, gained renewed attention as it proved to be effective against EV71, CAV16 and EV68 90, 91, 92, 93. These interesting and optimistic results put renewed focus on the development of antivirals that target viral proteases, and it is possible that one or several novel drug candidates may show efficacy in clinical trials and reach the market in the coming years.
Interactions between 2Apro and 3Cpro with host‐cell transcription and translation machinery
As mentioned in the preceding texts, the enterovirus proteases fulfil several other functions in addition to cleaving the viral polyprotein into mature viral proteins. For example, they cleave cellular proteins in order to favour viral propagation over cellular protein production. The protease 2Apro interferes with and shuts down host‐cell protein synthesis through cleavage of eukaryotic translation initiation factor 4 gamma 1 (eIF4G) 8, an essential component of the cap‐dependent RNA translation machinery. As enteroviruses are lacking a 7‐methylguanosine cap, the cleavage of eIF4G will not affect viral protein synthesis. Instead, the enteroviruses use a highly ordered secondary structure in the 5′ end of the viral RNA called the internal ribosome entry site (IRES) to achieve the initiation of translation 94, 95.
Host‐cell gene transcription is also affected by enterovirus infection. During infection, the 3CD precursor protein enters the nucleus and inhibits the transcription of cellular proteins by cleavage of the TATA box, cAMP response element‐binding protein, octamer binding transcription factor 1 (Oct‐1) and transcriptional activating factor p53 9, 96, 97, 98. Although the polymerase in 3CD contains a nuclear localization signal (NLS) 99, a recent study showed that 2Apro‐mediated proteolysis is required for the nuclear translocation of 3CD 100.
In addition to a direct cleavage of cellular proteins (Tables 1 and 2; for more complete list of published substrates, refer to Tables S1 and S2), the proteases can also indirectly affect cellular proteins to further promote viral replication. For example, 2Apro targets several nuclear pore complex (NPC) proteins like Nup62, ‐98 and ‐153 114, 115. This disrupts the NPC and results in the rearrangement of nuclear proteins into the cytoplasm, where viral replication occurs. An example of a protein that is redistributed in this process is cellular splicing factor (SRp20), which binds the cellular RNA‐binding protein poly(rC)‐binding protein (PCBP) and recruits ribosomes to the replicating viral RNA to promote IRES‐dependent initiation of the translation 114, 116. Thus, the relocation of cellular transcription factors is utilized to modulate both viral translation and at a later stage, the generation of a new viral RNA genome 117, 118, 119.
Table 1.
Examples of published enteroviral 2A substrates
| Target protein | Virus | Refseq/UniProtKB AC | Gene | Cleavage site (sequence) | Substrate's cellular localization/function | Consequence(s) of proteolytic cleavage | Ref. |
|---|---|---|---|---|---|---|---|
| Dystrophin | CVB3 | NP_000100/P11532 | DMD | PGLTTI2434‐GASP | Cytoplasmic/Connects the cytoskeleton of a muscle fibre to the surrounding extracellular matrix | Sarcolemmal disruption leading to myocarditis and cardiomyopathy | 14 |
| eIF4GI | CVB4 Polio | NP_886553/Q04637 | EIF4G1 | TTLSTR681‐GPPR | Cytoplasmic/Translation initiation | Decline of host‐cell protein synthesis | e.g. 101, 102 |
| Melanoma differentiation‐associated protein 5(MDA5) | EV71 | NP_071451/Q9BYX4 | IFIH1 | RTVATS53‐GNMQa | Nuclear, cytosolic/cellular processes involving translation initiation, nuclear and mitochondrial splicing and ribosome and spliceosome assembly | Inhibition of type I interferon response | 4, 10 |
| Interferon (α, β and ω) receptor 1 | EV71 | NP_000620/P17181 | IFNAR1 | RSDESV56‐GNVTa | Cell membrane/mediates type I interferon signalling | Antagonizes type I interferon signalling | 7 |
| RVQASD311‐GNNTa | |||||||
| Nucleoporin 62 | Polio RV16 | NP_001180286/P37198 | NUP62 | PATQTT72‐GFTFa | — | — | 103, 104 |
| ATITST217‐GPSLa | |||||||
| TPVTTA246‐GAPTa | |||||||
| EHLNTS461‐GAPAa | |||||||
| Nucleoporin 98 | Polio HRV2 HRV16 | NP_005378/P52948 | NUP98 | VGSTLF374‐GNNK | Nuclear membrane/traffic of biological molecules between the nucleus and the cytoplasm | Prevent mRNA trafficking from nucleus to cytoplasm. Relocation of cellular proteins and inhibition of nuclear import/export. | 103, 104, 105 |
| KALQTT552‐GTAKa | |||||||
| Nucleoporin 153 | Polio | NP_001265138/P49790 | NUP153 | SCTVTT781‐GTLGa | — | — | 103 |
| QTTSST1266‐GTAVa | |||||||
| NNTTTS1287‐GFGFa | |||||||
| Serum response factor | CVB3 | NP_003122/P11831 | SRF | TVLKST326‐GSGP | Nucleus/Cardiac‐enriched transcription factor | Impaired cardiac function by downregulation of cardiac‐specific contractile and regulatory genes | 60 |
Equal to UniprotKB sequence P52948 amino acid G569.
Predicted, unconfirmed cleavage site (Nurminen et al. Manuscript in preparation).
Table 2.
Examples of published enteroviral 3C substrates
| Target protein | Virus | Refseq/UniProtKB AC | Gene | Cleavage site (sequence) | Substrate's cellular localization/function | Consequence(s) of proteolytic cleavage | Ref. |
|---|---|---|---|---|---|---|---|
| Cleavage stimulation factor (Cst‐64) | EV71 | NP_001293138/P33240 | CSTF2 | LMQASM250‐QGGV one or more of glycines: 483, 496, 505, 510 and 515 | Nucleus/Recognizes the second polyadenylation sequence element on pre‐mRNA | Impairs cellular 3′‐end pre‐mRNA processing and polyadenylation. | 106 |
| CRE‐binding protein/cyclic AMP‐responsive element‐binding protein 1 | Polio | NP_004370/P16220 | CREB1 | YIAITQ187‐GGAI | CRE‐binding protein/cyclic AMP‐responsive element‐binding protein 1 | Inhibition of CREB‐activated transcription in host cells | 107 |
| Interferon regulatory factor 7, IRF7 | EV71 | NP_001563/Q92985 | IRF7 | LLQAVQQ189‐SCLA | Nucleus/Transcription factor | Inhibits IFN gene expression | 5 |
| Mitochondrial antiviral signalling protein (MAVS) | CVB3 | NP_065797/Q7Z434 | MAVS | PVQETQ0148‐APES | Mitochondrial antiviral‐signalling protein | Inhibition of types I and III interferon response — MAVS release from mitochondria, and morphological and functional changes of mitochondria | 13, 108 |
| Nucleoporin 62 | RV14 | NP_714941/P37198 | NUP62 | Many potential cleavage sites | Nuclear membrane/traffic of biological molecules between the nucleus and the cytoplasm | Relocation of cellular proteins and inhibition of nuclear import | 109 |
| Nucleoporin 153 | RV14 RV16 | NP_001265138/P49790 | NUP153 | Many potential cleavage sites | — | Prevent mRNA trafficking from nucleus to cytoplasm | 104, 109, 110 |
| Octamer binding transcription factor | Polio RV16 | NP_002688/P14859 | POU2F1 (OCT1) | KLGFTQ329‐GDVG | Nucleus/Transcription factor | Lose inhibition of transcriptional activation by the SV40 B enhancer | 79, 111 |
| Probable ATP‐dependent RNA helicase, RIG‐I | Polio echo1 RV16 | NP_055129/O95786 | DDX58 | KMIQTR728‐GRGRa | Cytoplasmic/Putative RNA helicase involved in viral RNA binding | Attenuate virus recognition and the innate immune response | 11 |
| p65‐RelA, transcription initiation factor TFIID subunit 4B | Polio | NP_001230913/Q04206 | RELA | QQLLNQ480‐GIPV | Nuclear factor of kappa light polypeptide gene enhancer in B‐cells NF‐κB complex | Suppression of NF‐κB response | 112 |
| TATA‐binding protein (TBP) | Polio | NP_003185/P20226 | TBP | GLASPQ18‐GAMT | Nucleus/Transcription factor | May inhibit RNA polymerase II | 9, 113 |
| TRIF, toll‐like receptor adaptor molecule 1 | CVB3 | NP_067681/Q86XR7 | TICAM1 (TRIF) | TPFALQ190‐TINA | Cytoplasm: signalosome | May suppress the types I and III IFN signalling and apoptosis | 13, 108 |
Predicted, unconfirmed cleavage site (Nurminen et al. Manuscript in preparation).
The role for 2Apro and 3Cpro in immune evasion
Infected cells have several intracellular receptors that recognize different types of viruses. The enteroviruses form a dsRNA structure during replication, and the main known receptors responsible for sensing enteroviruses are interferon induced with helicase C domain 1 (IFIH1) located in the cytoplasm and toll‐like receptor 3 (TLR3) in the endosomes. IFIH1 and the closely related PRR retinoic acid‐inducible gene I (RIG‐I) signal via a common adaptor protein called mitochondrial antiviral‐signalling protein 1 (MAVS), while TLR3 signals via TIR‐domain‐containing adapter‐inducing interferon‐B (TRIF). Signalling via MAVS and TRIF results in the phosphorylation of several transcription factors such as IRF3, IRF7 and NFkB, which then migrate into the nucleus and induce the expression of type I and III interferons (IFNs) as well as other inflammatory cytokines (Figure 1) 30, 108, 120, 121. Secreted IFNs act in an autocrine or paracrine manner to trigger the cells into entering an antiviral state by the induced expression of interferon‐stimulated genes (ISGs) 120, 122.
In addition to manipulating cellular proteins to favour viral replication, enteroviruses also utilize the proteases to escape recognition by the immune system. It has been shown that both 2Apro and 3Cpro cleave several proteins within the viral recognition pathway, thus inhibiting the induction of IFNs. For example, viral sensors like IFIH1 and RIG‐I are targets of the proteases 4, 10, 11, 12. In addition, TRIF and MAVS, both adaptor proteins for the two major RNA sensing pathways TLR3 and IFIH1/RIG‐I, are cleaved by 3Cpro and/ or 2Apro 4, 5, 13, 108, and downstream proteins like the transcription factor IRF7 can be targeted as well 5. It has also been shown that EV71 2Apro acts directly on the interferon receptor 1, reducing its expression and thereby impairing the efficacy of IFN as a treatment against infection 7.
Methods to Identify New Cellular Substrates for Enteroviral Proteases
Given that it has been noted that enteroviral proteases can contribute to disease pathology (e.g. cleavage of dystrophin in the heart muscle), it is possible that other enteroviral diseases are also associated with proteolytic activities of 2Apro and/or 3Cpro. The identification of additional host‐cell proteins that are targeted by the proteases may thus lead to the identification of novel disease mechanisms. Because enteroviruses are able to cause diverse diseases affecting different tissues and organs, it may also be of relevance to understand how these proteases act in specific tissues and cells.
There are number of approaches that have been used to study the cellular targets of 2Apro and 3Cpro. These include infection of cells or tissues, for example, 108, 118, 123, 124, 125, selective overexpression of viral proteases by transfection, for example, 7, 108, transgenic techniques 60, a variety of in vitro assays, in which the proteases have been incubated with cell lysates, for example, 108, 118, 126, and in silico prediction of the cleavage sites based on amino acid sequences and composition of potential target proteins 76. To analyse whether the experimental approaches result in cleavage by enteroviral enzymes, Western blotting is a frequently used method. With Western blotting, it is possible to observe the appearance of cleavage products and/or a decrease in the concentration of the potential target proteins (e.g. Figure 5). However, because antibodies may not recognize the produced fragments, this analysis can be cumbersome. Transfection studies have been used to reveal the protein responsible for the effects observed in the infected cells. Nevertheless, when conducting transfection studies to overexpress a selected viral protein, a caveat may be that the function of the viral protein might be dependent on other viral proteins, for example, 100. Also, it must be taken into account that the protease precursors could have different protein targets compared to mature proteases.
Figure 5.
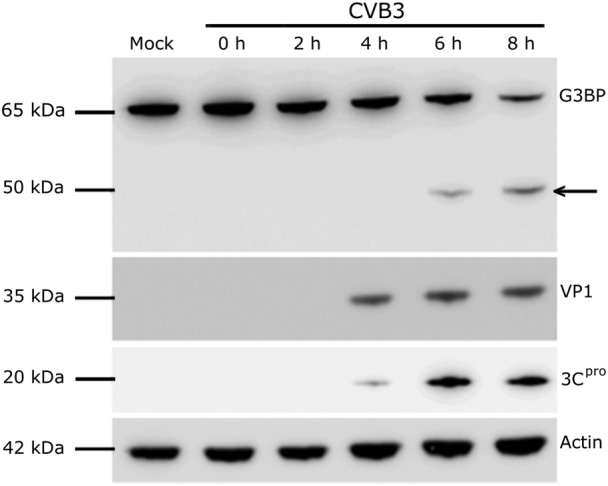
Infection of HeLa cells with coxsackievirus B3 (CVB3) results in the proteolytic cleavage of Ras GTPase‐activating protein‐binding protein 1 (G3BP1). HeLa cells were infected at a MOI of 20 with CVB3, (mock control sample treated with media alone). At each time‐point, the cells were lysed and the expression of G3BP1 and viral proteins VP1 and 3Cpro were analysed by Western blot. The arrow indicates an accumulation of G3BP1 cleavage product 6 h post infection. Actin was used as a loading control.
The technical limitations mentioned in the preceding texts may provide an explanation for the contradictory reports in the literature. One study indicated that the 3Cpro of coxsackievirus B3 can cleave MAVS 13, while other studies suggest that this cleavage is mediated by 2Apro 4, 108. Enterovirus infections can also activate endogenous proteases including caspases, which also cleave cellular proteins. Barral et al. 10 showed that poliovirus‐induced apoptosis during the course of infection correlates with the cleavage of MDA5. This cleavage also appeared after the cells were treated with puromycin, an inducer of apoptosis. Thus, activation of endogenous proteases may result in the erroneous identification of protein targets for 2Apro and 3Cpro.
Another drawback in the analyses described in the preceding text is that they are all hypothesis‐driven. Researchers identify a protein of interest and address whether it is affected by an enterovirus‐encoded protease. The outcome is that only one or a few cellular proteins are studied at the time. In order to overcome this shortage, a proteome‐wide approach was presented by Weng et al. 106, who identified new 3Cpro substrates using nuclear extracts that were treated with the 3Cpro in vitro. The treated lysates were analysed with the combination of 2D electrophoresis and mass spectrometry. They identified eight novel substrates for 3Cpro, out of which they analysed the cleavage of stimulation factor 64 in more detail. Newer methodologies in quantitative proteomics have recently been used to study how enteroviruses affect the host‐cell proteome 124, 125, and such methods may also be applied to identify new protease targets 127. A potential disadvantage with these type of analyses is that they are restricted to the proteins expressed by the infected cell, and will not provide a simultaneous analysis of the whole human proteome.
Cleavage Predictions Using in Silico Analysis Techniques, Bioinformatics
The sequences and structures of the viral polyproteins, as well as their identified cellular targets, may form the basis for the prediction of novel cellular protein substrates. The most comprehensive work completed to predict new cleavage sites of the enteroviral proteases 2Apro and 3Cpro has been performed by Blom et al. 76 through the use of a neural network algorithm for prediction. In their study, they used a collection of known cleavage sites to teach the algorithm how to predict the potential cleavage sites. The algorithm scores amino acid sequences for potential cleavage sites based on two calculated parameters, the first being sequence specificity, and the second being surface accessibility. The algorithm is published and available as a free tool on the Internet: NetPicoRNA Server (http://www.cbs.dtu.dk/services/NetPicoRNA/).
NetPicoRNA server seems, however, to underestimate the number of 2Apro cleavage targets, as for example, Nup98 is not recognized as a potential candidate, while the number of 3Cpro cleavage targets may be overestimated. At the time of the publication of the server (1996), only a limited number of cellular substrates were known, which may have caused a bias to certain kind of cleavage sites. In addition, the surface accessibility prediction was not based on resolved 3D structures, but on ab initio primary sequence analysis and the amino acid compositions of the proteins. Many new substrates have been identified since 1996 (Tables 1 and 2, Tables S1 and S3, and references therein), and a large amount of the human proteome 3D structural data is now available 128. Indeed, by the end of year 1996, the number of the structures reported in the PDB 129 was 5915, while from 1997 to 2014, this number increased to above 93 000. Therefore, it may be worth revising both the sequence specificity, as well as the surface accessibility predictions, with an aim to develop an improved algorithm that can be useful in identifying novel substrates. Such work is ongoing in our laboratories.
Concluding Remarks and Future Perspectives
Enteroviruses are important human pathogens whose manifestations range from subclinical infections to severe life‐threatening diseases. Hospital visits and symptomatic treatments for the severe infections are costly, and it is clear that a better understanding for the complex disease mechanisms underlying enterovirus‐mediated diseases would lead to more efficacious treatments. A few disease mechanisms have been identified, and in the near future, additional ones are likely to be discovered by research teams that integrate many scientific disciplines such as bioinformatics, molecular biology, proteomics and the use of patient materials. Several pathological mechanisms may be explained by the activity of the viral proteases 2Apro and 3Cpro. Some effects, such as the shutting down of host‐cell protein synthesis, immune evasion, as well as the hijacking of the cellular machinery to favour virus propagation, may be common to most enteroviruses. However, especially in the persistent types of infections when the production of viral proteins, including proteases 2Apro and 3Cpro, continues for months or years, the degradation of host proteins may be virus‐ and tissue‐specific and may lead to more selective pathological processes (e.g. myocarditis and the development of dilated cardiomyopathy). Currently, there are only a limited number of studies that have addressed the role of the proteases in a tissue‐specific manner. Better tools to globally identify and verify protease targets should assist in the identification of novel cellular protein substrates without limitations to particular cell types. Overall, this should provide a better understanding of how the proteases, in concert with other viral proteins, contribute to the induction of different diseases. Such information will also be of immediate importance for the development of novel drugs, including protease inhibitors, to prevent and treat diseases caused by enteroviruses. New prediction methods and proteome‐wide approaches are critical for the successful completion of this goal.
Conflict of Interest
The authors have no competing interests.
Supporting information
Table S1. Published enteroviral 2A substrates.
Table S2. Published enteroviral 3C substrates.
Supporting info item
Acknowledgements
The authors are very grateful to Ms Sabina Kapell for the artwork (Figure 1) and Dr Virginia Stone for the valuable comments of the manuscript. This work was supported by grants from Karolinska Institutet, Sweden; Novo Nordisk Foundation, Denmark; VINNOVA (grant number 2013‐01330), Sweden; and Tekes (grant number 1843/31/2014), Finland.
Laitinen, O. H. , Svedin, E. , Kapell, S. , Nurminen, A. , Hytönen, V. P. , and Flodström‐Tullberg, M. (2016) Enteroviral proteases: structure, host interactions and pathogenicity. Rev. Med. Virol., 26: 251–267. doi: 10.1002/rmv.1883.
References
- 1. Jacobs SE, Lamson DM, St George K, et al. Human rhinoviruses. Clinical Microbiology Reviews 2013; 26: 135–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tapparel C, Siegrist F, Petty TJ, et al. Picornavirus and enterovirus diversity with associated human diseases. Infection, Genetics and Evolution 2013; 14: 282–293. [DOI] [PubMed] [Google Scholar]
- 3. Hober D, Alidjinou EK. Enteroviral pathogenesis of type 1 diabetes: queries and answers. Current Opinion in Infectious Diseases 2013; 26: 263–269. [DOI] [PubMed] [Google Scholar]
- 4. Feng Q, Langereis MA, Lork M, et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. Journal of Virology 2014; 88: 3369–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lei X, Xiao X, Xue Q, et al. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. Journal of Virology 2013; 87: 1690–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang B, Xi X, Lei X, et al. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti‐viral type I interferon responses. PLoS Pathogens 2013; 9: e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu J, Yi L, Zhao J, et al. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. Journal of Virology 2012; 86: 3767–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lamphear BJ, Yan R, Yang F, et al. Mapping the cleavage site in protein synthesis initiation factor eIF‐4 gamma of the 2A proteases from human coxsackievirus and rhinovirus. Journal of Biological Chemistry 1993; 268: 19200–19203. [PubMed] [Google Scholar]
- 9. Clark ME, Lieberman PM, Berk AJ, et al. Direct cleavage of human TATA‐binding protein by poliovirus protease 3C in vivo and in vitro. Molecular and Cellular Biology 1993; 13: 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barral PM, Morrison JM, Drahos J, et al. MDA‐5 is cleaved in poliovirus‐infected cells. Journal of Virology 2007; 81: 3677–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barral PM, Sarkar D, Fisher PB, et al. RIG‐I is cleaved during picornavirus infection. Virology 2009; 391: 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lei X, Liu X, Ma Y, et al. The 3C protein of enterovirus 71 inhibits retinoid acid‐inducible gene I‐mediated interferon regulatory factor 3 activation and type I interferon responses. Journal of Virology 2010; 84: 8051–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mukherjee A, Morosky SA, Delorme‐Axford E, et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathogens 2011; 7: e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Badorff C, Berkely N, Mehrotra S, et al. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin‐based substrate analogue. Journal of Biological Chemistry 2000; 275: 11191–11197. [DOI] [PubMed] [Google Scholar]
- 15. Luo H, Wong J, Wong B. Protein degradation systems in viral myocarditis leading to dilated cardiomyopathy. Cardiovascular Research 2010; 85: 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barnabei MS, Sjaastad FV, Townsend D, et al. Severe dystrophic cardiomyopathy caused by the enteroviral protease 2A‐mediated C‐terminal dystrophin cleavage fragment. Science Translational Medicine 2015; 7: 294 ra106. [DOI] [PubMed] [Google Scholar]
- 17. Adams MJ, King AM, Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Archives of Virology 2013; 158: 2023–2030. [DOI] [PubMed] [Google Scholar]
- 18. Rossmann MG, He Y, Kuhn RJ. Picornavirus–receptor interactions. Trends in Microbiology 2002; 10: 324–331. [DOI] [PubMed] [Google Scholar]
- 19. Greve JM, Davis G, Meyer AM, et al. The major human rhinovirus receptor is ICAM‐1. Cell 1989; 56: 839–847. [DOI] [PubMed] [Google Scholar]
- 20. Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 1997; 275: 1320–1323. [DOI] [PubMed] [Google Scholar]
- 21. Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989; 56: 855–865. [DOI] [PubMed] [Google Scholar]
- 22. Hofer F, Gruenberger M, Kowalski H, et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor‐group common cold virus. Proceedings of the National Academy of Sciences of the United States of America 1994; 91: 1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hafenstein S, Bowman VD, Chipman PR, et al. Interaction of decay‐accelerating factor with coxsackievirus B3. Journal of Virology 2007; 81: 12927–12935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamayoshi S, Yamashita Y, Li J, et al. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nature Medicine 2009; 15: 798–80. [DOI] [PubMed] [Google Scholar]
- 25. Roivainen M, Hyypiä T, Piirainen L, Kalkkinen N, Stanway G, Hovi T. RGD‐dependent entry of coxsackievirus A9 into host cells and its bypass after cleavage of VP1 protein by intestinal proteases. Journal of Virology 1991; 65: 4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roivainen M, Piirainen L, Hovi T, et al. Entry of coxsackievirus A9 into host cells: specific interactions with alpha v beta 3 integrin, the vitronectin receptor. Virology 1994; 203: 357–365. [DOI] [PubMed] [Google Scholar]
- 27. Ohka S, Nomoto A. Recent insights into poliovirus pathogenesis. Trends in Microbiology 2001; 9: 501–506. [DOI] [PubMed] [Google Scholar]
- 28. Iwasaki A, Welker R, Mueller S, et al. Immunofluorescence analysis of poliovirus receptor expression in Peyer's patches of humans, primates, and CD155 transgenic mice: implications for poliovirus infection. Journal of Infectious Diseases 2002; 186: 585–592. [DOI] [PubMed] [Google Scholar]
- 29. Marjomaki V, Pietiainen V, Matilainen H, et al. Internalization of echovirus 1 in caveolae. Journal of Virology 2002; 76: 1856–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heikkila O, Susi P, Tevaluoto T, et al. Internalization of coxsackievirus A9 is mediated by {beta}2‐microglobulin, dynamin, and Arf6 but not by caveolin‐1 or clathrin. Journal of Virology 2010; 84: 3666–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ren J, Wang X, Hu Z, et al. Picornavirus uncoating intermediate captured in atomic detail. Nature Communications 2013; 4: 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guest S, Pilipenko E, Sharma K, et al. Molecular mechanisms of attenuation of the Sabin strain of poliovirus type 3. Journal of Virology 2004; 78: 11097–11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lind K, Huhn MH, Flodstrom‐Tullberg M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the innate immune response to enteroviruses and its possible role in regulating type 1 diabetes. Clinical and Experimental Immunology 2012; 168: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee YF, Nomoto A, Detjen BM, et al. A protein covalently linked to poliovirus genome RNA. Proceedings of the National Academy of Sciences of the United States of America 1977; 74: 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yogo Y, Wimmer E. Polyadenylic acid at the 3′‐terminus of poliovirus RNA. Proceedings of the National Academy of Sciences of the United States of America 1972; 69: 1877–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krausslich HG, Nicklin MJ, Lee CK, et al. Polyprotein processing in picornavirus replication. Biochimie 1988; 70: 119–130. [DOI] [PubMed] [Google Scholar]
- 37. Nicklin MJ, Krausslich HG, Toyoda H, et al. Poliovirus polypeptide precursors: expression in vitro and processing by exogenous 3C and 2A proteinases. Proceedings of the National Academy of Sciences of the United States of America 1987; 84: 4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Toyoda H, Nicklin MJ, Murray MG, et al. A second virus‐encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell 1986; 45: 761–770. [DOI] [PubMed] [Google Scholar]
- 39. Hober D, Sané F, Riedweg K, et al Viruses and type 1 diabetes: focus on the enteroviruses. Available online: http://www.intechopen.com/books/type-1-diabetes/viruses-and-type-1-diabetes-focus-on-the-enteroviruses (accessed on February 2013).
- 40. Basavappa R, Syed R, Flore O, et al. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9 a resolution. Protein Science 1994; 3: 1651–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suhy DA, Giddings TH Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy‐like origin for virus‐induced vesicles. Journal of Virology 2000; 74: 8953–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pathak HB, Arnold JJ, Wiegand PN, et al. Picornavirus genome replication: assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. Journal of Biological Chemistry 2007; 282: 16202–16213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Novak JE, Kirkegaard K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive‐strand encapsidation and the ratio of positive to negative strands in infected cells. Journal of Virology 1991; 65: 3384–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Plevka P, Perera R, Cardosa J, et al. Crystal structure of human enterovirus 71. Science 2012; 336: 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nugent CI, Johnson KL, Sarnow P, et al. Functional coupling between replication and packaging of poliovirus replicon RNA. Journal of Virology 1999; 73: 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pfister T, Pasamontes L, Troxler M, et al. Immunocytochemical localization of capsid‐related particles in subcellular fractions of poliovirus‐infected cells. Virology 1992; 188: 676–684. [DOI] [PubMed] [Google Scholar]
- 47. Inal JM, Jorfi S. Coxsackievirus B transmission and possible new roles for extracellular vesicles. Biochemical Society Transactions 2013; 41: 299–302. [DOI] [PubMed] [Google Scholar]
- 48. Robinson SM, Tsueng G, Sin J, et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathogens 2014; 10: e1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chehadeh W, Kerr‐Conte J, Pattou F, et al. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. Journal of Virology 2000; 74: 10153–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jartti T, Lehtinen P, Vuorinen T, et al. Persistence of rhinovirus and enterovirus RNA after acute respiratory illness in children. Journal of Medical Virology 2004; 72: 695–699. [DOI] [PubMed] [Google Scholar]
- 51. Klingel K, Hohenadl C, Canu A, et al. Ongoing enterovirus‐induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proceedings of the National Academy of Sciences of the United States of America 1992; 89: 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chia MY, Chiang PS, Chung WY, et al. Epidemiology of enterovirus 71 infections in Taiwan. Pediatrics and Neonatology 2014; 55: 243–249. [DOI] [PubMed] [Google Scholar]
- 53. Yip CC, Lau SK, Woo PC, et al. Human enterovirus 71 epidemics: what's next? Emerging Health Threats Journal 2013; 6: 19780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lang M, Mirand A, Savy N, et al. Acute flaccid paralysis following enterovirus D68 associated pneumonia, France, 2014. Euro Surveillance 2014; 19: 20952. [DOI] [PubMed] [Google Scholar]
- 55. Khan F. Enterovirus D68: acute respiratory illness and the 2014 outbreak. Emergency Medicine Clinics of North America 2015; 33: e19–e32. [DOI] [PubMed] [Google Scholar]
- 56. Stock I. Hand, foot and mouth disease—more than a harmless “childhood disease”. Medizinische Monatsschrift für Pharmazeuten 2014; 37: 4–10; quiz 11‐2. [PubMed] [Google Scholar]
- 57. Lim BK, Peter AK, Xiong D, et al. Inhibition of coxsackievirus‐associated dystrophin cleavage prevents cardiomyopathy. Journal of Clinical Investigation 2013; 123: 5146–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Massilamany C, Gangaplara A, Reddy J. Intricacies of cardiac damage in coxsackievirus B3 infection: implications for therapy. International Journal of Cardiology 2014; 177: 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nomoto A. Molecular aspects of poliovirus pathogenesis. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences 2007; 83: 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wong J, Zhang J, Yanagawa B, et al. Cleavage of serum response factor mediated by enteroviral protease 2A contributes to impaired cardiac function. Cell Research 2012; 22: 360–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Estrella NL, Naya FJ. Transcriptional networks regulating the costamere, sarcomere, and Other cytoskeletal structures in striated muscle. Cellular and Molecular Life Sciences 2014; 71: 1641–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim KS, Tracy S, Tapprich W, et al. 5′‐terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative‐strand viral RNA. Journal of Virology 2005; 79: 7024–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xiong D, Yajima T, Lim BK, et al. Inducible cardiac‐restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation 2007; 115: 94–102. [DOI] [PubMed] [Google Scholar]
- 64. Chapman NM, Kim KS, Drescher KM, et al. 5′ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 2008; 375: 480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Seipelt J, Guarne A, Bergmann E, et al. The structures of picornaviral proteinases. Virus Research 1999; 62: 159–168. [DOI] [PubMed] [Google Scholar]
- 66. Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 1983; 22: 2577–2637. [DOI] [PubMed] [Google Scholar]
- 67. Liebig HD, Ziegler E, Yan R, et al. Purification of two picornaviral 2A proteinases: interaction with eIF‐4 gamma and influence on in vitro translation. Biochemistry 1993; 32: 7581–7588. [DOI] [PubMed] [Google Scholar]
- 68. Wang QM, Johnson RB, Cox GA, et al. Enzymatic characterization of refolded human rhinovirus type 14 2A protease expressed in Escherichia coli . Journal of Virology 1998; 72: 1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cai Q, Yameen M, Liu W, et al. Conformational plasticity of the 2A proteinase from enterovirus 71. Journal of Virology 2013; 87: 7348–7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mu Z, Wang B, Zhang X, et al. Crystal structure of 2A proteinase from hand, foot and mouth disease virus. Journal of Molecular Biology 2013; 425: 4530–4543. [DOI] [PubMed] [Google Scholar]
- 71. Sun Y, Wang X, Yuan S, et al. An open conformation determined by a structural switch for 2A protease from coxsackievirus A16. Protein & Cell 2013; 4: 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shi J, Wei Z, Song J. Dissection study on the severe acute respiratory syndrome 3C‐like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. Journal of Biological Chemistry 2004; 279: 24765–24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen S, Zhang J, Hu T, et al. Residues on the dimer interface of SARS coronavirus 3C‐like protease: dimer stability characterization and enzyme catalytic activity analysis. Journal of Biochemistry 2008; 143: 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Costenaro L, Kaczmarska Z, Arnan C, et al. Structural basis for antiviral inhibition of the main protease, 3C, from human enterovirus 93. Journal of Virology 2011; 85: 10764–10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tan J, George S, Kusov Y, et al. 3C protease of enterovirus 68: structure‐based design of Michael acceptor inhibitors and their broad‐spectrum antiviral effects against picornaviruses. Journal of Virology 2013; 87: 4339–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Blom N, Hansen J, Blaas D, et al. Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Science 1996; 5: 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. UniProt Consortium . The Universal Protein Resource (UniProt). Nucleic Acids Research 2008; 36: D190–D195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Crooks GE, Hon G, Chandonia JM, et al. WebLogo: a sequence logo generator. Genome Research 2004; 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ramajayam R, Tan KP, Liang PH. Recent development of 3C and 3CL protease inhibitors for anti‐coronavirus and anti‐picornavirus drug discovery. Biochemical Society Transactions 2011; 5: 1371–1375. [DOI] [PubMed] [Google Scholar]
- 80. Thibaut HJ, De Palma AM, Neyts J. Combating enterovirus replication: state‐of‐the‐art on antiviral research. Biochemical Pharmacology 2012; 2: 185–192. [DOI] [PubMed] [Google Scholar]
- 81. Guo Y, Wang Y, Cao L, et al. A conserved inhibitory mechanism of a lycorine derivative against enterovirus and hepatitis C virus. Antimicrobial Agents and Chemotherapy 2015; 2: 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mandadapu SR, Weerawarna PM, Prior AM, et al. Macrocyclic inhibitors of 3C and 3C‐like proteases of picornavirus, norovirus, and coronavirus. Bioorganic and Medicinal Chemistry Letters 2013; 13: 3709–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dragovich PS, Prins TJ, Zhou R, et al. Structure‐based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L‐glutamine replacements. Journal of Medicinal Chemistry 1999; 7: 1213–1224. [DOI] [PubMed] [Google Scholar]
- 84. Rocha‐Pereira J, Nascimento MS, Ma Q, Hilgenfeld R, Neyts J, Jochmans D. The enterovirus protease inhibitor rupintrivir exerts cross‐genotypic anti‐norovirus activity and clears cells from the norovirus replicon. Antimicrobial Agents and Chemotherapy 2014; 8: 4675–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Patick AK, Brothers MA, Maldonado F, et al. In vitro antiviral activity and single‐dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrobial Agents and Chemotherapy 2005; 6: 2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hayden FG, Turner RB, Gwaltney JM, et al. Phase II, randomized, double‐blind, placebo‐controlled studies of ruprintrivir nasal spray 2‐percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrobial Agents and Chemotherapy 2003; 12: 3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hsyu PH, Pithavala YK, Gersten M, Penning CA, Kerr BM. Pharmacokinetics and safety of an antirhinoviral agent, ruprintrivir, in healthy volunteers. Antimicrobial Agents and Chemotherapy 2002; 2: 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. van der Linden L, Wolthers KC, van Kuppeveld FJ. Replication and inhibitors of enteroviruses and parechoviruses. Viruses 2015; 8: 4529–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. De Palma AM, Vliegen I, De Clercq E, Neyts J. Selective inhibitors of picornavirus replication. Medicinal Research Reviews 2008; 6: 823–884. [DOI] [PubMed] [Google Scholar]
- 90. Wang J, Fan T, Yao X, et al. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. Journal of Virology 2011; 19: 10021–10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang X1, Song Z, Qin B, et al. Rupintrivir is a promising candidate for treating severe cases of enterovirus‐71 infection: evaluation of antiviral efficacy in a murine infection model. Antiviral Research 2013; 3: 264–269. [DOI] [PubMed] [Google Scholar]
- 92. Lu G, Qi J, Chen Z, et al. Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti‐hand, foot, and mouth disease virus drug design. Journal of Virology 2011; 19: 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sun L, Meijer A, Froeyen M, et al. Antiviral activity of broad‐spectrum and enterovirus‐specific inhibitors against clinical isolates of enterovirus D68. Antimicrobial Agents and Chemotherapy 2015; 12: 7782–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jang SK, Krausslich HG, Nicklin MJ, et al. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. Journal of Virology 1988; 62: 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Pelletier J, Sonenberg N. Internal binding of eucaryotic ribosomes on poliovirus RNA: translation in HeLa cell extracts. Journal of Virology 1989; 63: 441–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Weidman MK, Yalamanchili P, Ng B, et al. Poliovirus 3C protease‐mediated degradation of transcriptional activator p53 requires a cellular activity. Virology 2001; 291: 260–271. [DOI] [PubMed] [Google Scholar]
- 97. Yalamanchili P, Weidman K, Dasgupta A. Cleavage of transcriptional activator Oct‐1 by poliovirus encoded protease 3Cpro. Virology 1997; 239: 176–185. [DOI] [PubMed] [Google Scholar]
- 98. Yalamanchili P, Harris K, Wimmer E, et al. Inhibition of basal transcription by poliovirus: a virus‐encoded protease (3Cpro) inhibits formation of TBP‐TATA box complex in vitro. Journal of Virology 1996; 70: 2922–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dingwall C, Laskey RA. Nuclear targeting sequences—a consensus? Trends in Biochemical Sciences 1991; 16: 478–481. [DOI] [PubMed] [Google Scholar]
- 100. Tian W, Cui Z, Zhang Z, et al. Poliovirus 2A(Pro) induces the nucleic translocation of poliovirus 3CD and 3C′ proteins. Acta Biochimica et Biophysica Sinica Shanghai 2011; 43: 38–44. [DOI] [PubMed] [Google Scholar]
- 101. Lamphear BJ, Rhoads RE. A single amino acid change in protein synthesis initiation factor 4G renders cap‐dependent translation resistant to picornaviral 2A proteases. Biochemistry 1996; 35: 15726–15733. [DOI] [PubMed] [Google Scholar]
- 102. Goldstaub D, Gradi A, Bercovitch Z, et al. Poliovirus 2A protease induces apoptotic cell death. Molecular and Cellular Biology 2000; 20: 1271–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Castello A, Izquierdo JM, Welnowska E, et al. RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. Journal of Cell Science 2009; 122: 3799–3809. [DOI] [PubMed] [Google Scholar]
- 104. Walker EJ, Younessi P, Fulcher AJ, et al. Rhinovirus 3C protease facilitates specific nucleoporin cleavage and mislocalisation of nuclear proteins in infected host cells. PLoS One 2013; 8: e71316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Park N, Schweers NJ, Gustin KE. Selective removal of FG repeat domains from the nuclear pore complex by enterovirus 2Apro. Journal of Virology 2015; 89: 11069–11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Weng KF, Li ML, Hung CT, et al. Enterovirus 71 3C protease cleaves a novel target CstF‐64 and inhibits cellular polyadenylation. PLoS Pathogens 2009; 5: e1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yalamanchili P, Datta U, Dasgupta A. Inhibition of host cell transcription by poliovirus: cleavage of transcription factor CREB by poliovirus‐encoded protease 3Cpro. Journal of Virology 1997; 71: 1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lind K, Svedin E, Domsgen E, et al. Coxsackievirus counters the host innate immune response by blocking type III interferon expression. Journal of General Virology 2016. DOI: 10.1099/jgv.0.000443. [DOI] [PubMed] [Google Scholar]
- 109. Gustin KE, Sarnow P. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. Journal of Virology 2002; 76: 8787–8796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ghildyal R, Jordan B, Li D, et al. Rhinovirus 3C protease can localize in the nucleus and alter active and passive nucleocytoplasmic transport. Journal of Virology 2009; 83: 7349–7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Amineva SP, Aminev AG, Palmenberg AC, et al. Rhinovirus 3C protease precursors 3CD and 3CD′ localize to the nuclei of infected cells. Journal of General Virology 2004; 85: 2969–2979. [DOI] [PubMed] [Google Scholar]
- 112. Neznanov N, Chumakov KM, Neznanova L, et al. Proteolytic cleavage of the p65‐RelA subunit of NF‐kappaB during poliovirus infection. Journal of Biological Chemistry 2005; 280: 24153–24158. [DOI] [PubMed] [Google Scholar]
- 113. Das S, Dasgupta A. Identification of the cleavage site and determinants required for poliovirus 3CPro‐catalyzed cleavage of human TATA‐binding transcription factor TBP. Journal of Virology 1993; 67: 3326–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fitzgerald KD, Chase AJ, Cathcart AL, et al. Viral proteinase requirements for the nucleocytoplasmic relocalization of cellular splicing factor SRp20 during picornavirus infections. Journal of Virology 2013; 87: 2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Park N, Skern T, Gustin KE. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. Journal of Biological Chemistry 2010; 285: 28796–28805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bedard KM, Daijogo S, Semler BL. A nucleo‐cytoplasmic SR protein functions in viral IRES‐mediated translation initiation. EMBO Journal 2007; 26: 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Belsham GJ, Sonenberg N. RNA–protein interactions in regulation of picornavirus RNA translation. Microbiological Reviews 1996; 60: 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Perera R, Daijogo S, Walter BL, et al. Cellular protein modification by poliovirus: the two faces of poly(rC)‐binding protein. Journal of Virology 2007; 81: 8919–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Svitkin YV, Imataka H, Khaleghpour K, et al. Poly(A)‐binding protein interaction with elF4G stimulates picornavirus IRES‐dependent translation. RNA 2001; 7: 1743–1752. [PMC free article] [PubMed] [Google Scholar]
- 120. Lind K, Richardson SJ, Leete P, et al. Induction of an antiviral state and attenuated coxsackievirus replication in type III interferon‐treated primary human pancreatic islets. Journal of Virology 2013; 87: 7646–7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lind K, Svedin E, Utorova R, et al. Type III interferons are expressed by coxsackievirus‐infected human primary hepatocytes and regulate hepatocyte permissiveness to infection. Clinical and Experimental Immunology 2014; 177: 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hultcrantz M, Huhn MH, Wolf M, et al. Interferons induce an antiviral state in human pancreatic islet cells. Virology 2007; 367: 92–101. [DOI] [PubMed] [Google Scholar]
- 123. Zaragoza C, Saura M, Padalko EY, et al. Viral protease cleavage of inhibitor of kappaBalpha triggers host cell apoptosis. Proceedings of the National Academy of Sciences of the United States of America 2006; 103: 19051–19056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Li HY, Zhang LK, Zhu XJ, et al. Analysis of EV71 infection progression using triple‐SILAC‐based proteomics approach. Proteomics 2015; 15: 3629–3643. [DOI] [PubMed] [Google Scholar]
- 125. Zhang LK, Lin T, Zhu SL, et al. Global quantitative proteomic analysis of human glioma cells profiled host protein expression in response to enterovirus type 71 infection. Proteomics 2015. [DOI] [PubMed] [Google Scholar]
- 126. Kuyumcu‐Martinez NM, Joachims M, Lloyd RE. Efficient cleavage of ribosome‐associated poly(A)‐binding protein by enterovirus 3C protease. Journal of Virology 2002; 76: 2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jagdeo JM, Dufour A, Fung G, et al. Heterogeneous nuclear ribonucleoprotein M facilitates enterovirus infection. Journal of Virology 2015; 89: 7064–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lane L, Argoud‐Puy G, Britan A, et al. neXtProt: a knowledge platform for human proteins. Nucleic Acids Research 2012; 40: D76–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bernstein FC, Koetzle TF, Williams GJ, et al. The protein data bank: a computer‐based archival file for macromolecular structures. Journal of Molecular Biology 1977; 112: 535–542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Published enteroviral 2A substrates.
Table S2. Published enteroviral 3C substrates.
Supporting info item